

Abstract
The most severe Primary Immune Deficiencies (PID) have been successfully treated with allogeneic hematopoietic stem cell transplantation for more than four decades. However, such transplants have the best outcomes when there is a well matched donor available, as immune complications such as graft versus host disease are higher without a matched sibling donor. Gene therapy has been developed as a method to perform autologous transplants of a patient’s own stem cells that are genetically corrected. Through an iterative bench-to-bedside-and-back process, methods to efficiently add new copies of the relevant gene to hematopoietic stem cells have led to safe and effective treatments for several PID, including forms of Severe Combined Immune Deficiency, Wiskott-Aldrich Syndrome, and Chronic Granulomatous Disease. New methods for gene editing may allow additional PID to be treated by gene therapy, as they will allow the endogenous gene to be repaired and expressed under its native regulatory elements, essential for genes involved in cell processes of signaling, activation and proliferation. Gene therapy is providing exciting new treatment options for patients with PID and advances are sure to continue.
Keywords: Hematopoietic stem cell transplantation, Gammaretroviral vector, Lentiviral vector, Gene Editing, Site-specific endonuclease, Zinc Finger Nuclease, CRISPR/Cas9
Gene therapy has developed from an attractive, but unrealized concept to a first licensed medicine over the past few decades. Much of the work on gene therapy for primary immune deficiency diseases (PID) has been based around the application of hematopoietic stem cell transplantation (HSCT) as a potentially lifelong, curative therapy. Since HSCT using bone marrow transplant (BMT) was first successfully performed for PID patients with Severe Combined Immune Deficiency (SCID) in the late 1960’s1, the power of replacing a patient’s HSC bearing a disease causing mutation that disables the immune system with HSC from a healthy donor has advanced greatly. HSCT has been successfully applied to many of the most severe PID, including SCID, Wiskott-Aldrich Syndrome (WAS), Chronic Granulomatous Disease (CGD), X-linked hyper IgM (X-HIM), Leukocyte Adhesion Deficiency (LAD) and others. The success of HSCT has relied on identification of a well-matched stem cell donor, ideally a human leukocyte antigen (HLA)-matched sibling or family member donor. For SCID, such matched sources of HSC may provide a graft that is accepted without conditioning to eliminate the patient’s endogenous stem cells or immune system. It was learned that for patients with less severe cellular-mediated immune defects (e.g. WAS, CGD), it was necessary to apply some regimen of cytoreductive conditioning to “make space” for donor HSC to engraft and often immune suppressive drugs to prevent residual immunity from rejecting the donor’s HSC.2 Increased pre-treatment conditioning increases risks of allogeneic HSCT due to direct toxic effects of the conditioning agents (often cytotoxic chemotherapy drugs), immune suppression in patients with pre-existing immune defects, and potential burdens of chronic infections.
Because the majority of PID patients will not have an eligible HLA matched family donor, techniques were developed to use HSC from less well-matched donors, either unrelated donors identified through increasingly large registries or from half-matched parental donors. These mismatched transplants carry higher risks of either graft rejection or the converse, graft versus host disease, in which donor T cells attack the patient’s body. Therefore, manipulation of the donor graft, such as T cell depletion, or more potent immune suppression may be needed peritransplant, with attendant morbidity. As experience with these more challenging transplants has increased, the outcomes have steadily improved.
Nevertheless, alternative approaches that may be safer with the same prospect of benefit are needed. The key concept of gene therapy for PID has been to provide an autologous HSCT option, using the patient’s own HSC that are corrected ex vivo by either adding a normal copy of the responsible disease-related gene or, in the near future, correcting the endogenous defective gene in situ in the HSC (Figure 1). The key technical challenge has been to effectively and non-toxically introduce the normal gene into the stem cells, without causing them to lose their stem cell capacity for life-long multi-lineage blood cell production. Over 2-3 decades, such techniques have been developed and used in clinical trials for a growing number of diseases (Table 1).
Figure 1.
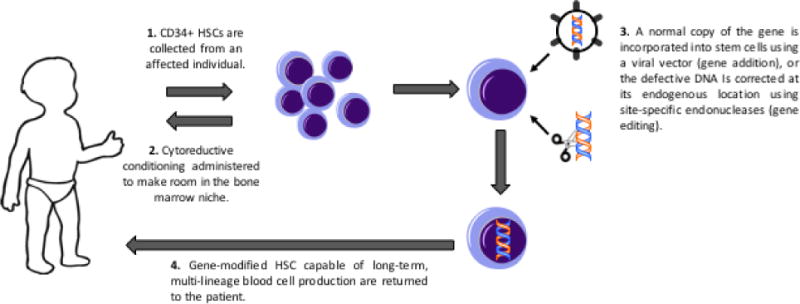
Clinical schema of gene therapy for PID.
Table 1.
Diseases treated by gene therapy using hematopoietic stem cells in clinical trials.
| Primary Immune Deficiencies |
| Adenosine deaminase (ADA)-deficient SCID |
| X-linked SCID (XSCID) |
| Wiskott-Aldrich Syndrome (WAS) |
| X-linked Chronic Granulomatous Disease (XCGD) |
| Leukocyte Adhesion Deficiency (LAD) |
| Lysosomal Storage and Metabolic Disorders |
| X-linked Adrenoleukodystrophy (XALD) |
| Metachromatic Leukodystrophy (MLD) |
| Hemoglobinopathies |
| Beta-thalassemia |
| Sickle Cell Disease |
| Stem Cell Defects |
| Fanconi’s anemia |
Technical advances that are enabling effective gene therapy of PID
The successes in gene therapy have been due to three sets of advances: better vectors, better HSC processing methods and better conditioning regimens. In the mid-1980’s murine retroviruses were developed that could transfer a foreign gene into mammalian cells (Figure 2).3 Several studies demonstrated their ability to introduce their gene into murine HSC, which could be transplanted and give rise to blood cells of both myeloid and lymphoid lineages containing and expressing the new gene.4,5 Initial attempts to use these vectors with human HSC were much less effective, but improvements in vector production methods to yield higher vector titers and changes in the viral envelope protein used to coat the virus and target it to cellular receptors led to higher levels of gene transfer to human HSC. Simultaneously, new hematopoietic growth factors were identified that could stimulate activation and proliferation of human HSC which greatly increased their ability to be take up the vectors.6 Additionally, it was learned that having an extracellular matrix protein, such as fibronectin or recombinant fragments thereof, could increase the uptake of virus by the stem cells and also better preserve their activity during the 2-4 days of ex vivo culture.7 Finally, the use of relatively low doses of the myelotoxic drug busulfan was shown to greatly enhance re-engraftment of the ex vivo-modified HSC by creating space in the bone marrow niche.8,9 Bringing these advances together in the late 1990’s, several investigators were able to provide significant benefit to patients with ADA-SCID and XSCID using gene therapy with patient autologous bone marrow HSC.8–19 Similar studies for WAS and XCGD followed and also led to restoration of the defective immune cell function.20–23
Figure 2. Gene delivery vectors used for gene therapy of PID.
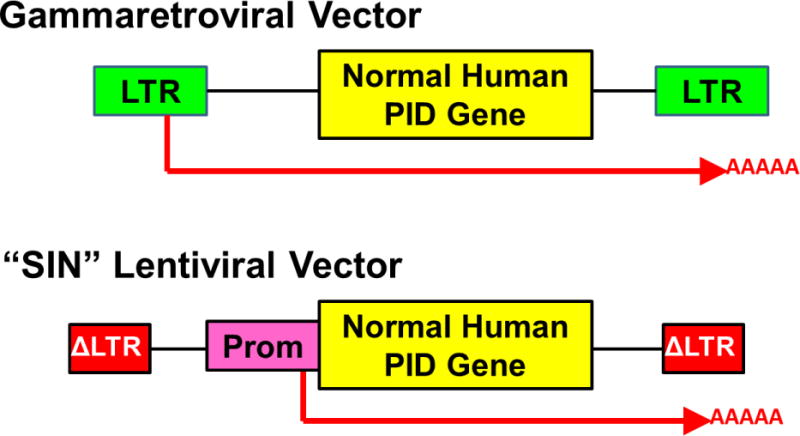
Upper: Gammaretroviral vectors have viral long terminal repeats (LTR) at each end with viral enhancers and promoters that drive transcription and termination/polyadenylation of a normal copy of the relevant human gene involved in the inherited PID. The messenger RNA produced from the vector is shown as a red arrow including the polyA tail. Lower: Self-inactivating (“SIN”) lentiviral vector has LTR with the enhancers deleted (Δ) and an internal promoter (Prom) to drive transcription of the human PID gene. In current clinical trials, lentiviral vectors being used for ADA SCID and XSCID are using the human Elongation Factor Alpha-1 gene short promoter (EFS) to drive the ADA and IL2Rg genes, respectively, the WASP gene endogenous promoter to drive a WASP gene, and a chimeric myeloid promoter to drive the gp91phox gene for XCGD.
However, severe complications occurred in many of these patients, with leukemia developing a few years after gene therapy.24–29 The retroviral vectors that were used randomly inserted into the chromosomal DNA of the target stem cells, and the potent enhancer elements they carried to drive expression of the therapeutic gene could trans-activate expression of nearby cellular proto-oncogenes (LMO2, MECOM). This eventually led to clonal outgrowth of transformed cells. While most of the patients were successfully treated for the leukemia, this was an unacceptable level of toxicity.
Fortunately, the remedy was already in hand. A new generation of gene delivery vectors, derived from the lentivirus class of retroviruses, had been developed that had markedly better safety profiles. Lentiviral vectors (LV) could be produced in a way in which the enhancer elements “self-inactivated” (so-called “SIN” vectors) so they had minimal, if any, ability to turn on cellular genes near their integration sites (Figure 2).30,31 Pre-clinical studies in murine cells showed SIN LV to have essentially no detectable transforming activity, whereas that of murine gammaretroviral vectors was readily demonstrable.32–36 Thus, most trials are currently using SIN LV and ongoing trials for ADA-SCID, XSCID, WAS and XCGD have shown effective immune reconstitution and no vector related complications to date, with more than 50 patients treated.37–40 Additionally, one study used a SIN gammaretroviral vector to treat XSCID with no complications of clonal expansions and good immune reconstitution in the majority of patients.41
One exception to the complications with murine gammaretroviral vectors has been in the setting of ADA SCID. More than 40 ADA SCID patients have been safely treated with no leukemia-like complications using gammaretroviral vectors since the landmark studies from The San Raffaele Telethon Institute for Gene Therapy (TIGET), Milan, Italy began in the late 1990’s, followed by studies at University College London/Great Ormond Street Hospital and at the University of California, Los Angeles.9,11,13,14 The work from TIGET led to the approval by the European Medicines Agency of their gammaretroviral vector gene therapy for ADA SCID, marketed by GSK as Strimvelis. It is only the second gene therapy product to receive licensure in the European Union (with none in the US or Canada to date). Why the gammaretroviral vectors have not had clinical safety problems in contrast to similar vectors for other PID remains unknown. Both the TIGET and UCLA groups have demonstrated vector integrations near some of the same genes implicated in leukoproliferation in other diseases, but there have been no clinical manifestations thus far.42
It should be emphasized that in all of these gene therapy trials, there has been no need to use immune suppressive drugs (e.g. cyclophosphamide, fludarabine, ATG, cyclosporine, corticosteroids) and there have been no problems with graft rejection or graft versus host disease. The amounts of pre-transplant conditioning used for the non-SCID disorders (WAS and CGD) were higher (8-12 mg/kg busulfan) than used for the ADA SCID (4 mg/kg) and XSCID (0-8 mg/kg), because of the perceived need for greater cytoablation for higher engraftment of the corrected stem cells to correct all blood cell lineages and not just lymphocytes as is needed for the SCID patients. While the dosages of these potent drugs are significant and have some risk, the results to date have been gratifyingly good, with only the expected marrow suppression and some mucositis and no severe transplant-related complications.
Future approaches and prospects for gene therapy of PID
One of the drawbacks of gene therapy is that each disease genotype entails a separate drug development project to develop the vector for that specific gene, perform pre-clinical activity and toxicity studies, etc. In contrast, in allogeneic HSCT, the donor cells have all of the corrective genes and it is more of a “one size fits all” approach. Nevertheless, there are ongoing projects at many centers to develop gene therapy for additional PIDs. Other forms of SCID, such as Artemis, Rag1 and Rag2 deficiency, autosomal recessive forms of CGD (p47, p67) and other PID (LAD, HLH) are under pre-clinical study with the goal of advancing to early phase clinical trials. Thus, these gene therapy approaches of gene addition will compete with the always improving methods of allogeneic HSCT to provide the best treatments for many of these diseases.
However, for other PID, gene addition therapies are not currently applicable. CVID, perhaps the most prevalent PID, may likely be caused by one of several genes, only a few of which are currently recognized. Each will again entail a separate development process and the efficacy and safety will need to be high enough to outweigh the results obtained currently with best medical therapy. Gain-of-function mutations, such as in STAT1 and STAT3, where the mutant allele may override proper expression of a new normal copy of the gene, poses additional challenges to traditional forms of gene therapy.
Additionally, genes that underlie some PID may not be amenable to the approach of gene addition using a viral vector. Genes that are involved in cell activation, intracellular signaling and other processes likely require precise temporal and physiologic control of their expression. For example, after the CD40 ligand gene was identified as a cause of X-HIM, studies were done to add that gene to bone marrow HSC from a gene knock-out mouse model.43 While initial immune restoration was achieved with class switching of B cells, many of the mice developed lymphoproliferative complications from constitutive, unregulated expression of CD40 ligand. Other genes involved in PID that may similarly require precise regulation are BTK, involved in X-linked agammaglobulinemia, FoxP3, causative of Immune dysregulation, Polyendocrinopathy, Enteropathy, X-linked syndrome (IPEX), as well as some of the B cell growth factors or their receptors that may be involved in some cases of CVID (e.g. TACI, BAFF-R). Efforts to derive vectors that display the precise gene regulation pattern of the endogenous gene have often been unsuccessful, as it may be difficult to include in the relative small genome size of a vector all of the DNA regulatory sequences that may surround and regulate these genes.
Gene editing to correct the genes causing PID
Once again, the answer is close at hand. New methods have been developed in the past few years for gene correction. If a disease-related gene could be repaired in its native chromosomal site, it should retain the normal expression pattern and restore the needed gene function. A real revolution of scientific advances has developed methods for gene editing that can either disrupt and turn-off a troublesome gene (e.g. dominant STAT2), repair a gene mutation (e.g. in BTK) or insert a new copy of the gene directly into the defective gene itself to use the gene expression regulatory elements to control the expression of the inserted gene (e.g. CD154). The critical component of this revolution in gene editing has been the recognition that such gene editing events can be greatly enhanced by introducing a precisely located break in the chromosomal DNA near the site of intended gene editing.44 A succession of designer site-specific endonucleases have been developed, including homing endonucleases, zinc finger nucleases (ZFN), TALENs and more importantly CRISPR/Cas9. Each of these systems can be engineered to recognize and introduce a DNA break at essentially one site out of the entire genome. If the DNA break is left to be resolved by the cell, it is often reannealed through a process called non-homologous end-joining (NHEJ) that often removes or adds a few base pairs at the break site and lead to disruption of the gene’s reading frame and gene silencing. However, if the cell is provided at the same time with copies of the target sequence as single stranded or double stranded DNA carrying the corrective DNA sequence, it may use the introduced information to repair the break by Homologous Directed Repair (HDR), “knocking-in” the corrective sequence and repairing the pathogenic mutation (Figure 3). This process of HDR may even be used to introduce new gene cassettes at a precise target site, such as a full length normal copy of the defective gene just downstream from the gene promoter sequences to maintain the gene’s normal expression patterns.
Figure 3. Gene editing using site-specific endonucleases.
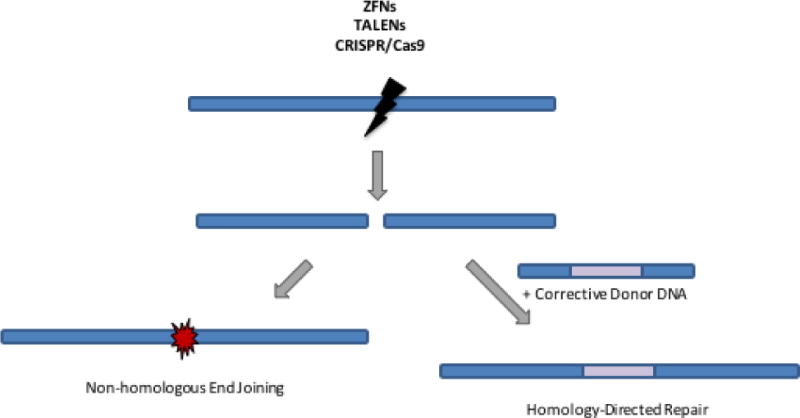
Site-specific endonucleases (ZFNs, TALENs, or CRISPR/Cas9) can be used to create targeted double stranded DNA breaks. These DNA breaks can be repaired by non-homologous end joining, which introduces small insertions and deletions and can be used for gene knockout strategies, as is employed in the CCR5 gene therapy trials for HIV. If a corrective donor DNA sequence is also provided to the cell, it can be used for homology-directed repair of the double-stranded DNA break, incorporating the therapeutic gene sequence and maintaining its expression under its endogenous regulatory elements.
These methods for gene editing have entered the clinic, with the first study using ZFN to disrupt the HIV CCR5 co-receptor in peripheral blood T cells from patients infected by HIV, rendering the cells relatively resistant to HIV infection.45 Other studies nearing clinical trials will seek to disrupt the CCR5 gene in HSC of HIV infected patients or disrupt the gene encoding a protein (Bcl11a) that represses expression of fetal globin to turn on the fetal hemoglobin as a way to treat beta-thalassemia and Sickle Cell Disease.
Gene correction in HSC for PID has been demonstrated in pre-clinical studies for XSCID, ADA SCID, XCGD, X-HIM and others.46–48 The methods are reaching levels of efficacy and non-toxicity to HSC to lead to clinical efficacy (Figure 4). The gene editing approaches may allow a much wider range of PID to be treated by gene therapy and autologous transplant, including XLA, XHIM, IPEX, CVID and others.
Figure 4. Site-specific Gene Editing of Autologous Hematopoietic Stem Cells for Gene Therapy of PID.
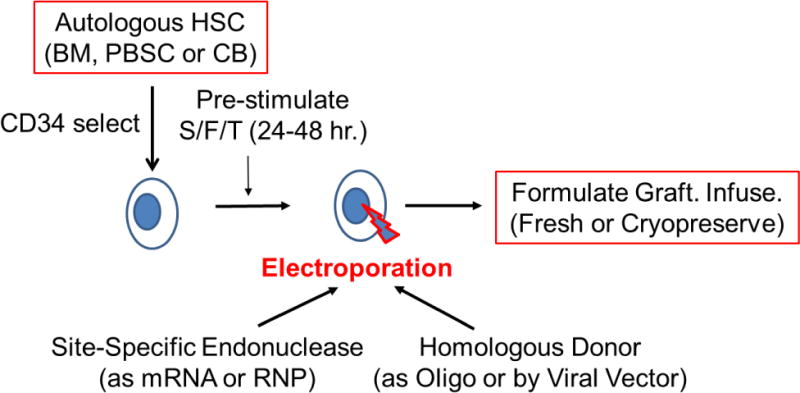
Autologous hematopoietic stem cells (HSC) can be obtained from bone marrow (BM), mobilized peripheral blood stem cells (PBSC) or umbilical cord blood (CB). Usually the HSC are enriched using CD34 immunoselection. The HSC are pre-stimulated to improve gene modification by culture for 24-48 hours with a combination of recombinant hematopoietic growth factors, such as ckit ligand/flt-3 ligand and thrombopoietin (S/F/T). The cells are then treated with electroporation with the site-specific endonuclease (zinc finger nuclease, homing endonuclease, TALEN or CRISPR), to introduce a double stranded break at the target gene, delivered as either in vitro transcribed messenger RNA (mRNA) or pre-formed ribonucleoprotein (RNP) containing the Cas9 protein and a short-guide RNA for CRISPR-mediated processes. The homologous donor containing the corrective sequences may be provided either as an oligonucleotide (Oligo) that is co-electroporated with the endonuclease or via a viral vector (e.g. adeno-associated virus {AAV} or integrase defective lentivirus {IDLV}). Following electroporation, the gene-modified HSC are formulated for intravenous administration and may either be infused into the patient fresh or after cryopreservation and thawing.
In addition to all of the cell based PID discussed above, there are other PID that are due to defects in blood proteins, such as Hereditary Angioedema (HAE). A recent report described studies in a mouse model of HAE where a different viral vector – adeno-associated virus (AAV) – was used to introduced a normal SERPING1 gene encoding the C1 inhibitor into cells of the liver that could then serve as a factory to produce the protein and release it into the bloodstream.49 This approach of a protein factory has also been shown to be effective for ADA SCID. In the mouse model, a LV carrying a normal human ADA gene was injected intravenously, targeted the liver, and served as a source of ADA sufficient to sustain survival and correct immunity. Similar approaches are under active study for non-PID protein disorders such as hemophilia, anti-trypsin inhibitor deficiency and others.
Remaining challenges for gene therapy for PID
Of course, there are challenges that remain to be fully overcome. Production of the gene addition vectors or gene editing reagents has been at relatively small scale for pilot studies and need to be advanced to pharmaceutical production methods at sufficient scale to treat many patients. Both gene addition with SIN LV as well as gene editing with the various endonucleases may still have some risks of genotoxicity if they disrupt important genes by either insertion or off-target cutting.
The conditioning regimens used to date have been based on chemotherapy regimens. While these have not shown significant toxicity in the short-term, they may have some late effects, even at the reduced dosages used. Once again, progress has been made in this essential area, by the development of methods to “make space” for transplanted gene corrected HSC using monoclonal antibodies to proteins on HSC, such as ckit or CD45.50–52 If effective, these new conditioning agents could greatly increase safety and allow applications of gene therapy to the somewhat milder PID such as XLA.
While current approaches of either gene addition or gene editing are done ex vivo with harvested bone marrow or mobilized stem cells, it may be advantageous, when possible, to deliver the needed gene or gene editing reagents in vivo and repair the HSC while remaining in their niches. And eventually, the use of pluripotent stem cells, such as induced pluripotent stem cells (iPSC), may allow autologous cell grafts to be produced, gene corrected, expanded and used for transplant with a much more abundant source of stem cells providing better immune reconstitution.53
It is an exciting time for science and a good time for patients as these new treatments using gene therapy may provide safer and more effective approaches that avoid the complications of allogeneic HSCT. Of course, it will take several years of comparative analyses to determine which approaches are best, but the existence of these various treatments for diseases that were once often fatal is indeed gratifying.
Key Concepts.
-
—
Severe PID can be treated by transplantation of hematopoietic stem cells from a healthy matched donor, but risks of immunologic complications exist and are heightened when the transplant is from an unrelated donor or haplo-identical donor.
-
—
Gene therapy for PID allows an autologous transplant in which the patient’s own hematopoietic stem cells are corrected and transplanted, essentially eliminating the immunologic complications.
-
—
Hematopoietic stem cells can be corrected by addition of a normal copy of the relevant gene using an integrating retroviral or lentiviral vector.
-
—
Initial studies with retroviral vectors that showed clinical efficacy, but some had leukoproliferative complications from the vectors (this has not been seen for ADA SCID).
-
—
Current trials using safer lentiviral vectors lacking strong enhancers have shown efficacy for ADA SCID, XSCID, WAS and CGD with no vector-related complications.
-
—
Recently, methods have been developed for gene editing that could allow the disease-causing mutation to be corrected in situ within a patient’s hematopoietic stem cells. This approach may be of particular value for disorders where the relevant PID-causing gene requires precise regulation of expression, such as CD40 ligand for X-HIM, BTK for XLA and FoxP3 for IPEX.
Therapeutic Implications.
Gene therapy provides a new therapeutic option for treatment of patients with severe PID that respond to allogeneic hematopoietic stem cell transplantation.
Abbreviations
- AAV
Adeno-associated virus
- BMT
Bone Marrow Transplant
- CGD
Chronic Granulomatous Disease
- CRISPR
Clustered Regularly Interspaced Short Palindromic Repeats
- CVID
Common Variable Immune Deficiency
- HAE
Hereditary angioedema
- HLA
Human Leukocyte Antigen
- HDR
Homology-directed repair
- HLH
Hereditary lymphohistiocytosis
- HSC
Hematopoietic Stem Cell
- HSCT
Hematopoietic Stem Cell Transplantation
- IDLV
Integration-Defective Lentiviral Vector
- IPEX
Immune dysregulation, Polyendocrinopathy, Enteropathy, X-linked syndrome
- iPSC
Induced pluripotent stem cell
- LAD
Leukocyte Adhesion Deficiency
- LV
Lentiviral Vector
- NHEJ
Non-homologous end-joining
- PID
Primary Immune Deficiency
- SCID
Severe Combined Immune Deficiency
- SIN
Self-inactivating
- TALEN
Transcription activator-like effector nucleases
- TIGET
The San Raffaele Telethon Institute for Gene Therapy
- WAS
Wiskott-Aldrich Syndrome
- XLA
X-linked agammaglobulinemia
- X-HIM
X-linked Hyper-IgM Syndrome
- ZFN
Zinc Finger Nuclease
References Cited
- 1.Gatti RA, Meuwissen HJ, Allen HD, Hong R, Good RA. Immunological reconstitution of sex-linked lymphopenic immunological deficiency. Lancet. 1968 Dec 28;2(7583):1366–9. doi: 10.1016/s0140-6736(68)92673-1. [DOI] [PubMed] [Google Scholar]
- 2.Parkman R, Rappeport J, Geha R, Belli J, Cassady R, Levey R, Nathan DG, Rosen FS. Complete correction of the Wiskott-Aldrich syndrome by allogeneic bone-marrow transplantation. N Engl J Med. 1978 Apr 27;298(17):921–7. doi: 10.1056/NEJM197804272981701. [DOI] [PubMed] [Google Scholar]
- 3.Miller AD, Rosman GJ. Improved retroviral vectors for gene transfer and expression. Biotechniques. 1989 Oct;7(9):980–2. 984–6, 989–90. [PMC free article] [PubMed] [Google Scholar]
- 4.Williams DA, Lemischka IR, Nathan DG, Mulligan RC. Introduction of new genetic material into pluripotent haematopoietic stem cells of the mouse. Nature. 1984 Aug 9-15;310(5977):476–80. doi: 10.1038/310476a0. [DOI] [PubMed] [Google Scholar]
- 5.Dick JE, Magli MC, Huszar D, Phillips RA, Bernstein A. Introduction of a selectable gene into primitive stem cells capable of long-term reconstitution of the hemopoietic system of W/Wv mice. Cell. 1985 Aug;42(1):71–9. doi: 10.1016/s0092-8674(85)80102-1. [DOI] [PubMed] [Google Scholar]
- 6.Nolta JA, Crooks GM, Overell RW, Williams DE, Kohn DB. Retroviral vector-mediated gene transfer into primitive human hematopoietic progenitor cells: effects of mast cell growth factor (MGF) combined with other cytokines. Exp Hematol. 1992 Oct;20(9):1065–71. [PubMed] [Google Scholar]
- 7.Moritz T, Patel VP, Williams DA. Bone marrow extracellular matrix molecules improve gene transfer into human hematopoietic cells via retroviral vectors. J Clin Invest. 1994 Apr;93(4):1451–7. doi: 10.1172/JCI117122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aiuti Alessandro, Slavin Shimon, Aker Memet, Ficara Francesca, Deola Sara, Mortellaro Alessandra, Morecki Shoshana, et al. Correction of ADA-SCID by Stem Cell Gene Therapy Combined with Nonmyeloablative Conditioning. Science (New York, NY) 2002;296(5577):2410–13. doi: 10.1126/science.1070104. [DOI] [PubMed] [Google Scholar]
- 9.Candotti Fabio, Shaw Kit L, Muul Linda, Carbonaro Denise, Sokolic Robert, Choi Christopher, Schurman Shepherd H, et al. Gene Therapy for Adenosine Deaminase – Deficient Severe Combined Immune Deficiency: Clinical Comparison of Retroviral Vectors and Treatment Plans. Blood. 2012;120(18):3635–46. doi: 10.1182/blood-2012-02-400937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aiuti Alessandro, Cattaneo Federica, Galimberti S, Benninghoff Ulrike, Cassani Barbara, Callegaro Luciano, Scaramuzza Samantha, et al. Gene Therapy for Immunodeficiency Due to Adenosine Deaminase Deficiency. The New England Journal of Medicine. 2009;360(5):447–58. doi: 10.1056/NEJMoa0805817. [DOI] [PubMed] [Google Scholar]
- 11.Cicalese MP, Ferrua F, Castagnaro L, Pajno R, Barzaghi F, Giannelli S, Dionisio F, Brigida I, Bonopane M, Casiraghi M, Tabucchi A, Carlucci F, Grunebaum E, Adeli M, Bredius RG, Puck JM, Stepensky P, Tezcan I, Rolfe K, De Boever E, Reinhardt RR, Appleby J, Ciceri F, Roncarolo MG, Aiuti A. Update on the safety and efficacy of retroviral gene therapy for immunodeficiency due to adenosine deaminase deficiency. Blood. 2016 Jul 7;128(1):45–54. doi: 10.1182/blood-2016-01-688226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gaspar H Bobby, Bjorkegren Emma, Parsley Kate, Gilmour Kimberly C, King Doug, Sinclair Joanna, Zhang Fang, et al. Successful Reconstitution of Immunity in ADA-SCID by Stem Cell Gene Therapy Following Cessation of PEG-ADA and Use of Mild Preconditioning. Molecular Therapy. 2006;14(4):505–13. doi: 10.1016/j.ymthe.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 13.Gaspar HB, Cooray S, Gilmour KC, Parsley KL, Zhang F, Adams S, Bjorkegren E, Bayford J, Brown L, Davies EG, Veys P, Fairbanks L, Bordon V, Petropoulou T, Kinnon C, Thrasher AJ. Hematopoietic stem cell gene therapy for adenosine deaminase-deficient severe combined immunodeficiency leads to long-term immunological recovery and metabolic correction. Sci Transl Med. 2011 Aug 24;3(97):97ra80. doi: 10.1126/scitranslmed.3002716. [DOI] [PubMed] [Google Scholar]
- 14.Shaw Kit L, Sokolic Robert, Davila Alejandra, Silvin Christopher, Garabedian Elizabeth, Oliveira Satiro De, Barman Provaboti, et al. Phase II Clinical Trial of Gene Therapy for Adenosine Deaminase-Deficient Severe Combined Immune Deficiency (ADA-SCID) Molecular Therapy. 2014;22(Supplement 1):S107. [Google Scholar]
- 15.Cavazzana-Calvo Marina. Gene Therapy of Human Severe Combined Immunodeficiency (SCID)-X1 Disease. Science. 2000;288(5466):669–72. doi: 10.1126/science.288.5466.669. [DOI] [PubMed] [Google Scholar]
- 16.Hacein-Bey-Abina Salima, Le Deist Françoise, Carlier Frédérique, Bouneaud Cécile, Hue Christophe, De Villartay Jean-Pierre, Thrasher Adrian J, et al. Sustained Correction of X-Linked Severe Combined Immunodeficiency by Ex Vivo Gene Therapy. The New England Journal of Medicine. 2002;346(16):1185–93. doi: 10.1056/NEJMoa012616. [DOI] [PubMed] [Google Scholar]
- 17.Hacein-Bey-Abina Salima, Hauer Julia, Lim Annick, Picard Capucine, Wang Gary P, Berry Charles C, Martinache Chantal, et al. Efficacy of Gene Therapy for X-Linked Severe Combined Immunodeficiency. The New England Journal of Medicine. 2010;363(4):355–64. doi: 10.1056/NEJMoa1000164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gaspar H Bobby, Parsley Kathryn L, Howe Steven, King Doug, Gilmour Kimberly C, Sinclair Joanna, Brouns Gaby, et al. Gene Therapy of X-Linked Severe Combined Immunodeficiency by Use of a Pseudotyped Gammaretroviral Vector. Lancet. 2004;364(9452):2181–87. doi: 10.1016/S0140-6736(04)17590-9. [DOI] [PubMed] [Google Scholar]
- 19.Gaspar HB, Cooray S, Gilmour KC, Parsley KL, Adams S, Howe SJ, Al Ghonaium A, et al. Long-Term Persistence of a Polyclonal T Cell Repertoire After Gene Therapy for X-Linked Severe Combined Immunodeficiency. Science Translational Medicine. 2011 doi: 10.1126/scitranslmed.3002715. [DOI] [PubMed] [Google Scholar]
- 20.Boztug Kaan, Schmidt Manfred, Schwarzer Adrian, Banerjee Pinaki P, Díez Inés Avedillo, Dewey Ricardo A, Böhm Marie, et al. Stem-Cell Gene Therapy for the Wiskott-Aldrich Syndrome. The New England Journal of Medicine. 2010;363(20):1918–27. doi: 10.1056/NEJMoa1003548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kang Elizabeth M, Choi Uimook, Theobald Narda, Linton Gilda, Long Priel Debra A, Kuhns Doug, Malech Harry L. Retrovirus Gene Therapy for X-Linked Chronic Granulomatous Disease Can Achieve Stable Long-Term Correction of Oxidase Activity in Peripheral Blood Neutrophils. Blood. 2010;115(4):783–91. doi: 10.1182/blood-2009-05-222760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kang Hyoung Jin, Bartholomae Cynthia C, Paruzynski Anna, Arens Anne, Kim Sujeong, Yu Seung Shin, Hong Youngtae, et al. Molecular Therapy: The Journal of the American Society of Gene Therapy. 11. Vol. 19. Nature Publishing Group; 2011. Retroviral Gene Therapy for X-Linked Chronic Granulomatous Disease: Results from Phase I/II Trial; pp. 2092–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ott Marion G, Schmidt Manfred, Schwarzwaelder Kerstin, Stein Stefan, Siler Ulrich, Koehl Ulrike, Glimm Hanno, et al. Correction of X-Linked Chronic Granulomatous Disease by Gene Therapy, Augmented by Insertional Activation of MDS1-EVI1, PRDM16 or SETBP1. Nature Medicine. 2006;12(4):401–9. doi: 10.1038/nm1393. [DOI] [PubMed] [Google Scholar]
- 24.Abina Salima Hacein-Bey, von Kalle Christof, Schmidt Manfred, Le Deist Françoise, Wulffraat Nicolas, McIntyre Elisabeth, Radford Isabelle, et al. A Serious Adverse Event after Successful Gene Therapy for X-Linked Severe Combined Immunodeficiency. The New England Journal of Medicine. 2003;348(3):255–56. doi: 10.1056/NEJM200301163480314. [DOI] [PubMed] [Google Scholar]
- 25.Hacein-Bey-Abina S. LMO2-Associated Clonal T Cell Proliferation in Two Patients after Gene Therapy for SCID-X1. Science. 2003;302(5644):415–19. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- 26.Fischer Alain, Abina Salima Hacein-Bey, Thrasher Adrian J, von Kalle Christof, Cavazzana-Calvo M. LMO2 and Gene Therapy for Severe Combined Immunodeficiency. The New England Journal of Medicine. 2004;350(24):2526–27. doi: 10.1056/NEJM200406103502422. [DOI] [PubMed] [Google Scholar]
- 27.Howe Steven J, Mansour Marc R, Schwarzwaelder Kerstin, Bartholomae Cynthia, Hubank Michael, Kempski Helena, Brugman Martijn H, et al. Insertional Mutagenesis Combined with Acquired Somatic Mutations Causes Leukemogenesis Following Gene Therapy of SCID-X1 Patients. Journal of Clinical Investigation. 2008;118(9):3143–50. doi: 10.1172/JCI35798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stein Stefan, Ott Marion G, Schultze-Strasser Stephan, Jauch Anna, Burwinkel Barbara, Kinner Andrea, Schmidt Manfred, et al. Genomic Instability and Myelodysplasia with Monosomy 7 Consequent to EVI1 Activation after Gene Therapy for Chronic Granulomatous Disease. Nature Medicine. 2010;16(2):198–204. doi: 10.1038/nm.2088. [DOI] [PubMed] [Google Scholar]
- 29.Braun Christian Jörg, Boztug Kaan, Paruzynski Anna, Witzel Maximilian, Schwarzer Adrian, Rothe Michael, Modlich Ute, et al. Gene Therapy for Wiskott-Aldrich Syndrome–Long-Term Efficacy and Genotoxicity. Science Translational Medicine. 2014;6(227):227ra33–227ra33. doi: 10.1126/scitranslmed.3007280. [DOI] [PubMed] [Google Scholar]
- 30.Naldini L, Blömer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, Trono D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996 Apr 12;272(5259):263–7. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- 31.Naldini L, Trono D, Verma IM. Lentiviral vectors, two decades later. Science. 2016 Sep 9;353(6304):1101–2. doi: 10.1126/science.aah6192. Modlich. [DOI] [PubMed] [Google Scholar]
- 32.Modlich U, Navarro S, Zychlinski D, Maetzig T, Knoess S, Brugman MH, Schambach A, Charrier S, Galy A, Thrasher AJ, Bueren J, Baum C. Insertional transformation of hematopoietic cells by self-inactivating lentiviral and gammaretroviral vectors. Mol Ther. 2009 Nov;17(11):1919–28. doi: 10.1038/mt.2009.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carbonaro Denise A, Zhang Lin, Jin Xiangyang, Montiel-Equihua Claudia, Geiger Sabine, Carmo Marlene, Cooper Aaron, et al. Preclinical Demonstration of Lentiviral Vector-Mediated Correction of Immunological and Metabolic Abnormalities in Models of Adenosine Deaminase Deficiency. Molecular Therapy. 2014;22(3):607–22. doi: 10.1038/mt.2013.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marangoni Francesco, Bosticardo Marita, Charrier Sabine, Draghici Elena, Locci Michela, Scaramuzza Samantha, Panaroni Cristina, et al. Evidence for Long-Term Efficacy and Safety of Gene Therapy for Wiskott-Aldrich Syndrome in Preclinical Models. Molecular Therapy: The Journal of the American Society of Gene Therapy. 2009;17(6):1073–82. doi: 10.1038/mt.2009.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Charrier S, Dupré L, Scaramuzza S, Jeanson-Leh L, Blundell MP, Danos O, Cattaneo F, et al. Lentiviral Vectors Targeting WASp Expression to Hematopoietic Cells, Efficiently Transduce and Correct Cells from WAS Patients. Gene Therapy. 2007;14(5):415–28. doi: 10.1038/sj.gt.3302863. [DOI] [PubMed] [Google Scholar]
- 36.Santilli Giorgia, Almarza Elena, Brendel Christian, Choi Uimook, Beilin Chiara, Blundell Michael P, Haria Sneha, et al. Molecular Therapy: The Journal of the American Society of Gene Therapy. 1. Vol. 19. Nature Publishing Group; 2011. Biochemical Correction of X-CGD by a Novel Chimeric Promoter Regulating High Levels of Transgene Expression in Myeloid Cells; pp. 122–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gaspar Hubert B, Buckland Karen, Carbonaro Denise A, Shaw Kit, Barman Provaboti, Davila Alejandra, Gilmour Kimberly C, et al. Immunological and Metabolic Correction After Lentiviral Vector Gene Therapy for ADA Deficiency. Molecular Therapy. 2015;23(Supplement 1):S102. [Google Scholar]
- 38.De Ravin SS, Wu X, Moir S, Anaya-OBrien S, Kwatemaa N, Littel P, Theobald N, et al. Lentiviral Hematopoietic Stem Cell Gene Therapy for X-Linked Severe Combined Immunodeficiency. Science Translational Medicine. 2016;8(335):335ra57–335ra57. doi: 10.1126/scitranslmed.aad8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aiuti Alessandro, Biasco Luca, Scaramuzza Samantha, Ferrua Francesca, Cicalese Maria Pia, Baricordi Cristina, Dionisio Francesca, et al. Lentiviral Hematopoietic Stem Cell Gene Therapy in Patients with Wiskott-Aldrich Syndrome. Science. 2013;341(6148):1233151. doi: 10.1126/science.1233151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abina Salima Hacein-Bey, Gaspar H Bobby, Blondeau Johanna, Caccavelli Laure, Charrier Sabine, Buckland Karen, Picard Capucine, et al. Outcomes Following Gene Therapy in Patients With Severe Wiskott-Aldrich Syndrome. Jama. 2015;313(15):1550–63. doi: 10.1001/jama.2015.3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hacein-Bey-Abina Salima, Pai Sung-Yun, Gaspar H Bobby, Armant Myriam, Berry Charles C, Blanche Stephane, Bleesing Jack, et al. A Modified γ-Retrovirus Vector for X-Linked Severe Combined Immunodeficiency. New England Journal of Medicine. 2014;371(15):1407–17. doi: 10.1056/NEJMoa1404588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aiuti Alessandro, Cassani Barbara, Andolfi Grazia, Mirolo Massimiliano, Biasco Luca, Recchia Alessandra, Urbinati Fabrizia, et al. Multilineage Hematopoietic Reconstitution without Clonal Selection in ADA-SCID Patients Treated with Stem Cell Gene Therapy. Journal of Clinical Investigation. 2007;117(8):2233–40. doi: 10.1172/JCI31666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brown Michael P, Topham David J, Sangster Mark Y, Zhao Jingfeng, Flynn Kirsten J, Surman Sherri L, Woodland David L, et al. Thymic Lymphoproliferative Disease after Successful Correction of CD40 Ligand Deficiency by Gene Transfer in Mice. Nature Medicine. 1998;4(11):1253–60. doi: 10.1038/3233. [DOI] [PubMed] [Google Scholar]
- 44.Porteus Matthew H, Baltimore David. Gene Targeting in Human Cells. Science. 2003 May;300:75390. doi: 10.1126/science.1078395. [DOI] [PubMed] [Google Scholar]
- 45.Tebas Pablo, Stein David, Tang Winson W, Frank Ian, Wang Shelley Q, Lee Gary, Spratt S Kaye, et al. Gene Editing of CCR5 in Autologous CD4 T Cells of Persons Infected with HIV. The New England Journal of Medicine. 2014;370(10):901–10. doi: 10.1056/NEJMoa1300662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lombardo A, Genovese P, Beausejour CM, Colleoni S, Lee YL, Kim KA, Ando D, Urnov FD, Galli C, Gregory PD, Holmes MC, Naldini L. Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat Biotechnol. 2007 Nov;25(11):1298–306. doi: 10.1038/nbt1353. [DOI] [PubMed] [Google Scholar]
- 47.Joglekar AV, Hollis RP, Kuftinec G, Senadheera S, Chan R, Kohn DB. Integrase-defective lentiviral vectors as a delivery platform for targeted modification of adenosine deaminase locus. Mol Ther. 2013 Sep;21(9):1705–17. doi: 10.1038/mt.2013.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.De Ravin Suk See, Reik Andreas, Liu Pei-Qi, Li Linhong, Wu Xiaolin, Su Ling, Raley Castle, et al. Nature Biotechnology. Nature Publishing Group; 2016. Feb, Targeted Gene Addition in Human CD34+ Hematopoietic Cells for Correction of X-Linked Chronic Granulomatous Disease; pp. 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pagovich Odelya E, Qui Ting, Whaley Adele S, Russo Anthony R, Rosenberg Jonathan, De Bishnu P, Russo Carlo, et al. One-Time Gene Therapy for Hereditary Angioedema. Molecular Therapy. 2016;24(Supplement 1):S298. [Google Scholar]
- 50.Czechowicz A, Kraft D, Weissman IL, Bhattacharya D. Efficient transplantation via antibody-based clearance of hematopoietic stem cell niches. Science. 2007 Nov 23;318(5854):1296–9. doi: 10.1126/science.1149726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chhabra A, Ring AM, Weiskopf K, Schnorr PJ, Gordon S, Le AC, Kwon HS, Ring NG, Volkmer J, Ho PY, Tseng S, Weissman IL, Shizuru JA. Hematopoietic stem cell transplantation in immunocompetent hosts without radiation or chemotherapy. Sci Transl Med. 2016 Aug 10;8(351):351ra105. doi: 10.1126/scitranslmed.aae0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Palchaudhuri R, Saez B, Hoggatt J, Schajnovitz A, Sykes DB, Tate TA, Czechowicz A, Kfoury Y, Ruchika F, Rossi DJ, Verdine GL, Mansour MK, Scadden DT. Non-genotoxic conditioning for hematopoietic stem cell transplantation using a hematopoietic-cell-specific internalizing immunotoxin. Nat Biotechnol. 2016 Jul;34(7):738–45. doi: 10.1038/nbt.3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Malech iPSC. Merling Randall K, Sweeney Colin L, Chu Jessica, Bodansky Aaron, Choi Uimook, Priel Debra Long, Kuhns Douglas B, et al. An AAVS1-Targeted Minigene Platform for Correction of iPSCs From All Five Types of Chronic Granulomatous Disease. Molecular Therapy. 2015;23(1):147–57. doi: 10.1038/mt.2014.195. [DOI] [PMC free article] [PubMed] [Google Scholar]