

Abstract
Reduced hepatic expression levels of bromodomain-containing protein 7 (BRD7) have been suggested to play a role in the development of glucose intolerance in obesity. However, the molecular mechanism by which BRD7 regulates glucose metabolism has remained unclear. Here, we show that BRD7 increases phosphorylation of glycogen synthase kinase 3β (GSK3β) in response to activation of the insulin receptor-signaling pathway shortly after insulin stimulation and the nutrient-sensing pathway after feeding. BRD7 mediates phosphorylation of GSK3β at the Serine 9 residue and this effect on GSK3β occurs even in the absence of AKT activity. Using both in vitro and in vivo models, we further demonstrate that BRD7 mediates phosphorylation of ribosomal protein S6 kinase (S6K) and leads to increased phosphorylation of the eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1) and, therefore, relieves its inhibition of the eukaryotic translation initiation factor 4E (eIF4E). However, the increase in phosphorylation of 4E-BP1 with BRD7 overexpression is blunted in the absence of AKT activity. In addition, using liver-specific BRD7 knockout (LBKO) mice, we show that BRD7 is required for mTORC1 activity on its downstream molecules. These findings show a novel basis for understanding the molecular dynamics of glucose metabolism and suggest the unique function of BRD7 in the regulation of glucose homeostasis.
Electronic supplementary material
The online version of this article (10.1007/s00018-017-2711-x) contains supplementary material, which is available to authorized users.
Keywords: Bromodomain-containing protein 7 (BRD7), Glycogen synthase kinase 3β (GSK3β), Insulin signaling, Type 2 diabetes
Introduction
Bromodomain-containing protein 7 (BRD7), a member of the bromodomain-containing protein family, was originally identified from nasopharyngeal carcinoma (NPC) cells [1, 2]. Since then, BRD7 has been reported to play a role in tumor suppression and regulation of the cell cycle and cell growth [1, 3, 4]. For example, previous studies have suggested that BRD7 binds to BRCA-1 and p53 [5–8]. Recent work has shown that BRD7 interacts with the regulatory subunits of phosphatidylinositol 3-kinase (PI3K), p85α and p85β [9, 10]. This interaction increases the nuclear translocation of p85s, as well as a transcription factor called the spliced form of X-box binding protein 1 (XBP1s) [10], which is a master regulator of the endoplasmic reticulum (ER) protein folding function. A disruption to ER homeostasis can cause ER stress, which has been previously implicated in multiple metabolic diseases, including diabetes [11–14]. Of pathophysiological importance, BRD7 protein levels are significantly reduced in the liver of obese mice [10]. Reinstating hepatic BRD7 levels in obese and type 2 diabetic mice enables the translocation of XBP1s to the nucleus, which leads to decreased ER stress and improved glucose tolerance [10]. However, understanding the exact role of BRD7 in the insulin-signaling pathway and how BRD7 is involved in the maintenance of glucose homeostasis needs further investigation.
The insulin-signaling pathway is activated by insulin binding to a tyrosine kinase receptor, insulin receptor (IR). IR phosphorylates insulin receptor substrate 1 and 2 (IRS1/2), which then recruits PI3K. Protein kinase B (AKT) is a key effector protein downstream of PI3K [15, 16]. Activation of AKT kinase leads to phosphorylation and inactivation of glycogen synthase kinase 3β (GSK3β) [17], which is a serine/threonine kinase that was originally discovered as a glycogen synthase (GS) regulator. It was previously reported that inhibiting GSK3β activity improves glucose tolerance in diabetic mice [18]; therefore, GSK3β has been suggested as a drug target for the treatment of type 2 diabetes [19]. However, the detailed mechanism by which GSK3β regulates glucose homeostasis is not fully understood.
Tuberous sclerosis protein 2 (TSC2) is another AKT substrate that acts as a GTPase-activating protein (GAP) for the Ras-related small G protein, Rheb. TSC2 negatively regulates the activity of the mammalian target of rapamycin (mTOR)–raptor complex 1 (mTORC1) by inhibiting Rheb-GTP to inactivate mTORC1 signaling [20]. Phosphorylation of TSC2 by AKT inhibits its GAP activity, allowing Rheb-GTP to activate mTORC1 [21] affecting cell growth, proliferation, and protein synthesis. Cells lacking TSC2 display constitutively high activation of mTORC1 activity and defective PI3K-AKT signaling [22]. The dysfunction of mTORC1 has been implicated in multiple diseases, including kidney disease [23], cancer, obesity, and type 2 diabetes [24].
Ribosomal protein S6 kinase (S6K) and eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1) are downstream effectors of mTORC1 [25]. S6K is an AGC family protein kinase that is activated upon insulin stimulation [26]. S6K is phosphorylated by raptor, a member of the mTOR complex, and plays a role in the assembly of the ribosomal translational complex. It has been shown that mice deficient in S6K share phenotypically similar features with type 2 diabetic patients; they both display hypoinsulinaemia and glucose intolerance [27]. Studies have shown that mTORC1 signaling can phosphorylate GSK3β through S6K in TSC1- or TSC2-deficient cells [28]. 4E-BP1 is a translational repressor that prevents cap-dependent translation by binding to eIF4E [29, 30]. Raptor also phosphorylates 4E-BP1, which releases eIF4E to form the translational complex [31].
Here, we report that BRD7 increases phosphorylation of GSK3β at the Serine 9 residue. We provide evidence that BRD7 can regulate GSK3β independently of AKT1/2 and that S6K signaling is involved in this regulation in the absence of AKT activity. These results suggest that BRD7 plays a significant role as a regulator in the insulin pathway and that this role circumvents the classic AKT-dependent signaling pathway. The classic insulin-signaling pathway is impaired in type 2 diabetes; therefore, the BRD7-mediated inhibition of GSK3β has the potential to be utilized in future treatments.
Materials and methods
Mice
C57BL/6J (wild-type) mice were obtained from The Jackson Laboratory. BRD7 Transgenic (BRD7Tg) mice were produced from our group as described in our results section. BRD7Tg mice were crossed with albumin–Cre+/− mice (The Jackson Laboratory) to generate a liver-specific transgenic line. Liver-specific BRD7 knockout (LBKO) mice were generated by first breeding heterozygous BRD7 whole body KO mice (BRD7+/−) [32] with a mouse line that carries Flp Recombinase, obtained from European Conditional Mouse Mutagenesis Program. The offspring were then bred with a mouse line that carries Cre recombinase under a modified human albumin promoter (The Jackson Laboratory). We determined genotypes by performing polymerase chain reaction (PCR) using genomic DNA isolated from tail biopsies at the age of 3 weeks, prior to weaning. The primers used for genotyping were as follows:
BRD7 (F): 5′-GTGACTTACATCCCCGGAGC-3′,
BRD7 (R): 5′-TTAGAGGAGTAGCCTTCCGTGAG-3′,
Cre (F): 5′-ACCAGCCAGCTATCAACTCG-3′,
Cre (R): 5′-TTACATTGGTCCAGCCACC-3′.
All experimental procedures were approved by, and adhered to the guidelines of, the Boston Children’s Hospital institutional animal care and use committee.
Cell culture
Mouse embryonic fibroblasts (MEFs) and TSC2−/− cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and antibiotics [1% penicillin (10,000 units/mL) and 1% streptomycin (10,000 μg/mL)] all purchased from Gibco. AKT1/2 KO cells were cultured in DMEM supplemented with 20% FBS and antibiotics. All cell lines were maintained in a 37 °C humidified incubator with 5% CO2. For infection with adenovirus, cells were incubated with DMEM supplemented with 1% FBS and antibiotics for 16 h. Cell lines were confirmed by western blot analysis for specific knockouts.
Cell lysate extraction
Cells were lysed on ice for 10 min with RIPA cell lysis buffer [25 mM Tris (pH 7.4), 10 mM NaF, 10 mM Na4P2O7, 1 mM EDTA, 1 mM EGTA, and 1% NP40] supplemented with protease and phosphatase inhibitors (Sigma-Aldrich). Lysates were centrifuged at 16,100×g for 5 min at 4 °C. Protein concentrations were determined using a DC protein assay (Bio-Rad). Samples were normalized and boiled for 5 min in 1× Laemmli buffer [2% SDS, 10% glycerol, 50 mM Tris (pH 6.8), 0.29 M β-mercaptoethanol, and 0.01% bromophenol blue].
Tissue lysate extraction
20 mg of tissue were lysed with tissue lysis buffer (25 mM Tris–HCl (pH 7.4), 10 mM Na3VO4, 100 mM NaF, 50 mM Na4P2O7, 10 mM EGTA, 10 mM EDTA, 1% NP40, and 2 mM PMSF) supplemented with protease and phosphatase inhibitors as described above. Tissues were homogenized in microcentrifuge tubes with a tungsten carbide bead by shaking at a frequency of 30 per second for 2 min on a tissue lyser (Qiagen). Homogenized tissue was then incubated with constant rotation for 1 h at 4 °C. Lysates were centrifuged at 16,100×g for 1 h at 4 °C. The lipid layer was carefully removed using a cotton-tipped applicator (Fisher Scientific). Protein concentrations were determined using a DC protein assay and samples were normalized as described above.
Nuclear protein extraction
Cells were lysed on ice for 5 min with cytoplasmic extraction buffer [10 mM HEPES (pH 7.5), 2 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 10 mM KCl, 10 mM NaF, and 0.1 mM Na3VO4], supplemented with protease and phosphatase inhibitors as described above. After incubation, 20 μL of 10% NP40 was added and samples were vortexed. After incubating on ice for 1 min, samples were centrifuged at 16,100×g for 1 min at 4 °C. The supernatant, containing the cytoplasmic portion, was transferred to a new tube and the pellet was resuspended using RIPA buffer supplemented with protease and phosphatase inhibitors as described above. The nuclear portion was then incubated for 10 min on ice, vortexed, and then centrifuged at 16,100×g for 5 min at 4 °C. Protein concentrations were determined using a DC protein assay and normalized as described above.
Western blotting
Proteins were resolved on an SDS-PAGE gel. Proteins were transferred to a polyvinylidene fluoride (PVDF) membrane (EMD Millipore) and then blocked in a blocking solution (1× Tris-buffered saline (TBS: 10 mM Tris base and 150 mM NaCl) with a pH of 7.4 and 10% blocking reagent (Roche BM Chemiluminescence Western Blotting Substrate)) for 1 h at room temperature. The membrane was then incubated in primary antibody in TBS with 0.01% Tween (TBST) and 5% blocking reagent overnight at 4 °C. The α-Tubulin, pGSK3β, GSK3β, β-actin, AKT, Lamin A/C, pS6K, S6K, p4E-BP1, 4E-BP1, and eIF4E antibodies were all purchased from cell-signaling technology. The BRD7 antibody was produced by Covance Inc. Next, the membrane was washed in TBST three times for 20 min followed by incubation in secondary antibody in TBST with 5% blocking reagent for 1 h at room temperature. Goat anti-mouse IgG-HRP and goat anti-rabbit IgG-HRP were purchased from Santa Cruz Biotechnology and anti-rabbit IgG-HRP-linked antibody was purchased from cell-signaling technology. The membranes were washed three times in TBST for 20 min each wash. Membranes were developed using either Thermo Scientific West Femto Maximum Sensitivity Substrate or Roche BM Chemiluminescence Western Blotting Substrate depending on the strength of the immunoblotted protein. Denville Scientific HyBlot CL Film was used to develop membranes.
Adenovirus production
Mouse BRD7 cDNA (NCBI reference sequence ID: NM_012047.2) and BRD7 shRNA (5′-CCGGGCCAAGATTACCCGTATGTTACTCGAGTAACATACGGGTAATCTTGGCTTTTT-3′) were cloned to pENTR3C (Invitrogen) and recombined to pAD/CMV/V5-DEST using a gateway recombination system from Invitrogen, following the manufacturer’s instructions. PacI (New England BioLabs) was used to digest the vector for transfection into 293A cells. 1 μg of vector was added to 30 μL of Opti-MEM (Gibco) and 3 μL of Lipofectamine 2000 (Invitrogen) was added to 30 μL of Opti-MEM, followed by 5 min of incubation at room temperature. The contents of the lipofectamine tube were then added to the tube containing the vector and incubated for 20 min at room temperature. Next, the mixture was added dropwise to a 60 mm cell culture plate of 293A cells containing 2 mL of DMEM supplemented with 10% FBS. After 16 h, medium was changed to DMEM supplemented with 10% FBS and antibiotics. 48 h after transfection, the cells were transferred to a 100 mm cell culture plate. The medium was replaced every 2–3 days until the cytopathic effect was observed. Cells were collected when the cytopathic effect was observed from 80% of cells, and virus was harvested by repeating three cycles of freezing and thawing in liquid nitrogen and at 37 °C. The product was centrifuged at 3300×g for 20 min at room temperature. The supernatant was then collected and stored at − 80 °C.
qPCR
RNA was extracted from 3 mg of tissue using Qiazol (Qiagen) and following the provided protocol. cDNA synthesis was done using an iScript cDNA Synthesis Kit (Bio-Rad), following their instructions. The reverse transcription conditions were 25 °C for 5 min, 46 °C for 20 min, and 95 °C for 1 min. qPCR was done using SYBR green master mix (Applied Biosystems) and the QuantStudio 6 Flex Real-Time PCR System. The primer sequences were as follows:
18S (F): 5′-AGTCCCTGCCCTTTGTACACA-3′,
18S (R): 5′-CGATCCGAGGGCCTCACTA-3′,
BRD7 (F): 5′-GAGGCTGAGGTGTTCCAGAG-3′,
BRD7 (R): 5′-TCACCTGGAGGCACTTGCTG-3′.
Quantification and statistical analysis
Quantification of western blotting was done using the ImageJ software’s gel analyzer. Results were normalized to loading controls. mRNA levels were determined by comparative Ct values normalized to 18S. Error bars are represented as mean ± SEM, and P values were determined by Student’s t test. *P < 0.05, **P < 0.01, ***P < 0.001.
Results
BRD7 increases phosphorylation of GSK3β
We have previously observed that the expression levels of gluconeogenic genes, such as glucose-6-phosphatase, fructose biphosphatase, and phosphoenolpyruvate carboxykinase, are reduced in the liver of obese and diabetic mice when BRD7 was overexpressed by tail vein injection of an adenovirus that carries BRD7 cDNA [10]. In light of the fact that these enzymes are negatively regulated by the inhibiting activity of GSK3β [33], we sought to investigate whether BRD7 affects the function of GSK3β. We first examined whether BRD7 modulates the activity of GSK3β by phosphorylation, since GSK3β activity is regulated by phosphorylation on its Serine 9 residue. To test this hypothesis, we infected mouse embryonic fibroblast (MEF) cells with Ad-BRD7 or an adenovirus that encodes eGFP (Ad-GFP) as a control. The western blot analysis from total lysates showed that BRD7 expression increases phosphorylation levels of GSK3β (Fig. 1a).
Fig. 1.
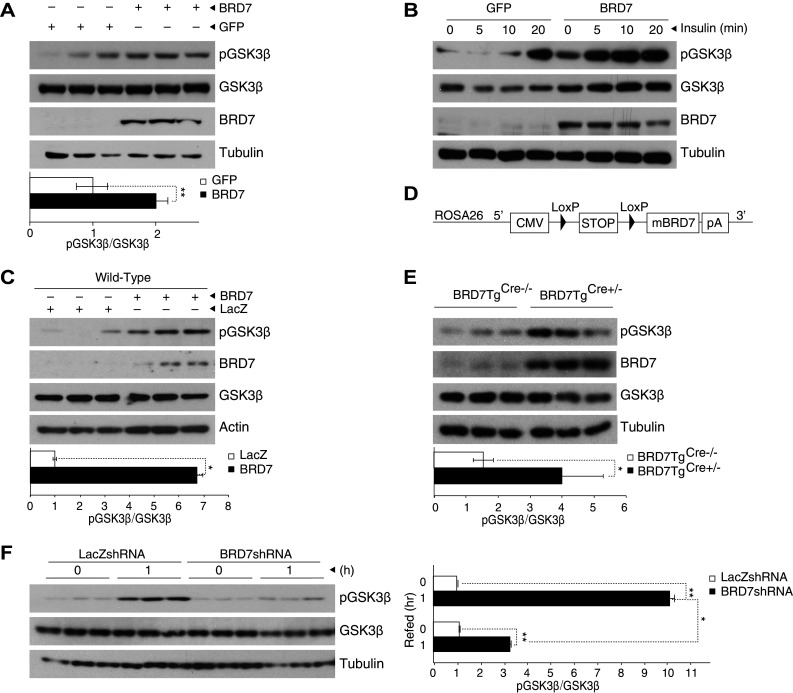
BRD7 increases phosphorylation of GSK3β. a MEFs were infected with Ad-BRD7 or Ad-GFP as a control and total lysates were blotted for pGSK3β, GSK3β, BRD7, and tubulin. The graph shows quantification of the western blot for pGSK3β normalized to total GSK3β. b MEFs were infected with Ad-BRD7 and Ad-GFP as a control, and then stimulated with insulin (500 nM) for the indicated timepoints. Protein lysates were blotted for the indicated antibodies. c 7-week-old C57/BL6J wild-type male mice were injected with Ad-BRD7 or Ad-LacZ as a control at a plaque forming unit (pfu) of 8 × 106 (n = 3 for each virus) through the tail vein. Western blots were performed from total lysate samples. d Schematic diagram of the construct used to generate BRD7 transgenic mouse model (BRD7Tg). e Liver was extracted from 7-week-old BRD7TgCre+/− mice and BRD7TgCre−/− as a control. Western blots were performed for the indicated antibodies. f 7-week-old wild-type mice were injected with Ad-LacZshRNA or Ad-BRD7shRNA (pfu of 8 × 106, n = 6 for each virus). Immunoblots show pGSK3β levels in total liver lysates during fast-refeeding (n = 3 for each condition). Error bars are represented as mean ± SEM. P values were determined by Student’s t test. (*P < 0.05, **P < 0.01, ***P < 0.001). Each experiment was independently repeated at least three times
Since GSK3β activity is modulated by insulin, we questioned whether this effect of BRD7 on GSK3β is also affected by insulin stimulation. To answer this, we expressed BRD7 in MEFs by infecting with Ad-BRD7 and Ad-GFP as a control, and stimulated with insulin at a concentration of 500 nM for 5, 10, or 20 min (Fig. 1b). The western blot results showed that BRD7 overexpression increases phosphorylation of GSK3β, which is further augmented by insulin stimulation from as early as the 5 min timepoint (Fig. 1b).
We have previously shown that under feeding conditions, mouse liver tissues express higher BRD7 levels. To isolate the function of BRD7 under refeeding conditions, we used BRD7 overexpression to mimic refeeding while reducing other potential confounding factors. To confirm this phenotype in an in vivo setting, we infected C57/BL6J wild-type mice with Ad-BRD7 and Ad-LacZ through the tail vein at a plaque forming unit (pfu) of 8 × 106 (n = 3 for each virus). The overexpression of BRD7 in the liver was confirmed at both mRNA and protein levels by performing qPCR (Figure S1A) and western blot analysis (Fig. 1c). The western blot results from the liver of Ad-BRD7 injected mice showed an increase in pGSK3β levels when compared to its control group. To further confirm this result in a mouse line that steadily expresses higher BRD7 levels, and in addition, to avoid any possible side effects caused by the use of an adenovirus injection, we produced a BRD7 transgenic mouse line (BRD7Tg). This line was generated by inserting a stop codon flanked by LoxP sites upstream of BRD7 cDNA. The sequence was then inserted at the ROSA26 site (Fig. 1d). The introduction of Cre recombinase (Cre) by breeding BRD7Tg with a mouse line that carries Cre under the albumin promoter deletes the stop sequence and initiates the expression of BRD7 (BRD7TgCre+/−). Induction of BRD7 expression was confirmed by qPCR (Figure S1B) and a western blot (Fig. 1e). The western blot results showed a significant increase in pGSK3β levels in the liver of BRD7TgCre+/− mice when compared to their control group (Fig. 1e).
Furthermore, to examine the effect of a lack of BRD7 on pGSK3β levels in mice, we generated an adenovirus that encodes shRNA specific for BRD7 (Ad-BRD7shRNA). We expressed BRD7shRNA in the liver of C57BL/6J mice by injecting Ad-BRD7shRNA through the tail vein at a pfu of 8 × 106 (n = 6 per each group). We injected Ad-LacZshRNA as a control (Figure S1C). 8 days after the injection, we fasted the mice for 24 h and refed ad libitum for 1 h. Western blots from total lysates showed an increase in pGSK3β levels under refeeding conditions with an Ad-LacZshRNA injection. This increased phosphorylation of GSK3β was diminished in the BRD7shRNA expressing group (Fig. 1f).
AKT1 and AKT2 are not required for BRD7-mediated increase in phosphorylation of GSK3β
Since AKT phosphorylates GSK3β upon insulin stimulation in the classic insulin-signaling pathway, we questioned whether AKT is involved in the BRD7-mediated phosphorylation of GSK3β. To answer this question, we examined whether BRD7 can increase phosphorylation levels of GSK3β in the absence of AKT1/2, or if the presence of AKT is required for this increase. For this, we used AKT1/2 knockout (KO) cells, where AKT1 and AKT2 isoforms have been silenced and corresponding control MEFs. We first checked total AKT levels in MEF and AKT1/2 KO cells using an antibody that recognizes all AKT isoforms (Fig. 2a). We infected AKT1/2 KO cells with Ad-BRD7 and Ad-GFP as a control. The western blot analysis from total lysates showed that AKT1/2 are not absolute requisites for BRD7-mediated phosphorylation of GSK3β (Fig. 2b). This result was additionally confirmed in nuclear fractionates (Fig. 2c). Furthermore, we observed that increased phosphorylation levels of GSK3β respond to insulin stimulation in AKT1/2 KO cells with BRD7 overexpression (Fig. 2d). To further confirm these results in comparison with MEFs, we infected MEF and AKT1/2 KO cells with Ad-BRD7 and compared the phosphorylation levels of GSK3β upon insulin stimulation. The results showed that BRD7 mediates the phosphorylation of GSK3β in the absence of AKT1/2 activity at a similar level to MEFs, where AKT1/2 is present (Fig. 2e).
Fig. 2.
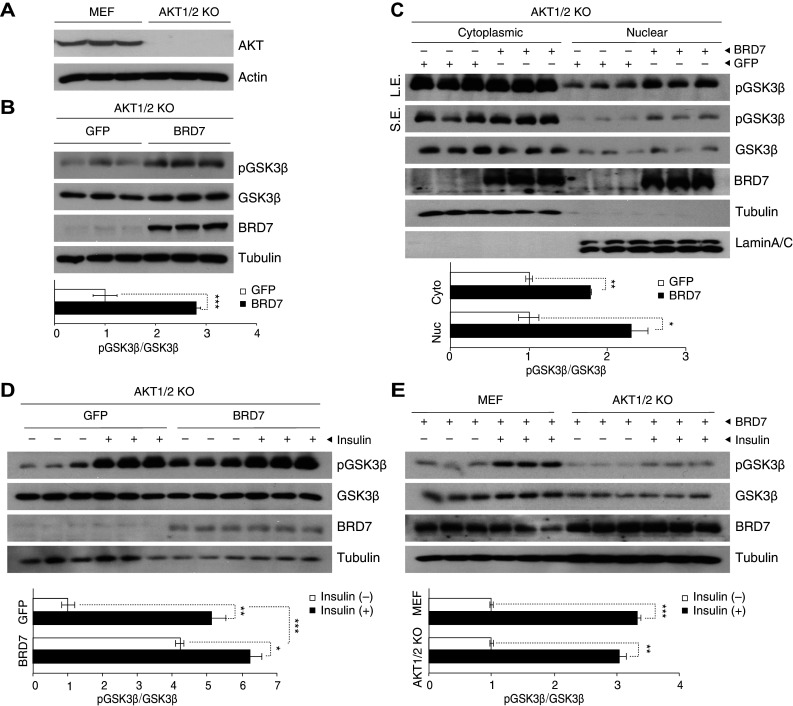
BRD7 increases phosphorylation of GSK3β in the absence of AKT1/2. a Total lysates from AKT1/2 knockout cells were immunoblotted for AKT and actin. b Immunoblots of pGSK3β, GSK3β, BRD7, and tubulin from AKT1/2 KO cells that were infected with Ad-GFP or Ad-BRD7. Quantification of the pGSK3β blot normalized to GSK3β is shown below. c Western blots from cytoplasmic and nuclear protein fractions of AKT1/2 KO cells that were infected with Ad-BRD7 or Ad-GFP as a control. Short exposure (SE) and long exposure (LE) shown for pGSK3β blot. d AKT1/2 KO cells were infected with Ad-GFP or Ad-BRD7, followed by insulin stimulation (500 nM) for 10 min. Total lysates were immunoblotted for the indicated antibodies. e MEF and AKT1/2 KO cells were infected with Ad-BRD7 and stimulated with insulin (500 nM, 10 min). Total lysates were immunoblotted for indicated antibodies. Error bars are represented as mean ± SEM. P values were determined by Student’s t test. *P < 0.05, **P < 0.01, ***P < 0.001. Each experiment was independently repeated at least three times
BRD7 increases phosphorylation of GSK3β through S6K
We sought to investigate which kinase is responsible for the phosphorylation of GSK3β in the absence of AKT activity. In light of the fact that mTORC1 plays a role when AKT is inhibited [34], we explored whether BRD7 increases the phosphorylation of GSK3β via pS6K. We overexpressed BRD7 in the liver of C57/BL6J wild-type mice by injecting Ad-BRD7 and Ad-LacZ through the tail vein at a pfu of 8 × 106 (n = 3 for each virus). The western blot analysis showed that phosphorylation of S6K at the Threonine 389 residue is increased with BRD7 regulation (Fig. 3a). We then confirmed the results in BRD7TgCre+/− mice and compared them to their control BRD7TgCre−/− mice (Fig. 3b). To understand whether BRD7 is needed for the activation of S6K, we generated a liver-specific BRD7 knockout mouse model (LBKO) and compared its pS6K levels to its control wild-type mice. The results showed that a lack of BRD7 leads to a significant decrease in pS6K levels (Fig. 3c). To observe the effect of BRD7 on pS6K in the absence of AKT1/2, we infected AKT1/2 KO cells with Ad-GFP or Ad-BRD7. Western blot results showed an increase in pS6K levels upon BRD7 overexpression in AKT1/2 KO cells (Fig. 3d).
Fig. 3.
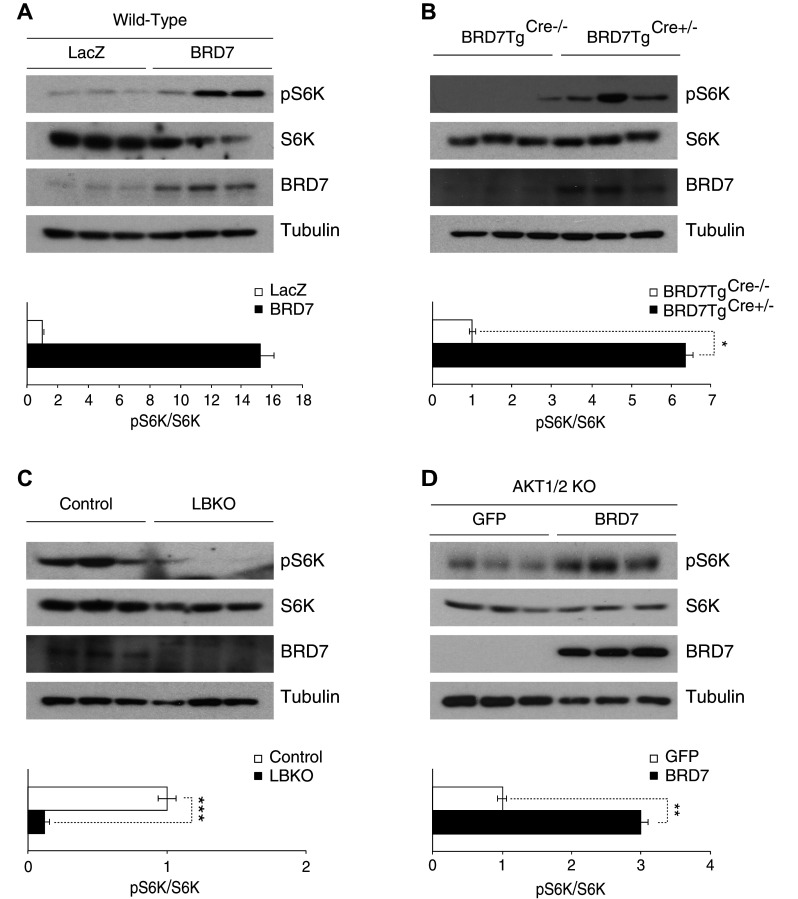
BRD7 increases phosphorylation of GSK3β through S6K. a 7-week-old wild-type male mice were injected with Ad-BRD7 or Ad-LacZ as a control and total lysates were blotted for pS6K, S6K, BRD7, and tubulin. The graph shows quantification of the western blot for pS6K normalized to total S6K. b Western blot analysis from the total lysate of the liver of 6-week-old BRD7TgCre+/− and BRD7TgCre−/− mice. c Western blots were performed for the indicated antibodies from the liver lysates of LBKO and control wild-type mice. d AKT1/2 KO cells were infected with Ad-BRD7 or Ad-GFP as a control and total lysates were blotted for indicated antibodies. Error bars are represented as mean ± SEM. P values were determined by Student’s t test. (*P < 0.05, **P < 0.01, ***P < 0.001). Each experiment was independently repeated at least three times
To determine the alternative mechanism of AKT1/2-independent BRD7-mediated phosphorylation of GSK3β, we tested the sensitivity of phosphorylation of GSK3β by treating with the mTOR inhibitor torin1. We utilized TSC2 KO cells, in which mTOR is highly active and AKT activation is defective. We treated TSC2 KO cells with torin1 at a concentration of 10 nM for 1 h, following infection with Ad-BRD7 and Ad-GFP as a control (Fig. 4a). The phosphorylation of GSK3β was increased in the Ad-BRD7-infected cells when compared to Ad-GFP infected cells. However, BRD7 did not mediate phosphorylation of GSK3β under torin1 treatment. Next, we treated TSC2 KO cells with rapamycin, an inhibitor of mTORC1, at a concentration of 200 μM for 30 min to specify if mTORC1 was responsible for this phenotype. In rapamycin treated TSC2 KO cells, phosphorylation of S6K was completely blunted and phosphorylation of GSK3β was not increased by BRD7 overexpression (Fig. 4b). Therefore, we show that S6K plays a role in BRD7-mediated phosphorylation of GSK3β through mTORC1 activity.
Fig. 4.
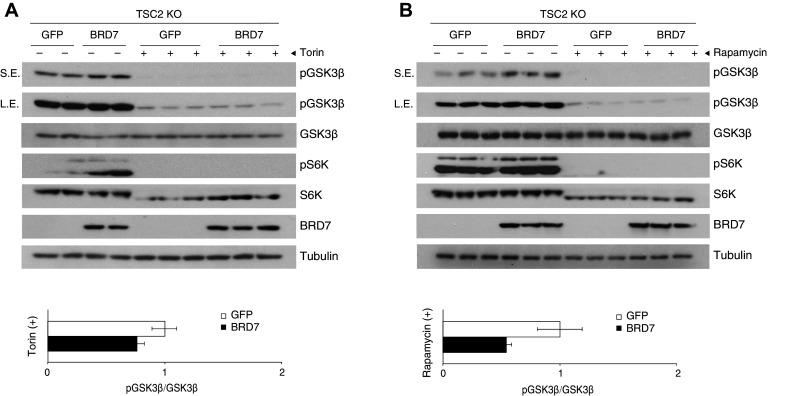
BRD7 does not increase phosphorylation of GSK3β in the absence of mTOR. a TSC2 KO cells were infected with either Ad-GFP or Ad-BRD7 and treated with 10 nM torin1 for 1 h. Western blots were performed for the indicated antibodies. Short exposure (SE) and long exposure (LE) shown for pGSK3β blot. b Immunoblots of TSC2 KO cells infected with Ad-BRD7 and Ad-GFP as a control and then treated with 200 μM rapamycin for 30 min. Error bars are represented as mean ± SEM. P values were determined by Student’s t test (*P < 0.05, **P < 0.01, ***P < 0.001). Each experiment was independently repeated at least three times
The BRD7-mediated phosphorylation of GSK3β leads to increased mTORC1 activity
Next, we examined the phosphorylation levels of 4E-BP1, another downstream effector protein of the PI3K pathway and mTOR kinase. We showed that increased BRD7 levels lead to a higher phosphorylation of 4E-BP1 in the liver of wild-type mice that were injected with Ad-BRD7 through the tail vein when compared to the Ad-LacZ-injected group (Fig. 5a). This increased phosphorylation of 4E-BP1 was confirmed in BRD7TgCre+/− mice when compared to BRD7TgCre−/− mice (Fig. 5b). We also showed that the phosphorylation levels of 4E-BP1 are significantly decreased in LBKO mice when compared to wild-type mice (Fig. 5c). However, this increase in phosphorylation levels of 4E-BP1 with BRD7 overexpression was not observed in AKT1/2 KO cells (Fig. 5d).
Fig. 5.
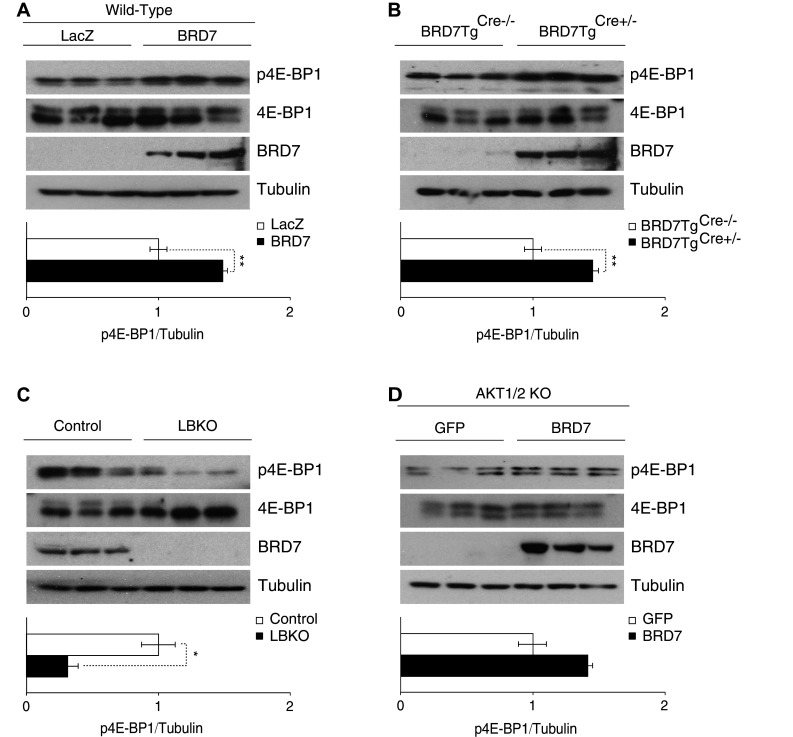
BRD7 increases the phosphorylation of 4E-BP1. a 7-week-old wild-type male mice were injected with Ad-LacZ or Ad-BRD7 through the tail vein at a pfu of 8 × 106 (n = 3 for each virus) and total lysates of liver tissue were blotted for p4E-BP1, 4E-BP1, BRD7, and tubulin. Quantification of the western blot shown below. b Western blots were performed for total lysates from 6-week-old BRD7TgCre−/− and BRD7Tg+Cre/− mice. c Immunoblots from the liver of LBKO mice and their control mice. d AKT1/2 knockout cells were infected with Ad-BRD7 or Ad-GFP as a control. Total lysates were blotted for the indicated antibodies. Error bars are represented as mean ± SEM. P values were determined by Student’s t test. *P < 0.05, **P < 0.01, ***P < 0.001. Each experiment was independently repeated at least three times
4E-BP1 binds to the eukaryotic translation initiation factor 4E (eIF4E) to prevent cap-dependent mRNA translation. To see the effect that BRD7 overexpression has on this segment of the mTORC1-signaling pathway, we analyzed total eIF4E levels in wild-type mice that were overexpressing BRD7 and compared them to the Ad-LacZ-injected group. We observed higher eIF4E levels in the tissues with higher BRD7 levels (Fig. 6a). This increase in eIF4E was also shown in the liver of BRD7TgCre+/− mice when compared to their control mice (Fig. 6b). We found that this pattern was not present in AKT1/2 KO cells infected with Ad-GFP or Ad-BRD7 (Fig. 6c).
Fig. 6.
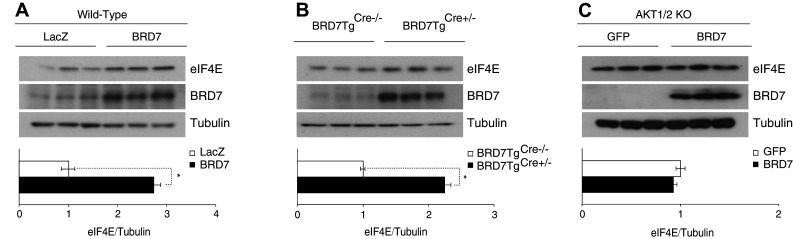
BRD7 increases total eIF4E levels. a Wild-type mice were injected with Ad-BRD7 or Ad-LacZ as a control and western blots were performed for eIF4E, BRD7, and tubulin. b Western blot analysis from BRD7TgCre−/− and BRD7TgCre+/− mice with indicated antibodies. c Immunoblots for AKT1/2 knockout cells infected with Ad-GFP or Ad-BRD7. Error bars are represented as mean ± SEM. P values were determined by Student’s t test. *P < 0.05, **P < 0.01, ***P < 0.001. Each experiment was independently repeated at least three times
Discussion
Glycogen synthase kinase 3β (GSK3β) was initially discovered as a regulator of glycogen synthase [35]. More recently, GSK3β has been shown to have a broad range of functions, including in bone resorption of osteoclasts [36] and tumor suppression in prostate carcinoma cells [37]. Increased GSK3β activity has been implicated in type 2 diabetes, and reducing GSK3β activity has been suggested as a possible therapeutic target [18, 19, 38, 39]. However, inhibiting its activity by means of continuous activation of AKT would not be an ideal course of treatment, because altering AKT activity has been implicated in the development of various cancers [40, 41]. In this report, we show that an increase in BRD7 protein levels leads to an increase in phosphorylation of GSK3β and that this occurs even in the absence of AKT1/2 kinase activity.
The existence of an AKT-independent pathway that controls glucose homeostasis has recently been discussed in the field. For example, it was previously thought that AKT is absolutely required in insulin signaling to inactivate Forkhead box O1 (FoxO1) activity and reduce glucose production. However, it has been shown that AKT is dispensable for hepatic glucose production in the absence of FoxO1 [42]. This suggests that there could be another AKT-independent pathway that controls glucose metabolism. Our results show that BRD7 mediates phosphorylation of GSK3β in an AKT-independent manner, which suggests that it could be involved in an alternative mechanism for controlling glucose homeostasis.
In an attempt to understand how BRD7 increases phosphorylation of GSK3β in the absence of AKT, we determined whether mTOR is involved. To investigate whether BRD7 increases phosphorylation of GSK3β through mTORC1, we used TSC2 KO cells, in which AKT activity is defective, and we further inhibited mTORC1 activity by treating cells with torin1. When both AKT and mTOR activity was blocked, increased phosphorylation of GSK3β with BRD7 overexpression was not observed.
Even though AKT and mTOR share some common effector proteins, a key regulator that controls the traffic of AKT and mTORC signaling pathways has yet to be determined. In our previous work, we showed that BRD7 overexpression in an obese condition increases phosphorylation of AKT through the insulin receptor-signaling pathway [10]. In our current report, we show that BRD7 can also induce GSK3β phosphorylation through an AKT-independent mechanism that requires S6K. Furthermore, under certain circumstances where AKT is dysregulated, such as cancerous conditions [43], BRD7 overexpression leads to decreased phosphorylation of AKT at the Serine 473 residue [9]. Considering these previous reports and our current findings, this suggests that BRD7 acts as a central molecule that switches from one kinase to another to regulate activation depending on the source of stimuli and basal activation levels.
What would be the benefit of having BRD7 induce phosphorylation of GSK3β both by AKT, and also independently of AKT? Under normal insulin-stimulated conditions, such as after a meal, BRD7 induces phosphorylation of GSK3β through AKT. Phosphorylation of GSK3β leads to the activation of glycogen synthase (GS) activity, which in turn increases glycogen content. After a meal, however, sustained activation of AKT would not be ideal. AKT is involved in a complex network that integrates a multitude of signals, which can also promote oncogenic signaling pathways. At this point, BRD7 perhaps establishes S6K as the predominant regulator of GSK3β to maintain GS activity, while nutrients are still abundant (Fig. 7a). This agrees with a previous report, which showed that AKT is phosphorylated within a few minutes of insulin or amino acid stimulation, and then returns to its basal state within an hour time period. Alternatively, S6K is activated at a later timepoint than AKT and remains active for more than an hour [44]. We have previously reported that obese and type 2 diabetic mice have decreased hepatic BRD7 levels [10]. In an obese state, it is possible that low BRD7 protein levels are a contributing factor to the dysregulation of GSK3β activity. Under cancerous conditions where AKT activation levels are high, increasing BRD7 works to decrease AKT activity and acts as a tumor suppressor.
Fig. 7.

BRD7 acts as a regulator of the insulin-signaling pathway. a Schematic diagram of BRD7 activity in the insulin-signaling pathway
mTORC1 is regulated by growth factors and nutrients, and controls protein translation by regulating the downstream targets of S6K and 4E-BP1, which are the rate-limiting factors of translational initiation. AKT indirectly regulates mTOR activity by phosphorylating and inactivating the TSC1/2 complex, which is an inhibitor of mTOR activity. In this report, we show that BRD7 also increases the downstream effector proteins of mTORC1, S6K, and 4E-BP1. Meanwhile, it is interesting to note that the increased phosphorylation levels of 4E-BP1 with BRD7 overexpression were not observed in the absence of AKT. However, phosphorylation levels of S6K were increased with BRD7 even without the presence of AKT. This implies that BRD7 increases phosphorylation of GSK3β through S6K in the absence of AKT activity, but AKT is required for more global actions of mTORC1, such as inhibiting 4E-BP1 to control translation. In other words, BRD7 acts as a nutrient-sensing molecule that signals which effector protein should be active to phosphorylate GSK3β without globally affecting translation after each meal. Furthermore, there is a reduction in targets downstream of mTORC1 in liver tissue lacking BRD7. This suggests that BRD7 is required for the activity of mTORC1.
There have been tremendous efforts to understand the physiology of obesity, which often involves insulin resistance, and to find a way to overcome this insulin resistant state. However, under obese conditions, many kinases are hyperactivated; in such a condition, further activation of the insulin-signaling pathway would be challenging and may not even be appropriate. A strategy to restore reduced BRD7 activity in obesity could establish glucose homeostasis by decreasing hepatic glucose production through GSK3β while preventing the body from having hyperactivated signaling pathways.
In summary, our work here partially unravels the complexity of the crosstalk network in the insulin-signaling pathway, which is triggered upon insulin stimulation, but also responds to nutrient availability. Considering the anti-diabetic effects of BRD7 and its role in gluconeogenesis, our current work suggests that BRD7 could serve as an attractive therapeutic target for the treatment of type 2 diabetes.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary material 1 (EPS 2395 kb) Figure S1. Quantification of BRD7 levels in liver tissues by qPCR analysis. (A–C) BRD7 mRNA levels in the liver were quantified through qPCR and normalized to 18S. (A) 7-week-old C57/BL6 J wild-type male mice were injected with Ad-BRD7 or Ad-LacZ as a control at a plaque forming unit (pfu) of 8 x 106 (n = 3 for each virus) through the tail vein. (B) 7-week-old BRD7 TgCre+/− mice and BRD7 TgCre−/−. (C) 7-week-old wild-type mice were injected with Ad-LacZshRNA or Ad-BRD7shRNA at a pfu of 8 x 106 (n = 6 for each virus) through the tail vein. Error bars are represented as mean ± SEM; P values were determined by Student’s t test. (*p < 0.05, **p < 0.01, ***p < 0.001)
Acknowledgements
We are indebted to Dr. U. Ozcan for reagents and helpful discussions. Research reported in this publication was supported by the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health under Award number R00DK093788. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. This work was also supported by the American Diabetes Association/Innovative Basic Science Grant (1-17-IBS-104), Boston Children’s Hospital Office of Faculty Development/BTREC/CTREC Faculty Career Development fellowship, and the Division of Endocrinology, Boston Children’s Hospital/Faculty Start up fund provided to S. W. P.
Abbreviations
- BRD7
Bromodomain-containing protein 7
- GSK3β
Glycogen synthase kinase 3β
- 4E-BP1
Eukaryotic translation initiation factor 4E-binding protein 1
- eIF4E
Eukaryotic translation initiation factor 4E
- NPC
Nasopharyngeal carcinoma
- BRCA-1
Breast cancer susceptibility gene 1
- PI3K
Phosphatidylinositol 3-kinase
- XBP1s
The spliced form of X-box binding protein 1
- ER
Endoplasmic reticulum
- IR
Insulin receptor
- IRS1
Insulin receptor substrate 1
- IRS2
Insulin receptor substrate 2
- AKT
Protein kinase B
- GS
Glycogen synthase
- TSC2
Tuberous sclerosis protein 2
- GAP
GTPase-activating protein
- mTORC
Mammalian target of rapamycin (mTOR)-raptor complex
- S6K
Ribosomal protein S6 kinase
- Pfu
Plaque forming unit
- MEF
Mouse embryonic fibroblasts
- KO
Knockout
- LBKO
Liver-specific BRD7 knockout mouse model
- FoxO1
Forkhead box O1
References
- 1.Zhou J, Ma J, Zhang BC, Li XL, Shen SR, Zhu SG, Xiong W, Liu HY, Huang H, Zhou M, Li GY. BRD7, a novel bromodomain gene, inhibits G1-S progression by transcriptionally regulating some important molecules involved in ras/MEK/ERK and Rb/E2F pathways. J Cell Physiol. 2004;200(1):89–98. doi: 10.1002/jcp.20013. [DOI] [PubMed] [Google Scholar]
- 2.Liu H, Zhang L, Niu Z, Zhou M, Peng C, Li X, Deng T, Shi L, Tan Y, Li G. Promoter methylation inhibits BRD7 expression in human nasopharyngeal carcinoma cells. BMC Cancer. 2008;8:253. doi: 10.1186/1471-2407-8-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaeser MD, Aslanian A, Dong MQ, Yates JR, 3rd, Emerson BM. BRD7, a novel PBAF-specific SWI/SNF subunit, is required for target gene activation and repression in embryonic stem cells. J Biol Chem. 2008;283(47):32254–32263. doi: 10.1074/jbc.M806061200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peng C, Liu HY, Zhou M, Zhang LM, Li XL, Shen SR, Li GY. BRD7 suppresses the growth of nasopharyngeal carcinoma cells (HNE1) through negatively regulating beta-catenin and ERK pathways. Mol Cell Biochem. 2007;303(1–2):141–149. doi: 10.1007/s11010-007-9466-x. [DOI] [PubMed] [Google Scholar]
- 5.Harte MT, O’Brien GJ, Ryan NM, Gorski JJ, Savage KI, Crawford NT, Mullan PB, Harkin DP. BRD7, a subunit of SWI/SNF complexes, binds directly to BRCA1 and regulates BRCA1-dependent transcription. Cancer Res. 2010;70(6):2538–2547. doi: 10.1158/0008-5472.CAN-09-2089. [DOI] [PubMed] [Google Scholar]
- 6.Mantovani F, Drost J, Voorhoeve PM, Del Sal G, Agami R. Gene regulation and tumor suppression by the bromodomain-containing protein BRD7. Cell Cycle. 2010;9(14):2777–2781. doi: 10.4161/cc.9.14.12309. [DOI] [PubMed] [Google Scholar]
- 7.Drost J, Mantovani F, Tocco F, Elkon R, Comel A, Holstege H, Kerkhoven R, Jonkers J, Voorhoeve PM, Agami R, Del Sal G. BRD7 is a candidate tumour suppressor gene required for p53 function. Nat Cell Biol. 2010;12(4):380–389. doi: 10.1038/ncb2038. [DOI] [PubMed] [Google Scholar]
- 8.Burrows AE, Smogorzewska A, Elledge SJ. Polybromo-associated BRG1-associated factor components BRD7 and BAF180 are critical regulators of p53 required for induction of replicative senescence. Proc Natl Acad Sci USA. 2010;107(32):14280–14285. doi: 10.1073/pnas.1009559107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chiu YH, Lee JY, Cantley LC. BRD7, a tumor suppressor, interacts with p85alpha and regulates PI3K activity. Mol Cell. 2014;54(1):193–202. doi: 10.1016/j.molcel.2014.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Park SW, Herrema H, Salazar M, Cakir I, Cabi S, Basibuyuk Sahin F, Chiu YH, Cantley LC, Ozcan U. BRD7 regulates XBP1s’ activity and glucose homeostasis through its interaction with the regulatory subunits of PI3K. Cell Metab. 2014;20(1):73–84. doi: 10.1016/j.cmet.2014.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Unanue ER, Urano F. Endoplasmic reticulum: an interface between the immune system and metabolism. Diabetes. 2014;63(1):48–49. doi: 10.2337/db13-1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oakes SA, Papa FR. The role of endoplasmic reticulum stress in human pathology. Annu Rev Pathol. 2015;10:173–194. doi: 10.1146/annurev-pathol-012513-104649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park SW, Ozcan U. Potential for therapeutic manipulation of the UPR in disease. Semin Immunopathol. 2013;35(3):351–373. doi: 10.1007/s00281-013-0370-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park SW, Zhou Y, Lee J, Ozcan U. Sarco(endo)plasmic reticulum Ca2+-ATPase 2b is a major regulator of endoplasmic reticulum stress and glucose homeostasis in obesity. Proc Natl Acad Sci USA. 2010;107(45):19320–19325. doi: 10.1073/pnas.1012044107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fruman DA. Regulatory subunits of class IA PI3K. Curr Top Microbiol Immunol. 2010;346:225–244. doi: 10.1007/82_2010_39. [DOI] [PubMed] [Google Scholar]
- 16.Yu J, Zhang Y, McIlroy J, Rordorf-Nikolic T, Orr GA, Backer JM. Regulation of the p85/p110 phosphatidylinositol 3′-kinase: stabilization and inhibition of the p110alpha catalytic subunit by the p85 regulatory subunit. Mol Cell Biol. 1998;18(3):1379–1387. doi: 10.1128/MCB.18.3.1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Birney DM, Cole DC, Crosson CE, Kahl BF, Neff BW, Reid TW, Ren K, Walkup RD. Use of beta-methylphenylalanine (beta MeF) residues to probe the nature of the interaction of substance P with its receptor: effects of beta MeF-containing substance P analogs on rabbit iris smooth muscle contraction. J Med Chem. 1995;38(13):2478–2482. doi: 10.1021/jm00013a024. [DOI] [PubMed] [Google Scholar]
- 18.Jope RS, Yuskaitis CJ, Beurel E. Glycogen synthase kinase-3 (GSK3): inflammation, diseases, and therapeutics. Neurochem Res. 2007;32(4–5):577–595. doi: 10.1007/s11064-006-9128-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rayasam GV, Tulasi VK, Sodhi R, Davis JA, Ray A. Glycogen synthase kinase 3: more than a namesake. Br J Pharmacol. 2009;156(6):885–898. doi: 10.1111/j.1476-5381.2008.00085.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307(5712):1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 21.Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4(9):648–657. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- 22.Manning BD, Logsdon MN, Lipovsky AI, Abbott D, Kwiatkowski DJ, Cantley LC. Feedback inhibition of Akt signaling limits the growth of tumors lacking Tsc2. Genes Dev. 2005;19(15):1773–1778. doi: 10.1101/gad.1314605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ising C, Koehler S, Brahler S, Merkwirth C, Hohne M, Baris OR, Hagmann H, Kann M, Fabretti F, Dafinger C, Bloch W, Schermer B, Linkermann A, Bruning JC, Kurschat CE, Muller RU, Wiesner RJ, Langer T, Benzing T, Brinkkoetter PT. Inhibition of insulin/IGF-1 receptor signaling protects from mitochondria-mediated kidney failure. EMBO Mol Med. 2015;7(3):275–287. doi: 10.15252/emmm.201404916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149(2):274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol. 2009;10(5):307–318. doi: 10.1038/nrm2672. [DOI] [PubMed] [Google Scholar]
- 26.Jefferies HB, Fumagalli S, Dennis PB, Reinhard C, Pearson RB, Thomas G. Rapamycin suppresses 5′TOP mRNA translation through inhibition of p70s6k. EMBO J. 1997;16(12):3693–3704. doi: 10.1093/emboj/16.12.3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pende M, Kozma SC, Jaquet M, Oorschot V, Burcelin R, Le Marchand-Brustel Y, Klumperman J, Thorens B, Thomas G. Hypoinsulinaemia, glucose intolerance and diminished beta-cell size in S6K1-deficient mice. Nature. 2000;408(6815):994–997. doi: 10.1038/35050135. [DOI] [PubMed] [Google Scholar]
- 28.Zhang HH, Lipovsky AI, Dibble CC, Sahin M, Manning BD. S6K1 regulates GSK3 under conditions of mTOR-dependent feedback inhibition of Akt. Mol Cell. 2006;24(2):185–197. doi: 10.1016/j.molcel.2006.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Raught B, Gingras AC. eIF4E activity is regulated at multiple levels. Int J Biochem Cell Biol. 1999;31(1):43–57. doi: 10.1016/S1357-2725(98)00131-9. [DOI] [PubMed] [Google Scholar]
- 30.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18(16):1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 31.Nojima H, Tokunaga C, Eguchi S, Oshiro N, Hidayat S, Yoshino K, Hara K, Tanaka N, Avruch J, Yonezawa K. The mammalian target of rapamycin (mTOR) partner, raptor, binds the mTOR substrates p70 S6Kinase and 4E-BP1 through their TOR signaling (TOS) motif. J Biol Chem. 2003;278(18):15461–15464. doi: 10.1074/jbc.C200665200. [DOI] [PubMed] [Google Scholar]
- 32.Kim Y, Andres Salazar Hernandez M, Herrema H, Delibasi T, Park SW. The role of BRD7 in embryo development and glucose metabolism. J Cell Mol Med. 2016;20(8):1561–1570. doi: 10.1111/jcmm.12907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lochhead PA, Coghlan M, Rice SQ, Sutherland C. Inhibition of GSK-3 selectively reduces glucose-6-phosphatase and phosphatase and phosphoenolypyruvate carboxykinase gene expression. Diabetes. 2001;50(5):937–946. doi: 10.2337/diabetes.50.5.937. [DOI] [PubMed] [Google Scholar]
- 34.Zhang X, Zhao F, Si Y, Huang Y, Yu C, Luo C, Zhang N, Li Q, Gao X. GSK3beta regulates milk synthesis in and proliferation of dairy cow mammary epithelial cells via the mTOR/S6K1 signaling pathway. Molecules. 2014;19(7):9435–9452. doi: 10.3390/molecules19079435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Embi N, Rylatt DB, Cohen P. Glycogen synthase kinase-3 from rabbit skeletal muscle. Separation from cyclic-AMP-dependent protein kinase and phosphorylase kinase. Eur J Biochem. 1980;107(2):519–527. doi: 10.1111/j.1432-1033.1980.tb06059.x. [DOI] [PubMed] [Google Scholar]
- 36.Matsumoto T, Nagase Y, Hirose J, Tokuyama N, Yasui T, Kadono Y, Ueki K, Kadowaki T, Nakamura K, Tanaka S. Regulation of bone resorption and sealing zone formation in osteoclasts occurs through protein kinase B-mediated microtubule stabilization. J Bone Miner Res. 2013;28(5):1191–1202. doi: 10.1002/jbmr.1844. [DOI] [PubMed] [Google Scholar]
- 37.Salas TR, Reddy SA, Clifford JL, Davis RJ, Kikuchi A, Lippman SM, Menter DG. Alleviating the suppression of glycogen synthase kinase-3beta by Akt leads to the phosphorylation of cAMP-response element-binding protein and its transactivation in intact cell nuclei. J Biol Chem. 2003;278(42):41338–41346. doi: 10.1074/jbc.M302972200. [DOI] [PubMed] [Google Scholar]
- 38.Eldar-Finkelman H. Glycogen synthase kinase 3: an emerging therapeutic target. Trends Mol Med. 2002;8(3):126–132. doi: 10.1016/S1471-4914(01)02266-3. [DOI] [PubMed] [Google Scholar]
- 39.Song G, Ouyang G, Bao S. The activation of Akt/PKB signaling pathway and cell survival. J Cell Mol Med. 2005;9(1):59–71. doi: 10.1111/j.1582-4934.2005.tb00337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carnero A, Blanco-Aparicio C, Renner O, Link W, Leal JF. The PTEN/PI3K/AKT signalling pathway in cancer, therapeutic implications. Curr Cancer Drug Targets. 2008;8(3):187–198. doi: 10.2174/156800908784293659. [DOI] [PubMed] [Google Scholar]
- 41.Altomare DA, Testa JR. Perturbations of the AKT signaling pathway in human cancer. Oncogene. 2005;24(50):7455–7464. doi: 10.1038/sj.onc.1209085. [DOI] [PubMed] [Google Scholar]
- 42.Lu M, Wan M, Leavens KF, Chu Q, Monks BR, Fernandez S, Ahima RS, Ueki K, Kahn CR, Birnbaum MJ. Insulin regulates liver metabolism in vivo in the absence of hepatic Akt and Foxo1. Nat Med. 2012;18(3):388–395. doi: 10.1038/nm.2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marte BM, Downward J. PKB/Akt: connecting phosphoinositide 3-kinase to cell survival and beyond. Trends Biochem Sci. 1997;22(9):355–358. doi: 10.1016/S0968-0004(97)01097-9. [DOI] [PubMed] [Google Scholar]
- 44.Dalle Pezze P, Sonntag AG, Thien A, Prentzell MT, Godel M, Fischer S, Neumann-Haefelin E, Huber TB, Baumeister R, Shanley DP, Thedieck K. A dynamic network model of mTOR signaling reveals TSC-independent mTORC2 regulation. Sci Signal. 2012;5(217):ra25. doi: 10.1126/scisignal.2002469. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material 1 (EPS 2395 kb) Figure S1. Quantification of BRD7 levels in liver tissues by qPCR analysis. (A–C) BRD7 mRNA levels in the liver were quantified through qPCR and normalized to 18S. (A) 7-week-old C57/BL6 J wild-type male mice were injected with Ad-BRD7 or Ad-LacZ as a control at a plaque forming unit (pfu) of 8 x 106 (n = 3 for each virus) through the tail vein. (B) 7-week-old BRD7 TgCre+/− mice and BRD7 TgCre−/−. (C) 7-week-old wild-type mice were injected with Ad-LacZshRNA or Ad-BRD7shRNA at a pfu of 8 x 106 (n = 6 for each virus) through the tail vein. Error bars are represented as mean ± SEM; P values were determined by Student’s t test. (*p < 0.05, **p < 0.01, ***p < 0.001)