

Abstract
Recently, an innovative paradigm has been proposed in macromolecular therapeutics for treatment of B-cell lymphomas that can specifically kill cancer cells without a drug. The design rationale of this drug-free macromolecular therapeutic (DFMT) system is crosslinking the cell surface receptor to initiate apoptosis. However, how the apoptosis signal is triggered after receptor hyper-crosslinking remains to be elucidated. Here, two pathways, calcium influx dependent pathway and mitochondrial signal pathway, are identified to play major roles in triggering the programmed cell death. With the first step pretargeting and second step multiple binding, receptor hyper-crosslinking is achieved in a highly specific, time-dependent manner and largely mediated by multivalence. As a consequence, extracellular calcium influx is triggered, which subsequently decreases the mitochondrial membrane potential and induces apoptosis. The mitochondrial depolarization also stems from the Bcl-2 inhibition mediated by DFMT, followed by the cytochrome c release that activates caspase signaling. With the participation of the two-pronged mechanism, a programmed apoptosis is induced in response to DFMT treatment. The current findings can offer important implications to optimize the anti-CD20 strategies to treat B-cell non-Hodgkin lymphomas.
Keywords: apoptosis, cancer therapy, drug-free macromolecular therapeutics, lymphoma, receptor crosslinking
1. Introduction
Although rituximab (anti-CD 20) has improved the outcome for patients with non-Hodgkin’s lymphoma (NHL), its Fc-mediated events (e.g., complement activation) can sometimes contribute to clinical side effects.[1–3] Still, a portion of patients does not respond to rituximab treatment, due to the lack of Fc–Fc receptor interaction with immune effector cells to hypercrosslink rituximab.[4,5] Moreover, studies have shown rituximab-mediated apoptosis signaling is independent of its Fc functions.[6] Consequently, a less toxic and more potent rituximab-based therapeutics that induces direct apoptosis without Fc fragments might hold great promise for clinical use.
Recently, inspired by the direct initiation of apoptosis after cell-surface receptor clustering, we have developed a two-step pretargeted nanotherapy for CD20 crosslinking (Figure 1).[7–12] In this system, the Fc fragment of rituximab was depleted and CD20 crosslinking was achieved based on the biorecognition of (1) a pretargeting component Fab′-MORF1 (anti-CD20 Fab′ conjugated with an morpholino oligonucleotide and (2) a crosslinking component P-(MORF2)X N-(2-hydroxypropyl)meth-acrylamide (HPMA) copolymer grafted with multiple copies of complementary morpholino oligonucleotide. Results have demonstrated the superior antilymphoma efficacy to rituximab in vivo. Because no small molecular drug or Fc region is involved and each individual component in this system is biocompatible, we hence named it “drug-free macromolecular therapeutics (DFMT).”
Figure 1.

Structures of complementary MORF1 and MORF2, and schematic illustration of the preparation of two pairs of DFMT nanoconjugates: A) Fab′-MORF1 and Fab′-MORF1-Cy5; B) P-(MORF2)x and P-(MORF2)x-Cy3; C) 3′-end modification of MORF1; D) 3′-end modification of MORF2.
With the consecutive processes of specific CD20 binding and multimeric crosslinking, our bioinspired system can generate apparent onset of apoptosis. However, how the apoptosis signal is triggered after CD20 hyper-crosslinking remains to be elucidated. A better understanding of apoptotic stimulation machinery can have broad implications to optimize the rituximab-mimetic platform or modulate augmented/downregulated intracellular signal combined with other agents. Here, in the present study, the following objectives were investigated: (1) As an indication for CD20 clustering,[13–15] could the self-assembly of DFMT at the cell surface via oligonucleotide hybridization trigger the accelerated endocytosis? (2) If internalized, would DFMT follow the well-defined recycling pathway of CD20[16] or redirect the intracellular trafficking to specific pathway? (3) How would the valence of the crosslinking polymer affect CD20 clustering? (4) Since CD20 functions as a store-operated calcium channel,[17,18] could DFMT induce calcium influx and is it essential for apoptosis induction? (5) Could DFMT mediate programmed cell death using the mitochondrial pathway where a variety of key events reside?[19] (6) Could DFMT maintain the ability to downregulate the Bcl-2, antiapoptosis proteins that contribute to drug resistance, given that rituximab was reported to sensitize chemotherapeutics by inhibiting Bcl-2?[20–22]
2. Experimental Section
2.1. Materials
The 3′-amine-derivatized 25-mer phosphorodiamidate morpholino oligonucleotide (MORF1) and its complementary 3′-disulfide amide 25-mer phosphorodiamidate morpholino oligomer (MORF2) were customized and purchased from Gene Tools (Philomath, OR). Rituximab (Genentech) was obtained from Hunstman Cancer Hospital, University of Utah at a stock concentration of 10 mg mL−1. N-(3-Aminopropyl)methacrylamide hydrochloride (APMA) was purchased from Polysciences (Warrington, PA). Cy3-/Cy5-NHS (N-hydroxysuccinimide) esters were purchased from Lumiprobe (Hallandale Beach, FL). The hetero-bifunctional linker succinimidyl-4-(N-maleimidomethyl) cyclohexane-1-carboxylate (SMCC) and succinimidyl-(N-maleimidopropionamido)-diethyleneglycol]ester (SM(PEG)2) were purchased from Highfine Biotech (Suzhou, China) and Thermo Fisher Scientific, respectively. Tris(2-carboxyethyl) phosphine (TCEP), carbonyl cyanide m-chlorophenylhydrazone (CCCP), and JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenz imidazolylcarbocyanine iodide) were also purchased from Thermo Fisher Scientific. 4,4-Azobis(4-cyanopentanoic acid) (V-501), pepsin, ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), and β-cyclodextrin (β-CD) were from Sigma-Aldrich (St. Louis, MO). All solvents were purchased from Fisher Scientific as the highest purity available (Pittsburgh, PA). HPMA[23] and 4-cyanopentanoic acid dithiobenzoate[24] were prepared as previously described. The initiator 2,2-azobis(2,4-dimethyl valeronitrile) (V-65) was purchased from Wako Chemicals (Richmond, VA).
2.2. Synthesis and Characterization of Nanoconjugates
2.2.1. Fab′-MORF1/Fab′-Cy5-MORF1
The conjugate Fab′-MORF1 and its fluorescent dye labeled conjugate were synthesized similarly as previously described.[7] Monoclonal antibody Rituximab was first digested into F(ab′)2 with 10% (w/w) pepsin in citric buffer (pH 4.0). F(ab′)2 was reduced to Fab′-SH using 20 × 10−3 M TCEP in 10 × 10−3 M PBS (pH 6) immediately before conjugation with MORF1. Each step was monitored using size-exclusion chromatography (SEC) to ensure the reaction was completed. An ÄKTA FPLC system equipped with Superdex 200 10/300 GL column (GE Healthcare) eluted with phosphate buffered saline (PBS, pH 7.2) at flow rate of 0.4 mL min−1 was used.
To conjugate MORF1 with 3′ primary amine to Fab′-SH, the end amino group of MORF1 was converted to maleimide group by reaction with 40× excess of hetero-bifunctional crosslinker SM(PEG)2. The reaction was performed at room temperature (RT) for 2 h. Excess SM(PEG)2 was removed by ultrafiltration (3000 Da molecular weight cut off (MWCO)) and product washed with deionized (DI) H2O four times. The MORF1-mal was lyophilized to yield a white powder. Maleimide content was measured using modified Ellman’s assay, and >90% conversion was achieved.
The conjugation of MORF1-mal to Fab′-SH was performed in 10 × 10−3 M PBS (pH 6) at RT for 2 h with the ratio of [mal]:[SH] = 1.1:1. The Fab′-MORF1 conjugate was purified using SEC to remove free, unconjugated Fab′ and MORF1. An ÄKTA FPLC system equipped with Sephacryl S-100 HR16/60 column (GE Healthcare) eluted with PBS (pH 7.2) was used. Fab′ equivalent concentration and MORF1 content in Fab′-MORF1 conjugate were determined using bicinchoninic acid protein assay (Pierce) and UV–vis spectra (solution in 0.1 N HCl at 260 nm on a Varian Cary 400 Bio UV–visible spectro-photometer, respectively.
To prepare Fab′-MORF1-Cy5, F(ab′)2 was labeled with Cy5-NHS by 2 h reaction with lysine side-chains at RT. F(ab′)2-Cy5 was purified using a PD 10 column to remove the unreacted dye, then followed above procedures.
2.2.2. Synthesis of P-(MORF2)x/P-(MORF2)x-Cy3
HPMA copolymer grafted with multiple copies of MORF2 was prepared via thiol-ene reaction (Figure 1B). The synthesis contains three steps: HPMA copolymer containing pendent amino groups (P-NH2) was first synthesized by reversible addition-fragmentation chain transfer (RAFT) copolymerization of HPMA with APMA; Polymer precursor (P-mal) was then obtained by reaction of P-NH2 with a hetero-bifunctional agent SMCC. Conjugation was achieved by attaching freshly reduced MORF2 (3′-primary terminated with thiol group) to P-mal.
As an example, HPMA (1 g, 7 mmol) and APMA (66 mg, 0.37 mmol) were weighed into a one-neck flask equipped with a magnetic stir bar. After purge with nitrogen, the monomers were dissolved with 5 mL DI H2O containing 0.1% (v/v) acetic acid. 4-cyanopentanoic acid dithiobenzoate as RAFT agent (100 μL with conc. 27 mg mL−1 in methanol) and 4,4′-azobiscyanovaleric acid (V501) as initiator (100 μL with conc. 9 mg mL−1 in methanol) were added via septum using a syringe. The flask was bubbled with N2 in ice bath for 30 min then put into 70 °C oil bath for 20 h. The copolymer was precipitated into acetone. After filtration, the copolymer was dissolved in methanol and reprecipitated into acetone to remove unreacted monomers. Over 900 mg pink powder was obtained (yield ≈85%). The average molecular weight and the polydispersity of the conjugates were determined by SEC on an AKTA FPLC system equipped with a UV detector (GE Healthcare), miniDAWN TREOS, and OptilabrEX (refractive index, RI) detector (Wyatt Technology) using a Superose 6 HR10/30 column with sodium acetate buffer containing 30% acetonitrile (pH 6.5) as mobile phase. The weight average molecular weight Mw was 99 kDa with polydispersity index (PDI) 1.14.
The dithiobenzoate end group was removed by radical-induced end-modification using excess of V-65 in methanol at 55 °C for 2 h. After precipitation into acetone and filtration, white powder was obtained. The polymer was further purified by dialysis (MWCO 6000–8000) against water over 16 h followed by lyophilization. The amine content in the copolymer P-NH2 was 366 nmol mg−1 (32 molecules of NH2 group per chain) as determined by ninhydrin test.
To conjugate 3′-thio-modified MORF2 to polymer backbone, the pendant amino group was further converted to maleimide group by reaction with SMCC in dimethylformamide (DMF) in the presence of a tertiary amine N,N-diisopropylethylamine (DIPEA) at RT for 2 h (molar ratio of [NH2]:[SMCC]:[DIPEA] = 1:1.5:3). The maleimide content of the precursor was 294 nmol mg−1 (26 molecules of maleimide groups per chain) as measured by modified Ellman’s assay.
To fluorescently label copolymer conjugate, 20 μL Cy3-NHS stock solution in dimethyl sulfoxide (DMSO) was added into copolymer P-NH2 in 0.1 M NaHCO3 (pH 8.2) with molar ratio of [Cy3]:[NH2] = 1:16 and stirred in dark at RT for 2 h. After reaction, the sample was first precipitated into acetone and then purified using PD-10 Sephadex G25 column (GE Healthcare) to remove free dye. To quantify Cy3 substitution, P-NH2-Cy3 was dissolved in PBS and the absorbance at 547 nm was determined on a UV–vis spectrophotometer (Agilent Cary Bio 400). The amount of Cy3 per chain was calculated based on the Cy3 standard curve. The rest of amino groups at the side chain were converted to maleimide groups as above described to yield P-mal-Cy3.
3′-disulfide amide derived MORF2 was reduced with TCEP (10 × 10−3 M, pH 7.0) in 10 × 10−3 M PBS (pH 5.1) below 40 °C for 30 min to generate free thiol end group. Then the resultant fragment 4-thiolbutanamide and excess TCEP were removed by ultrafiltration (3000 Da MWCO) and washed three times with 10 × 10−3 M NaH2PO4 (pH 5.1). The freshly prepared MORF2-SH was added into P-mal/P-mal-Cy3 solution in 10 × 10−3 M PBS and kept stirring at RT for 3 h. At the end, unreacted MORF2-SH was removed by ultrafiltration (30 000 Da MWCO), washed four times with DI water and freeze-dried.
Graft copolymers with different valences of MORF2 were obtained by varying the feed ratio of thiol with maleimide group. The content of MORF2 in the conjugates P-(MORF2)X or P-(MORF)X-Cy3 was determined by UV–visible spectrophotometry at 260 nm in 0.1 N HCl, and calculated based on the molar absorptivity of MORF2 (252120 M−1 cm−1, provided by the manufacturer Gene Tools). Four conjugates (with/without Cy3 labeling) were prepared; they are listed in Table 1.
Table 1.
Preparation and characterization of conjugates P-(MORF2)x/P-(MORF2)x-Cy3.
| Conjugate | Mw [kDa] | PDI | Valence | Cy3 content/chain |
|---|---|---|---|---|
| P-mal | 113 | 1.20 | 0 | N/A |
| P-MORF2-Low | 141 | 1.21 | 3.0 | N/A |
| P-MORF2-High | 197 | 1.18 | 9.5 | N/A |
| P-mal-Cy3 | 121 | 1.18 | 0 | 1.62 |
| P-MORF2-Cy3-Low | 149 | 1.21 | 3.1 | 1.29 |
| P-MORF2-Cy3-High | 214 | 1.18 | 11.2 | 1.52 |
2.3. Cell Culture
Human Burkitt’s B-cell non-Hodgkin’s lymphoma Raji cells (ATCC) were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS) and a mixture of antibiotics (100 units mL−1 penicillin, 0.1 mg mL−1 streptomycin) at 37 °C in a humidified atmosphere containing 5% CO2.
2.4. Flow Cytometry
2 × 105 Raji cells were treated with 1 × 10−6 M Fab′-MORF1-Cy5 for 1 h, then the cells were washed by PBS to remove the unbounded Fab′-MORF1-Cy5 and further incubated with 1 × 10−6 M P-(MORF2)x or P-(MORF2)x-Cy3 with different valences (0, 3, 10) for 1, 3, and 6 h, prior to the flow cytometry quantification using 488 nm excitation with 585/42 nm bandpass filter (BLUFL2 channel, for Cy3 detection) and 640 nm excitation with 670/30 nm bandpass filter (REDFL1 channel, for Cy5 detection). For Förster resonance energy transfer (FRET) signal measurement, the flow cytometry was applied with the laser excitation at 488 nm and emission at 675 nm. All experiments were carried out in triplicate.
2.5. Confocal Microscopy
For confocal microscopy imaging, 2 × 105 Raji cells were treated with 1 × 10−6 M Fab′-MORF1-Cy5 for 1 h, then the cells were washed with PBS to remove unbound Fab′-MORF1-Cy5 and further incubated with 1 × 10−6 M P-(MORF2)x or P-(MORF2)x-Cy3 for 6 h. Afterward, the lysosomes of the cells were stained with the lysotracker green DND-26 (Thermo Scientific) for 15 min and the nuclei of the cells were stained with 5 μg mL−1 Hoechst 33392 (Thermo Scientific) for 5 min. Then the cells were washed with PBS and resuspended in PBS in 4 well chambers prior to confocal visualization. For CD 20 blocking, Raji cells were pretreated with 20-fold excess of rituximab for 1 h and then coincubated with Fab′-MORF1-Cy5 for 1 h followed by the same procedures.
2.6. Calcium Influx Investigation
4 × 105 Raji cells were loaded with Fluo-3AM (5 × 10−6 M, Thermo Scientific), an intracellular calcium indicator in 100 μL 1640 cell culture medium (containing 2.5 × 10−3 M Ca2+) for 30 min at 37 °C. Then cells were further treated with Fab′-MORF1 (1 × 10−6 M) for 30 min. Afterward, cells were excited at 488 nm and emission was measured at 530 nm by flow cytometry. A baseline was obtained for 60 s before stimulation with P-(MORF2)x crosslinking. Immediately after the addition of P-(MORF2)x, quantitative changes in the intracellular Ca2+ were monitored by flow cytometry for 10 min with 20 s intervals. To determine the extracellular calcium influx or intracellular calcium release, extracellular calcium free condition was created by adding 10 × 10−3 M EGTA to 1640 medium.[25] To deplete cholesterol, cells were preincubated 1 h at 37 °C in the presence of β-CD (beta cyclodextrin; 2%).[17,18]
2.7. Mitochondrial Membrane Potential Study
JC-1 mitochondrial membrane potential sensor (Thermo Scientific) was used. 2 × 105 Raji cells were consecutively treated with Fab′-MORF1 (1 × 10−6 M, 1 h)/P-(MORF2)x (1 × 10−6 M, 24 h) in the presence or absence of various inhibitors. Then cells were washed by PBS and resuspended in 100 μL PBS. 2 μL of 200 × 10−6 M JC-1 was added and incubated the cells at 37 °C for 30 min. For the positive control group, 1 μL of 50 × 10−3 M CCCP was added and incubated simultaneously with JC-1 for 30 min. After washed by PBS, cells were resuspended in PBS and analyzed by flow cytometry using 488 nm excitation with 530/30 nm and 585/42 nm band-pass filters or observed under confocal microscopy. All experiments were carried out in triplicate.
2.8. Bcl-2 Detection
2.5 × 105 Raji cells were treated with cell culture medium (control) or consecutive treatment Fab′-MORF1 (1 × 10−6 M, 1 h)/P-(MORF2)x (1 × 10−6 M, 24 h). Then cells were sequentially fixed by 4% paraformaldehyde for 15 min at room temperature, permeabilized by 90% methanol for 30 min on ice, and immunostained by Alexa Fluor 488 conjugated Bcl-2 mAb (1:500, Santa Cruz Biotechnology) in 0.5% bovine serum albumin (BSA) buffer for 1 h at room temperature. After washing by PBS, the fluorescence was quantified by flow analysis. All experiments were carried out in triplicate.
2.9. Cytochrome c Release Measurement
After treatments with PBS or Fab-MORF1/P-(MORF2)x, Raji cells were collected by centrifugation. Then cells were washed twice by cold PBS and cell pellet was lysed by cell extraction buffer (with 1 × 10−3 M phenylmethanesulfonyl fluoride (PMSF) and 1:100 protease inhibitor cocktail) at 100 μL of 2.0 × 105 cells, for 30 min on ice. Then the extract was transferred to microcentrifuge tube and centrifuged at 13 000 rmp for 10 min at 4 °C. The activity was measured using an ELISA kit (R&D Systems) according to manufacturer’s instructions. All experiments were carried out in triplicate.
2.10. Apoptosis Assay
FITC Annexin V (also written as Annexin V-FITC, fluorochrome-labeled Annexin V, Biolegend) and propidium iodide (PI) staining were performed following the RAPID protocol provided by the manufacturer (Oncogene Research Products, Boston, MA). 2.5 × 105 Raji cells were treated with cell culture medium (control) or consecutive treatment Fab′-MORF1 (1 × 10−6 M, 1 h)/P-(MORF2)x (1 × 10−6 M, 24 h). Then cells were washed by PBS and stained with Annexin V-FITC and PI in dark for 15 min, followed by flow cytometry analysis.[10,26] All experiments were carried out in triplicate.
2.11. Caspase 3 Activity Measurement
A Phi-PhiLux kit (OncoImmunin, Gaithersburg, MD) was used. 2.5 × 105 Raji cells were treated with cell culture medium (control) or consecutive treatment Fab′-MORF1 (1 × 10−6 M, 1h)/P-(MORF2)x (1 × 10−6 M, 24 h). Then cells were washed by PBS and analyzed for caspase-3 activity following the manufacturer’s protocol. All experiments were carried out in triplicate.
3. Results and Discussion
3.1. Preparation of Two Biorecognizable Nanoconjugates
In this study, we changed conjugation chemistry from previously used attachment of MORF2-NH2 to polymer backbone via pendant thiazolidine-2-thione (TT) groups[7] to a thiol-ene reaction (Figure 1B). The advantage of this revised approach is multifold, in particular the reproducibility of controlled polymerization, the reaction efficiency of MORF2, and high commercial potential of DFMT. Copolymerization of HPMA with comonomer containing TT-group usually can only reach 50% yield, whereas preparation of P-NH2 has usually a 80–90% yield. SEC profiles of P-NH2 and P-mal indicated high Mw with narrow polydispersity (Figure 2B). The stability and reactivity of maleimide groups are superior to TT. In addition, the comonomer APMA is commercially available. It permits to scale-up the synthesis of polymer precursor P-NH2/P-mal without problems. The shortcomings compared with the original approach are two additional steps needed: MORF2-SH needs to be prepared freshly from its disulfide compound, and polymer precursor P-NH2 has to be reacted with a hetero-bifunctional linker SMCC.
Figure 2.

Physicochemical characterization of synthesized nanoconjugates A,B) Fab′-MORF1 and C,D) P-(MORF2)x. A) Size-exclusion chromatography (SEC) profiles of mAb rituximab, digested F(ab′)2, labeled F(ab′)2-Cy5, reduced Fab′-Cy5, and final conjugate Fab′-MORF1-Cy5 as determined on Superdex 200 10/300 GL column eluted with PBS (pH 7.2) at flow rate 0.4 mL min−1. B) UV spectra of F(ab′)2-Cy5 and Fab′-MORF1-Cy5. C) SEC profiles of precursors P-mal and P-mal-Cy3 and final conjugates P-(MORF2)x and P-(MORF2)x-Cy3 determined on Superose 6 10/300 GL column eluted with PBS (pH 7.2) at flow rate 0.4 mL min−1. D) UV spectra of P-(MORF2)x and P-(MORF2)x-Cy3.
3.2. DFMT Triggers CD 20 Clustering through Two-Step Consecutive Process
Using direct stochastic optical reconstruction microscopy we previously demonstrated the assembly/hybridization of Fab′-MORF1 and P-(MORF2)x conjugates at CD20 antigens on the cell surface.[27,28] It is still necessary to validate whether or not the CD20 is crosslinked after this two-step pretargeted process. CD20 clustering could be indicated by the CD20 redistribution from cell surface to intracellular compartments.[13,15] In this way, the Fab′ targeting and binding to the CD20 antigens is supposed to be followed by endocytosis into the cell. Thus, we conducted flow cytometry and confocal microscopy studies to investigate the internalization of Fab′-MORF1-Cy5 in the presence and absence of P-(MORF2)x multimeric binding, using CD20-expressing human NHL Raji B-cell line (Figure 3).
Figure 3.

Investigation of Fab′-MORF1-Cy5 internalization using flow cytometry and confocal microscopy. A) Flow cytometric histogram, B) relative fluorescence intensity, and C) confocal microscopy imaging of Fab′-MORF1-Cy5 alone (top) and Fab′-MORF1-Cy5 followed by P-(MORF2)10 (bottom). D) Z-stack confocal images of colocalization between Fab′-MORF1-Cy5/P-(MORF2)10 system and lysosome. Yellow color denotes the overlay of Cy5 fluorescence (red) and lysosome staining (green).
Exposure of Raji cells to Fab′-MORF1-Cy5 alone for 5 h incubation only resulted in a surface-bound ring pattern around the targeted cell, due to the Fab′-CD20 biorecognition, but with minimal internalization (Figure 3A). This is because CD20 is noninternalizing/slow-internalizing receptor when it is not hyper-crosslinked.[9,29] By contrast, the sequential addition of P-(MORF2)10 could greatly facilitate the cell uptake of Fab′-MORF1-Cy5 which increased by 2.5-fold, as quantified by flow cytometry (Figure 3B). This could be explained by the resultant crosslinking of CD20 with subsequent internalization, as further confirmed by confocal microscopy (Figure 3C).
Next, we evaluated the intracellular fate of the DFMT system after its endocytosis. Mounting evidence has indicated surface antigen crosslinking triggered by antibodies shifts the endosomal localization of the protein from a recycling pathway to a degradative pathway (lysosome).[15] Hence, 3D confocal microscopy was applied to observe the colocalization between DFMT and lysosome. As shown in Figure 3D, a large proportion of Cy5 fluorescence from DFMT that entered into the cell overlapped with the lysosome. The substantial lysosomal localization of DFMT indicated a redirected pathway distinct from the normal CD20 recycling pathway, which provided additional support that CD20 has been hyper-crosslinked after the two-step DFMT treatment.
3.3. DFMT Internalization is CD20 Specific, Time-Dependent, and Mediated by P-(MORF2)x Valence
To visualize the cell surface biorecognition of two-step DFMT system, Raji cells were consecutively treated with Fab′-MORF1-Cy5 and P-(MORF2)10-Cy3. Confocal microscopy results showed a substantial colocalization of Cy5 signal and Cy3 in the absence of rituximab (Figure 4A), whereas the fluorescence of both signals was barely observable when the cells were pretreated with rituximab. Flow cytometry results also indicated a substantially reduced amount of cells in Cy3/Cy5 double positive quadrant when CD20 was blocked with rituximab (Figure 4B). These results demonstrated the two-step DFMT system specifically assembled at cell surface CD20 antigens.
Figure 4.

A) Flow cytometric analysis and confocal microscopy imaging of Raji cells consecutively treated with Fab′-MORF1-Cy5 (1 h) and P-(MORF2)10-Cy3 (6 h). B) For blocking purpose, the Raji cells were pretreated with excess (20-fold) rituximab for 1 h and then coincubated with the Fab′-MORF1-Cy5 (1 h) followed by P-(MORF2)10-Cy3 (6 h). The nucleus of the Raji cells was stained using Hoechst 33392. Cy3, green; Cy5, red; nucleus, blue. C) The internalization kinetics of the consecutive treatment. Raji cells were treated with Fab′-MORF1-Cy5 for 1 h, and then treated with P-(MORF2)10-Cy3 for 1, 3, or 6 h prior to flow cytometry analysis. D) The effect of multivalence on CD20 crosslinking (internalization kinetics) of DFMT systems. Raji cells were incubated with the mixture of Fab′-MORF1-Cy5 and P-(MORF2)x-Cy3 with different valence (0, 3, 10) for 1, 3, and 6 h prior to flow cytometry analysis. E) Intracellular FRET measurement at different time intervals after Raji cells were treated with the mixture of Fab′-MORF1-Cy5 and P-(MORF2)10-Cy3 for 1h (Prem); or consecutively treated with Fab′-MORF1-Cy5 for 1 h followed by P-(MORF2)10-Cy3 for 1 h (Cons). Flow cytometry was used with the laser excitation at 488 nm and emission at 675 nm.
We next investigated the internalization kinetic of the two-step DFMT system (Figure 4C). After 1 h incubation of Fab′-MORF1-Cy5, almost 80% of Raji cells were decorated with Fab′-MORF1-Cy5 and this remained unchanged (two Cy5+ quadrants on the right) as time extended after removal of media containing Fab′-MORF1-Cy5. Meanwhile, upon the addition of P-(MORF2)10-Cy3, nearly no cell dot appeared in single Cy3+ quadrant (upper quadrant on the left), indicating that CD20 binding by Fab′-MORF1 was prerequisite for the second-step P-MORF2 binding. As the incubation time progressed from 1 to 6 h, the number of cell dots shifted substantially from Cy3–Cy5+ quadrant (lower quadrant on the right) to Cy3+Cy5+ quadrant (upper quadrant on the right) where the double positive fluorescence intensity increased gradually, indicating the two-step hybridization between P-(MORF2)10-Cy3 and Fab′-MORF1-Cy5 that eventually crosslinked CD20 and triggered internalization.
We also investigated whether the DFMT internalization correlates with the multivalence (Figure 4D). Raji cells were treated with the mixture of Fab′-MORF1-Cy5 and P-(MORF2)x-Cy3 with different valence (x = 0, 3, 10) for 1, 3, and 6 h prior to flow cytometry analysis. Results showed that the high valence of P-(MORF2)10-Cy3 triggers the fastest and highest internalization of DFMT system while the absence of MORF2 (using P-Cy3—an HPMA copolymer labeled with Cy3) led to modest internalization, as indicated by the cell population in the Cy3/Cy5 double positive quadrant. As the internalization is an indicator for the CD20 crosslinking, this result demonstrated that the multiple binding of P-(MORF2)10 to Fab′-MORF1 that was bound to CD20 facilitated the CD 20 crosslinking, which eventually resulted in elevated level of apoptosis. These data are in agreement with our previous study[10] that demonstrated that the higher the valence of the P-(MORF2)x conjugate the higher the apoptotic response, due to the enhanced biorecognition.
To investigate the cell surface self-assembly efficiency of the two-step conjugates, the intracellular FRET signal was measured by flow cytometery after Raji cells were first treated with Fab′-MORF1-Cy5 for 1 h, followed by the removal by PBS washing and secondary treatment of P-(MORF2)10-Cy3 for 1, 3, 6 h (Figure 4E). Results have shown that the FRET efficiency increased as incubation time increased. This result further confirmed that after Fab′-MORF1 binds to CD20 antigen on cell surface, P-(MORF2)x gradually binds to Fab′-MORF1, which can subsequently trigger the CD20 crosslinking and endocytosis into the cell.
To investigate the intracellular stability of DFMT system, the FRET signal was detected 1, 3, 6 h after Raji cells were incubated for 1 h with the premixure of Fab′-MORF1-Cy5 and P-(MORF2)10-Cy3 (Figure 4E). The FRET signal only decreased negligibly indicating the DFMT internalized into the cell as a whole rather than separately as Fab′-MORF1 and P-(MORF2)x and had relative stability inside the cell.
3.4. DFMT Triggers Extracellular Calcium Influx via CD20 Crosslinking to Induce Apoptosis
As many previous reports described,[17,18,30] CD20 functions as a store-operated calcium channel and hyper-crosslinking of CD20 by antibodies bound to the cell surface results in an increase in calcium conductivity. We then investigated the effect of DFMT on the induction of calcium-associated apoptosis (Figure 5). After DFMT treatment, a rapid rise in the intracellular Ca2+ concentration took place in the beginning and then maintained at a relative high level compared with the control (Figure 5A). We also demonstrated that extracellular Ca2+ influx prevailed over the release of Ca2+ from intracellular stores and became the major source of this intracellular Ca2+ increment, as cells pretreated with EGTA, an extracellular chelating agent of Ca2+, showed very small Ca2+ elevation. Furthermore, the pretreatment with β-CD, the inhibitor of lipid rafts essential for CD20 crosslinking, completely blocked the Ca2+ influx, revealing that CD20 crosslinking is required for the DFMT triggered calcium influx. Confocal microscopy imaging results in Figure 5B displayed the same trend, further confirming DFMT triggered extracellular calcium influx through CD20 crosslinking.
Figure 5.

A) Flow cytometric analysis of time-dependent change in intracellular Ca2+ represented by Fluo-3AM fluorescence intensity. Raji cells pretreated with Fab′-MORF1 and loaded with Fluo-3AM were excited at 488 nm and emission was measured at 530 nm on flow cytometry. A baseline was obtained for 60 s before stimulation with P-(MORF2)x crosslinking (indicated by black arrow). B) Confocal microscopy visualization of Raji cells stained with Fluo-3AM (green fluorescence) after DFMT treatment for 1 h. C) Apoptosis evaluation of DFMT with or without various inhibitors by Annexin V/PI method. To determine the extracellular calcium influx or intracellular calcium release, extracellular calcium free condition was created by adding 10 × 10−3 M EGTA to 1640 medium. To deplete cholesterol and inhibit CD20 crosslinking, cells were preincubated 1 h at 37 °C in the presence of β-CD.
Numerous findings revealed a crucial role for Ca2+ in apoptosis induction.[31] Consistently, our results in Figure 4C showed the apoptosis induced by DFMT was remarkably blocked by both β-CD and EGTA. This result demonstrated the apoptosis induced by DFMT required CD20 crosslinking and involved Ca2+ influx dependent signaling pathway.
3.5. DFMT Triggers Mitochondrial Depolarization
One consequence of rapid calcium influx is a loss of mitochondrial membrane potential.[31,32] Thus we further exploited the impact of DFMT induced Ca2+ influx on mitochondrial depolarization using JC-1 staining (Figure 6). DFMT treatment resulted in a significant decrease in mitochondrial membrane potential, whereas the EGTA pretreatment group decreases the mitochondrial membrane potential to a lesser extent. It is worthy to note that EGTA did not completely prevent the mitochondrial depolarization induced by DFMT. This suggests the calcium influx might partially contribute to the mitochondrial depolarization after DFMT treatment. Other pathways, such as Bcl-2 phosphorylation, might also be involved. Meanwhile, β-CD pretreatment could also significantly inhibit the mitochondrial depolarization, suggesting CD20 crosslinking is the pre-requisite for initiating apoptosis pathways including the mitochondrial signal pathway and calcium-dependent signal pathway.
Figure 6.

Mitochondrial membrane potential measurement in Raji cells after DFMT treatment in the presence and absence of extracellular calcium chelating agent EGTA or CD20 crosslinking inhibitor β-CD, using JC-1 staining method. JC-1 dye exhibits potential-dependent accumulation in mitochondria, indicated by a fluorescence emission shift from green (≈529 nm) to red (≈590 nm). Consequently, mitochondrial depolarization is indicated by a decrease in the red/green fluorescence intensity ratio.
3.6. DFMT Triggers Programmed Cell Death via Mitochondrial Signal Pathway
Rituximab was reported to inhibit the p38 MAPK, NF-kB, ERK1/2, and Akt signaling pathways and led to the inhibition of Bcl-2 or Bcl-XL expression, thus resulting in chemosensitization.[ 20–22] More importantly, this sensitization should be distinguished from antibody-dependent cellular cytotoxicity (ADCC), and may be due to a direct effect of rituximab binding to CD20. Therefore, it would be interesting to investigate whether the DFMT system could also reduce the Bcl-2 expression, thereby reducing its protective effect against apoptosis. Impressively, results in Figure 7A showed DFMT resulted in strong inhibition of Bcl-2 protein compared with control (no treatment). This finding suggests that DFMT maintained Bcl-2 downregulation function of rituximab and could reset the susceptibility to apoptosis. In addition, the Bcl-2 inhibition could also partially account for the loss of mitochondrial membrane potential as observed above, because when the decrease in the ratio of Bcl-2/Bax is of sufficient magnitude in the mitochondrial outer membrane, the membrane permeability will collapse causing commitment to programmed cell death.[33]
Figure 7.

A) Bcl-2 inhibition, B) cytochrome c release, C) caspase 3 activation, and D) apoptosis induction in Raji cells after DMFT treatment.
Accompanied with the mitochondrial depolarization and membrane disruption, apoptogenic molecules such as cytochrome c could be released into cytosol to activate the caspase-dependent apoptosis.[34] We then evaluated this cascade process of apoptosis signal (mitochondrial signal pathway). Correlated well with the postulation, our results verified that DFMT treatment facilitates the release of cytochrome c (Figure 7B), which subsequently activated the caspase-3 (Figure 7C) and initiated apoptosis (Figure 6D).
3.7. Perspective and Conclusions
Rituximab is currently the mainstay for the treatment of B-cell NHLs. However, the clinically low response rate (<50%) and Fc-related side effects largely restrict its therapeutic value. Three major independent mechanisms were demonstrated to be involved in the antitumor efficacy of rituximab: (1) complement-dependent cytotoxicity (CMC), (2) ADCC, and (3) apoptosis.[16,20] Among these mechanisms, the degree that CMC contributes to clinical therapeutic response is not definite, but it is evident that the Fc-mediated complement activation is responsible for the severe adverse effects such as lung injuries[3] and cytokine-release syndrome[16] after rituximab infusion. ADCC is the most important effector mechanism for rituximab to eradicate cancer cells through either phagocytosis or cytotoxic granules release, but it requires the presentation by natural killer (NK) cells, granulocytes, and macrophages expressing Fc receptors.[20] Unfortunately, resistance of rituximab to ADCC is frequently observed because of conformational alterations in the CD20/rituximab complex, overexpression of HLA class I or downregulation of vitamin D that facilitates escape from NK cells, Fc receptor polymorphisms that result in decreased affinity between Fc–Fc receptor interaction, and deficient effector cells in patient.[16] Direct apoptosis induction by rituximab requires surface CD20 crosslinking, which also depends on the participation of Fc receptor bearing effector cells.
DFMT system presents a new paradigm for safe and effective NHL treatment. First, the absence of rituximab Fc fragment might potentially remove the Fc-related side effects. Second, the inactivity of immune effector cells to crosslink rituximab can be overcome and replaced by our biomimetic two-step system that biorecognizes and crosslinks surface CD20. Third, the apoptosis level can be amplified because of the high valence and multivalent CD20 binding in the DFMT system. Indeed, our results have demonstrated that Fab′-MORF1 pre-targets the CD20 via antigen-Fab′ biorecognition; P-(MORF2)x functions as the effector conjugate that binds to Fab′-MORF1, which facilitates the intracellular endocytosis of the system to lysosome as the result of efficient CD20 crosslinking. More-over, the CD20 crosslinking effect and subsequent apoptosis induction are also enhanced as the valence of P-(MORF2)x increases. This can be ascribed to the fact that multivalence leads to enhanced interaction with multiple CD20 receptors, thus producing dramatically augmented receptor ligation and consequent apoptotic response. This is advantageous over rituximab, as one effector cell (macrophages, NK cells) can only cluster a limited number of rituximab, thus rendering rituximab hyper-crosslinking in vivo impractical. Also the recruitment of effector cells and the Fc–Fc receptor interaction are likely to take more time.[13,16,20]
In the present study, two major pathways, calcium influx dependent pathway and mitochondrial signal pathway, were identified to induce DFMT mediated apoptosis. Both pathways are directed by the CD20 crosslinking. After CD20 crosslinking, DFMT triggers extracellular calcium influx, which subsequently decreases the mitochondrial membrane potential and initializes apoptosis. The mitochondrial depolarization also stems from the Bcl-2 inhibition mediated by DFMT, followed by the cytochrome c release and caspase activation. With the participation of the two-pronged mechanism, a programmed cell death is induced in response to DFMT treatment (Figure 8). A better understanding of how DFMT functions to induce apoptosis offers new opportunity to tailor the anti-CD20 strategies. For instance, the overexpression of antiapoptotic Bcl-2 proteins accounts for the multidrug resistance in many cancer cell lines. DFMT downregulates Bcl-2, thus resetting the susceptibility to apoptotic stimuli and resulting in the chemosensitization, when used in combination with chemotherapeutic drugs. In summary, direct apoptosis initiated by DFMT system is validated as evidenced by the induction of calcium influx and mitochondrial signaling pathways, and DFMT provides a potent approach to treat B-cell non-Hodgkin lymphomas.
Figure 8.
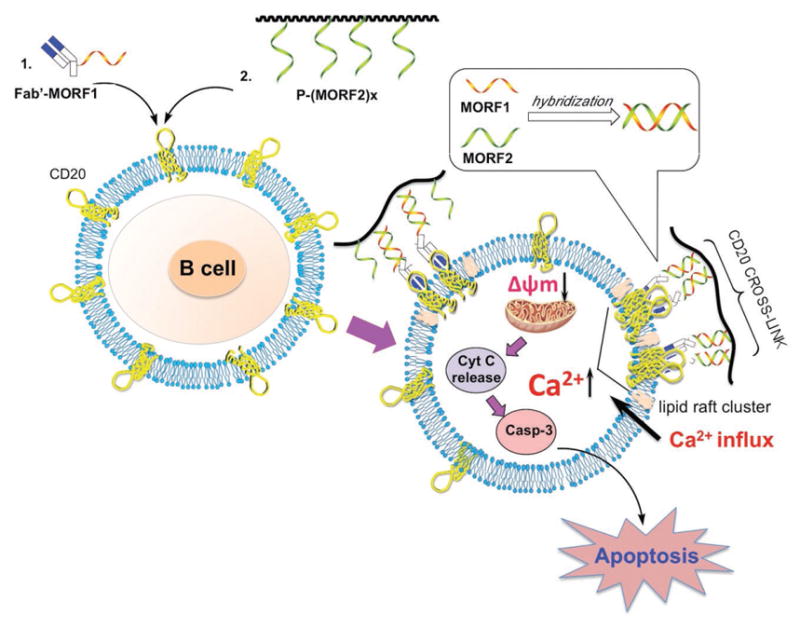
Schematic illustration of mechanisms of apoptosis induction. The rational for DFMT to induce apoptosis in B lymphoma cells is a consecutive two-step process: (1) CD20 pretargeting by Fab′-MORF1 and (2) CD20 crosslinking by P-(MORF2)x. Following biorecognition, CD20 antigens on cell surface were crosslinked. Two major pathways, calcium influx dependent pathway and mitochondrial signal pathway, were identified to induce DFMT mediated apoptosis.
The CD20 antigen is one of the most reliable biomarkers and ideal target for B-lymphocytes. Monoclonal antibodies (mAbs) targeted to the B-cell surface antigen CD20 have revolutionized the treatment of B-cell NHLs.[35] Currently, several anti-CD20 mAbs including rituximab, ofatumumab, obinutuzumab, tositumomab have been approved by the Food and Drug Administration (FDA) and applied clinically either alone or in combination therapy. Although CD20 is expressed on both normal and malignant B-cells from late pro-B-cells through memory cells, it is absent on the first and last stages of B-cell development: haematopoietic progenitor cells or stem cells and mature plasma cells do not express CD20.[36,37] Hence, B-cell depletion therapeutic strategy inevitably destroys normal B-cells; normal numbers of B-cells can be restored from the stem cells after treatment.[35,38] Therefore, these facts indicate the drug-free macromolecular therapeutic system can be considered relatively safe.
In summary, the answers to tasks mentioned in the Introduction are as follows:
Self-assembly of two nanoconjugates at the cell surface triggers accelerated endocytosis of crosslinked CD20 receptors.
Evaluation of subcellular trafficking of CD20 receptors demonstrated a shift from the recycling pathway to the lysosomal pathway.
The multivalent effect is operative in DFMT. The higher the valency of P-(MORF2)x, the more efficient is the CD20 crosslinking.
DFMT mediated CD20 clustering triggers the extracellular calcium influx into cytoplasm, which considerably contributes to apoptosis induction.
DFMT induces mitochondrial depolarization and initiates caspase-3 dependent apoptosis.
DFMT causes Bcl-2 downregulation enhancing the susceptibility to apoptosis.
Acknowledgments
The research was supported in part by NIH grant GM095606 and by Bastion Biologics.
Footnotes
Conflict of Interest
J.Y. and J.K. are co-inventors on a pending US patent application (PCT/US2014/023784; assigned to the University of Utah) related to this work. J.K. is chief Scientific Advisor and J.Y. is Scientific Advisor for Bastion Biologics. Otherwise, the authors declare no competing financial interests.
Contributor Information
Dr. Lian Li, Department of Pharmaceutics and Pharmaceutical Chemistry/CCCD, University of Utah, Salt Lake City, UT 84112, USA
Prof. Jiyuan Yang, Department of Pharmaceutics and Pharmaceutical Chemistry/CCCD, University of Utah, Salt Lake City, UT 84112, USA
Jiawei Wang, Department of Pharmaceutics and Pharmaceutical Chemistry/CCCD, University of Utah, Salt Lake City, UT 84112, USA.
Prof. Jindřich Kopeček, Department of Pharmaceutics and Pharmaceutical Chemistry/CCCD, University of Utah, Salt Lake City, UT 84112, USA. Department of Bioengineering, University of Utah, Salt Lake City, UT 84112, USA
References
- 1.Van der Kolk LE, Grill-López AJ, Baars JW, Hack CE, Van Oers MHJ. Br J Haematol. 2001;115:807. doi: 10.1046/j.1365-2141.2001.03166.x. [DOI] [PubMed] [Google Scholar]
- 2.Allison M. Nat Biotechnol. 2010;28:105. doi: 10.1038/nbt0210-105. [DOI] [PubMed] [Google Scholar]
- 3.Kamei K, Ito S, Iijima K. Pediatr Nephrol. 2010;25:1193. doi: 10.1007/s00467-009-1408-2. [DOI] [PubMed] [Google Scholar]
- 4.Smith MR. Oncogene. 2003;22:7359. doi: 10.1038/sj.onc.1206939. [DOI] [PubMed] [Google Scholar]
- 5.Cartron G, Dacheux L, Salles G, Celigny PS, Bardos P, Colombat P, Watier H. Blood. 2002;99:754. doi: 10.1182/blood.v99.3.754. [DOI] [PubMed] [Google Scholar]
- 6.Vega MI, Huerta-Yepez S, Martinez-Paniagua M, Martinez-Miguel B, Hernandez-Pando R, González-Bonilla CR, Chinn P, Hanna N, Hariharan K, Jazirehi AR, Bonavida B. Clin Cancer Res. 2009;15:6582. doi: 10.1158/1078-0432.CCR-09-1234. [DOI] [PubMed] [Google Scholar]
- 7.Chu TW, Yang J, Zhang R, Sima M, Kopeček J. ACS Nano. 2014;8:719. doi: 10.1021/nn4053827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chu TW, Zhang R, Yang J, Chao MP, Shami PJ, Kopeček J. Theranostics. 2015;5:834. doi: 10.7150/thno.12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chu TW, Kopeček J. Biomater Sci. 2015;3:908. doi: 10.1039/C4BM00442F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang L, Fang Y, Yang J, Kopeček J. J Controlled Release. 2017 doi: 10.1016/j.jconrel.2016.12.025. https://doi.org/10.1016/j.jconrel.2016.12.025. [DOI] [PMC free article] [PubMed]
- 11.Wu K, Liu J, Johnson RN, Yang J, Kopeček J. Angew Chem, Int Ed. 2010;49:1451. doi: 10.1002/anie.200906232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu K, Yang J, Liu J, Kopeček J. J Controlled Release. 2012;157:126. doi: 10.1016/j.jconrel.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang N, Khawli L, Hu P, Epstein A. Clin Cancer Res. 2005;11:5971. doi: 10.1158/1078-0432.CCR-05-0554. [DOI] [PubMed] [Google Scholar]
- 14.Chu TW, Kosak KM, Shami PJ, Kopeček J. Drug Delivery Transl Res. 2014;4:389. doi: 10.1007/s13346-014-0209-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moody PR, Sayers EJ, Magnusson JP, Alexander C, Borri P, Watson P, Jones AT. Mol Ther. 2015;23:1888. doi: 10.1038/mt.2015.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seyfizadeh N, Seyfizadeh N, Hasenkamp J, Huerta-Yepez S. Crit Rev Oncol Hematol. 2016;97:275. doi: 10.1016/j.critrevonc.2015.09.001. [DOI] [PubMed] [Google Scholar]
- 17.Janas E, Priest R, Wilde JI, White JH, Malhotra R. Clin Exp Immunol. 2005;139:439. doi: 10.1111/j.1365-2249.2005.02720.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Unruh TL, Li H, Mutch CM, Shariat N, Grigoriou L, Sanyal R, Brown CB, Deans JP. Immunology. 2005;116:223. doi: 10.1111/j.1365-2567.2005.02213.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spierings D, McStay G, Saleh M, Bender C, Chipuk J, Maurer U, Green DR. Science. 2005;310:66. doi: 10.1126/science.1117105. [DOI] [PubMed] [Google Scholar]
- 20.Cartron G, Watier H, Golay J, Solal-Celigny P. Blood. 2004;104:2635. doi: 10.1182/blood-2004-03-1110. [DOI] [PubMed] [Google Scholar]
- 21.Jazirehi AR, Bonavida B. Oncogene. 2005;24:2121. doi: 10.1038/sj.onc.1208349. [DOI] [PubMed] [Google Scholar]
- 22.Emmanouilides C, Jazirehi AR, Bonavida B. Cancer Biother Radiopharm. 2002;17:621. doi: 10.1089/108497802320970226. [DOI] [PubMed] [Google Scholar]
- 23.Kopeček J, Bažilová H. Eur Polym J. 1973;9:7. [Google Scholar]
- 24.Mitsukami Y, Donovan MS, Lowe AB, McCormick CL. Macro-molecules. 2001;34:2248. [Google Scholar]
- 25.Yao H, Zhang Y, Liu L, Xu Y, Liu X, Lin J, Zhou W, Wei P, Jin P, Wen LP. Biomaterials. 2016;108:143. doi: 10.1016/j.biomaterials.2016.08.036. [DOI] [PubMed] [Google Scholar]
- 26.Li L, Sun W, Zhang Z, Huang Y. J Controlled Release. 2016;232:62. doi: 10.1016/j.jconrel.2016.04.021. [DOI] [PubMed] [Google Scholar]
- 27.Hartley JM, Chu TW, Peterson EM, Zhang R, Yang J, Harris J, Kopeček J. ChemBioChem. 2015;16:1725. doi: 10.1002/cbic.201500278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hartley JM, Zhang R, Gudheti M, Yang J, Kopeček J. J Controlled Release. 2016;231:50. doi: 10.1016/j.jconrel.2016.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Michel RB, Mattes MJ. Clin Cancer Res. 2002;8:2701. [PubMed] [Google Scholar]
- 30.Deans JP, Li H, Polyak MJ. Immunology. 2002;107:176. doi: 10.1046/j.1365-2567.2002.01495.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Orrenius S, Zhivotovsky B, Nicotera P. Nat Rev Mol Cell Biol. 2003;4:552. doi: 10.1038/nrm1150. [DOI] [PubMed] [Google Scholar]
- 32.Duchen MR. J Physiol. 2000;529:57. doi: 10.1111/j.1469-7793.2000.00057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fletcher JI, Meusburger S, Hawkins CJ, Riglar DT, Lee EF, Fairlie WD, Huang DC, Adams JM. Proc Natl Acad Sci USA. 2008;105:18081. doi: 10.1073/pnas.0808691105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim R, Emi M, Tanabe K. Cancer Chemother Pharmacol. 2006;57:545. doi: 10.1007/s00280-005-0111-7. [DOI] [PubMed] [Google Scholar]
- 35.Cheson BD, Leonard JP. N Engl J Med. 2008;359:613. doi: 10.1056/NEJMra0708875. [DOI] [PubMed] [Google Scholar]
- 36.Stashenko P, Philip LM, Hardy R, Schlossman SF. Proc Natl Acad Sci USA. 1981;78:3848. doi: 10.1073/pnas.78.6.3848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stashenko P, Nadler LM, Hardy R, Schlossman SF. J Immunol. 1980;125:1678. [PubMed] [Google Scholar]
- 38.Maloney DG. N Engl J Med. 2012;366:2008. doi: 10.1056/NEJMct1114348. [DOI] [PubMed] [Google Scholar]