

Summary
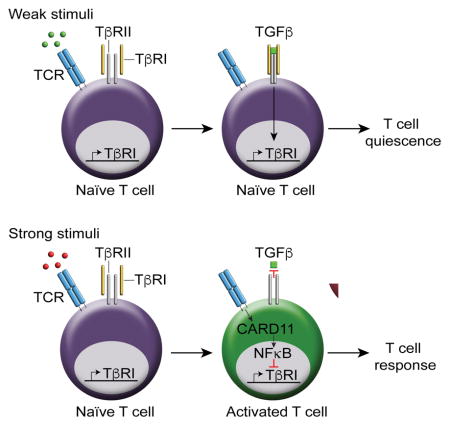
It is unclear how quiescence is enforced in naïve T cells, but activation by foreign antigens and self-antigens is allowed, despite the presence of inhibitory signals. We showed that active transforming growth factor-beta (TGFβ) signaling was present in naïve T cells and T cell receptor (TCR) engagement reduced TGFβ signaling during T cell activation by downregulating TGFβ type 1 receptor (TβRI) through activation of caspase recruitment domain-containing protein 11 (CARD11) and nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB). TGFβ prevented TCR-mediated TβRI downregulation but this was abrogated by interleukin-6 (IL-6). Mitigation of TCR-mediated TβRI downregulation through overexpression of TβRI in naïve and activated T cells rendered T cells less responsive and suppressed autoimmunity. Naïve T cells in autoimmune patients exhibited reduced TβRI expression and increased TCR-driven proliferation compared to healthy subjects. Thus, TCR-mediated regulation of TβRI-TGFβ signaling acts as a crucial criterion to determine T cell quiescence and activation.
eTOC Blurb
It is unclear how quiescence is enforced in naïve T cells. Tu et al. show that TGFβ signaling maintains T cell quiescence, preventing aberrant responses to self-antigens. Strong TCR stimuli reduce TβRI expression and consequently abolish TGFβ signaling in T cells. TCR-mediated TβRI downregulation acts as a “third criterion” to fully activate T cells in addition to the “two-signal” model.
Introduction
The initiation and magnitude of the T cell response is dependent on the balance of stimulatory and inhibitory signals. Naïve T cells are present in blood and peripheral lymphoid organs in their quiescent state, characterized by small cell size and reduced metabolic activity. The quiescent state of naïve T cells was thought to occur by default due to the lack of activation signals. However, accumulating studies have shown that survival of naïve T cells in the steady state requires TCR tickling by self-MHC molecules (Takada and Jameson, 2009). TCR tickling does not lead to autoimmunity in healthy individuals as T cell quiescence is actively reinforced by extrinsic factors such as regulatory T (Treg) cells, and intrinsic mechanisms such as transcription factors Peli1, TRIM28, Foxp1, Tsc1, and Tob (Chang et al., 2011; Chikuma et al., 2012; Feng et al., 2011; Sakaguchi et al., 2008; Tzachanis et al., 2001; Yang et al., 2011).
However, a few unresolved issues have arisen from these studies. First, it is not understood how T cell activation can still occur upon antigen stimulation when these mechanisms are in place to maintain T cell quiescence and tolerance. The “two-signal” model of T cell activation has been widely accepted: the first signal provided by the engagement of TCR to peptide-MHC complexes on antigen presenting cells (APCs) and the second signal provided by co-stimulation (Smith-Garvin et al., 2009). It is plausible that an additional signal is required to release T cells from quiescence programs to achieve T cell activation. Secondly, although hyperactivation and hyperproliferation of T cells were observed in mice with deletion of any of the quiescence-associated factors, none of these mice developed early onset lethal autoimmune diseases like mice with deficiency in forkhead box P3 (Foxp3), cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) or TGFβ (Fontenot et al., 2003; Hori et al., 2003; Shull et al., 1992; Waterhouse et al., 1995). However, Foxp3 and CTLA-4 are unlikely to regulate quiescence in naïve T cells intrinsically as they are not expressed in naïve T cells (Egen and Allison, 2002; Josefowicz et al., 2012). These findings collectively suggest that there must be other mechanism(s) that play a major role in governing quiescence of naïve T cells, and TGFβ signaling is one such candidate. TGFβ is involved in the development, survival and function of various immune cells, especially T cells (Tu et al., 2014). Bioactive TGFβ binds to TGFβ type II receptor (TβRII) and induces the assembly of a tetrameric TGFβ receptor complex (TβR) composed of TβRII and TβRI, which phosphorylates transcription factors mothers against decapentaplegic (SMAD)2 and SMAD3. Phosphorylated SMAD2 and/or SMAD3 form complexes with SMAD4 and are translocated into the nucleus, where they associate with DNA-binding cofactors to regulate the transcription of target genes (Shi and Massague, 2003). In addition, SMAD-independent pathways are also involved in mediating TGFβ signaling (Derynck and Zhang, 2003). The roles of TGFβ in suppressing activation of T cells have been well demonstrated by either addition of exogenous TGFβ to T cells in vitro (Ruegemer et al., 1990) or by genetic mutation of TGFβ ligands or receptors in T cells (Li et al., 2006; Liu et al., 2008; Marie et al., 2006; Shull et al., 1992). However, few studies have investigated the impact and mechanisms of TCR stimulation in TGFβ signaling and the consequential effects on the balance between T cell quiescence and T cell activation.
Here we showed that active TGFβ signaling was present in naïve T cells and strong TCR stimulation abolished TGFβ signaling to overcome its ongoing inhibition, through downregulation of TβRI expression. TβRII did not play an important role in the process. Accordingly, overexpression of TβRI in naïve T cells and restoration of TβRI in activated T cells constrained T cell responses. TCR drove the downregulation of TβRI through activation of the CARD11 and NFκB pathway. We demonstrated that TβRI expression was significantly lower in naïve CD4+ T cells of systemic lupus erythematosus (SLE) patients compared to healthy subjects. These results reveal a TCR-NFκB-TβRI axis that acts as a switch between T cell quiescence and T cell activation.
Results
TCR signals downregulate TGFβ signaling and TβR expression in naïve T cells
We first examined TGFβ signaling in naïve CD4+ T cells isolated from normal mice and humans. The amount of phosphorylated SMAD2 (p-SMAD2) protein was used as a readout for the degree of TGFβ signaling in T cells as SMAD2 is phosphorylated when TβR is activated by TGFβ. Freshly isolated naïve CD4+ T cells (0 hr) exhibited p-SMAD2 protein, indicating the presence of active TGFβ signal in naïve CD4+ T cells (Figure 1A). We next stimulated naïve CD4+ T cells in vitro with anti-CD3 and anti-CD28 to determine TGFβ signaling during T cell activation. We found that the amount of p-SMAD2 but not total SMAD2 protein decreased rapidly when T cells were activated (Figure 1A). A similar phenomenon was observed when human naïve and/or resting CD4+ T cells were stimulated (Figure 1A). To exclude the possibility that the reduction of p-SMAD2 in TCR-stimulated T cells might be caused by lack of sufficient amounts of TGFβ in cultures, we treated naïve and previously-activated (24 hr stimulation) T cells with a high concentration of TGFβ (2 ng/ml). We still detected higher levels of p-SMAD2 in naïve T cells than that in TCR-activated T cells in the presence of exogenous TGFβ, indicating an intrinsic feature of TCR stimulation in reducing TGFβ signals (Figure 1B). These results altogether indicate that active TGFβ signaling is present in naïve T cells but is reduced during T cell activation.
Figure 1. T cell activation induces TβR downregulation.

(A) Western blot of p-SMAD2 and SMAD2 in naïve CD4+ T cells stimulated for indicated time periods with anti-CD3 and anti-CD28. (B) Western blot of p-SMAD2 and SMAD2 in naïve (Naïve + TGFβ) or previously-activated CD4+ T cells (Activated + TGFβ) stimulated for 24 hr with anti-CD3 and anti-CD28 plus TGFβ. (C, D) Tgfbr1 and Tgfbr2 mRNA in naïve (C) mouse and (D) human CD4+ T cells stimulated for indicated time with anti-CD3 and anti-CD28. (E) Tgfbr1 and Tgfbr2 mRNA in naïve CD4+ T cells stimulated for 24 hr with anti-CD3 plus anti-CD28 or anti-CD3 alone. (F) IL2, Tgfbr1 and Tgfbr2 mRNA in T cells from DO11.10xRag2−/− mice treated with PBS (Ctrl) or OVA peptide (pOVA). (G) Western blot of TβRI, TβRII and GAPDH in naïve or CD4+ T cells stimulated for 24 hr with anti-CD3 and anti-CD28. (H) Tgfbr1 and Tgfbr2 mRNA in naïve CD4+ T cells stimulate for 24 hr with 1 μg/ml anti-CD28 plus indicated concentration of anti-CD3. (I) Tgfbr1 and Tgfbr2 mRNA in T cells from DO11.10xRag2−/− mice treated with indicated doses of pOVA. (J) Tgfbr1 and Tgfbr2 mRNA in naïve 5CC7 T cells cultured for 24 hr with APCs and indicated peptides. Data are pooled from two (D, I), three (C, F, H, J) or four (B, E) independent experiments or are representative of three (A, G) independent experiments. In (F, I), each circle represents the data from one mouse. Student’s t-test was used; bars (mean), error bars (SD), *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001. See also Figure S1.
We next examined the components of the TGFβ signaling pathway affected upon TCR stimulation. We showed that activation of naïve CD4+ T cells by anti-CD3 and anti-CD28 led to downregulation of TβRI and TβRII mRNA expression. This was observed in both mouse (Figure 1C) and human (Figure 1D) CD4+ T cells, as well as in mouse CD8+ T cells (data not shown). There was a time dependency in TβRI and TβRII downregulation, as indicated by much lower TβRI and TβRII expression in T cells that received prolonged stimulation (24 hr) compared to T cells that received short stimulation (1 hr). The downregulation of TβRI and TβRII was ascribed to TCR signals since stimulation of naïve CD4+ T cells with anti-CD3 alone was sufficient to induce the downregulation (Figure 1E), whereas stimulation with anti-CD28 alone did not cause TβR downregulation (data not shown). We further demonstrated downregulation of TβRI and TβRII in T cells activated in vivo by challenging DO11.10 TCR transgenic mice with OVA peptide that was specifically recognized by T cells in these mice, in which downregulation of TβRI and TβRII correlated with IL-2 mRNA upregulation (Figure 1F). Thus, TCR stimulation downregulates TβRI and TβRII transcription in naïve CD4+ T cells.
We next investigated whether TβRI and TβRII proteins were also regulated by TCR signals. We showed that TCR stimulation reduced the amount of TβRI protein (Figure 1G). However, the level of TβRII protein was not affected by TCR stimulation (Figure 1G), despite reduced TβRII mRNA expression. The data indicate that the reduction of TβRI, but not TβRII, correlates with the downregulation of TGFβ signaling in T cells upon T cell activation.
Strength of TCR stimuli determines TβRI expression
As the change in TβRI reflects the change in TGFβ signaling upon TCR stimulation, we next sought to determine the effect of TCR stimulation strength on TβR expression. When naïve T cells were stimulated with different concentrations of anti-CD3, it was evident that the degree of TβRI and TβRII downregulation was regulated by the strength of TCR signals, with a high concentration of anti-CD3 causing more TβRI and TβRII mRNA downregulation (Figure 1H). In contrast, weak TCR stimulation (e.g. 0.01 μg/ml anti-CD3) failed to reduce, and instead, upregulated TβRI and TβRII expression compared to naïve T cells. The strength of TCR signals also had a similar effect on TβRI and TβRII expression in vivo when DO11.10 T cells were stimulated with different doses of OVA peptide (Figure 1I). Furthermore, we showed that the TCR affinity to antigens also differentially regulated the expression of TβR. When 5CC7 TCR transgenic T cells were stimulated with their natural ligand moth cytochrome C (MCC) or the superagonist K5 (Krogsgaard et al., 2003), significantly lower TβRI and TβRII expression was detected compared to 5CC7 T cells stimulated with weak agonist peptide, 102S (Figure 1J). We also extended this finding to CD8+ T cells by stimulating OT-I TCR transgenic T cells with OVA peptide variants (Daniels et al., 2006). Significant T cell activation and TβRI downregulation were detected when OT-I T cells were stimulated with their high-affinity cognate antigen, N4 peptide (Figure S1A and S1B). In comparison, T4 peptide which has intermediate affinity for OT-I TCR, induced T cell activation and TβRI downregulation only at a high dose and the weak peptide E1 was unable to induce TβRI downregulation even at a high dose.
As TβRI expression in T cells negatively correlated with the strength of TCR stimuli, we hypothesized that strength of TCR stimuli could regulate T cell differentiation, especially the T cell subsets that require TGFβ for their generation (Tu et al., 2014). We adoptively transferred naïve 5CC7 T cells to the syngeneic B10.A mice and challenged the mice with MCC peptide (pMCC) at different doses (Figure 2A). Consistent with our findings in vitro, low-dose pMCC (0.1 μg) stimulation led to a higher TβRI expression in 5CC7 T cells compared to high-dose (10 μg) (Figure 2B), whereas TβRII was not affected regardless of dose. Low-dose antigen stimulation also induced a higher frequency of Foxp3+ Treg cells but a lower frequency of IFNγ-producing T helper 1 (Th1) cells compared to high-dose antigen stimulation (Figure 2C and 2D). In contrast, a high dose of pMCC led to more Th1 cells, but fewer Treg cells (Figure 2C and 2D). We next investigated if reduction of TβRI expression with a high dose of antigen stimulation could affect Th17 cell differentiation in vivo. When 5CC7 T cells were challenged with a high dose (10 μg) or a low dose (0.1 μg) of pMCC with complete Freund’s adjuvant (CFA), a decrease in TβRI but not TβRII was observed in high-dose antigen-challenged transgenic T cells compared to low-dose antigen-challenged T cells (Figure 2E). Indeed, an increase in the frequency of Th17 cells in these T cells correlated with decreased TβRI expression in T cells that were challenged with a high dose of pMCC (Figure 2F). These data altogether indicate that the strength of TCR stimulation regulates TβRI expression, which correlates with T cell lineage decisions.
Figure 2. Antigen stimulation strength affects T cell differentiation and TβRI expression.

(A) Experimental workflow of naïve 5CC7 T cells challenged with pMCC. (B) Tgfbr1 and Tgfbr2 mRNA and, (C) Treg cell and (D) Th1 cell frequencies in 5CC7 T cells from mice challenged with indicated doses of pMCC. (E) Tgfbr1 and Tgfbr2 mRNA and, (F) Th17 cell frequency in 5CC7 T cells from mice challenged with indicated doses of pMCC plus CFA. Data are pooled from three independent experiments. In (C, D, F), each circle represents one mouse. Student’s t-test was used; bars (mean), error bars (SEM), *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001.
TCR signals downregulate TβRI expression through the NFκB pathway
TCR-mediated downregulation of TβR was likely to depend on transcriptional regulation since both total and nascent TβRI and TβRII mRNA were significantly lower in activated T cells compared to naïve T cells (Figure 3A). We next investigated the underlying molecular mechanisms by which TCR signals regulate TβR expression. SMAD7 has been previously reported to act as an inhibitor for TGFβ signaling and TβR expression in epithelial cells and cancer cell lines (Nakao et al., 1997). However, we found that CD4+ T cells from Smad7f/fCd4-cre and Smad7+/+Cd4-cre mice showed similar degrees of TβR downregulation upon TCR activation (Figure S2A), suggesting non-involvement of SMAD7. Furthermore, TGFβ signaling in T cells was not significantly affected by the absence of SMAD7 when T cells were stimulated in the presence or absence of TGFβ (Figure S2B). Thus, downregulation of TβR during T cell activation is independent of SMAD7.
Figure 3. NFκB downregulates TβRI expression during T cell activation.

(A) Total and nascent Tgfbr1 and Tgfbr2 mRNA in naïve or CD4+ T cells stimulated for 24 hr with anti-CD3 and anti-CD28 (Activated). (B) Tgfbr1, Tgfbr2 and IL2 mRNA in naïve CD4+ T cells stimulated for indicated time periods with anti-CD3 and anti-CD28 plus DMSO (Control), NFκB or NFAT inhibitor. (C) Tgfbr1 and Tgfbr2 mRNA in wild-type (WT), Card11−/− or NfkbiaΔN-Tg naïve or CD4+ T cells stimulated for 24 hr with anti-CD3 and anti-CD28 (Activated). (D) Tgfbr1 mRNA in WT, Nfkb1−/−, Nfkb2−/− or Rel−/− naïve or CD4+ T cells stimulated for 24 hr with anti-CD3 and anti-CD28 (Activated). (E) Tgfbr1 mRNA in naïve CD4+ T cells stimulated for indicated time periods with anti-CD3 and anti-CD28 plus DMSO (Control) or CAPE. (F) ChIP-coupled real-time PCR analysis of p65 enrichment in the promoter region of Tgfbr1 gene of naïve or T cells stimulated for 24 hr with anti-CD3 and anti-CD28 (Activated). p65 enrichment was assessed using p65 antibody and presented relative to input and compared with control IgG. Data are pooled from two (A, B, D), three (E) or four (C) independent experiments or are representative of three (F) independent experiments. In (A, C, D, F), Student’s t-test was used. In (B, E), two-way ANOVA was used; bars (mean), error bars (SEM), *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001. See also Figure S2 and S3.
Given that downregulation of TβRI is mediated by TCR signals, it is likely that transcription factors that are downstream of TCR act as repressors for TβRI expression during T cell activation. Indeed, predicted binding sites for transcription factors, such as NFκB and nuclear factor of activated T cells (NFAT), were detected in the promoter region of TβRI using bioinformatics tools (BIOBASE, ALGGEN-PROMO). NFκB and NFAT play important roles in TCR signaling (Smith-Garvin et al., 2009). As expected, we found that chemical inhibitors for NFκB (IKK inhibitor VII), and NFAT (cyclosporin A) were both able to suppress the upregulation of IL-2 mRNA in activated T cells. However, only NFκB inhibition prevented TCR-mediated TβRI downregulation, but not inhibition of NFAT (Figure 3B). Neither NFκB nor NFAT inhibitors had any effect on the downregulation of TβRII mRNA induced by TCR stimulation (Figure 3B). Despite not finding any transcription factor that regulates TβRII mRNA during T cell activation, we found that TCR signals significantly accelerated TβRII mRNA decay (Figure S3A), providing a possible mechanism for TCR-mediated TβRII mRNA downregulation.
We further confirmed the role of NFκB as the repressor of TβRI expression using T cells from Card11−/− mice and NfkbiaΔN-Tg mice. The NFκB pathway is inhibited in T cells from Card11−/− mice since TCR mediates NFκB activation through CARD11 (Egawa et al., 2003). NfkbiaΔN-Tg mice express, selectively in T cells, a super-repressor form of IκBα, which constitutively inactivates NFκB (Boothby et al., 1997). Naïve CD4+ T cells from Card11−/− and NfkbiaΔN-Tg mice did not show downregulation of TβRI expression after TCR stimulation (Figure 3C). Inhibition of NFκB activity in Card11−/− and NfkbiaΔN-Tg mice could not rescue TβRII mRNA during T cell activation (Figure 3C). We next determined which NFκB subunits were required for the reduction of TβRI. p50, p52 and cRel were not required for the downregulation of TβRI by TCR signals as TCR stimulation induced a similar degree of TβRI downregulation in T cells from mice that lacked these NFκB subunits compared to wild-type mice (Figure 3D). As p65 (RelA) deletion is embryonically lethal, we inhibited p65 activity in T cells using caffeic acid phenethyl ester (CAPE) that prevented the translocation of p65 to the nucleus (Natarajan et al., 1996). CAPE prevented TβRI downregulation in stimulated T cells (Figure 3E). Using chromatin immunoprecipitation (ChIP) assays, we also detected significantly higher binding of p65 at the predicted NFκB binding site in the promoter region of TβRI in activated T cells compared to naïve T cells (Figure 3F). In contrast, there was no significant enrichment of other NFκB subunits at the same region of TβRI gene in activated T cells (Figure S3B). Together, these experiments indicate that NFκB is essential for TCR-mediated downregulation of TβRI.
TGFβ prevents TCR-mediated TβRI downregulation
In addition to TCR stimuli, we also investigated the role of cytokines on TβR expression in CD4+ T cells. We added a panel of cytokines to TCR-stimulated naïve CD4+ T cell cultures and determined TβRI and TβRII mRNA expression. Only TGFβ prevented TCR-mediated downregulation of TβRI, but not TβRII mRNA (Figure 4A and 4B). This was also reflected in TβRI protein levels (Figure 4C). In contrast, TβRII protein was detected at similar levels with or without TGFβ (Figure 4C). Similarly, TGFβ treatment of naïve T cells without TCR stimulation also led to an increase in TβRI, but not TβRII protein (Figure S4A). The TGFβ-mediated maintenance of TβRI expression in activated T cells corresponded to substantial amounts of p-SMAD2 in these T cells (Figure 4D). We further confirmed the effect of TGFβ on TβRI expression using Tgfbr2f/fEsr1-cre mice (Nakatsukasa et al., 2015). These mice express a mutated form of the mouse estrogen receptor and administration of tamoxifen induce efficient cre-mediated deletion of TβRII. T cells that were deficient in TβRII failed to upregulate their TβRI expression when stimulated in the presence of TGFβ (Figure 4E). We also determined that the upregulation of TβRI by TGFβ was, in part, through a SMAD-dependent pathway. Smad3−/− T cells showed lower TβRI expression than wild-type T cells when stimulated in the presence of TGFβ. Nevertheless, increased TβRI expression was still observed in TGFβ-treated Smad3−/− T cells compared to untreated Smad3−/− T cells (Figure 4F), suggesting involvement of SMAD3-independent pathways. Additionally, we showed that SMAD7 did not affect TGFβ-mediated TβRI upregulation since similar degrees of TβRI upregulation were observed between T cells from Smad7+/+Cd4-cre and Smad7f/fCd4-cre mice (Figure S2C).
Figure 4. TGFβ upregulates TβRI expression.

(A) Tgfbr1 and Tgfbr2 mRNA in naïve or CD4+ T cells stimulated for 24 hr with anti-CD3 and anti-CD28 plus different cytokines as indicated. (B) Tgfbr1 and Tgfbr2 mRNA in naïve CD4+ T cells stimulated for indicated time periods with anti-CD3 and anti-CD28 in the presence or absence of TGFβ. (C) Western blot of TβRI, TβRII and GAPDH in CD4+ T cells stimulated with anti-CD3 and anti-CD28 for 24 hr in the presence or absence of TGFβ. (D) Western blot of p-SMAD2 and SMAD2 in T cells stimulated for indicated time periods with anti-CD3 and anti-CD28 plus TGFβ. (E) Tgfbr1 mRNA in naïve Tgfbr2f/fEsr1-cre CD4+ T cells stimulated for indicated time periods with anti-CD3 and anti-CD28 in the presence or absence of TGFβ. Tgfbr2+/+; untreated Tgfbr2f/fEsr1-cre mice, Tgfbr2−/−; tamoxifen-treated Tgfbr2f/fEsr1-cre mice. (F) Tgfbr1 mRNA in naïve Smad3−/− or Smad3+/+ CD4+ T cells stimulated for indicated time periods with anti-CD3 and anti-CD28 in the presence or absence of TGFβ. (G) Tgfbr1 and Tgfbr2 mRNA in CD4+CD25− non-Treg cells and CD4+CD25+ Treg cells that were unstimulated or stimulated with anti-CD3 and anti-CD28 for 24 hr. Data are pooled from two (A, E, F, G) or three (B) independent experiments or are representative of three (C, D) independent experiments. In (B, E, F), two-way ANOVA was used. In (G), Student’s t-test was used; bars (mean), error bars (SD in A, E–G; SEM in B), *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001. See also Figure S4.
As TGFβ signaling in T cells is essential for the development of CD4+CD25+Foxp3+ Treg cells (Chen et al., 2003; Liu et al., 2008), we hypothesized that Treg cells, due to their exposure to TGFβ, would express a higher level of TβRI than non-Treg cells (CD4+CD25−). Indeed, a significantly higher level of TβRI was detected in Treg cells compared to non-Treg cells before and after anti-CD3 and anti-CD28 stimulation. However, lower TβRII expression was detected in freshly-isolated Treg cells and no subsequent changes occurred upon TCR stimulation (Figure 4G). Overall, these data indicate that TCR-mediated reduction of TβRI is prevented by TGFβ.
IL-6 abolishes TGFβ-mediated prevention of TβRI reduction
As cytokines alone (with the exception of TGFβ) did not influence TCR-mediated TβRI downregulation in naïve T cells (Figure 4A), we next investigated whether proinflammatory cytokines could interfere with the effect of TGFβ on TβRI expression. We stimulated naïve CD4+ T cells with anti-CD3 and anti-CD28 in the presence of both TGFβ and proinflammatory cytokines: IL-1β, IL-2, IL-4, IL-6, IL-12, IL-23, IFNγ and TNFα. Only IL-6 blocked the upregulation of TβRI by TGFβ (Figure S4B; data not shown). However, IL-6 had no effect on TβRII expression (Figure S4B).
We further confirmed the inhibitory effects of IL-6 on TGFβ-mediated TβRI upregulation in vivo. Since bacteria antigens in CFA enhance IL-6 production in innate immune cells, we analyzed myelin oligodendrocyte glycoprotein (MOG)-specific T cells in mice that were immunized with MOG peptide with either CFA or incomplete Freund’s adjuvant (IFA) (Figure S4C). The hypothesis was that IL-6 induced by CFA would reduce TβRI levels in MOG-specific T cells, compared to T cells from IFA-treated mice that received the same TCR stimulation in a TGFβ-present microenvironment. Indeed, MOG-specific T cells in mice treated with MOG and CFA showed lower TβRI expression but not TβRII, compared to T cells in mice treated with MOG and IFA (Figure S4D). MOG and CFA also increased IL-17 production in MOG-specific T cells (Figure S4E), consistent with the previous notion that reduction of TGFβ signaling favors Th17 cell differentiation (Zhou et al., 2008). These results suggest that IL-6 may affect TGFβ signaling in T cells by antagonizing the effect of TGFβ on TβRI expression.
TβRI downregulation promotes T cell responses
We next determined the effect of TCR-mediated downregulation of TβRI on T cell responses. Since the majority of T cells are hyperactivated in T cell-specific TβR-deficient mice, we used naïve CD4+ T cells from Tgfbr1f/fEsr1-cre mice to address this question. Similar CD4+ T cell frequencies were detected in the lymphoid organs of Tgfbr1f/fEsr1-cre mice compared to wild-type mice, and TβRI was deleted in Tgfbr1f/fEsr1-cre mice upon completion of tamoxifen treatment (data not shown). Naïve CD4+ T cells from tamoxifen-treated Tgfbr1f/fEsr1-cre mice (Tam) and untreated control Tgfbr1f/fEsr1-cre mice (Ctrl) were cultured with different concentrations of soluble anti-CD3 and APCs, and levels of T cell proliferation and IFNγ production were measured. In response to TCR stimulation, greater cell proliferation was detected in TβRI-deficient T cells than those in control T cells. Furthermore, we detected a greater difference between TβRI-deficient and control T cells when the cells were stimulated with a low concentration of anti-CD3 compared to a high concentration (Figure S5A). When T cells were stimulated with plate-bound anti-CD3, which provides stronger TCR stimulation than soluble anti-CD3, a difference in cell proliferation was only detected when the cells were stimulated with a low, but not a high concentration of anti-CD3 between TβRI-deficient and control T cells (Figure S5B). This was most likely due to depletion of TβRI in control T cells by strong TCR stimulation. We also detected a similar pattern in IFNγ production: weak anti-CD3 stimulation led to a more dramatic difference in IFNγ production between TβRI-deficient and control T cells, compared to strong anti-CD3 stimulation (Figure S5C). In addition, TβRI-deficient T cells were more resistant to suppression by Treg cells (Figure S5D), consistent with findings that TGFβ signaling in responder cells is required for Treg cell-mediated immunosuppression (Fahlen et al., 2005). These results demonstrate an increase in the magnitude of the T cell response to TCR stimulation in the absence of TβRI expression.
We next investigated whether T cell responses would be affected if we override the inherent TβR downregulation by overexpressing TβR in naïve T cells. We generated retrogenic mice using bone marrow cells infected with retrovirus for TβRIGFP, or TβRIICherry or controls (CtrlGFP, CtrlCherry). Naïve CD4+ T cells expressing TβRIGFP or TβRIICherry were isolated from the retrogenic mice based on their respective fluorescence. Elevated levels of TβRI or TβRII expression were detected in TβRIGFP+ and TβRIICherry+ T cells, respectively, compared with their controls (Figure 5A and S6A). TGFβ was not able to upregulate transgenic expression of TβRI, as TβRIGFP expression was driven by a CMV promoter that was subject to different regulatory mechanisms compared to the endogenous TβRI. TβRIGFP+CD4+ T cells showed a significant decrease in IL-2 expression and increase in Foxp3 expression in response to TCR stimulation, even in the absence of exogenous TGFβ (Figure 5A), and these changes were further enhanced by TGFβ. Consistently, TβRIGFP+CD4+ T cells exhibited less proliferation compared to CtrlGFP+CD4+ T cells when stimulated with anti-CD3 and APCs (Figure 5B). In contrast, IL-2 was expressed at a similar level between TβRIICherry and CtrlCherry T cells upon stimulation (Figure S6A).
Figure 5. TβRI Overexpression in naïve T cells suppresses T cell proliferation and autoimmunity.

(A) Tgfbr1, Il2 and Foxp3 mRNA in naïve TβRIGFP+ or CtrlGFP+ CD4+ T cells stimulated for 24 hr with anti-CD3 and anti-CD28 in the presence or absence of TGFβ. (B) Frequency of Ki-67+ in naïve TβRIGFP+ or CtrlGFP+ CD4+ T cells stimulated for 3 days with anti-CD3 and anti-CD28 plus TGFβ. (C–G) Naïve CD4+ T cells from TβRIGFP+ or CtrlGFP+ retrogenic mice were transferred into Rag1−/− mice. (C) Weight gain of recipient mice after T cell transfer. (D) Quantitation of colon pathology. (E) Representative histology images of colon sections. Scale bars, 200 μm. (F) Frequencies of the donor TβRIGFP+ or CtrlGFP+ CD4+ T cells in recipient mice. (G) Frequencies of Foxp3+ cells in the donor CD4+ T cell population. Data are pooled from two (A, B) or three (C–G) independent experiments. In (D, F, G), each circle represents the data from one mouse. In (A, B, D, F, G), Student’s t-test was used. In (C), two-way ANOVA was used; bars (mean), error bars (SD in A, B; SEM in C), *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001. See also Figure S5 and S6.
We also tested the response of these transgenic T cells in vivo using a colitis model based on T cell responses to commensal bacteria in the gut. Rag1−/− mice that received naïve TβRIGFP+CD4+ T cells were better able to maintain their body weight than mice that received naïve CtrlGFP+CD4+ T cells, which showed dramatic weight loss (Figure 5C), and the severity of colitis in mice was significantly lower in the former group (Figure 5D and 5E). Significantly fewer effector CD4+ T cells were detected in mice that received TβRIGFP+CD4+ T cells compared to those in the controls (Figure 5F). More Treg cells were also found in mice that received TβRIGFP+CD4+ T cells (Figure 5G), suggesting an enhancement of TGFβ signaling in these T cells. In contrast, naïve TβRIICherry+CD4+ T cells induced weight loss and colitis to a similar degree as naïve CtrlCherry+CD4+ T cells (Figure S6B–S6D), although less effector T cells were observed in the colon of mice that received TβRIICherry+CD4+ T cells (Figure S6E). There was also no difference in the frequencies of Treg cells between mice that received TβRIICherry+ or CtrlCherry+ T cells (Figure S6F). These data further suggest that it is the level of TβRI, but not TβRII, that determines TGFβ signaling in T cells. Taken together, these depletion and overexpression experiments establish that downregulation of TβRI allows for efficient T cell responses.
Restoration of TβRI in activated T cells reduces T cell proliferation
We also investigated the effect of restoration of TβRI expression on activated T cells. Our hypothesis was that if TCR-mediated TβRI downregulation was indeed required for optimal T cell responses, restoration of TβRI in activated T cells would restrain this. To address this question, activated CD4+ T cells were infected with retrovirus for either TβRIGFP or TβRIICherry, followed by TCR re-stimulation. As expected, activated T cells that expressed either TβRIGFP or TβRIICherry maintained high levels of TβRI or TβRII, respectively, upon TCR re-stimulation (Figure 6A and S7A). However, expression of TβRIGFP but not TβRIICherry in activated T cells downregulated the expression of IL-2 when the T cells were re-stimulated (Figure 6A and S7A), suggesting an inhibitory role of TβRI but not TβRII even in activated T cells.
Figure 6. TβRI restoration in activated T cells reduces T cell proliferation.

(A) Tgfbr1, and Il2 mRNA in CtrlGFP+ or TβRIGFP+ activated T cells re-stimulated with anti-CD3 and anti-CD28 for 24 hr. (B) Experimental workflow of TβRIGFP+- or TβRIICherry+-DO11.10 T cells challenged with pOVA. (C) Cell proliferation of CtrlGFP+- or TβRIGFP+-DO11.10 T cells challenged with pOVA. (D) Experimental workflow of TβRIGFP+- or TβRIICherry+-DO11.10 T cells challenged with OVA protein in an airway inflammation model. (E) Frequencies of CtrlGFP+- or TβRIGFP+-DO11.10 T cells in the lungs and draining lymph nodes (dLN) of recipient mice. (F) Frequencies of Ki-67+ cells in CtrlGFP+- or TβRIGFP+-DO11.10 T cells in dLN of recipient. (G) Western blot of phosphorylated p65 (p-p65) and p65 in CtrlGFP+ or TβRIGFP+ activated T cells re-stimulated with anti-CD3 and anti-CD28 for 24 hr. Data are pooled from two (A, E, F) or three (C) independent experiments or are representative of two (G) independent experiments. In (C, E, F), each circle represents one mouse. Student’s t-test was used; bars (mean), error bars (SEM), *P < 0.05, **P < 0.01. See also Figure S7.
We next examined the effect of TβRIGFP and TβRIICherry on activated T cells in vivo by expressing TβRIGFP or TβRIICherry in activated DO11.10 transgenic T cells (TβRIGFP+-DO11.10, TβRIICherry+-DO11.10) (Figure 6B). TβRIGFP+-DO11.10 T cells were less responsive to challenge with OVA peptide compared to CtrlGFP+-DO11.10 T cells (Figure 6C). Conversely, TβRIICherry had no effect on the proliferative response of activated DO11.10 T cells when challenged with OVA peptide (Figure S7B).
When TβRIGFP+- and TβRIICherry+-DO11.10 T cells were tested in a mouse model of airway inflammation (Figure 6D), significantly fewer TβRIGFP+-DO11.10 T cells were found in the lungs and the draining lymph nodes compared to CtrlGFP+-DO11.10 T cells (Figure 6E). This was consistent with reduced Ki-67 expression in TβRIGFP+-DO11.10 T cells (Figure 6F). Although none of the mice developed prominent pathological changes in their lungs upon completion of intratracheal challenge with OVA protein (data not shown), which was likely due to very low frequencies of both TβRIGFP+- and CtrlGFP+-DO11.10 T cells in the lung tissue, significantly fewer TβRIGFP+-DO11.10 T cells were found in the lungs (Figure 6E). Moreover, we found that restoration of TβRI in activated T cells led to reduced NFκB activity, as indicated by a decreased level of phosphorylated p65 compared to the control T cells (Figure 6G), which may account for reduced T cell responses in TβRIGFP+ activated T cells. In contrast, expression of TβRIICherry did not affect the frequency and proliferation of activated DO11.10 T cells (Figure S7C and S7D). These results altogether demonstrate that restoration of TβRI in activated T cells reduced T cell responses to antigens.
TβRI expression is reduced in naïve T cells of autoimmune patients
Since strong TCR stimulation is required for downregulation of TβRI, we hypothesized that there would be a difference in the level of TβRI between healthy individuals and patients with autoimmunity. We tested this hypothesis in recently diagnosed and untreated SLE patients. We detected significantly lower TβRI expression in naïve CD4+CD45RA+ T cells from SLE patients compared to those from healthy individuals (Figure 7A). Consistently, upon anti-CD3 stimulation, naïve T cells from SLE patients further reduced their TβRI (Figure 7A) and exhibited significantly more proliferation than healthy control T cells (Figure 7B). There was an increase in the proliferation of naïve T cells from healthy individuals when anti-CD3 stimulation was increased from 1 to 10 μg/ml. However, naïve T cells from SLE patients already reached the plateau for proliferation at a low concentration of anti-CD3 (1 μg/ml) and failed to proliferate further when simulated with a high concentration (10 μg/ml) (Figure 7B), indicating a significant decrease in TCR signaling threshold in SLE naïve T cells. Thus, the data suggest that intrinsic differences such as TβRI expression in naïve T cells between healthy individual and autoimmunity patients may contribute to increased T cell responses.
Figure 7. TβRI expression is reduced in naïve T cells of SLE patients.

Naïve CD4+ T cells were isolated from healthy individuals and SLE patients. (A) TGFBR1 mRNA in naïve or T cells stimulated for 24 hr with anti-CD3 and anti-CD28 (Activated). (B) Cell proliferation of naïve T cells from healthy individuals or SLE patients stimulated for 3 days with indicated concentration of anti-CD3 and APCs. Each circle represents one individual. Student’s t-test was used; bars (mean), *P < 0.05, **P < 0.01 and ****P < 0.0001.
Discussion
A mounting body of evidence suggests that the quiescent state of naïve T cells is actively maintained by extrinsic and intrinsic mechanisms. We here revealed active TGFβ signaling in naïve T cells, suggesting that T cells are controlled by TGFβ signaling in their naïve state before they receive sufficient TCR stimulation. This notion is supported by the fact that mice with T-cell specific deletion of TβR exhibit systemic T cell activation and responses (Li et al., 2006; Liu et al., 2008; Marie et al., 2006). It was unclear as to how T cells achieve T cell activation despite the omnipresence of TGFβ in tissues and blood. We showed that strong TCR stimulation induced downregulation of TβRI, which reduced sensitivity of T cells to TGFβ. Furthermore, TCR-mediated downregulation of TβRI is essential for T cells to induce optimal T cell responses, as evidenced by the data from TβRI-overexpressing retrogenic mice, as well as restoration of TβRI expression in activated T cells. Therefore, our results demonstrate that in addition to the two signals that are required for T cell activation, TCR-mediated downregulation of TβRI acts as “third criterion” for T cells to achieve the full array of immune responses.
When naïve CD4+ T cells were TCR-stimulated, a significant decrease in TβRI expression was observed in both mRNA and protein levels, which was associated with reduction in TGFβ signaling in activated T cells. Despite a rapid decrease in TβRII mRNA during T cell activation, the amount of TβRII protein surprisingly remained relatively unchanged in activated T cells. Although we found that TCR signals accelerated decay of TβRII mRNA, this did not lead to a decrease in the protein level. One possibility of this non-correlation may be the long half-life of TβRII protein in T cells, but this remains to be elucidated. Alternatively, it is possible that we detected the decrease in TβRII mRNA during a short window of time when it was transiently downregulated. Indeed, significant downregulation of TβRI and TβRII mRNA was seen in T cells that were stimulated in vivo for 24 hours by strong antigen stimulation. However, only the reduction of TβRI, but not TβRII mRNA was detected seven days after stimulation, suggesting that the change in TβRII mRNA is not sustained.
Our data also strongly suggest that TβRI and TβRII might be fundamentally different with regard to their correlation with TGFβ signaling in T cells. Previous studies showed that both TβRI and TβRII were important in TGFβ-mediated T cell tolerance since mice with T cell-specific deletion of either TβRI or TβRII die from lethal autoimmune disease (Li et al., 2006; Liu et al., 2008; Marie et al., 2006). This was expected, as TβRII is required to bind to TGFβ ligands and then activates TβRI. However, multiple lines of evidence show that TβRI plays a more important role than TβRII in TCR-mediated downregulation of TGFβ signaling: 1) TβRI protein, but not TβRII protein, was reduced upon TCR stimulation; 2) No difference in colitis severity was seen between mice that received TβRIICherry overexpressing naïve T cells and those that received control T cells, whereas naïve T cells overexpressing TβRIGFP were unable to induce severe colitis; 3) Mice that received TβRIGFP+ T cells displayed increased Treg cell frequencies, but not mice that received TβRIICherry+ T cells; 4) Restoration of TβRI alone in activated T cells was sufficient to reduce T cell proliferation upon antigen challenge whereas TβRII-overexpression had no effect. Although mechanistically elusive, these results do suggest that TβRI serves as the accurate reflection of TGFβ signaling that controls the development of T cell responses. This conclusion however, does not eliminate a role for TβRII in TGFβ signaling; only, that the changes in levels of TβRII do not reflect and control TGFβ signaling in T cells in response to TCR stimulation. A recent study on the conversion of natural killer (NK) cells into type 1 innate lymphoid cells (ILC1s) has indicated that TβRI can achieve TGFβ signaling independently of TβRII, in these cells (Cortez et al., 2017). However, the non-redundant role of TβRII in TGFβ signaling in T cells has been shown by other groups (Li et al., 2006; Marie et al., 2006). Thus, it is likely that TβR and its downstream targets may function differently among different immune cell subsets.
We found that TβRI expression is tightly regulated by the strength of TCR stimulation. T cells that were stimulated with weak or low dose TCR stimuli showed no reduction and, very likely, higher TβRI expression compared to naive T cells. In marked contrast, T cells that were activated with strong or high dose TCR stimuli exhibited rapid and profound reduction of TβRI compared to naïve T cells. This finding has important implications in understanding the quiescence program and general lack of autoimmunity in healthy individuals, despite the existence of self-reactive T cells that regularly encounter self-antigens. Moreover, our findings may help reconcile how T cell quiescence is maintained while T cell activation and proliferation are allowed during encounter with pathogens, even in the presence of TGFβ. Low levels of active TGFβ in blood and tissues are normally sufficient to prevent unwanted T cell response to self-antigens, despite the fact that the majority of TGFβ is present in a latent form. This is because, following establishment of central and peripheral tolerance, T cells that are normally present in the body recognize self-antigens with low affinity. “Tickling” by the low affinity self-antigens is not sufficient to induce TβRI downregulation, and allows for ongoing TGFβ signaling. On the other hand, engagement of TCR with high affinity antigens or foreign antigens is able to induce TβRI downregulation and promote T cell responses. This was demonstrated when N4 peptide induced T cell activation and TβRI downregulation in OT-I T cells, whereas E1 peptide was not able to induce TβRI downregulation, even at a high peptide dose. It was previously shown that E1 peptide induced positive selection of OT-I thymocytes (Daniels et al., 2006), and we show in our experiments that this peptide did not induce T cell activation and TβRI downregulation in peripheral OT-I T cells, which reflects the interaction between naïve T cells and self-antigens. Thus, in addition to the amount of endogenous TGFβ in the environment, the strength of TCR stimulation also determines the inhibitory effects of TGFβ on T cells. Given that TGFβ signaling in T cells is essential for Treg cell generation, our data may also explain the phenomenon that weak TCR stimulation favors the generation of Treg cells (Gottschalk et al., 2010). We found higher TβRI expression in T cells, greater Treg cell frequencies and lower Th1 and Th17 frequencies in mice that received low-dose antigen stimulation compared to those that received high dose.
The findings in this study may provide an explanation as to why activated T cells are more resistant to suppression by Treg cells compared to naïve T cells (Korn et al., 2007; Tu et al., 2013). It can be envisioned that TβRI downregulation in activated T cells contributes to their resistance to Treg cells. Supporting this notion, we have shown that T cells that lack TβRI are more resistant to Treg cell suppression. Furthermore, our findings may provide insight as to why Treg cells are much more effective in preventing the development of autoimmunity than in treating ongoing diseases (Tu et al., 2013). Since pathogenic T cells in ongoing autoimmunity are already activated by autoantigens, reduced TβRI expression and TGFβ signaling in these T cells may render Treg cells ineffective.
Our finding that TGFβ prevents TCR-mediated TβRI downregulation in T cells has important implications in our understanding of the development and pathogenesis of autoimmunity and cancer. The preservation and even upregulation of TβRI by TGFβ present in blood and tissues may be critical in enforcing quiescence in naïve T cells and preventing T cells from abnormal activation in response to TCR stimulation, thus avoiding autoimmunity. This may also account for insufficient anti-tumor T cell responses in tumor patients, despite the presence of tumor antigen-specific T cells within tumors. Since TGFβ is produced in large amounts in the tumor microenvironment, TGFβ may inhibit anti-tumor immunity by upregulation of TβRI in T cells, thereby increasing their threshold for T cell activation. This conclusion is supported by previous studies that demonstrate anti-tumor immunity and tumor clearance after reduction of TGFβ signaling in T cells (Gorelik and Flavell, 2001; Thomas and Massague, 2005). Furthermore, poor immunogenicity of tumor antigens may further impede anti-tumor responses since weak antigen stimulation does not induce TβRI downregulation, especially in the presence of substantial amounts of TGFβ. Therapeutic strategies to deliver strong antigen stimulation to downregulate TβRI expression in T cells may be critical in achieving better anti-tumor immunity. We have recently shown that TGFβ signaling to Treg cells is required for Treg cell activity (Konkel et al., 2017), which suggests that Treg cells rely heavily on TGFβ for the maintenance of their phenotype. This may be facilitated by the high expression of TβRI in Treg cells. Furthermore, Treg cells express higher levels of membrane-bound TGFβ compared to non-Treg cells (Nakamura et al., 2001), and it is possible that membrane-bound TGFβ is required to keep Treg cells active and increase sensitivity of Treg cells to TGFβ.
The surprising finding that IL-6 was able to reduce the positive effect of TGFβ on TβRI expression in T cells provides insights into our understanding of the mechanisms at play in anti-infection immunity and the development of autoimmunity. Since IL-6 is secreted by innate immune cells during infection, it is possible that IL-6 induced by bacterial or viral pathogens suppresses upregulation of TβRI by TGFβ and therefore reduces the threshold of TCR activation. Furthermore, this may help explain why autoimmunity can occur in individuals following infection: IL-6 induced by infection may lower the ability of TGFβ to suppress self-reactive T cells by reducing TβRI expression on T cells and facilitate the differentiation of autoimmune pro-inflammatory T cells. Our findings also raise the possibility that, in addition to promoting expression of IL-23 receptor (Zhou et al., 2008), IL-6 facilitates Th17 cell generation by restraining TβRI expression. Although mechanistically elusive, it is likely that the NFκB pathway is also involved: we showed herein that NFκB negatively regulates TβRI and IL-6 was previously reported to induce NFκB activation (Wang et al., 2003).
Indeed, TCR-mediated NFκB activation provides a molecular mechanism that links TCR signaling to the regulation of TGFβ signaling. We showed that NFκB and NFAT inhibitors were both able to suppress the upregulation of IL-2 transcription in stimulated T cells. Nevertheless, only NFκB inhibitor was able to prevent TβRI downregulation. This finding suggests that the maintenance of TβRI expression by NFκB inhibitor was not the result of inhibition of T cell activation, but due to specific inhibition of the NFκB pathway. This is in line with a previous report showing that strong TCR stimulation dampens Treg cell generation through the TCR-CARD11-NFκB pathway (Molinero et al., 2011). We have observed direct binding of CARD11 and TβRI proteins in activated T cells (data not shown), suggesting a possibility for post-translational regulation of TβRI by NFκB. It should be noted that, in addition to the TCR-NFκB-TβRI axis, T cells might employ other mechanisms to regulate TGFβ signaling during T cell activation, such as the TCR-protein kinase Cθ pathway (Giroux et al., 2010). Although SMAD7 has been shown in other cell types to be an inhibitor of TGFβ signaling, it did not play a role in the regulation of TβRI expression during T cell activation.
In contrast to previous reports that mainly focused on the inhibitory role of TGFβ signaling in T cell activation, we here reveal that the cross-talk between TGFβ and TCR is not unidirectional. TCR signals, through NFκB, reciprocally reduce TGFβ signaling by downregulating TβRI expression. We thus propose a model in which TGFβ signaling is regulated by TCR signals, which in turn controls the switch between T cell quiescence and activation: when T cells encounter weak TCR stimuli such as self-antigens or tumor antigens, TβRI expression allows for TGFβ signaling which inhibits T cell activation and/or promotes Treg cell generation, further reinforcing T cell quiescence and tolerance. However, when T cells encounter strong TCR stimuli such as pathogens, an adequate T cell response is generated due to reduced sensitivity of activated T cells to TGFβ as a result of TβRI downregulation. In addition, proinflammatory cytokines such as IL-6 may interfere with the balance of T cell quiescence and activation enforced by TGFβ. This may promote anti-infection immunity or trigger the onset of autoimmunity. In sum, this model may provide deeper insights into many fundamental immunology questions, including how naïve T cells are kept quiescent and how T cell activation occurs despite the perpetual presence of inhibitory signals.
STAR Methods
Experimental Model and Subject Details
Mice
C57BL/6, CD45.1 (C57BL/6) and Rag1−/− mice were purchased from The Jackson Laboratory. DO11.10xRag2−/−, 5CC7.CD45.1xRag2−/−, BALB/c and B10.A mice were obtained from Taconic. Tgfbr2f/fEsr1-cre and Smad3−/− mice were previously described (Nakatsukasa et al., 2015; Yang et al., 1999). Tgfbr1f/fEsr1-cre mice were generated in house by crossing Esr1-cre mice with Tgfbr1f/f mice. Smad7f/fCd4-cre mice were generated by crossing Cd4-cre mice with Smad7f/f mice obtained from M. Diaz (National Institutes of Health). Tgfbr1f/fEsr1-cre and Tgfbr2f/fEsr1-cre mice were treated with tamoxifen (1 μg/mouse) per day for 5 days to delete TβRI or TβRII, respectively. Lymphoid tissue from Card11−/− and NfkbiaΔN-Tg mice were obtained from M. Alegre (University of Chicago). Lymphoid tissues from Nfkb1−/− and Nfkb2−/− mice were obtained from U. Siebenlist (National Institutes of Health). Lymphoid tissues from Rel−/− mice were obtained from R. Sen (National Institutes of Health). Lymphoid tissues from OT-IxRag1−/− mice were obtained from G. Altan-Bonnet (National Institutes of Health). All animal studies were approved by the Animal Care and Use Committees of National Institute of Dental and Craniofacial Research, National Institutes of Health. 6 to 10 weeks old, male and female sex-matched mice were used for all experiments.
Sources of human T cells
Human peripheral blood mononuclear cells (PBMC) were provided by healthy volunteers and obtained from the NIH Department of Transfusion Medicine through their approved protocol. PBMC from SLE patients and healthy controls were harvested at The Affiliated Drum Tower Hospital of Nanjing University Medical School, Nanjing Medical University (Nanjing, China) through their approved protocol. Donors from 20 to 58 years old, of both genders, were recruited in this study. All samples were provided on a de-identified basis. A signed informed consent was obtained from all donors.
Method Details
Real-time PCR
Total RNA was derived from cells using RNeasy Mini Kit (Qiagen) and reversed transcribed using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Quantitative real-time PCR was performed according to the protocol of TaqMan Gene Expression Master Mix (Applied Biosystems) with the following TagMan primers: Hprt, Mm00446968_m1; Tgfbr1, Mm00436964_m1, Hs00610320_m1, Rn00688966_m1; Tgfbr2, Mm03024091_m1, Hs00234253_m1; Il2, Mm00434256_m1; 18s, Mm03928990_g1; Gapdh, Hs02758991_m1; and Foxp3, Mm00475162_m1.
Flow cytometry analysis
Intranuclear staining was carried out using the Fixation/Permeabilization buffer solution (eBioscience) according to manufacturer’s instruction. For intracellular cytokine staining, cells were stimulated with PMA (50 ng/ml), Ionomycin (250 ng/ml) and Golgi-Plug (1:1000 dilution, BD PharMingen) at 37 °C for 3 hr, followed by fixation with the Fixation/Permeabilization buffer solution (BD Biosciences) according to manufacturer’s instruction. The staining procedures for MOG38–49-specific tetramer were previously described (Kasagi et al., 2014). Stained cells were analyzed on a LSRFortessa (BD Biosciences) and data was analyzed with FlowJo software.
In vitro T cell activation
Naïve mouse (CD4+CD62L+CD44−) and human (CD4+CD45RA+) T cells were purified by magnetic cell sorting (Miltenyi Biotec) and stimulated with indicated concentration of plate-bound anti-CD3 and anti-CD28, with or without addition of various recombinant cytokines at 37 °C for the indicated durations. In some experiments, cells were treated with IKK inhibitor VII (1 μM), Cyclosporin A (100 ng/ml) or caffeic acid phenethyl ester (20 μM) for 1 hr before stimulation with anti-CD3 and anti-CD28. Naïve 5CC7 T cells were stimulated with irradiated APCs plus 102S, MCC or K5 peptide at 10 ng/ml for 24 hr. Naïve OT-I T cells were stimulated with irradiated APCs plus N4, T4 or E1 peptide at either 0.1 or 10 ng/ml for 24 hr.
In vivo T cell activation
DO11.10xRag2−/− mice were treated with either PBS or indicated doses of OVA323–339 peptide for 24 hr before T cells were analyzed. In adoptive transfer experiments, naïve 5CC7.CD45.1 T cells (2 × 106) were transferred into B10.A mice and the mice were immunized the next day with indicated doses of MCC88–103 peptide. 5CC7.CD45.1 T cells were purified by flow cytometric cell sorting based on their CD45.1 expression 7 days after the immunization. In some experiments, the recipient B10.A mice were immunized with MCC88–103 peptide plus CFA (IFA supplemented with 300 μg/mouse M. Tuberculosis) at tail base and 5CC7 T cells in the draining inguinal lymph nodes were harvested for analysis. For the generation of MOG-specific T cells, C57BL/6 mice were immunized with MOG35–55 peptide (200 μg/mouse) plus either IFA or CFA at tail base. 7 days after pMOG immunization, MOG-specific T cells in the draining inguinal lymph nodes were identified using MOG38–49-specific tetramer and purified by flow cytometric cell sorting for analysis.
Isolation of nascent RNA
0.5 mM 5-ethynyl uridine (5-EU) was added into the T cell culture and was incorporated into the cells for 30 min. Total RNA was isolated using RNeasy Mini Kit (Qiagen) and 5-EU-labeled nascent RNA was captured using Click-iT Nascent RNA Capture Kit (Life Technologies) according to manufacturer’s instruction. The captured nascent RNA was then compared with total RNA in real-time PCR.
Analysis of RNA decay during T cell stimulation
Naïve CD4+ T cells were stimulated with plate-bound anti-CD3 and anti-CD28 for indicated period of time. Cell were washed with PBS and incubated with actinomycin D (10 μg/ml). Total RNA was isolated at discrete time points over a 4 hr period using TRIzol reagent (Invitrogen) to determine mRNA half-life.
ChIP assay
Naïve and activated CD4+ T cells (1 × 107) were prepared for ChIP assay using HighCell# ChIP Kit (Diagenode), according to manufacturer’s instruction. Cell lysates were sonicated, followed by precipitation with anti-p50, anti-p52, anti-p65, anti-RelB, anti-cRel, control rabbit IgG or control goat IgG. ChIP DNA was analyzed by real-time PCR using Power SYBR Green PCR Master Mix with the following primers: forward (5′-TCTGGAGCTCATTTTGGCGT) and reverse (5′-AGCGGGAGCAGTCATAGGT A).
T cell proliferation and Treg cell suppression assays
Freshly isolated naïve T cells were labeled with CellTrace CFSE according to manufacturer’s instruction. CFSE-labeled mouse T cells were stimulated with indicated concentration of anti-CD3 and irradiated APCs or plate-bound anti-CD3 and anti-CD28 (1 μg/ml). CFSE-labeled human T cells were stimulated with indicated concentration of plate-bound anti-CD3 and anti-CD28 (1 μg/ml). After 3 days of culture, dilution of CFSE fluorescence was used to determine cell division. In Treg cell suppression assay, CD4+CD25+ Treg cells were purified by magnetic cell sorting (Miltenyi Biotec) and cultured with CFSE-labeled CD4+ T cells (Teff), at indicated Treg:Teff ratios, together with irradiated APCs and anti-CD3 (1 μg/ml) for 3 days. Treg cell suppression was determined by the percentage of T cells that were inhibited by Treg cells compared with T cells cultured alone.
TβR retroviral constructs
Expression plasmids for Tgfbr1 and Tgfbr2 were previously described (Yu et al., 2002). cDNA encoding full-length Tgfr1 and Tgfbr2 were subcloned into pMSCV-IRES-GFP and pMSCV-IRES-Cherry retroviral vectors, respectively. Hek293T cells were transfected with pCL-Eco packaging plasmid and retroviral plasmids using Turbofect (Thermofisher). Retroviral supernatant was harvest 24–48 hr after transfection.
Generation of TβR retrogenic mice
TβRIGFP, TβRIICherry and corresponding control (CtrlGFP, CtrlCherry) retrogenic mice were generated via retroviral-transduced bone marrow reconstitution. Briefly, bone marrow was harvested from 8-week old donor C57BL/6 mice, 72 hr after administration of 5-fluorouracil (0.2 mg per gram of body weight). Bone marrow cells were cultured in C-DMEM supplemented with 20% FBS, mIL-3 (20 ng/ml), hIL-6 (50 ng/ml) and mSCF (50 ng/ml). Bone marrow cells were spinfected with retroviral supernatant and polybrene (5 μg/ml) at 2500 rpm for 2 hr at 32°C. Cells were harvested 48 hr later and sorted for GFP or Cherry expression, and injected into sublethally-irradiated Rag1−/− mice together with wild-type bone marrow cells at 1:1 ratio. GFP+ or Cherry+ naïve CD4+ T cells were harvested from these mice by flow cytometric cell sorting 8–10 weeks after transplantation.
Adoptive transfer model of colitis
GFP+CD4+CD45RBhighCD25− or Cherry+CD4+CD45RBhighCD25− T cells were harvested from TβRIGFP, TβRIICherry and their corresponding control mice by flow cytometric cell sorting. The sorted T cells (3 × 105) were transferred into Rag1−/− mice. Recipient mice were weighed twice weekly for 5–7 weeks, after which they were euthanized. The presence and severity of colitis in recipient mice were determined by their body weight loss and histological examination of colon tissue. The histopathological assessment of colitis was as followed: score 0, normal colon tissue; score 1, very mild, scattered mononuclear cell infiltration; score 2, mild submucosal mononuclear cell infiltration, sometimes accompanied by focal aggregates of mononuclear cells into the mucosal area, with minimal epithelial hyperplasia and mucin depletion from goblet cells; score 3, moderate mononuclear cell infiltration, accompanied by moderate epithelial hyperplasia and mucin depletion; score 4, marked mononuclear cell infiltration throughout the glandular mucosa and often transmural, with ulceration and marked epithelial hyperplasia and mucin depletion; score 5, as for score 4 except loss of intestinal glands. Lamina propria lymphocytes were isolated from colon tissue as previously described (Zanvit et al., 2015).
In vivo challenge of TβR-overexpressing activated T cells
Naïve CD4+ T cells from DO11.10xRag2−/− mice were activated overnight with plate bound anti-CD3 and anti-CD28. Cells were spinfected with retroviral supernatant and polybrene at 2500 rpm for 90 min at 32°C. Activated DO11.10 T cells that expressed TβRIGFP, TβRIICherry or control (CtrlGFP, CtrlCherry) were harvested 48 hr later and labeled with CellTrace Violet according to manufacturer’s instruction. The labeled cells (1 × 106) were transferred into BALB/c mice and the recipient mice were immunized the next day with OVA323–339 peptide plus CFA at tail base and the draining inguinal lymph nodes were harvested for analysis 2 days later. In the model of airway inflammation, BALB/c mice that received activated DO11.10 T cells (0.5 × 106) expressing TβRIGFP, TβRIICherry or control (CtrlGFP, CtrlCherry) were challenged intratracheally with OVA protein (100 μg) once a day for 4 days before the mice were sacrificed for analysis. T cells were isolated from lung tissue by digesting lung tissue in DMEM medium supplemented with DNase (2 mg/ml) and collagenase (4 mg/ml) for 40 min at 37°C.
Quantification and Statistical Analysis
Statistical analysis
Statistical analysis was performed using either unpaired two-tailed Student’s t-tests or two-way ANOVA in GraphPad Prism.
Supplementary Material
Highlights.
Naïve T cells show active TGFβ signaling and TβR expression
Strong TCR stimulation decreases TβRI and TGFβ signaling by CARD11 and NFκB
Overexpression of TβRI suppresses T cell response and autoimmunity
TβRI expression is reduced in naïve T cells of SLE patients
Acknowledgments
We thank Drs. M. Diaz, U. Siebenlist, R. Sen and G. Altan-Bonnet, NIH for providing gene mutated mice and reagents. We thank the FACS core of NIDCR for their technical assistance. This research was supported by the Intramural Research Program of NIDCR, NIH and by the National Science Foundation of China Project #81120108021 and #81720108020 (L.S.) and the Jiangsu Province Major Research and Development Program, China #BE2015062 (L.S.). C.P.Z. C was supported in part by the A*STAR International Fellowship, Singapore. S.A. P was supported in part by the Korean Biomedical Scientist Fellowship Program.
Footnotes
Author contributions
E. T designed and performed experiments, analyzed data and wrote the manuscript; C.P.Z. C, D.Z, S.A. P, and W. J designed and performed experiments; W.W. C and D. W designed and performed experiments and analyzed data for human studies; M-L. A and Y.E. Z provided critical materials and scientific input; L. S designed and supervised human studies and provided critical scientific input; W.J. C conceived, initiated and supervised the whole study, designed experiments and wrote the manuscript.
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Boothby MR, Mora AL, Scherer DC, Brockman JA, Ballard DW. Perturbation of the T lymphocyte lineage in transgenic mice expressing a constitutive repressor of nuclear factor (NF)-kappaB. J Exp Med. 1997;185:1897–1907. doi: 10.1084/jem.185.11.1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang M, Jin W, Chang JH, Xiao Y, Brittain GC, Yu J, Zhou X, Wang YH, Cheng X, Li P, et al. The ubiquitin ligase Peli1 negatively regulates T cell activation and prevents autoimmunity. Nat Immunol. 2011;12:1002–1009. doi: 10.1038/ni.2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, Wahl SM. Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chikuma S, Suita N, Okazaki IM, Shibayama S, Honjo T. TRIM28 prevents autoinflammatory T cell development in vivo. Nat Immunol. 2012;13:596–603. doi: 10.1038/ni.2293. [DOI] [PubMed] [Google Scholar]
- Cortez VS, Ulland TK, Cervantes-Barragan L, Bando JK, Robinette ML, Wang Q, White AJ, Gilfillan S, Cella M, Colonna M. SMAD4 impedes the conversion of NK cells into ILC1-like cells by curtailing non-canonical TGF-beta signaling. Nat Immunol. 2017;18:995–1003. doi: 10.1038/ni.3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels MA, Teixeiro E, Gill J, Hausmann B, Roubaty D, Holmberg K, Werlen G, Hollander GA, Gascoigne NR, Palmer E. Thymic selection threshold defined by compartmentalization of Ras/MAPK signalling. Nature. 2006;444:724–729. doi: 10.1038/nature05269. [DOI] [PubMed] [Google Scholar]
- Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- Egawa T, Albrecht B, Favier B, Sunshine MJ, Mirchandani K, O’Brien W, Thome M, Littman DR. Requirement for CARMA1 in antigen receptor-induced NF-kappa B activation and lymphocyte proliferation. Curr Biol. 2003;13:1252–1258. doi: 10.1016/s0960-9822(03)00491-3. [DOI] [PubMed] [Google Scholar]
- Egen JG, Allison JP. Cytotoxic T lymphocyte antigen-4 accumulation in the immunological synapse is regulated by TCR signal strength. Immunity. 2002;16:23–35. doi: 10.1016/s1074-7613(01)00259-x. [DOI] [PubMed] [Google Scholar]
- Fahlen L, Read S, Gorelik L, Hurst SD, Coffman RL, Flavell RA, Powrie F. T cells that cannot respond to TGF-beta escape control by CD4(+)CD25(+) regulatory T cells. J Exp Med. 2005;201:737–746. doi: 10.1084/jem.20040685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng X, Wang H, Takata H, Day TJ, Willen J, Hu H. Transcription factor Foxp1 exerts essential cell-intrinsic regulation of the quiescence of naive T cells. Nat Immunol. 2011;12:544–550. doi: 10.1038/ni.2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- Giroux M, Delisle JS, O’Brien A, Hebert MJ, Perreault C. T cell activation leads to protein kinase C theta-dependent inhibition of TGF-beta signaling. J Immunol. 2010;185:1568–1576. doi: 10.4049/jimmunol.1000137. [DOI] [PubMed] [Google Scholar]
- Gorelik L, Flavell RA. Immune-mediated eradication of tumors through the blockade of transforming growth factor-beta signaling in T cells. Nat Med. 2001;7:1118–1122. doi: 10.1038/nm1001-1118. [DOI] [PubMed] [Google Scholar]
- Gottschalk RA, Corse E, Allison JP. TCR ligand density and affinity determine peripheral induction of Foxp3 in vivo. J Exp Med. 2010;207:1701–1711. doi: 10.1084/jem.20091999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–1061. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol. 2012;30:531–564. doi: 10.1146/annurev.immunol.25.022106.141623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasagi S, Zhang P, Che L, Abbatiello B, Maruyama T, Nakatsukasa H, Zanvit P, Jin W, Konkel JE, Chen W. In vivo-generated antigen-specific regulatory T cells treat autoimmunity without compromising antibacterial immune response. Sci Transl Med. 2014;6:241ra278. doi: 10.1126/scitranslmed.3008895. [DOI] [PubMed] [Google Scholar]
- Konkel JE, Zhang D, Zanvit P, Chia C, Zangarle-Murray T, Jin W, Wang S, Chen W. Transforming Growth Factor-beta Signaling in Regulatory T Cells Controls T Helper-17 Cells and Tissue-Specific Immune Responses. Immunity. 2017;46:660–674. doi: 10.1016/j.immuni.2017.03.015. [DOI] [PubMed] [Google Scholar]
- Korn T, Reddy J, Gao W, Bettelli E, Awasthi A, Petersen TR, Backstrom BT, Sobel RA, Wucherpfennig KW, Strom TB, et al. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat Med. 2007;13:423–431. doi: 10.1038/nm1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogsgaard M, Prado N, Adams EJ, He XL, Chow DC, Wilson DB, Garcia KC, Davis MM. Evidence that structural rearrangements and/or flexibility during TCR binding can contribute to T cell activation. Mol Cell. 2003;12:1367–1378. doi: 10.1016/s1097-2765(03)00474-x. [DOI] [PubMed] [Google Scholar]
- Li MO, Flavell RA. TGF-beta: a master of all T cell trades. Cell. 2008;134:392–404. doi: 10.1016/j.cell.2008.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MO, Sanjabi S, Flavell RA. Transforming growth factor-beta controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity. 2006;25:455–471. doi: 10.1016/j.immuni.2006.07.011. [DOI] [PubMed] [Google Scholar]
- Liu Y, Zhang P, Li J, Kulkarni AB, Perruche S, Chen W. A critical function for TGF-beta signaling in the development of natural CD4+CD25+Foxp3+ regulatory T cells. Nat Immunol. 2008;9:632–640. doi: 10.1038/ni.1607. [DOI] [PubMed] [Google Scholar]
- Marie JC, Liggitt D, Rudensky AY. Cellular mechanisms of fatal early-onset autoimmunity in mice with the T cell-specific targeting of transforming growth factor-beta receptor. Immunity. 2006;25:441–454. doi: 10.1016/j.immuni.2006.07.012. [DOI] [PubMed] [Google Scholar]
- Molinero LL, Miller ML, Evaristo C, Alegre ML. High TCR stimuli prevent induced regulatory T cell differentiation in a NF-kappaB-dependent manner. J Immunol. 2011;186:4609–4617. doi: 10.4049/jimmunol.1002361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K, Kitani A, Strober W. Cell contact-dependent immunosuppression by CD4(+)CD25(+) regulatory T cells is mediated by cell surface-bound transforming growth factor beta. J Exp Med. 2001;194:629–644. doi: 10.1084/jem.194.5.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakao A, Afrakhte M, Moren A, Nakayama T, Christian JL, Heuchel R, Itoh S, Kawabata M, Heldin NE, Heldin CH, ten Dijke P. Identification of Smad7, a TGFbeta-inducible antagonist of TGF-beta signalling. Nature. 1997;389:631–635. doi: 10.1038/39369. [DOI] [PubMed] [Google Scholar]
- Nakatsukasa H, Zhang D, Maruyama T, Chen H, Cui K, Ishikawa M, Deng L, Zanvit P, Tu E, Jin W, et al. The DNA-binding inhibitor Id3 regulates IL-9 production in CD4(+) T cells. Nat Immunol. 2015;16:1077–1084. doi: 10.1038/ni.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan K, Singh S, Burke TR, Jr, Grunberger D, Aggarwal BB. Caffeic acid phenethyl ester is a potent and specific inhibitor of activation of nuclear transcription factor NF-kappa B. Proc Natl Acad Sci U S A. 1996;93:9090–9095. doi: 10.1073/pnas.93.17.9090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruegemer JJ, Ho SN, Augustine JA, Schlager JW, Bell MP, McKean DJ, Abraham RT. Regulatory effects of transforming growth factor-beta on IL-2- and IL-4-dependent T cell-cycle progression. J Immunol. 1990;144:1767–1776. [PubMed] [Google Scholar]
- Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133:775–787. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, Allen R, Sidman C, Proetzel G, Calvin D, et al. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–699. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith-Garvin JE, Koretzky GA, Jordan MS. T cell activation. Annu Rev Immunol. 2009;27:591–619. doi: 10.1146/annurev.immunol.021908.132706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephen TL, Payne KK, Chaurio RA, Allegrezza MJ, Zhu H, Perez-Sanz J, Perales-Puchalt A, Nguyen JM, Vara-Ailor AE, Eruslanov EB, et al. SATB1 Expression Governs Epigenetic Repression of PD-1 in Tumor-Reactive T Cells. Immunity. 2017;46:51–64. doi: 10.1016/j.immuni.2016.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada K, Jameson SC. Naive T cell homeostasis: from awareness of space to a sense of place. Nat Rev Immunol. 2009;9:823–832. doi: 10.1038/nri2657. [DOI] [PubMed] [Google Scholar]
- Thomas DA, Massague J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell. 2005;8:369–380. doi: 10.1016/j.ccr.2005.10.012. [DOI] [PubMed] [Google Scholar]
- Tu E, Bourges D, Gleeson PA, Ang DK, van Driel IR. Pathogenic T cells persist after reversal of autoimmune disease by immunosuppression with regulatory T cells. Eur J Immunol. 2013;43:1286–1296. doi: 10.1002/eji.201242771. [DOI] [PubMed] [Google Scholar]
- Tu E, Chia PZ, Chen W. TGFbeta in T cell biology and tumor immunity: Angel or devil? Cytokine Growth Factor Rev. 2014;25:423–435. doi: 10.1016/j.cytogfr.2014.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzachanis D, Freeman GJ, Hirano N, van Puijenbroek AA, Delfs MW, Berezovskaya A, Nadler LM, Boussiotis VA. Tob is a negative regulator of activation that is expressed in anergic and quiescent T cells. Nat Immunol. 2001;2:1174–1182. doi: 10.1038/ni730. [DOI] [PubMed] [Google Scholar]
- Wang L, Walia B, Evans J, Gewirtz AT, Merlin D, Sitaraman SV. IL-6 induces NF-kappa B activation in the intestinal epithelia. J Immunol. 2003;171:3194–3201. doi: 10.4049/jimmunol.171.6.3194. [DOI] [PubMed] [Google Scholar]
- Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, Thompson CB, Griesser H, Mak TW. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995;270:985–988. doi: 10.1126/science.270.5238.985. [DOI] [PubMed] [Google Scholar]
- Yang K, Neale G, Green DR, He W, Chi H. The tumor suppressor Tsc1 enforces quiescence of naive T cells to promote immune homeostasis and function. Nat Immunol. 2011;12:888–897. doi: 10.1038/ni.2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Letterio JJ, Lechleider RJ, Chen L, Hayman R, Gu H, Roberts AB, Deng C. Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF-beta. EMBO J. 1999;18:1280–1291. doi: 10.1093/emboj/18.5.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, Hebert MC, Zhang YE. TGF-beta receptor-activated p38 MAP kinase mediates Smad-independent TGF-beta responses. EMBO J. 2002;21:3749–3759. doi: 10.1093/emboj/cdf366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanvit P, Konkel JE, Jiao X, Kasagi S, Zhang D, Wu R, Chia C, Ajami NJ, Smith DP, Petrosino JF, et al. Antibiotics in neonatal life increase murine susceptibility to experimental psoriasis. Nat Commun. 2015;6:8424. doi: 10.1038/ncomms9424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Lopes JE, Chong MM, Ivanov II, Min R, Victora GD, Shen Y, Du J, Rubtsov YP, Rudensky AY, et al. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature. 2008;453:236–240. doi: 10.1038/nature06878. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.