

Abstract
The Fanconi anemia pathway is an important coordinator of DNA repair pathways and is particularly relevant to repair of DNA inter-strand crosslinks. Central to the pathway is monoubiquitination of FANCD2, requiring the function of multiple proteins in an upstream Fanconi core complex. We present development and analytical characterization of a novel assay for quantification of unmodified and monoubiquitinated FANCD2 proteoforms, based on peptide immunoaffinity enrichment and targeted multiple reaction monitoring mass spectrometry (immuno-MRM). The immuno-MRM assay is analytically characterized using fit-for-purpose method validation. The assay linear range is >3 orders of magnitude with total repeatability <16% CV. In proof-of-principle experiments, we demonstrate application of the multiplex assay by quantifying the FANCD2 proteoforms following mitomycin-c treatment in an isogenic pair of FancA-corrected and uncorrected cell lines, as well as primary peripheral blood mononuclear cells from Fanconi Anemia patients. Additionally, we demonstrate detection of endogenous FANCD2 monoubiquitination in human breast cancer tissue. The immuno-MRM assay provides a potential functional diagnostic for patients with Fanconi Anemia with defects in the upstream FA complex or FANCD2, and a potential test for predicting sensitivity to DNA cross-linking agents in human cancers.
Keywords: Fanconi anemia, multiple reaction monitoring, DNA damage response, immuno-MRM
Graphical Abstract
1. Introduction
Fanconi anemia (FA) is an inherited disorder characterized by progressive bone marrow failure and an increased risk of cancers, especially during early adulthood. FA patients harbor mutations in a set of genes encoding the FA pathway, a network of 21 proteins that is specialized for repairing DNA inter-strand cross-links [1]. Activation of the FA core complex results in the monoubiquitination of FANCD2 and FANCI, which interact with several downstream components (e.g. BRCA1, Rad51, BRCA2) for DNA repair [2]. Cells with mutations in any of the core complex proteins lack the ability to monoubiquitinate FANCD2, making FANCD2 ubiquitination a convergence point in the pathway, with an estimation of >90% FA patients defective in this step [1,3]. FA patients experience excessive toxicities with chemotherapy or radiation, so early diagnosis is essential to inform therapeutic decisions [4]. Additionally, somatic mutations In FA genes render tumor cells sensitive to DNA crosslinking agents, so identification of FA pathway defects provides an opportunity for therapeutic targeting [5–7].
Diagnosis of FA based on presenting symptoms alone is difficult and unreliable due to the broad phenotypic spectrum of FA and overlapping features with other genetic (and nongenetic) diseases. A significant subset of FA patients lack characteristic physical findings [8]. Currently, the standard for FA diagnostic testing is manual enumeration of chromosomal breakage following exposure to clastogens [9,10]. In this test, T lymphocytes in peripheral blood are stimulated with phytohaemagglutinin (PHA) to proliferate in the presence or absence of DNA cross-linking agents, typically mitomycin C (MMC) or diepoxybutane (DEB), followed by counting chromosomal breaks and fusions in metaphase spreads. Chromosomal breakage may also be assessed in skin fibroblasts, since somatic mosaicism may result in a falsely negative blood test. The approach is laborious and difficult to standardize, partly because results are dependent on several variables (e.g. culture time, exposure time, concentration, threshold for positive/negative). Molecular subtyping by DNA sequencing or complementation analysis is increasingly utilized clinically, though interpretation is complicated by variants of unknown significance [11], ascertainment of biallelic mutation distribution, and distinguishing constitutional versus somatic mutations [12,13].
Since pathogenic mutations in FA genes functioning upstream of the FANCD2 complex impair FANCD2 monoubiquitination, an assay to directly quantify the level of monoubiquitinated FANCD2, and thus determine defects in the pathway, would provide a complementary functional readout to existing assays. However, testing of FANCD2 monoubiquitination with traditional immunoassays or intracellular localization assays are low throughput and semi-quantitative [3,14], establishing a need for a more effective quantitative assay.
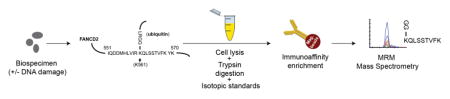
Targeted, multiple reaction monitoring mass spectrometry (MRM) is a quantitative approach capable of quantifying analyte peptides with high sensitivity and specificity [15,16]. MRM has been used in clinical laboratories for decades to measure small molecules [17], and is increasingly applied in protein and peptide measurement [18–21]. The targeted MRM mode of proteomic analysis is different from untargeted (“shotgun”) discovery proteomics, where methods are designed to profile as many peptides as possible by matching MS/MS spectra to protein databases. In contrast, for MRM, protein lysates from biospecimens are proteolyzed (typically with trypsin), and proteotypic peptide sequences unique to the protein of interest (“proteotypic peptides”) are targeted for detection by MRM mass spectrometry (Figure 1A). Instrument and method parameters are optimized to enable the most sensitive and specific measurement of the targeted peptide as a surrogate for the protein of interest. Furthermore, a synthetic, HPLC-purified, stable isotope-labeled version of the analyte peptide is spiked into the biospecimen at a known concentration as an internal standard. The internal standard peptide serves several functions, including: i) to control for much of the preanalytical and all of the analytical variation in the assay, ii) to enable precise, relative quantification of the endogenous analyte, and iii) to help confirm specificity of the assay. The high specificity of MRM-based assays is based on detection of multiple MRM transitions (i.e. combinations of precursor/fragment ions), the chromatographic co-elution of analyte and internal standard, and the consistency of the relative transition ratios between the endogenous and standard peptides. In addition to being highly specific, MRM is standardizable and transferrable across laboratories [22–25], is capable of highly multiplexed measurements [25,26], and is capable of quantifying cell signaling events through chromatographic enrichment of low abundance analytes (e.g., ubiquitinated or phosphorylated peptides) [27–30]. A public repository of highly characterized targeted MS assays has been created [31], and the assay platform is beginning to gain acceptance in clinical laboratories [32,33]. For example, an MRM-based assay targeting a single proteotypic peptide sequence unique to the human thyroglobulin protein is used by clinical laboratories as a surrogate to quantify thyroglobulin (2768 amino acids, ~300kDa) in patients that make autoantibodies that interfere with conventional immunoassays [34,35]. Other examples include similar assays developed for apolipoproteins [36] and troponin [37].
Fig. 1. Development and characterization of the multiplex assay.

(A) Description of the assay workflow: A variety of sample types can be analyzed by MRM, with previous applications demonstrated in plasma, serum, cell line lysates, and tissue specimens. Proteins are extracted from the biological sample using standard procedures amenable to downstream mass spectrometry. The protein lysates are enzymatically digested, typically using trypsin, and an HPLC-purified, stable isotope-labeled standard (SIS) peptide corresponding to each analyte is spiked-in at a known concentration to act as an internal standard. Although each pair of spiked-in SIS peptide and its endogenous analyte peptide share the same amino acid sequence (and hence the same chemical and physical properties), they are separately detected by the mass spectrometer due to the m/z difference introduced by the stable isotope label. The analyte peptides, along with their stable-isotope analogs, are enriched from the complex sample using anti-peptide antibodies. Measurement of the enriched analytes and standards is performed using quantitative mass spectrometry in a technique called multiple reaction monitoring (MRM). MRM focuses the analytical capability of the instrument on the specific analyte of interest by using a combination of mass filters in a ‘transition’ (i.e. a specific combination of precursor and fragment ions). Transitions can be cycled through sequentially to allow multiplexing. Specificity is confirmed through orthogonal biophysical properties of the analyte peptides: detection of multiple m/z transitions per peptide, chromatographic co-elution (i.e. identical retention time) of the analyte and internal standard, and identical relative abundance of transitions in the analyte and internal standard. The peak area ratio (light endogenous analyte peptide to heavy SIS peptide) is used for precise, relative quantification. Synthetic peptide standards allow the assays to be highly standardized across laboratories. (B) Design of the assay for the unmodified and monoubiquitinated FANCD2 proteoforms. Ubiquitination of FANCD2 at lysine 561 (depicted in the uppermost figure) blocks the lysine from proteolysis during trypsin digestion. This results in formation of the peptide sequence from amino acids 561–569 containing a –GG remnant on the modified lysine. Peptides containing the modified remnant and the unmodified tryptic peptide were selected for assay development. (C) Characterization of the immuno-MRM assay by response curves. Curves are plotted on linear and log(base 10) scales. Error bars are the standard deviation of technical triplicates.
MRM-based assays have several advantages over conventional protein measurement technologies (e.g., Western blotting, immunohistochemistry, ELISA), such as direct detection of the analyte by a mass spectrometer (contributing to high specificity), the use of internal standards (enabling reproducibility and inter-laboratory standardization), the capability to readily multiplex analysis of many target peptides, a large linear range (typically ≥103), and relatively less time and cost required for assay development [38]. In MRM, cross reactivity of antibodies usually does not affect specificity, because the mass spectrometer directly detects the target of interest, and interferences are detected based on analytical criteria [39]. In contrast, Western blots (WB) can feature multiple nonspecific bands and uncertainties about which band represents the analyte of interest. While WB is semi-quantitative at best and has not been standardized across laboratories, MRM uses HPLC-purified, synthetic, internal stable isotope-labeled standard peptides (spiked into the biospecimen at a known concentration) for standardization across laboratories [22–24].
We recently demonstrated the utility of peptide immuno-MRM (coupling immuno-affinity enrichment of peptides to MRM) to quantify the pharmacodynamics of post-translationally modified (e.g. phosphorylated and ubiquitinated) peptides in response to DNA damage [28,30]. Here, we present a multiplex immuno-MRM assay for quantifying the unmodified and monoubiquitinated proteoforms of a proteotypic peptide unique to FANCD2 (K561). The assay is analytically characterized using fit-for-purpose method validation, including linearity, repeatability, and stability. Proof-of-principle application of the assay is demonstrated using human immortalized cell lines (congenic FancA+/−) and Fanconi Anemia patient primary human T cells treated with the DNA cross-linking agent MMC. We also demonstrate detection of monoubiquitinated FANCD2 in breast tumor tissue. The assay can be multiplexed with other immuno-MRM assays [28,30] to profile more comprehensively the DNA damage response in a variety of sample types and conditions, making it useful for a range of biomedical research studies.
2. Materials and Methods
See supplementary material for full materials and methods.
3. Results and discussion
3.1 Method development and parameter optimization
An overview of the workflow for the immuno-MRM assay is shown in Figure 1A. Development of the assay requires identification of a proteotypic peptide sequence (i.e. unique to the protein of interest) for measurement, generation of affinity reagents for enrichment of the targeted peptide, and optimization of analytical parameters for detection of the endogenous and synthetic peptide standards. To develop an assay, we first identified the target peptide analyte as the tryptic fragment surrounding the K561 site of ubiquitination on FANCD2. A blast search confirmed that this sequence is unique to the FANCD2 protein. Ubiquitination covalently attaches the ubiquityl moiety to the lysine at position 561, blocking the site from enzymatic digestion. Additionally, there is a trypsin cleavage site at the lysine residing on the ubiquityl moiety. Thus, following trypsin digestion, the modified protein produces a peptide encompassing amino acids 561–569 containing a –GG remnant on K561, and the unmodified protein produces a fully tryptic peptide of amino acids 562–569 (see Figure 1B). We sought to develop an immuno-MRM assay capable of measuring both forms of the peptide (monoubiquitinated and unmodified).
We followed established protocols to generate affinity reagents for use in peptide immunoaffinity enrichment [38,40]. The modified peptide containing the ubiquitin (i.e. –GG) remnant was used as the immunogen sequence in generating an anti-peptide rabbit monoclonal antibody. Using established protocols [40], monoclonal supernatants were screened against both forms of the peptide (modified and unmodified) to obtain a clone that was capable of immuno-precipitating both forms. Purified antibodies were subjected to quality control by BCA assay for protein concentration, SDS gel to confirm intact antibody, and recovery efficiency in immuno-MRM enrichments to confirm antibody activity (data not shown).
Synthetic analyte peptides (i.e., HPLC-purified, heavy stable isotope-labeled versions of the modified and unmodified tryptic peptides) were used to optimize the mass spectrometer parameters [41]. Tandem mass spectrometry was used to analyze the HPLC-purified synthetic peptides to confirm the peptide identity and select the best combinations of precursor and fragment ions (i.e. transitions) to be used for identification and quantification of the peptide during MRM. Following best practices [16,42], the top 4–6 transitions were identified for testing in preliminary response curves in a background matrix of pooled lysates prepared from LCLs +/− 10 Gy ionizing radiation. For the final assay, we selected the 3 best transitions that were associated with the highest intensity and free from background noise and interferences. Collision energy was optimized for each transition to produce the most complete fragmentation (i.e. the highest intensities) [41]. Optimized transition ions and collision energies are reported in Supplementary Table 1. Consistent with best practices [39,43], the specificity for detection of the analyte was confirmed in all runs by detection of multiple transitions, co-elution of the analyte peptide and internal standard, and identical relative abundances of their transition ions.
3.2 Fit-for-purpose characterization: Response curves
The immuno-MRM assay targeting the FANCD2 analyte peptides was characterized using fit-for-purpose method validation to determine the assay performance and analytical figures of merit [21,44,45]. Response curves were generated in a background matrix consisting of an equal mixture of four components: LCL GM07057 + 10 Gy ionizing radiation (IR) (1 hr), LCL GM07057 + mock IR (1 hr), LCL GM01526 + 10 Gy IR (1 hr), LCL GM01526 + mock IR (1 hr) The curves were used to characterize the linear range, lower limits of quantification (LLOQ), and upper limits of quantification (ULOQ). All points were analyzed in triplicate with nine replicates for the blank samples. The linear range was > 3 orders of magnitude for both peptides (Figure 1C), with LLOQs of 1.4 and 0.7 fmol/mg for the ubiquitinated and unmodified peptides, respectively. Curves were linear at the highest concentration point measured, thus ULOQs were a minimum estimate of > 4000 fmol/mg, as were the linear ranges. Assay performance in the response curves is reported in Supplementary Table 2, and data are provided in Supplementary Table 3.
3.3 Fit-for-purpose characterization: Repeatability
Repeatability experiments were used to determine the expected intra-assay (within day) and inter-assay (between day) repeatability. Heavy peptides were spiked into 500 μg aliquots of pooled protein lysates from LCLs (GM07057/GM01526) +/− ionizing radiation (same pool of lysates used in response curves) at three concentrations (1.5, 150, 1500 fmol/mg) with constant concentration of light peptide (200 fmol/mg). Complete process triplicates (including tryptic digestion) were prepared and analyzed on five independent days. For the ubiquitinated peptide assay, the mean intra-assay variability was 6%, 7%, and 4% CV, and the mean inter-assay variability was 17%, 7%, and 10% for the low, medium, and high concentration samples, respectively. For the unmodified peptide assay, the mean intra-assay variability was 8%, 8%, and 7% CV, and the mean inter-assay variability was 8%, 9%, and 6% for the low, medium, and high concentration samples, respectively. Repeatability performance is reported in Supplementary Table 2 and data are provided in Supplementary Table 3.
3.4 Fit-for-purpose characterization: Peptide stability
Peptide stability in the processed samples was evaluated to determine safe handling conditions of the samples. Heavy peptides were spiked into 500 μg aliquots of pooled protein lysates from LCLs (GM07057/GM01526) +/− IR (same pool of lysates used in response curves) at 150 fmol/mg, and light peptides were added at 200 fmol/mg. Aliquots of the sample were enriched in duplicate. Control samples were analyzed immediately, and test samples were stored at 4°C in the autosampler for approximately 6 hours and 24 hours, with additional aliquots subjected to 1 freeze-thaw, 2 freeze-thaws, and storage at −80°C for approximately five months. Variation and percent difference relative to the fresh sample were evaluated (Supplementary Table 2 and data in Supplementary Table 3). All points fell within the expected range except for the ubiquitinated peptide in the sample with two freeze-thaws (35% CV and percent difference 24%). These results demonstrate that the samples are sufficiently preserved under all conditions up to a single freeze-thaw.
3.5 Repeatability of endogenous analyte detection
The final assay was validated by measuring the amount of endogenous analyte expression in aliquots of a common sample prepared and analyzed over multiple days. Individual aliquots of 500 μg of protein lysate from pooled LCLs (GM07057/GM01526) +/− IR were analyzed over five days. For the ubiquitinated assay, the mean intra-assay and inter-assay variability was 13% and 10% CV. For the unmodified peptide, the mean intra-assay and inter-assay variability was 8% and 11% CV. The total assay variability was calculated by taking the square root of the sum of squares for the intra- and inter-assay variability. Total assay variability was 16% and 13% CV for detection of the endogenous ubiquitinated and unmodified analyte peptides, respectively. Supplementary Table 2 shows the repeatability for measurement of endogenous analytes and data are provided in Supplementary Table 3.
3.6 Quantification of FANCD2 proteoforms in immortalized human cell lines
To demonstrate the utility of the assay in determining defects in the FA pathway, we measured the ubiquitination of FANCD2 in response to DNA damage in human cells. As described previously [46], an isogenic uncorrected/corrected pair of lymphoblast cell lines was prepared from the FANCA-deficient HSC72 cell line, by introducing a wild type FANCA cDNA. We compared the ability of HSC72 and FANCA-corrected HSC72 cells to ubiquitinate FANCD2 in response to the DNA crosslinking agent mitomycin-C (MMC). An additional wildtype lymphoblast cell line, GM07057, was included as a positive control. Cells were harvested following 2 hours and 24 hours of incubation in MMC, and mock-treated negative controls were harvested at 2 hours. Protein lysates were generated, and 500 μg aliquots were analyzed using the multiplexed immuno-MRM assay targeting monoubiquitinated and unmodified FANCD2 peptides. Experiments were performed in biological triplicate and analyzed in a blinded fashion (data are provided in Supplementary Table 4).
As shown in Figure 2A (upper panel), all three cell lines express FANCD2 (using results from the non-modified QSLSSTVFK peptide). The GM07057 (wildtype) and FANCA-corrected HSC72 cells express FANCD2 ubK561 at baseline and show an increase in the amount of FANCD2 ubK561 at the 2 and 24 hour time-points compared to the mock-treated cells. However, no FANCD2 ubK561 is detected in the FANCA-deficient HSC72 cells (Figure 2A, lower panel), due to disruption of the Fanconi Core complex [3,47]. These data demonstrate the utility of the assay to detect defects in the Fanconi Core complex in immortalized cells.
Fig. 2.

Quantification of total and monoubiquitinated FANCD2 in human-derived cell lines and primary human cells and tissue. (A) Concentration of monoubiquitinated FANCD2 peptide and unmodified FANCD2 peptide in human LCLs treated with MMC. Error bars are the standard deviation of biological triplicates or the expected error based on the characterized repeatability of the assay. ‘ND’ is not detected (i.e. below the lower limit of quantification). The FA patient complementation group assignment, where known, is indicated in parentheses. (B) Concentration of monoubiquitinated FANCD2 peptide and unmodified FANCD2 peptide in primary human PBMCs treated with MMC. Error bars are the expected error based on the characterized total CV of the assays. ‘ND’ is not detected (i.e. below the lower limit of quantification). (C) MRM transition ion chromatograms for light (endogenous) and heavy (internal standard) peptides detected in human breast tumor tissue. Specificity is confirmed by detection of multiple transitions from each of the endogenous and heavy-labeled standard peptides, as well as equivalent retention times and relative intensities of transitions for light and heavy peptides.
3.6 Quantifiation of ubiquitinated FANCD2 in primary human cells
To determine if the assay could quantify defects in the DNA damage response in primary human cells, we isolated and expanded peripheral blood mononuclear cells (PBMCs) from three FA patients and a healthy control. PBMCs were exposed to MMC for a period of 2 hours and 24 hours. Negative controls were mock-treated. Protein lysates were proteolyzed with trypsin, and 500 μg aliquots were analyzed by the multiplex immuno-MRM assay.
As shown in Figure 2B (upper panel), FANCD2 protein is expressed at much higher levels in the control and FA-A patients compared to the FA-D2 patient, as has been described [3]. Additionally, the assay confirms induction of FANCD2 ubK561 in the control PBMC, while no FANCD2 ubK561 signal above the limits of detection was observed in the FA patient cells (Figure 2B, lower panel). Genomic analysis confirms that patient 1 harbored biallelic mutations in FANCA, while patient 3 had mutations in the FANCD2 gene. Patient 2 was diagnosed with FA by chromosomal breakage, but had an undetermined subtype (as mentioned above, molecular subtyping by DNA sequencing is complicated by variants of unknown significance [11], ascertainment of biallelic mutation distribution, and distinguishing constitutional versus somatic mutations [12,13]). Patient 2 is an example where the immuno-MRM assay provides a complementary method of assessing a molecular pathway to inform diagnosis.
Finally, we tested the feasibility of quantifying the monoubiquitinated and unmodified FANCD2 peptides in human tumor tissue (Figure 2C). An aliquot of 500 μg of protein lysate from a frozen human breast cancer biospecimen was analyzed using the immuno-MRM assay. The endogenous expression of FANCD2 and the monoubiquitinated FANCD2 were detected in the cancer sample, showing the utility for application of the assay in tissue biopsies. Given the signal-to-noise ratio in the specimen tested, we estimate the amount of input material could be decreased as much as five-fold and still maintain detection of the modification, compatible with analysis of core needle biopsies.
4. Conclusion
We describe a novel, multiplexed quantitative assay for unmodified and monoubiquitinated proteoforms of FANCD2. The assay is highly reproducible, provides good sensitivity, and can be run at moderate throughput. The specificity of the assay is confirmed based on an abundance of evidence, including nine criteria that exceed widely recognized best practices for targeted peptide measurements [39,44]: (i) The sequence (QLSSTVFK) is unique to the FANCD2 protein, based on a blast search of the uniprot human protein database; (ii) The proteotypic peptide analyte QLSSTVFK (±monoubiquitination) and its synthetic, stable isotope-labeled internal standards co-elute (i.e. have the same retention time and peak shape) during the liquid chromatography separation; (iii) Three independent transitions are detected for each peptide, consistent with best practices [39,43]; (iv) The transitions from the analyte peptides and internal standards have the same relative abundances, again consistent with best practices [39,43]; (v) The linear response of the assay follows expected behavior using spiked analytes and standards; (vi) The unmodified FANCD2 analyte peptide is not detected in the biological samples (i.e., Fanconi patient PBMCs) known to lack functional FANCD2 protein (Figure 2B); (vii) The monoubiquitinated FANCD2 analyte peptide is not detected in the biological samples (lymphoblast cell lines, patient PBMCs) known to lack expression of the FANCD2 or FANCA proteins (Figure 2A–B); (viii) MRM-based detection of the monoubiquitinated FANCD2 analyte peptide is restored in the FANCA-deficient cell line when the wild type FANCA gene is added back to the cells (Figure 2A); (ix) The quantity of the monoubiquitinated proteoform detected by the assay is increased following treatment of cells with a DNA cross-linking agent (MMC) (Figure 2A–B), consistent with known biology. The assay is currently configured to perform precise, relative quantification of a surrogate peptide for the FANCD2 protein (in both its unmodified and ubiquitinated forms). Because several factors can affect peptide recovery during sample processing (e.g. trypsin digestion efficiency and peptide solubility), the accuracy of the assay for quantifying the full length protein could be improved by incorporating protein standards and potentially incorporating multiple representative peptides for the unmodified protein.
The assay can be further multiplexed with other immuno-MRM assays to build upon a panel of analytes capable of measuring the activation of the DNA damage response [28,30]. Thus, the immuno-MRM assay provides a potential functional diagnostic for patients with Fanconi Anemia with defects in the upstream FA complex or FANCD2, and a potential test for predicting sensitivity to DNA cross-linking agents in human cancers. This test provides a useful functional analysis of variants of unknown clinical significance in FA genes upstream of and including FANCD2 by quantitatively assessing FANCD2 monoubiquitination and FANCD2 levels. Clinical diagnosis is critical to inform clinical management, choice of therapy, and family counseling. Additionally, this assay identifies somatic acquired defects in the Fanconi anemia pathway, a functional vulnerability that could inform therapeutic targeting using DNA cross-linking agents (e.g. platinum compounds) in oncology. Since this is a functional assay, mechanisms that would be missed by genetic sequencing, such as epigenetic silencing of FA genes in cancer cells, would be flagged by this method [5].
Supplementary Material
Highlights.
Ubiquitination of FANCD2 is central to the Fanconi Anemia DNA repair pathway.
We developed and analytically validated a novel multiplex immuno-MRM assay to quantify unmodified and monoubiquitinated FANCD2 proteoforms.
We demonstrate detection and quantification of signaling deficiencies in Fanconi Anemia cell lines and primary cells, as well as quantification in human breast tumor tissue.
Acknowledgments
Funding
Research was supported by the National Cancer Institute (U24CA160034, U01CA214114, and R50CA211499) and the National Heart Lung and Blood Institute (P01HL048546) of the National Institutes of Health (NIH). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Abbreviations
- FA
Fanconi anemia
- MRM
multiple reaction monitoring
- PTMs
post-translational modifications
- MS
mass spectrometry
- DDR
DNA damage response
- MMC
mitomycin-C
- LCL
lymphoblast cell line
- WB
Western blot
- VUS
variants of undetermined significance
- IR
ionizing radiation
Footnotes
Conflict of Interest statement
A.G.P. is the founder of Precision Assays.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mamrak NE, Shimamura A, Howlett NG. Recent discoveries in the molecular pathogenesis of the inherited bone marrow failure syndrome Fanconi anemia. Blood Rev. 2017;31:93–99. doi: 10.1016/j.blre.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ceccaldi R, Sarangi P, D’Andrea AD. The Fanconi anaemia pathway: new players and new functions. Nat Rev Mol Cell Biol. 2016;17:337–349. doi: 10.1038/nrm.2016.48. [DOI] [PubMed] [Google Scholar]
- 3.Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M, D’Andrea AD. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell. 2001;7:249–262. doi: 10.1016/s1097-2765(01)00173-3. [DOI] [PubMed] [Google Scholar]
- 4.Ebens CL, MacMillan ML, Wagner JE. Hematopoietic cell transplantation in Fanconi anemia: current evidence, challenges and recommendations. Expert Rev Hematol. 2017;10:81–97. doi: 10.1080/17474086.2016.1268048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taniguchi T, Tischkowitz M, Ameziane N, Hodgson SV, Mathew CG, Joenje H, Mok SC, D’Andrea AD. Disruption of the Fanconi anemia-BRCA pathway in cisplatin-sensitive ovarian tumors. Nat Med. 2003;9:568–574. doi: 10.1038/nm852. [DOI] [PubMed] [Google Scholar]
- 6.Chirnomas D, Taniguchi T, de la Vega M, Vaidya AP, Vasserman M, Hartman AR, Kennedy R, Foster R, Mahoney J, Seiden MV, D’Andrea AD. Chemosensitization to cisplatin by inhibitors of the Fanconi anemia/BRCA pathway. Mol Cancer Ther. 2006;5:952–961. doi: 10.1158/1535-7163.MCT-05-0493. [DOI] [PubMed] [Google Scholar]
- 7.Jacquemont C, Simon JA, D’Andrea AD, Taniguchi T. Non-specific chemical inhibition of the Fanconi anemia pathway sensitizes cancer cells to cisplatin. Mol Cancer. 2012;11:26. doi: 10.1186/1476-4598-11-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Giampietro PF, Adler-Brecher B, Verlander PC, Pavlakis SG, Davis JG, Auerbach AD. The need for more accurate and timely diagnosis in Fanconi anemia: a report from the International Fanconi Anemia Registry. Pediatrics. 1993;91:1116–1120. [PubMed] [Google Scholar]
- 9.Oostra AB, Nieuwint AWM, Joenje H, de Winter JP. Diagnosis of fanconi anemia: chromosomal breakage analysis. Anemia. 2012;2012:238731. doi: 10.1155/2012/238731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Auerbach AD. Diagnosis of Fanconi anemia by diepoxybutane analysis. Curr Protoc Hum Genet. 2015;85:8.7.1–17. doi: 10.1002/0471142905.hg0807s85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Easton DF, Deffenbaugh AM, Pruss D, Frye C, Wenstrup RJ, Allen-Brady K, Tavtigian SV, Monteiro ANA, Iversen ES, Couch FJ, Goldgar DE. A systematic genetic assessment of 1,433 sequence variants of unknown clinical significance in the BRCA1 and BRCA2 breast cancer-predisposition genes. Am J Hum Genet. 2007;81:873–883. doi: 10.1086/521032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chandrasekharappa SC, Lach FP, Kimble DC, Kamat A, Teer JK, Donovan FX, Flynn E, Sen SK, Thongthip S, Sanborn E, Smogorzewska A, Auerbach AD, Ostrander EA. NISC Comparative Sequencing Program, Massively parallel sequencing, aCGH, and RNA-Seq technologies provide a comprehensive molecular diagnosis of Fanconi anemia. Blood. 2013;121:e138–148. doi: 10.1182/blood-2012-12-474585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang MY, Keel SB, Walsh T, Lee MK, Gulsuner S, Watts AC, Pritchard CC, Salipante SJ, Jeng MR, Hofmann I, Williams DA, Fleming MD, Abkowitz JL, King MC, Shimamura A. Genomic analysis of bone marrow failure and myelodysplastic syndromes reveals phenotypic and diagnostic complexity. Haematologica. 2015;100:42–48. doi: 10.3324/haematol.2014.113456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shimamura A, Montes de Oca R, Svenson JL, Haining N, Moreau LA, Nathan DG, D’Andrea AD. A novel diagnostic screen for defects in the Fanconi anemia pathway. Blood. 2002;100:4649–4654. doi: 10.1182/blood-2002-05-1399. [DOI] [PubMed] [Google Scholar]
- 15.Shi T, Song E, Nie S, Rodland KD, Liu T, Qian WJ, Smith RD. Advances in targeted proteomics and applications to biomedical research. Proteomics. 2016;16:2160–2182. doi: 10.1002/pmic.201500449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vidova V, Spacil Z. A review on mass spectrometry-based quantitative proteomics: Targeted and data independent acquisition. Anal Chim Acta. 2017;964:7–23. doi: 10.1016/j.aca.2017.01.059. [DOI] [PubMed] [Google Scholar]
- 17.Chace DH, Kalas TA. A biochemical perspective on the use of tandem mass spectrometry for newborn screening and clinical testing. Clin Biochem. 2005;38:296–309. doi: 10.1016/j.clinbiochem.2005.01.017. [DOI] [PubMed] [Google Scholar]
- 18.Whiteaker JR, Lin C, Kennedy J, Hou L, Trute M, Sokal I, Yan P, Schoenherr RM, Zhao L, Voytovich UJ, Kelly-Spratt KS, Krasnoselsky A, Gafken PR, Hogan JM, Jones LA, Wang P, Amon L, Chodosh LA, Nelson PS, McIntosh MW, Kemp CJ, Paulovich AG. A targeted proteomics-based pipeline for verification of biomarkers in plasma. Nat Biotechnol. 2011;29:625–634. doi: 10.1038/nbt.1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gillette MA, Carr SA. Quantitative analysis of peptides and proteins in biomedicine by targeted mass spectrometry. Nat Methods. 2013;10:28–34. doi: 10.1038/nmeth.2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Picotti P, Bodenmiller B, Aebersold R. Proteomics meets the scientific method. Nat Methods. 2013;10:24–27. doi: 10.1038/nmeth.2291. [DOI] [PubMed] [Google Scholar]
- 21.Song E, Gao Y, Wu C, Shi T, Nie S, Fillmore TL, Schepmoes AA, Gritsenko MA, Qian WJ, Smith RD, Rodland KD, Liu T. Targeted proteomic assays for quantitation of proteins identified by proteogenomic analysis of ovarian cancer. Sci Data. 2017;4:170091. doi: 10.1038/sdata.2017.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Addona TA, Abbatiello SE, Schilling B, Skates SJ, Mani DR, Bunk DM, Spiegelman CH, Zimmerman LJ, Ham AJ, Keshishian H, Hall SC, Allen S, Blackman RK, Borchers CH, Buck C, Cardasis HL, Cusack MP, Dodder NG, Gibson BW, Held JM, Hiltke T, Jackson A, Johansen EB, Kinsinger CR, Li J, Mesri M, Neubert TA, Niles RK, Pulsipher TC, Ransohoff D, Rodriguez H, Rudnick PA, Smith D, Tabb DL, Tegeler TJ, Variyath AM, Vega-Montoto LJ, Wahlander A, Waldemarson S, Wang M, Whiteaker JR, Zhao L, Anderson NL, Fisher SJ, Liebler DC, Paulovich AG, Regnier FE, Tempst P, Carr SA. Multi-site assessment of the precision and reproducibility of multiple reaction monitoring-based measurements of proteins in plasma. Nat Biotechnol. 2009;27:633–641. doi: 10.1038/nbt.1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prakash A, Rezai T, Krastins B, Sarracino D, Athanas M, Russo P, Zhang H, Tian Y, Li Y, Kulasingam V, Drabovich A, Smith CR, Batruch I, Oran PE, Fredolini C, Luchini A, Liotta L, Petricoin E, Diamandis EP, Chan DW, Nelson R, Lopez MF. Interlaboratory reproducibility of selective reaction monitoring assays using multiple upfront analyte enrichment strategies. J Proteome Res. 2012;11:3986–3995. doi: 10.1021/pr300014s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuhn E, Whiteaker JR, Mani DR, Jackson AM, Zhao L, Pope ME, Smith D, Rivera KD, Anderson NL, Skates SJ, Pearson TW, Paulovich AG, Carr SA. Interlaboratory evaluation of automated, multiplexed peptide immunoaffinity enrichment coupled to multiple reaction monitoring mass spectrometry for quantifying proteins in plasma. Mol Cell Proteomics MCP. 2012;11:M111.013854. doi: 10.1074/mcp.M111.013854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kennedy JJ, Abbatiello SE, Kim K, Yan P, Whiteaker JR, Lin C, Kim JS, Zhang Y, Wang X, Ivey RG, Zhao L, Min H, Lee Y, Yu MH, Yang EG, Lee C, Wang P, Rodriguez H, Kim Y, Carr SA, Paulovich AG. Demonstrating the feasibility of large-scale development of standardized assays to quantify human proteins. Nat Methods. 2014;11:149–155. doi: 10.1038/nmeth.2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huttenhain R, Soste M, Selevsek N, Rost H, Sethi A, Carapito C, Farrah T, Deutsch EW, Kusebauch U, Moritz RL, Nimeus-Malmstrom E, Rinner O, Aebersold R. Reproducible quantification of cancer-associated proteins in body fluids using targeted proteomics. Sci Transl Med. 2012;4:142ra94. doi: 10.1126/scitranslmed.3003989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wolf-Yadlin A, Hautaniemi S, Lauffenburger DA, White FM. Multiple reaction monitoring for robust quantitative proteomic analysis of cellular signaling networks. Proc Natl Acad Sci U S A. 2007;104:5860–5865. doi: 10.1073/pnas.0608638104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Whiteaker JR, Zhao L, Yan P, Ivey RG, Voytovich UJ, Moore HD, Lin C, Paulovich AG. Peptide Immunoaffinity Enrichment and Targeted Mass Spectrometry Enables Multiplex, Quantitative Pharmacodynamic Studies of Phospho-Signaling. Mol Cell Proteomics MCP. 2015;14:2261–2273. doi: 10.1074/mcp.O115.050351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kennedy JJ, Yan P, Zhao L, Ivey RG, Voytovich UJ, Moore HD, Lin C, Pogosova-Agadjanyan EL, Stirewalt DL, Reding KW, Whiteaker JR, Paulovich AG. Immobilized Metal Affinity Chromatography Coupled to Multiple Reaction Monitoring Enables Reproducible Quantification of Phospho-signaling. Mol Cell Proteomics MCP. 2016;15:726–739. doi: 10.1074/mcp.O115.054940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Whiteaker JR, Zhao L, Saul RG, Kaczmarczyk JA, Schoenherr RM, Moore HD, Jones-Weinert CE, Ivey RG, Lin C, Hiltke T, Reding KW, Whiteley GR, Wang P, Paulovich AG. A multiplexed mass spectrometry-based assay for robust quantification of phospho-signaling in response to DNA damage. Radiation Research. 2018 doi: 10.1667/RR14963.1. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Whiteaker JR, Halusa GN, Hoofnagle AN, Sharma V, MacLean B, Yan P, Wrobel JA, Kennedy J, Mani DR, Zimmerman LJ, Meyer MR, Mesri M, Rodriguez H, Paulovich AG Clinical Proteomic Tumor Analysis Consortium (CPTAC) CPTAC Assay Portal: a repository of targeted proteomic assays. Nat Methods. 2014;11:703–704. doi: 10.1038/nmeth.3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hoofnagle AN, Becker JO, Oda MN, Cavigiolio G, Mayer P, Vaisar T. Multiple-reaction monitoring-mass spectrometric assays can accurately measure the relative protein abundance in complex mixtures. Clin Chem. 2012;58:777–781. doi: 10.1373/clinchem.2011.173856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Annesley TM, Cooks RG, Herold DA, Hoofnagle AN. Clinical Mass Spectrometry-Achieving Prominence in Laboratory Medicine. Clin Chem. 2016;62:1–3. doi: 10.1373/clinchem.2015.251272. [DOI] [PubMed] [Google Scholar]
- 34.Kushnir MM, Rockwood AL, Roberts WL, Abraham D, Hoofnagle AN, Meikle AW. Measurement of thyroglobulin by liquid chromatography-tandem mass spectrometry in serum and plasma in the presence of antithyroglobulin autoantibodies. Clin Chem. 2013;59:982–990. doi: 10.1373/clinchem.2012.195594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hoofnagle AN, Roth MY. Clinical review: improving the measurement of serum thyroglobulin with mass spectrometry. J Clin Endocrinol Metab. 2013;98:1343–1352. doi: 10.1210/jc.2012-4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Agger SA, Marney LC, Hoofnagle AN. Simultaneous Quantification of Apolipoprotein A-I and Apolipoprotein B by Liquid Chromatography–Multiple Reaction Monitoring/Mass Spectrometry. Clin Chem. 2010 doi: 10.1373/clinchem.2010.152264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schneck NA, Phinney KW, Lee SB, Lowenthal MS. Quantification of cardiac troponin I in human plasma by immunoaffinity enrichment and targeted mass spectrometry. Anal Bioanal Chem. 2018 doi: 10.1007/s00216-018-0960-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Whiteaker JR, Zhao L, Abbatiello SE, Burgess M, Kuhn E, Lin C, Pope ME, Razavi M, Anderson NL, Pearson TW, Carr SA, Paulovich AG. Evaluation of large scale quantitative proteomic assay development using peptide affinity-based mass spectrometry. Mol Cell Proteomics MCP. 2011;10(4):M110.005645. doi: 10.1074/mcp.M110.005645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hoofnagle AN, Whiteaker JR, Carr SA, Kuhn E, Liu T, Massoni SA, Thomas SN, Townsend RR, Zimmerman LJ, Boja E, Chen J, Crimmins DL, Davies SR, Gao Y, Hiltke TR, Ketchum KA, Kinsinger CR, Mesri M, Meyer MR, Qian WJ, Schoenherr RM, Scott MG, Shi T, Whiteley GR, Wrobel JA, Wu C, Ackermann BL, Aebersold R, Barnidge DR, Bunk DM, Clarke N, Fishman JB, Grant RP, Kusebauch U, Kushnir MM, Lowenthal MS, Moritz RL, Neubert H, Patterson SD, Rockwood AL, Rogers J, Singh RJ, Van Eyk JE, Wong SH, Zhang S, Chan DW, Chen X, Ellis MJ, Liebler DC, Rodland KD, Rodriguez H, Smith RD, Zhang Z, Zhang H, Paulovich AG. Recommendations for the Generation, Quantification, Storage, and Handling of Peptides Used for Mass Spectrometry-Based Assays. Clin Chem. 2016;62:48–69. doi: 10.1373/clinchem.2015.250563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schoenherr RM, Zhao L, Whiteaker JR, Feng LC, Li L, Liu L, Liu X, Paulovich AG. Automated screening of monoclonal antibodies for SISCAPA assays using a magnetic bead processor and liquid chromatography-selected reaction monitoring-mass spectrometry. J Immunol Methods. 2010;353:49–61. doi: 10.1016/j.jim.2009.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maclean B, Tomazela DM, Abbatiello SE, Zhang S, Whiteaker JR, Paulovich AG, Carr SA, Maccoss MJ. Effect of collision energy optimization on the measurement of peptides by selected reaction monitoring (SRM) mass spectrometry. Anal Chem. 2010;82:10116–10124. doi: 10.1021/ac102179j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lange V, Picotti P, Domon B, Aebersold R. Selected reaction monitoring for quantitative proteomics: a tutorial. Mol Syst Biol. 2008;4:222. doi: 10.1038/msb.2008.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Clinical and Laboratory Standards Institute, C62-A. Liquid Chromatography-Mass Spectrometry Methods; Approved Guideline. 2014
- 44.Carr SA, Abbatiello SE, Ackermann BL, Borchers C, Domon B, Deutsch EW, Grant RP, Hoofnagle AN, Hüttenhain R, Koomen JM, Liebler DC, Liu T, MacLean B, Mani DR, Mansfield E, Neubert H, Paulovich AG, Reiter L, Vitek O, Aebersold R, Anderson L, Bethem R, Blonder J, Boja E, Botelho J, Boyne M, Bradshaw RA, Burlingame AL, Chan D, Keshishian H, Kuhn E, Kinsinger C, Lee JSH, Lee SW, Moritz R, Oses-Prieto J, Rifai N, Ritchie J, Rodriguez H, Srinivas PR, Townsend RR, Van Eyk J, Whiteley G, Wiita A, Weintraub S. Targeted peptide measurements in biology and medicine: best practices for mass spectrometry-based assay development using a fit-for-purpose approach. Mol Cell Proteomics MCP. 2014;13:907–917. doi: 10.1074/mcp.M113.036095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grant RP, Hoofnagle AN. From lost in translation to paradise found: enabling protein biomarker method transfer by mass spectrometry. Clin Chem. 2014;60:941–944. doi: 10.1373/clinchem.2014.224840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Becker PS, Taylor JA, Trobridge GD, Zhao X, Beard BC, Chien S, Adair J, Kohn DB, Wagner JE, Shimamura A, Kiem HP. Preclinical correction of human Fanconi anemia complementation group A bone marrow cells using a safety-modified lentiviral vector. Gene Ther. 2010;17:1244–1252. doi: 10.1038/gt.2010.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Taniguchi T, Garcia-Higuera I, Xu B, Andreassen PR, Gregory RC, Kim ST, Lane WS, Kastan MB, D’Andrea AD. Convergence of the fanconi anemia and ataxia telangiectasia signaling pathways. Cell. 2002;109:459–472. doi: 10.1016/s0092-8674(02)00747-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.