

Abstract
Clostridium difficile PCR ribotype 106 (also identified as restriction endonuclease analysis [REA] group DH) recently emerged as the most common strain causing C. difficile infection (CDI) among US adults. We previously identified this strain predominating our pediatric cohort. Pediatric clinical CDI isolates previously characterized by REA underwent antibiotic resistance testing and whole genome sequencing. Of 134 isolates collected from children, 31 (23%) were REA group DH. We performed a comparative genomics analysis to identify DH-associated accessory genes. We identified five DH-associated genes that are associated with virulence in other bacterial species but not previously known to contribute to CDI. These genes are associated with intestinal mucosal adhesion (collagen-binding surface protein), sporulation (sporulation integral membrane protein YtvI), and protection from oxidative stress and foreign DNA (DNA phosphorothioation-dependent restriction proteins, sulfurtransferase, and DNA sulfur modification proteins). The association of these genes was validated in a cohort of 623 publicly available C. difficile sequences, 10 (1.6%) of which were monophyletic to REA group DH through in silico multilocus sequence typing and core genome phylogenetic analysis. Further investigation is required to determine the contribution of these genes to the emergence and virulence of this epidemic strain.
Keywords: Clostridium difficile, pediatric, DH, 106, comparative genomics
1. Introduction
The clinical and molecular epidemiology of Clostridium difficile infections (CDI) has recently received considerable attention because of the emergence of highly virulent strains. These include strain BI/NAP1/027 (identified as BI by restriction endonuclease analysis [REA], NAP1 by pulsed-field gel electrophoresis), and 027 by polymerase chain reaction [PCR] ribotyping) [1], as well as BK/NAP7,8,9/078 [2] and AF/244 [3]. The recent global dissemination of epidemic strain BI/NAP1/027 was associated with emergence of fluoroquinolone resistance,[4] and changes in BI/NAP1/027 infection phenotype may be related to binary toxin production and a loss-of-function mutation in tcdC, a negative regulator for toxins A and B [1]. Because of increased CDI morbidity, mortality, and associated healthcare costs and the predominance of antibiotic-resistant strains such as BI/NAP1/027, the U.S. Centers for Disease Control and Prevention (CDC) classified C. difficile among the most serious antibiotic resistant “public health threats that require urgent and aggressive action”[5].
Recent CDC Emerging Infections Program CDI surveillance data [6, 7] from ten US states suggest that BI/NAP1/027 prevalence declined between 2012 and 2014. Ribotype 106, also identified as group DH by REA and NAP11 by pulsed-field gel electrophoresis [8], emerged as the predominant strain causing CDI among US adults [6, 7]. While ribotype 106 was previously uncommonly reported among US adults, ribotype 106 was the second-most prevalent ribotype in the UK until 2009 [9], after which yearly declines in incidence were noted [10]. Until recently, for unknown reasons, ribotype 106 was uncommon outside of the UK [9]. We previously reported the predominance of strain DH in children [11].
The molecular epidemiology and pathogenesis of strain DH/NAP11/106 are poorly understood. Because strain DH/NAP11/106 is now the most common cause of CDI among US adults, the primary study objective was to describe the microbiologic and genotypic characteristics of this epidemic subclade. Specifically, we explored whether BI/NAP1/027-associated virulence factors and antibiotic resistance patterns contributed to the emergence of strain DH/NAP11/106. Additionally, using a comparative genomics approach, we identified several accessory genomic elements (AGEs) associated with this epidemic subclade that may represent candidate virulence factors. These data provide an important framework for further exploration of the pathogenesis of this epidemic subclade.
2. Materials and Methods
2.1 Clinical Microbiology
This study included patients diagnosed with CDI (i.e., unformed stool that tested positive by tcdB PCR) between March 2011 and November 2013 at the Ann & Robert H. Lurie Children’s Hospital of Chicago. The Institutional Review Board approved this study and waived informed consent.
Clinical stool specimens were stored at −70°C and batch processed for isolation of C. difficile by anaerobic culture, as previously described [11, 12], at the Microbiology Research Laboratory at Edward Hines Jr. VA Hospital. All C. difficile isolates underwent REA, as previously described[11, 13]. REA group was assigned to each isolate based on comparison of the DNA band pattern to a library of reference REA groups.
Antibiotic susceptibility data were derived from a previous study [14]. Mean inhibitory concentrations (MIC) of the following antibiotics were measured at the Loyola University Chicago Stritch School of Medicine: metronidazole, vancomycin, rifaximin, fidaxomicin, surotomycin, clindamycin, and moxifloxacin (i.e., high-level fluoroquinolone resistance). Agar dilution method was used for susceptibility testing [15, 16]. MIC breakpoints were set for metronidazole (≥32 ug/ml), clindamycin (≥8 ug/ml), and moxifloxacin (≥8 ug/ml) based on Clinical and Laboratory Standards Institute breakpoints[16] and for vancomycin (≥4 ug/ml) based on the European Committee on Antimicrobial Susceptibility Testing epidemiological cutoff value [17]. The rifaximin-resistant breakpoint (≥32 ug/ml) was previously described [18]. Susceptibility breakpoints have not been established for surotomycin and fidaxomicin.
2.2 Whole Genome Sequencing
C. difficile isolates previously characterized by REA underwent whole genome sequencing (WGS). Genomic DNA was extracted using the BiOstic Bacteremia DNA Isolation Kit (MO BIO Laboratories, Carlsbad, CA). Paired-end sequencing libraries were prepared using the Nextera XT DNA Library Prep Kit (Illumina, San Diego, CA), and WGS was performed using the Illumina MiSeq system to produce paired 300 base pair (bp) reads. De novo genome assembly was performed using SPAdes (v3.6.2; http://cab.spbu.ru/software/spades/) [19]. Contigs ≥ 200 bp were annotated using Prokka (v1.11; http://www.vicbioinformatics.com/software.prokka.shtml) [20]. The nucleotide sequences for the 134 sequenced genomes have been deposited at DDBJ/ENA/GenBank under the accession numbers listed in the Supplementary Materials. Sequence types (STs) were assigned to each isolate based on the allelic patterns of 7 housekeeping genes[21] using the C. difficile multilocus sequence typing (MLST) database (http://pubmlst.org/cdifficile).
Illumina reads for each sequenced isolate were quality trimmed using Trimmomatic[22] (v0.36; http://www.usadellab.org/cms/?page=trimmomatic) and aligned to the strain 630 chromosomal sequence (GenBank accession number: AM180355.1) using bwa (v0.7.6a-r433; http://bio-bwa.sourceforge.net/). The mpileup function of SAMtools and bcftools (v0.1.19-44428cd; http://www.htslib.org/) were used to identify nucleotide variants relative to the reference. The default options for mpileup were used with the following exceptions: extended BAQ values were calculated, indel calling was not performed, and BCF output was generated. The view function of bcftools was used to call variants using default options with the following exceptions: single nucleotide variant (SNV) calling was requested, likelihood based analyses were performed, and genotype calling was performed at variant sites. The following criteria were used to filter variant positions: a variant must be supported by ≥ 75% of covering reads; a position must be covered by a minimum of 5 reads and a maximum of 3×4 the median read coverage of the entire alignment; there must be at least one read in both directions covering a position; the variant is determined to be homozygous under the diploid model; and the position did not fall within a region in the reference genome determined to be low complexity using NCBI dustmaker (BLAST+; https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE_TYPE=BlastDocs&DOC_TYPE=Download) [23]. A multiple alignment was produced by replacing variant positions passing the above filters with the variant base in the reference sequence. Variant positions not passing the above filters and non-variant positions covered by fewer than 5 reads were replaced with ambiguous bases. To produce a core genome alignment, all positions with ambiguous bases in one or more of the strain alignments were replaced with the reference base in the sequences for all of the strains. A phylogenetic tree omitting regions of likely recombination was produced using Gubbins (v. 2.2.1; https://sanger-pathogens.github.io/gubbins/) [24]. Evolview (http://www.evolgenius.info/evolview/) [25] and Interactive tree of life (iTOL; http://itol.embl.de/)[26] were used to visualize and annotate phylogenetic trees.
Genomes were screened for various genes whose association with virulence or antibiotic resistance was previously described in other C. difficile strains. These genes were identified in draft genome sequences using an in silico PCR program developed by one of the study authors (E.A.O.; https://github.com/egonozer/in_silico_pcr/releases) using previously published primer sequences (Table 1) [27]. Up to one base mismatch and one base insertion or deletion in primer sequences were permitted. SNVs, insertions, and deletions were manually identified using CLC Sequence Viewer (v7.7; https://www.qiagenbioinformatics.com/products/clc-sequence-viewer/). Non-synonymous SNVs were assessed for their potential impact on protein function by the program SIFT (http://sift.bii.a-star.edu.sg/) [28]. Using ResFinder (https://cge.cbs.dtu.dk//services/ResFinder/) [29], draft genomes were screened for 73 genes associated with macrolide-lincosamide-streptogramin B (MLSB) resistance.
Table 1.
Primer Sequences of Known Virulence Factors for which Were Screened by in silico PCR Among 31 C. difficile REA Group DH Strains
| Gene | Protein | Primer sequence (5’ ➔ 3’) [27] |
|---|---|---|
| tcdA | Toxin A | GCATGATAAGGCAACTTCAGTGGTA AGTTCCTCCTGCTCCATCAAATG |
| tcdB | Toxin B | CCAAARTGGAGTGTTACAAACAGGTG GCATTTCTCCATTCTCAGCAAAGTA GCATTTCTCCGTTTTCAGCAAAGTA |
| tcdC | Negative regulator of toxins A and B | AAAAGGGAGATTGTATTATGTTTTC CAATAACTTGAATAACCTTACCTTCA |
| cdtA | Binary toxin subunit A | GGGAAGCACTATATTAAAGCAGAAGC GGGAAACATTATATTAAAGCAGAAGC CTGGGTTAGGATTATTTACTGGACCA |
| cdtB | Binary toxin subunit B | TTGACCCAAAGTTGATGTCTGATTG CGGATCTCTTGCTTCAGTCTTTATAG |
2.3 Comparative Genomics Analysis
Core genome, defined as nucleotide sequences of genomic regions present in ≥ 95% (i.e., ≥ 128 of 134) of the isolates in our pediatric cohort, was determined using Spine (v0.2; http://vfsmspineagent.fsm.northwestern.edu/cgi-bin/spine.cgi) [30]. Accessory genomic elements (AGEs) were designated as those sequences in each isolate not identified as core genome by Spine. AGEs may contain zero, one, or more than one complete or incomplete genes. The distributions of individual AGEs among the 134 pediatric isolates genomic sequences were determined using ClustAGE (v0.7.2; https://sourceforge.net/projects/clustage/). Briefly, ClustAGE identifies representative contiguous AGEs within the input data set of all AGEs from each of the 134 strain genomic sequences and delineates the distribution of discrete AGEs among those genomes. A minimum of 85% nucleotide sequence identity was set as a cutoff for AGE similarity between any two strains. The distribution of AGEs (at least 200 bp in length) in DH strains versus non-DH strains was determined.
The strength of association of each AGE with strain DH was determined by calculating Cramer’s V, and statistical significance was determined by chi square test, using Stata/IC statistical software, v12.1 (StataCorp, College Station, TX). All AGEs with strong correlation (i.e., Cramer’s V > 0.75, P < 1×10−25) underwent manual validation with nucleotide and protein BLAST alignments against strain genomic sequences and predicted coding sequences. Of note, the cutoff of Cramer’s V > 0.75 in this analysis was associated with P < 1×10−25. When strength of association is measured for 6000 AGEs and alpha = 0.05, the Bonferroni correction for statistical significance is P < 8 ×10−6. Thus, all AGEs with Cramer’s V > 0.75 are strongly statistically significant.
To externally validate the strain association of these candidate virulence factors in a larger and more diverse cohort, additional comparative genomics analyses were performed. All complete and draft C. difficile genomes publicly available in the National Center for Biotechnology Information (NCBI) Genome database (as of January 2017) were downloaded and grouped into an NCBI cohort against which candidate virulence factors in the DH strains were compared. In silico MLST was performed on all 626 downloaded sequences identified as C. difficile by NCBI, and a core genome phylogenetic tree was generated from the downloaded NCBI sequences and our 134 newly sequenced pediatric isolate sequences using kSNP (v3.021; https://sourceforge.net/projects/ksnp/). A core genome definition of 95% (722 of 750 genomes) was used. NCBI isolates underwent BLAST analysis for the previously identified novel candidate virulence factors, and the strain-specific association of these virulence factors was assessed.
Of note, NCBI isolates were not available for REA typing. Thus, the NCBI isolates belonging to this epidemic subclade were identified by a variety of methods. As described above, we collected in silico MLST data from our pediatric isolates that had previously been characterized by REA. In addition, we confirmed the ST-associations with other typing methods through a review of the literature, which demonstrated previous classification of ribotype 106 strains as ST-42 [21, 31]. To provide validation of our definition of DH-associated STs, we performed in silico MLST on 626 C. difficile NCBI sequences, and also identified previously published typing data, when available, for those downloaded sequences classified as a DH-associated ST. Ribotyping data were available for two of these ST-42 sequences, and those isolates had been previously classified as ribotype 106 [32]. Finally, core genome phylogenetic analysis permitted visualization of the genetic similarity of all sequences included in this study irrespective of strain typing results. We confirmed that all isolates identified as a DH-associated ST belonged to the same subclade on the core genome phylogenetic tree. Accession numbers and MLST data for these 626 NCBI sequences are listed in the Supplementary Materials.
To determine the similarities in the accessory genomes of our pediatric and the NCBI DH isolates, pairwise comparisons of AGE presence or absence among all DH isolate sequences were performed. Bray-Curtis distances based on total lengths of shared AGEs ≥ 100 bp in size were calculated for each pair [33]. A neighbor-joining tree was generated from the pairwise Bray-Curtis distance matrix using Phylip (v3.695; http://evolution.gs.washington.edu/phylip). Bootstrap values were calculated from 100 resamplings and branches with less than 50 bootstrap support were collapsed. Tree and heatmap data were visualized using iTOL (http://itol.embl.de/).
3. Results
3.1 Whole Genome Sequencing
This study included 134 pediatric CDI cases. Of these, 31 (23.1%) C. difficile isolates had been previously identified as REA group DH [11]. WGS was performed on 134 C. difficile isolates from the pediatric cohort. Median read coverage was 72× (range 25×-393×); median contig number (from de novo assembly of the reads) was 173 (range 51–1,183); and median N50 (i.e., a statistic indicating the value at which 50% of the entire assembly is contained in contigs of at least this length) was 113,353 bp (range 5,540–519,873). WGS statistics for each pediatric isolate are listed in the Supplementary Materials.
3.2 Sequence Typing and Phylogenetics
In silico MLST was performed on each isolate, and sequence types (STs) were assigned based on allelic patterns of 7 housekeeping genes [21]. The pediatric cohort contained a highly diverse group of isolates represented by 46 REA groups (18 defined REA groups and 28 unique non-specific REA groups) and 35 STs; core genome phylogenetic analysis (Fig. 1) separated pediatric isolates into 4 of the 5 C. difficile clades (https://pubmlst.org/cdifficile/) [34]. The DH isolates in our pediatric cohort formed a monophyletic group containing two STs: ST-42 (30/31 [96.8%]) and ST-28 (1/31 [3.2%]). Compared to ST-42, ST-28 differs only in its allele for the sodA housekeeping gene (3 SNVs: A234G, A364G, C381G). All DH strains were in clade 1 and appear to be most closely related to the strains belonging to endemic REA group Y (ST-2 and ST-110). Of note, all ST-42 and ST-28 isolates in this cohort were previously identified as REA group DH. This suggests that all DH strains were correctly identified by REA and no additional DH isolates were mistakenly excluded.
Fig. 1. Core genome phylogenetic tree of 134 pediatric CDI isolates.
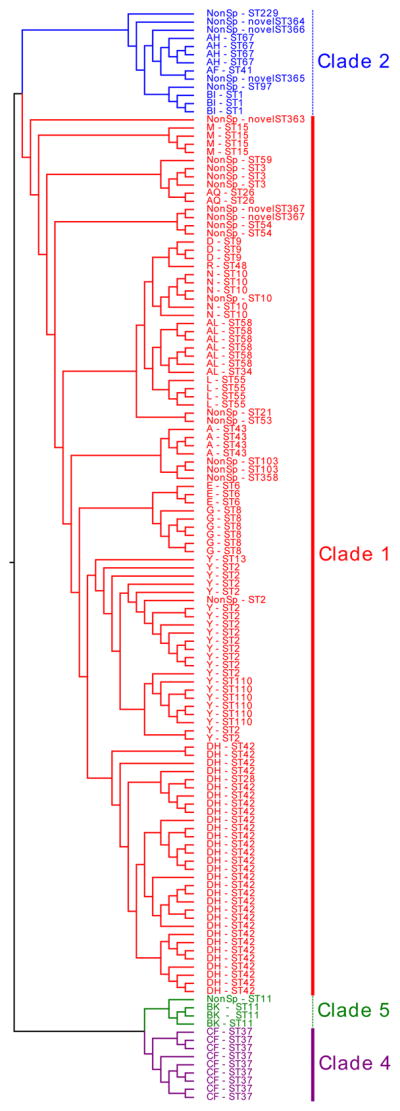
Isolates are grouped among 4 of the previously classified C. difficile clades (clades 1, 2, 4, and 5 per the MLST scheme; https://pubmlst.org/cdifficile/ [34]) and identified by their REA group and ST. Five novel STs were identified. NonSp: non-specific REA group.
3.3 Screening for Established Virulence Factors
DH strains were screened for genes of various known virulence factors (Table 1). All DH strains were positive for tcdA and tcdB (toxins A and B) and negative for cdtA and cdtB (binary toxin). The sequences of tcdB were compared among various C. difficile reference strains by BLAST alignment. The closed genome of our DH (clade 1) reference strain (GenBank accession number: CP022524.1) was compared to both reference strains 630 (clade 1) and R20291 (an epidemic BI/NAP1/027 clade 2 strain). The tcdB gene shared 99% sequence identity between our DH reference strain and strain 630, but only shared 93% sequence identity between our DH reference strain and strain R20291.
Among the 31 REA group DH strains, tcdC was identical in all 30 of the ST-42 isolates. The single ST-28 isolate contained an 18 bp deletion at positions 330–347 similar to the well described tcdC deletion in epidemic strain BI/NAP1/027 [1]. However, the single base deletion at position 117 that results in a truncated protein and confers tcdC loss-of-function in BI/NAP1/027 was not identified in any of the DH strains. In addition to the 18 bp deletion in the DH/ST-28 strain as described above, all DH strains had a non-synonymous SNV (T21G) relative to reference strain 630. This SNV was not predicted to impact protein function by SIFT analysis.
3.4 Antibiotic Susceptibility
Antibiotic susceptibility data were available for 28/31 (90.3%) of the DH strains. All DH isolates were susceptible to metronidazole, vancomycin, and moxifloxacin and had favorable MICs to both fidaxomicin (MIC range 0.03–0.25 ug/ml) and surotomycin (MIC range 0.125–2 ug/ml). Rifaximin and clindamycin resistance were noted among 6/28 (21.4%) and 11/28 (39.3%) DH isolates, respectively. The six rifaximin-resistant isolates all contained a non-synonymous SNV (G1514A) in rpoB that is reported to be associated with rifaximin resistance [35]. The 11 clindamycin-resistant DH isolates were screened for MLSB resistance determinants; an MLSB efflux pump gene (mefAE) and an rRNA methylase gene (ermB) were identified in one and six isolates, respectively. Thus, no clear clindamycin resistance mechanism was identified in 4/11 (36.4%) clindamycin-resistant DH strains.
3.5 Comparative Genomics: Pediatric Isolates
Amongst the combined accessory genomes of 134 C. difficile isolates from our pediatric cohort, 5,710 unique AGEs (≥ 200 bp) were identified. The prevalence of each AGE among DH and non-DH isolates was compared. AGEs were ranked by their associations with strain DH. Cramer’s V of AGEs ranged between 1 (i.e., present in all DH and lacking in all non-DH strains) and -0.89 (i.e., lacking in all DH strains and present in 97/103 [94.2%] of non-DH strains). Annotated genes among 10 AGEs with strong correlation (i.e., Cramer’s V > 0.75; P < 1×10−25) with strain DH are listed in Table 2. Between 1 and 6 annotated proteins were identified among each of the 10 DH-associated AGEs. Predicted open reading frames that remained hypothetical proteins after manual validation of automatic annotation results were excluded. The genomic context of each AGE is illustrated in Figure 2.
Table 2.
Annotated Proteins of C. difficile Strain DH-Associated Accessory Genomic Elements
| Protein Annotation of Genes within Each DH-associated AGE | Cramer’s V-Pediatric Cohort (% DH / Non-DH Strains) (n=134) | Cramer’s V-Validation Cohort (% DH / Non-DH Strains) (n=757) |
|---|---|---|
|
DH-AGE-1 DNA phosphorothioation-dependent restriction protein DptF DNA phosphorothioation-dependent restriction protein DptG DNA phosphorothioation-dependent restriction protein DptH type IV secretion-system coupling DNA-binding domain protein |
1 (100/0) | 1 (100/0) |
|
DH-AGE-2 multidrug resistance protein pyruvate phosphate dikinase PEP/pyruvate-binding protein TetR family transcriptional regulator |
0.92 (100/4) | 0.76 (100/4) |
|
DH-AGE-3 acetyltransferase |
0.92 (100/4) | 0.77 (100/3) |
|
DH-AGE-4 putative sulfurtransferase DndC DNA sulfur modification protein DndD DNA sulfur modification protein DndE |
0.89 (100/6) | 0.60 (100/9) |
|
DH-AGE-5 bifunctional 5'-methylthioadenosine/S- adenosylhomocysteine nucleosidase/phosphatase |
0.86 (100/8) | 0.73 (100/5) |
|
DH-AGE-6 putative transcriptional regulator type II restriction enzyme, methylase subunit |
0.84 (100/9) | 0.57 (100/10) |
|
DH-AGE-7 restriction modification system DNA specificity domain protein |
0.84 (100/9) | 0.34 (100/29) |
|
DH-AGE-8 conjugative transposon FtsK/SpoIIIE-like protein |
0.83 (100/10) | 0.16 (98/65) |
|
DH-AGE-9 sporulation integral membrane protein YtvI peptidylarginine deiminase acetyltransferase drug/sodium antiporter cytidylate kinase 2 MerR family transcriptional regulator |
0.78 (100/13) | 0.45 (100/18) |
|
DH-AGE-10 collagen-binding surface protein |
0.78 (100/13) | 0.61 (98/8) |
Fig. 2. Genomic context of 10 DH-associated AGEs.

The location of 10 AGEs (Table 2), in relation to the C. difficile pathogenicity locus (PaLoc) that includes tcdA and tcdB, are illustrated in the closed genome of our DH reference strain (LC5624; GenBank accession number: CP022524.1).
3.6 Comparative Genomics: External Validation of Candidate Virulence Factors
In silico MLST was performed on 626 downloaded NCBI C. difficile isolate sequences. Of these, 60 unique STs (distributed among all 5 MLST clades) were identified in this cohort. Of note, 72 (11.9%) could not be assigned a ST for various reasons; 6 had a novel pattern of previously characterized MLST alleles; 26 had a novel MLST allele; 12 had at least 1 allelic sequence that was truncated; 20 were missing at least 1 allele; 7 had both missing and novel alleles; and 1 had both a truncated and novel allele. A phylogenetic tree was generated from the core genome alignment of the NCBI sequences and the 134 newly sequenced pediatric isolate sequences (Fig. 3). The tree incorporated 176,115 variant positions among the group of strains. Three of the 626 downloaded sequences (GCF_000450985.2, GCF_900011355.1, and GCF_900012755.1) were omitted because MLST, the constructed phylogenetic tree, and core genome analysis all indicated that the 3 sequences had been misidentified as C. difficile. This assessment was confirmed by BLAST alignment of the genomic sequences of these three isolates against the NCBI 16S rRNA gene sequence database. One of these isolates (GCF_000450985.2) was identified as Clostridium innocuum, and the other two (GCF_900011355.1 and GCF_900012755.1) were identified as Terrisporobacter species. Thus, 623 NCBI isolates were included in the analyses.
Fig. 3. Core genome circular cladogram of 757 C. difficile isolates (134 pediatric CDI isolates and 623 downloaded NCBI isolates).

The 41 isolates (31 REA group DH isolates from the pediatric cohort and 10 monophyletic isolates from the NCBI cohort) that are monophyletic with REA group DH are delineated in red branching and shading along with the red portion of the outer colored ring. Of note, with the exception of one closely related ST-28 strain in the pediatric cohort, all strains on that DH branch were classified as ST-42 and there were no other ST-42 strains on other non-DH branches in the phylogenetic tree. Thus, the phylogenetic tree validates the definition of DH-associated STs in this study. All non-DH strains are delineated in blue along the outer colored ring. The pediatric C. difficile isolates are labeled LC and their unique identifier (delineated in yellow along the inner colored ring), and the downloaded NCBI isolates are labeled by their NCBI GenBank assembly accession numbers (delineated in black along the inner colored ring).
Of the 623 NCBI sequences included in the analysis, 10 (1.6%) were ST-42 and 0 were ST-28 (i.e., the DH-associated STs in our cohort) and 613 (98.4%) were other non-DH-associated STs. Core genome phylogenetic analysis (Fig. 3) confirmed that these 10 NCBI strains formed a monophyletic group with our 31 REA group DH isolates from the pediatric cohort. Of note, zero non-ST-42 strains from the NCBI cohort clustered with our DH strains on the core genome phylogenetic tree. Thus, between both our pediatric cohort and the NCBI cohort, 757 sequences were available: 41 strains belonging to this epidemic subclade and 716 strains outside of this subclade.
BLAST alignment was performed for all annotated proteins identified among the 10 DH-associated AGEs against all 757 C. difficile sequences. All of the DH-associated AGEs had a similar association with this epidemic subclade in this larger and more diverse cohort (Table 2), with the exception of conjugative transposon FtsK/SpoIIIE-like protein (DH-AGE-8) that was identified in 65% of the non-DH strains in the validation cohort.
Fig. 4 demonstrates the similarities in accessory genome content among the 41 strains belonging to the epidemic subclade; reference strains 630 and R20291 are included for comparison. The accessory genomes of the monophyletic group of pediatric and NCBI strains were generally highly similar to each other, with a few exceptions. Notably, the accessory genomes (Fig. 4) of the two NCBI strains isolated from adults in Ireland and the UK were relatively dissimilar to other strains in this subclade, while the two strains from Canada were highly similar to our pediatric isolates in this subclade, as well as the other NCBI strains sequenced in the US. The accessory genomes of all strains within this subclade were highly dissimilar to that of the two reference strains outside of this subclade (630 and R20291).
Fig 4. Relative amount of shared accessory genome content among 41 C. difficile strains monophyletic with REA group DH.

The 31 REA group DH isolates from the pediatric cohort are labeled LC and their unique identifier, and the 10 isolates from the NCBI cohort that are monophyletic with REA group DH are labeled by their NCBI GenBank assembly accession numbers. Based on review of NCBI data linked to each sequence accession number, all isolates were sequenced in the US with the exception of two sequences from Quebec, Canada (labels italicized in red font) and one sequence each from Ireland and the UK (both labels italicized in blue font). For comparison, reference strains 630 and R20291 (an epidemic BI/NAP1/027 strain) are included. Bray-Curtis distances (d) were calculated for every pairwise comparison of shared AGE content between strains. Neighbor-joining tree (left) is a consensus across 100 bootstrap resamplings of AGE distributions. Branches with support < 50 were collapsed. Heatmap (right) shows relative pairwise AGE content similarity (1 - d) between strains.
4. Discussion
According to recent CDC Emerging Infections Program data, the incidence of CDI caused by C. difficile strain DH/NAP11/106 has surpassed BI/NAP1/027, and DH/NAP11/106 is now the most common strain causing CDI among US adults [6, 7]. Interestingly, strain DH/NAP11/106 had been the second most common strain (after strain BI/NAP1/027) causing CDI in the UK [9], but recent declines in incidence have been noted [10]. Strain DH/NAP11/106 had been previously uncommonly reported outside of the UK.
Host and pathogen characteristics that account for this shift in C. difficile molecular epidemiology and the clinical consequences of infection with strain DH/NAP11/106 are poorly understood. A previous UK study demonstrated that strain DH/NAP11/106 generally caused less severe CDI than BI/NAP1/027 [36]. The infection phenotype of DH/NAP11/106 requires additional investigation in US adults.
Genomic analyses of our large cohort of isolates demonstrate that strain DH/NAP11/106 lacks several virulence factors previously identified in BI/NAP1/027 strains [1], specifically binary toxin and the single nucleotide deletion in tcdC that results in a loss-of-function mutation of this negative regulator of toxins A and B. Widespread use of fluoroquinolones in adult patients likely contributed to emergence and dissemination of BI/NAP1/027 strains, which are fluoroquinolone resistant [1]. In contrast, DH strains in the present study had a more favorable antibiotic susceptibility pattern; all were susceptible to moxifloxacin and most were susceptible to clindamycin. Thus, a similar competitive advantage related to antibiotic use was not identified among our DH strains. Furthermore, although clindamycin resistance in our DH isolates approached 50%, our previous study indicated that only 8% of patients in this pediatric cohort had been exposed to clindamycin in the previous 30 days [11]. Thus, antibiotic exposure seemed to play a limited role in the emergence of strain DH/NAP11/106 in our cohort. Of note, a similar favorable antibiotic susceptibility pattern of DH/NAP11/106 isolates in adult patients was reported in North America[18], while DH/NAP11/106 strains among adults in Ireland[37] and Scotland[38] were generally fluoroquinolone-resistant. Additional investigation of antibiotic susceptibility patterns of strain DH/NAP1/106 among US adults is needed.
Using a comparative genomics analysis, we identified several genes that are strongly associated with strain DH requiring further investigation to assess their role in C. difficile pathogenesis. Although many of these genes and the functions of their putative translated proteins have not yet been well delineated in C. difficile, several have been described in other bacterial species. Many of these proteins play a role in intestinal mucosal adhesion, sporulation, and protection from oxidative stress and foreign DNA. These particular physiologic processes could potentially provide a bacterial competitive advantage that could explain the clinical associations with strain DH that we have previously described, namely an association with multiply recurrent CDI and long time intervals between CDI relapses [11].
The DH-associated AGEs included a diverse array of genes likely involved in a multitude of cellular functions, including metabolism and antibiotic resistance. However, the specific role in C. difficile virulence is not entirely clear for many of these AGEs. Several AGEs associated with the DH epidemic subclade contained genes whose role in virulence have been described in other bacterial species. For example, the operon involved in sulfur modification and phosphorothioation of nucleic acids (i.e., DNA phosphorothioation-dependent restriction proteins, sulfurtransferase, and DNA sulfur modification proteins) has been well characterized. In Streptomyces lividans [39], phosphorothioation of DNA enhanced survival upon exposure to oxidative stress. In Salmonella enterica, phosphorothioation permitted restriction of foreign DNA, which is hypothesized to provide protection from bacteriophages [40]. Bacteriophages are highly prevalent in the gastrointestinal tract, and resistance to killing by bacteriophages could provide a competitive advantage to C. difficile in this niche. Another AGE strongly associated with DH strains contained a gene important for sporulation, which could also potentially provide a survival advantage. In Bacillus subtilis, targeted mutagenesis of the gene encoding sporulation integral membrane protein YtvI resulted in sporulation defects [41]. In addition, other orthologs of this gene are present in other genera of spore-forming bacteria, supporting the role of this protein in sporulation of C. difficile strain DH [41].
The ability of cell surface proteins to mediate mucosal adherence have been well described, and enhanced mucosal adherence could in turn prevent clearance and facilitate recurrent CDI [42, 43]. Cell wall-anchored collagen-binding proteins are important for gut mucosal adhesion. Specifically, collagen binding protein A, a microbial surface component recognizing adhesive matrix molecules (MSCRAMM) in reference strain 630, mediates adherence to types I and IV collagen on gut mucosal tissue [42, 44]. Of note, the DH-associated collagen-binding surface protein identified in the present study is distinct from collagen binding protein A in strain 630 (CD3145). Interestingly, this AGE shares 85% sequence identity with the LPxTG-domain-containing cell wall anchor domain found in E. faecium (GenBank accession: WP_010776652.1), which contributes to enterococcal virulence via mucosal adhesion and biofilm formation [45].
Strengths of this study include the large and diverse cohort of isolates that underwent WGS. Because greater number and diversity of isolates being analyzed more reliably distinguishes core and accessory genome [30], the comparative genomics analysis included a large number of AGEs that may not have been recognized in a smaller and/or less diverse collection of isolates. Furthermore, the association of these AGEs with the DH subclade was externally validated in a larger and more diverse cohort of publicly available isolate sequences. It is important to note that we identified genes in the accessory genome, rather than expressed proteins, that are associated with strain DH. Because observed AGE associations do not definitively indicate causality, AGEs of interest require further investigation to determine their patterns of expression and contributions to virulence in C. difficile. Core genome changes may be present that were not identified through our analysis.
In summary, C. difficile strain DH/NAP11/106, which surpassed BI/NAP1/027 as the most common strain among US adults, lacks several pathogen characteristics that contributed to BI/NAP1/027 dissemination and virulence. Strain DH/NAP11/106 possesses several genes that encode proteins that have been previously described in other bacterial species to be involved in intestinal mucosal adhesion, sporulation, and protection from oxidative stress and foreign DNA. Further investigation is needed to delineate the contribution of these candidate virulence factors to C. difficile pathogenesis. This knowledge may improve our understanding of the emergence, dissemination, and virulence of this epidemic strain.
Supplementary Material
Acknowledgments
The authors acknowledge James Osmolski at Loyola University Chicago Stritch School of Medicine for his assistance with performance of antibiotic susceptibility testing on bacterial isolates, and Katherine Murphy and the NUSeq Core at Northwestern University Feinberg School of Medicine for their assistance with performance of whole genome sequencing.
This work was supported by grants from the Thrasher Research Fund [Early Career Award number 11854 to L.K.K.], the National Institute of Allergy and Infectious Diseases at the National Institutes of Health [K23 AI123525 to L.K.K.], and the American Cancer Society [MRSG-13-220-01 to E.A.O.]. Research reported in this publication was supported, in part, by the National Institutes of Health's National Center for Advancing Translational Sciences, Grant Number UL1TR001422. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Conflicts of Interest
L.K.K. is a scientific advisor for Actelion, has received research supplies from Alere, and received research grants from Merck and Cubist. D.N.G. holds patents for the prevention of Clostridium difficile infection; is a consultant for Sanofi Pasteur, DaVolterra, MGB, and Pfizer; and advisory board member of Merck, Rebiotix, Summit, and Actelion. A.R.H, D.W.H., and E.A.O. report no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kelly CP, Lamont JT. Clostridium difficile- More difficult than ever. N Engl J Med. 2008;359:1932–40. doi: 10.1056/NEJMra0707500. [DOI] [PubMed] [Google Scholar]
- 2.Freeman J, Bauer MP, Baines SD, Corver J, Fawley WN, Goorhuis B, et al. The changing epidemiology of Clostridium difficile infections. Clin Microbiol Rev. 2010;23:529–49. doi: 10.1128/CMR.00082-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lim SK, Stuart RL, Mackin KE, Carter GP, Kotsanas D, Francis MJ, et al. Emergence of a ribotype 244 strain of Clostridium difficile associated with severe disease and related to the epidemic ribotype 027 strain. Clin Infect Dis. 2014;58:1723–30. doi: 10.1093/cid/ciu203. [DOI] [PubMed] [Google Scholar]
- 4.He M, Miyajima F, Roberts P, Ellison L, Pickard DJ, Martin MJ, et al. Emergence and global spread of epidemic healthcare-associated Clostridium difficile. Nat Genet. 2013;45:109–13. doi: 10.1038/ng.2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.United States Centers for Disease Control and Prevention. Antibiotic resistance threats in the United States. 2013 http://www.cdc.gov/drugresistance/pdf/ar-threats-2013-508.pdf.
- 6.Paulick A, Karlsson M, Albreght V, Granade M, Guh A, et al. EIP CDI Pathogen Group. The Role of Ribotype 106 as a Cause of Clostridium difficile Infection in the United States 2012–2014; Abstr 13th Biennial Congress of the Anaerobe Society of the Americas, abstr PIII-7; 2016. http://www.anaerobe.org/2016/2016abBook.pdf. [Google Scholar]
- 7.Karlsson M, Paulick A, Albreght V, Granade M, Guh A, Rasheed JK, et al. EIP CDI Pathogen Group. Molecular Epidemiology of Clostridium difficile Isolated in the United States, 2014; Abstr 13th Biennial Congress of the Anaerobe Society of the Americas, abstr PIII-4; 2016. http://www.anaerobe.org/2016/2016abBook.pdf. [Google Scholar]
- 8.Tenover FC, Akerlund T, Gerding DN, Goering RV, Bostrom T, Jonsson AM, et al. Comparison of strain typing results for Clostridium difficile isolates from North America. J Clin Microbiol. 2011;49:1831–7. doi: 10.1128/JCM.02446-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilcox MH, Shetty N, Fawley WN, Shemko M, Coen P, Birtles A, et al. Changing Epidemiology of Clostridium difficile infection following the introduction of a national ribotyping-based surveillance scheme in England. Clin Infect Dis. 2012;55:1056–63. doi: 10.1093/cid/cis614. [DOI] [PubMed] [Google Scholar]
- 10.Public Health England. Clostridium difficile Ribotyping Network (CDRN) for England and Northern Ireland, Biennial report (2013–2015) https://www.gov.uk/government/uploads/system/uploads/attachment_data/file/491253/CDRN_2013-15_Report.pdf.
- 11.Kociolek LK, Patel SJ, Shulman ST, Gerding DN. Molecular epidemiology of Clostridium difficile infections in children: A retrospective cohort study. Infect Control Hosp Epidemiol. 2015;36:445–51. doi: 10.1017/ice.2014.89. [DOI] [PubMed] [Google Scholar]
- 12.Wilson KH, Kennedy MJ, Fekety FR. Use of sodium taurocholate to enhance spore recovery on a medium selective for Clostridium difficile. J Clin Microbiol. 1982;15:443–6. doi: 10.1128/jcm.15.3.443-446.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clabots CR, Johnson S, Bettin KM, Mathie PA, Mulligan ME, Schaberg DR, et al. Development of a rapid and efficient restriction endonuclease analysis typing system for Clostridium difficile and correlation with other typing systems. J Clin Microbiol. 1993;31:1870–5. doi: 10.1128/jcm.31.7.1870-1875.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kociolek LK, Gerding DN, Osmolski JR, Patel SJ, Snydman DR, McDermott LA, et al. Differences in the molecular epidemiology and antibiotic susceptibility of Clostridium difficile isolates in pediatric and adult patients. Antimicrob Agents Chemother. 2016;60:4896–900. doi: 10.1128/AAC.00714-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clinical and Laboratory Standards Institute. Methods for antimicrobial susceptibility testing of anaerobic bacteria; Approved Standard. 8. Wayne, PA: Clinical and Laboratory Standards Institute; 2012. [Google Scholar]
- 16.Clinical and Laboratory Standards Institute. Performance standards for antimicrobial susceptibility testing; Twenty-fifth Informational Supplement. Wayne, PA: Clinical and Laboratory Standards Institute; 2015. [Google Scholar]
- 17.European Committee on Antimicrobial Susceptibility Testing. Breakpoint tables for interpretation of MICs and zone diameters. http://www.eucast.org/fileadmin/src/media/PDFs/EUCAST_files/Breakpoint_tables/v_6.0_Breakpoint_table.pdf.
- 18.Tenover FC, Tickler IA, Persing DH. Antimicrobial-resistant strains of Clostridium difficile from North America. Antimicrob Agents Chemother. 2012;56:2929–32. doi: 10.1128/AAC.00220-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19:455–77. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–9. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 21.Griffiths D, Fawley W, Kachrimanidou M, Bowden R, Crook DW, Fung R, et al. Multilocus sequence typing of Clostridium difficile. J Clin Microbiol. 2010;48:770–8. doi: 10.1128/JCM.01796-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morgulis A, Gertz EM, Schaffer AA, Agarwala R. A fast and symmetric DUST implementation to mask low-complexity DNA sequences. J Comput Biol. 2006;13:1028–40. doi: 10.1089/cmb.2006.13.1028. [DOI] [PubMed] [Google Scholar]
- 24.Croucher NJ, Page AJ, Connor TR, Delaney AJ, Keane JA, Bentley SD, et al. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 2015;43:e15. doi: 10.1093/nar/gku1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.He Z, Zhang H, Gao S, Lercher MJ, Chen WH, Hu S. Evolview v2: an online visualization and management tool for customized and annotated phylogenetic trees. Nucleic Acids Res. 2016;44:W236–41. doi: 10.1093/nar/gkw370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Letunic I, Bork P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016;44(W1):W242–5. doi: 10.1093/nar/gkw290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Persson S, Torpdahl M, Olsen KE. New multiplex PCR method for the detection of Clostridium difficile toxin A (tcdA) and toxin B (tcdB) and the binary toxin (cdtA/cdtB) genes applied to a Danish strain collection. Clin Microbiol Infect. 2008;14:1057–64. doi: 10.1111/j.1469-0691.2008.02092.x. [DOI] [PubMed] [Google Scholar]
- 28.Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11:863–74. doi: 10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zankari E, Hasman H, Cosentino S, Vestergaard M, Rasmussen S, Lund O, et al. Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother. 2012;67:2640–4. doi: 10.1093/jac/dks261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ozer EA, Allen JP, Hauser AR. Characterization of the core and accessory genomes of Pseudomonas aeruginosa using bioinformatic tools Spine and AGEnt. BMC Genomics. 2014;15:737–54. doi: 10.1186/1471-2164-15-737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou Y, Burnham CA, Hink T, Chen L, Shaikh N, Wollam A, et al. Phenotypic and genotypic analysis of Clostridium difficile isolates: a single center study. J Clin Microbiol. 2014;52:4260–6. doi: 10.1128/JCM.02115-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kurka H, Ehrenreich A, Ludwig W, Monot M, Rupnik M, Barbut F, et al. Sequence similarity of Clostridium difficile strains by analysis of conserved genes and genome content is reflected by their ribotype affiliation. PloS One. 2014;9:e86535. doi: 10.1371/journal.pone.0086535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shapiro BJ, Friedman J, Cordero OX, Preheim SP, Timberlake SC, Szabo G, et al. Population genomics of early events in the ecological differentiation of bacteria. Science. 2012;336:48–51. doi: 10.1126/science.1218198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Knight DR, Elliott B, Chang BJ, Perkins TT, Riley TV. Diversity and evolution in the genome of Clostridium difficile. Clin Microbiol Rev. 2015;28:721–41. doi: 10.1128/CMR.00127-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pecavar V, Blaschitz M, Hufnagl P, Zeinzinger J, Fiedler A, Allerberger F, et al. High-resolution melting analysis of the single nucleotide polymorphism hot-spot region in the rpoB gene as an indicator of reduced susceptibility to rifaximin in Clostridium difficile. J Med Microbiol. 2012;61:780–5. doi: 10.1099/jmm.0.041087-0. [DOI] [PubMed] [Google Scholar]
- 36.Sundram F, Guyot A, Carboo I, Green S, Lilaonitkul M, Scourfield A. Clostridium difficile ribotypes 027 and 106: clinical outcomes and risk factors. J Hosp Infect. 2009;72:111–8. doi: 10.1016/j.jhin.2009.02.020. [DOI] [PubMed] [Google Scholar]
- 37.Solomon K, Fanning S, McDermott S, Murray S, Scott L, Martin A, et al. PCR ribotype prevalence and molecular basis of macrolide-lincosamide-streptogramin B (MLSB) and fluoroquinolone resistance in Irish clinical Clostridium difficile isolates. J Antimicrob Chemother. 2011;66:1976–82. doi: 10.1093/jac/dkr275. [DOI] [PubMed] [Google Scholar]
- 38.Mutlu E, Wroe AJ, Sanchez-Hurtado K, Brazier JS, Poxton IR. Molecular characterization and antimicrobial susceptibility patterns of Clostridium difficile strains isolated from hospitals in south-east Scotland. J Medical Microbiol. 2007;56:921–9. doi: 10.1099/jmm.0.47176-0. [DOI] [PubMed] [Google Scholar]
- 39.Dai D, Du A, Xiong K, Pu T, Zhou X, Deng Z, et al. DNA phosphorothioate modification plays a role in peroxides resistance in Streptomyces lividans. Front Microbiol. 2016;7:1380. doi: 10.3389/fmicb.2016.01380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu T, Yao F, Zhou X, Deng Z, You D. A novel host-specific restriction system associated with DNA backbone S-modification in Salmonella. Nucleic Acids Res. 2010;38:7133–41. doi: 10.1093/nar/gkq610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eichenberger P, Jensen ST, Conlon EM, van Ooij C, Silvaggi J, Gonzalez-Pastor JE, et al. The SigmaE regulon and the identification of additional sporulation genes in Bacillus subtilis. J Mol Biol. 2003;327:945–72. doi: 10.1016/s0022-2836(03)00205-5. [DOI] [PubMed] [Google Scholar]
- 42.Kirk JA, Banerji O, Fagan RP. Characteristics of the Clostridium difficile cell envelope and its importance in therapeutics. Microb Biotechnol. 2016;10:76–90. doi: 10.1111/1751-7915.12372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Merrigan MM, Venugopal A, Roxas JL, Anwar F, Mallozzi MJ, Roxas BA, et al. Surface-layer protein A (SlpA) is a major contributor to host-cell adherence of Clostridium difficile. PloS One. 2013;8:e78404. doi: 10.1371/journal.pone.0078404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tulli L, Marchi S, Petracca R, Shaw HA, Fairweather NF, Scarselli M, et al. CbpA: a novel surface exposed adhesin of Clostridium difficile targeting human collagen. Cell Microbiol. 2013;15:1674–87. doi: 10.1111/cmi.12139. [DOI] [PubMed] [Google Scholar]
- 45.Hendrickx AP, Willems RJ, Bonten MJ, van Schaik W. LPxTG surface proteins of enterococci. Trends Microbiol. 2009;17:423–30. doi: 10.1016/j.tim.2009.06.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.