

Abstract
The plant metabolite salvinorin A potently and selectively agonizes the human kappa-opioid receptor, an emerging target for next-generation analgesics. Here we review analogs of the salvinorin chemotype and their effects on selectivity, affinity and potency. Extensive peripheral modifications using isolated salvinorin A have delivered a trove of SAR information. More deep-seated changes are now possible by advances in chemical synthesis.
Graphical abstract
Acute pain is an important learning device to discourage harmful behavior and allow injuries to heal. Chronic pain is generally detrimental to health and quality of life. Pain is a subjective experience and what qualifies as “chronic” is ill-defined, so that estimates of chronic pain prevalence vary. In the United States, between 301 and 100 million Americans suffer from chronic pain2; at either extreme, this is a dramatic proportion of the population (323 million total in 2016). Sufferers cannot merely ‘grin and bear it.’ The long-term sequelae associated with untreated chronic pain include sleep disruption, mental health deterioration and cardiovascular events. For instance, chronic pain intensity tracks with probability of hypertension and risk of cardiovascular morbidity.3
Both chronic and acute pain are treated with opioid analgesics, use of which has risen steadily since 1999.4 By 2016, the total number of prescriptions written in the US for opioid pain medications had surpassed 210 million, with over 60 million unique recipients5 (the per capita prescription rate should not escape the reader’s attention). These stark numbers actually represent a decrease in prescription rates from their peak in the years 2010–12.6 Nevertheless, as licit opioid use has grown, illicit use, abuse, and overdose fatalities have followed. Drug overdose is now the leading cause of death for Americans under the age of 55.7
Traditional opioids target the mu-opioid receptor (µ-OR), a transmembrane, G protein-coupled receptor (GPCR) named for its most famous agonist, morphine. This metabolite of the opium poppy plant Papaver somniferum was isolated in the early 1800s, an event that ushered in the beginnings of the pharmaceutical industry.8 Opium (the crude extract from P. somniferum) had been used for the entirety of recorded human history.9 But the diversification of the morphine structure into a broad class of analgesics,10,11 and the discovery of the opioid receptors,12 paved the way to a deeper appreciation—and capitalization—of neuroscience and pain. Morphinans have been developed to include selective agonists, selective antagonists, nonselective opioid agonists (multiple opioid subtypes bound, see below), mixed agonist-antagonists, peripherally restricted compounds, and agonists of other brain receptors like the N-methyl-D-aspartate (NMDA) receptor.13
Opioid Receptor Biology
Four major opioid receptor subtypes have been identified to date: in addition to µ-OR, there exist the kappa-opioid receptor (κ-OR), the delta-opioid receptor (δ-OR), and the nociceptin opioid peptide receptor (NOP). All four opioid receptors (including µ-OR) are G protein-coupled—they convert an extracellular signal into intracellular G protein activation. And all four are associated with G protein Gi/Go (although NOP is also associated with several other G proteins).14
When an agonist binds to an opioid receptor (Figure 1B), conformational changes of the receptor induce activation of the associated heterotrimeric G protein complex: GTP is exchanged for GDP and the Gα subunit disassociates from the Gβγ subunits. Activated Gαi/o inhibits adenylyl cyclases and cAMP production, while the freed β and γ subunits (which are permanently tethered together) can interact with ion channels (among them, pre- and postsynaptic Ca2+ channels, G protein-coupled inwardly rectifying K+ channels, and Na+ channels).15 The net cellular effect of opioid receptor activation is inhibitory—it hyperpolarizes the neuron. However, opioid receptors are heterogeneously expressed throughout the central and peripheral sensory neurons, and their variable locations are thought to partly explain the different, unwanted side effects observed between receptor types.11 For example, agonism of the µ-OR, κ-OR, or δ-OR can result in analgesia. However, µ-OR agonism infamously results in tolerance, dependence, reduced bowel motility, and respiratory depression. Agonism of κ-OR can result diuresis, dysphoria, hallucinations, and sedation. δ-OR agonism can cause physical dependence and convulsion.15
Figure 1.

A. Opioid receptors are membrane proteins with 7 transmembrane domains, associated with G protein Gi/o. B. Agonism of an opioid receptor can activate the associated G protein by exchanging GDP for GTP. The activated Gα subunit inhibits adenylyl cyclases, while Gβγ regulate various ion channels. C. Alternatively, another opioid active-state conformation can induce phosphorylation by a GPCR Kinase (GRK) and βarrestin recruitment.
Biased signaling further complicates this class. Opioid receptors are known to activate at least two distinct signaling pathways: G protein signaling and βarrestin recruitment. An alternative opioid receptor active-state conformation may lead to βarrestin recruitment,16,17 which is correlated with receptor phosphorylation, internalization,18 and desensitization (Figure 1C).15 Many of the non-analgesic effects of opioid agonists (but not all) have been correlated with βarrestin recruitment. For example, βarrestin recruitment has been correlated with developed tolerance and respiratory depression at µ-OR, and with dysphoria and stress at κ-OR.19 A biased agonist imparts GPCR signaling that favors one pathway over another—G protein-biased versus βarrestin-biased. Such ligands could help disentangle the multiple effects of opioid receptor activation.
Salvinorin A and the κ-OR
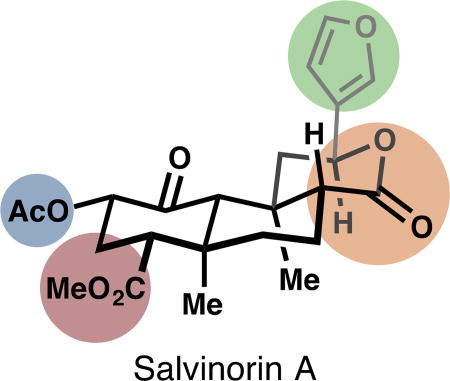
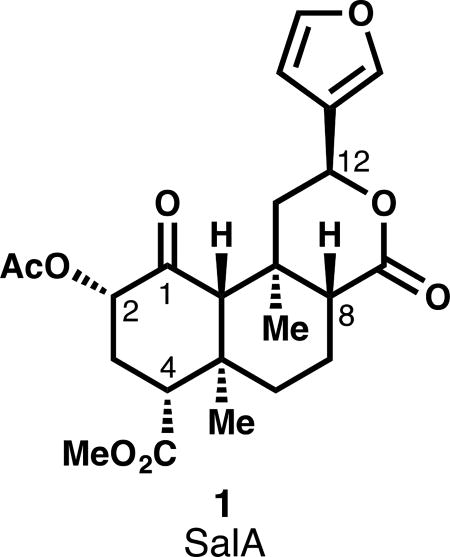
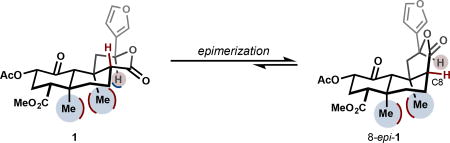
An opioid-strength analgesic devoid of side effects, especially addiction, continues to elude pain researchers. Recent research efforts in this area have focused on developing subtype-selective and G protein-biased agonists,20 as well as subtype-selective antagonists.21 Such compounds are desirable for their potential analgesia, anti-pruritis, and anti-depressive effects, among others.19 This review will center on analog development around the κ-OR agonist salvinorin A (SalA, 1, see inset), the active metabolite in the Mexican sage bush and recreational hallucinogen Salvia divinorum.22 Unlike traditional hallucinogens that exert their effects primarily through activation of the serotonin 5-HT2A receptor, SalA agonizes κ-OR with very high selectivity against other opioid receptors.23 Also unlike conventional CNS-active secondary metabolites, SalA contains no basic amines or nitrogen atoms at all, and therefore is not ionized at cellular pH. It passes through the blood-brain barrier quickly, achieving a max concentration at 40 seconds in primates.24 It clears the primate brain quickly, with a half-life of 8 minutes, which tracks nicely with the duration of hallucinogenic effects in humans.24 Its metabolism is still not completely understood. Ex vivo, the primary metabolite is salvinorin B, or the product of hydrolysis of the C2 acetate (15, Fig. 2).25,26 In vivo, Sal B also appears to be a major metabolite,27 but other metabolites have escaped characterization.28 In vivo PET imaging in baboons suggests at least two routes of excretion (renal and biliary), which suggests at least two modes of metabolism.24 In vitro studies of rat serum metabolism identified lactone hydrolysis as another viable metabolic pathway, catalyzed by calcium-dependent lactonases.29 Another source of instability is configurational: treatment of SalA with acid or base causes epimerization of the C8 carbon to favor a less active isomer (8-epi-1, see inset).30 This instability has required semi-synthetic medicinal chemistry efforts to discover mild enough conditions to avoid epimerization. In some cases, separation of the diastereomers is difficult, and for every new transformation, analysis of epimerization is necessary.
Figure 2.

Selected examples of C2-modified SalA analogs. Ki and EC50 are represented logarithmically: red bars represent greater than 1000-fold loss in affinity compared to SalA, whereas orange bars represent between 100- and 1000-fold loss, yellow between 10- and 100-fold loss, green between 1- and 10-fold loss, and blue represents an improvement vs. SalA.
Nearly all SalA analogs prepared to date are a result of semi-synthetic modification of the isolated natural product.31 This has resulted in a set of analogs that, while numerous, is not particularly diverse. Of about 600 analogs, a near-majority (295) involve substitution of the C2 acetate group, and 42% involve only a change at C2. An additional 33% of analogs involve a change to the furan ring; most of these are more sterically demanding than a one-for-one aryl ring replacement. Structure–activity relationships have been difficult to generalize, in part because a consensus binding model has not been established. Alanine mutagenesis has provided evidence of important contacts between κ-OR and SalA,32 but the docking model remains unconfirmed. A recent report detailed the first known active-state conformation of κ-OR17; the docked pose of SalA differs from poses derived from the inactive-state κ-OR structure, but requires experimental validation.
Comparison of the analogs is complicated by variation of biological assays between reports. Different studies include different types of data, sometimes omitting Ki, EC50, or κ-OR/µ-OR/δ-OR selectivity. Incomplete data sets paint an incomplete picture. Binding affinity (Ki) alone does not indicate potency: a ligand can bind well without stabilizing an active conformation. Also within each type of assay are additional sources of inconsistency. Ki has been measured with different radioligands, which can produce different binding constants for the same analog. Expression levels differ from experiment to experiment, which can result in different observed potencies. And EC50 has been measured by Ca2+ mobilization assays, GTP binding assays, or cAMP production inhibition assays, which demonstrate different responses of ligand binding. Even within the same study, SalA has shown a 2-fold change in Ki and EC50 from experiment to experiment.33 Some studies neglect measurement of a reference compound altogether (typically SalA or another known agonist, like U50,488H) but instead include a previously determined value for SalA. In other cases, the reported value for SalA or an analog identically matches those previously reported, but the authors do not explicitly note whether they are relaying the previous data or a new data point. Few studies include G protein versus βarrestin bias. Our review will present binding and activity data of the analogs relative to the reported SalA binding or activity, in an attempt to standardize these data. But differences still remain. Raw data from the original literature and a more comprehensive summary of all the analogs that have been reported are available in the Supporting Information.
Analogs at C2
As mentioned above, C2 acetate replacement is the most common alteration to SalA. Conditions for hydrolysis of the acetate group with minimal C8 epimerization, lactone hydrolysis, or methyl ester hydrolysis have been developed, which has afforded easy access to a broad sampling of substitution patterns here.34 Carbonates, carbamates, alternative ester groups, ethers, amines, amides, sulfonic esters, sulfonamides, thioesters, and halides have all been prepared and evaluated.
Some of the earliest analogs include alkyl esters. Propionate 2 demonstrates up to a 5-fold loss in affinity (one report shows its Ki as comparable to SalA) and a 3.5–7.5-fold drop in activity.33,35–37 Further increases in size result in further losses: isopropyl ester 3 loses affinity 10-fold37 while tert-butyl ester 4 is inactive.36 In the opposite direction, the decreased bulk of formic ester 5 results in around a 5-fold loss in affinity and a 7–11-fold loss in activity.38–40 The compound with inverted stereochemistry at C2 (2-epi-1) has produced widely discrepant data: reports range from 8.6- to 2800-fold less potent.33,41–43 Trifluoroacetate 644 and cyclopropyl ester 7 are inactive compounds.36,44 Analogs designed to be covalent binders like thiocyanate 8 or bromoacetate 9 improve on SalA’s affinity (3-fold and 1.2-fold better, respectively) and potency (250-fold and 1.5-fold better, respectively), but no verification of irreversible binding has been reported (see below).45
Bulkier aryl esters like 10–14 result in a loss of affinity and potency at κ-OR, but in many cases the scaffold gains µ-OR activity (discussion below).37,42,46–48 Substitution introduced to the benzene ring of 10, regardless of electronic effect (bromides, nitro groups, methoxy groups, a fluoride, carboxylic acid) or placement, is unable to recover κ-OR activity.46,47 The only aryl ester yet developed that falls within an order of magnitude of SalA’s affinity is the styrenyl ester 14 (3.3-fold worse affinity, 19-fold worse potency).48
Several non-ester derivatives at C2 have been prepared. The corresponding alcohol SalB (15) has had a range of values ascribed to it: complete loss of affinity36,49 versus an 85–150-fold decrease compared to SalA.33,35,44,47,50 One recent report even described it as 11-fold less potent, with a raw EC50 of 2.4 ± 0.8 nM.43 This single result stands in contrast to past reports and should be corroborated. Complete removal of the acetate group (16) has provided similarly conflicting reports—one paper describes a 35-fold loss in potency,39 while the other one reports complete inactivity at 10 µM.43 A series of halides (as in bromides 17 and 2-epi-17) experienced dramatic losses in potency: a 32-fold drop was the smallest decrease in the series.39 Halides are one of the few substituents with better affinity or potency as the unnatural C2 epimer. Equatorial bromide 17 has a Ki 10 times higher than that of axial bromide 2-epi-17.
Replacing the acetate ester with alkyl ethers has a deleterious effect: methyl ether 18 loses 120–170-fold affinity,33,35 while ethyl ether 19 binds 6–23-fold less efficiently.33,35,51 Larger alkyl ethers see worse losses (see SI). Similarly, introducing equatorial amine substituents results in compounds with 15–250-fold less affinity for κ-OR, but in contrast to the ether series (not shown), axial amine substituents are more active than their equatorial counterparts. For example, axial isopropylamine 2-epi-20 is similar in affinity (1.8-fold loss) and potency (1.6-fold loss) to SalA, while the equatorial version 20 binds 14–16-fold less efficiently and is 4.2-fold less potent.33,51 Introducing basic nitrogen atoms at other positions in the acetate chain was deleterious in general—a variety of protected and unprotected amino acids installed at C2 resulted in complete loss of activity.35,46,52 The only compounds in this series to demonstrate affinity at 1 µM, glycine 21 and valine 22, still lose around 50-fold affinity vs. SalA.35,46
Introduction of H-bond donors at this A-ring site generally hurts binding. Acetamide 23 binds to κ-OR 16–110-fold worse than SalA,33,47 whereas the corresponding N-methylacetamide 24 largely retains SalA’s binding (1.3–2.5-fold drop) and was reported to improve upon its potency (1.9-fold improvement).33,51 This trend holds in other comparisons of secondary and tertiary amides (see SI). Similarly, the H-bond donor in sulfonamides like 25 is problematic: this compound binds to κ-OR with 140-fold worse affinity,47 whereas the corresponding methyl sulfonic ester 26 is similar in affinity (1.2-fold worse) and potency (1.3-fold better) to SalA.37 The phenyl sulfonic ester 27 experiences a similar loss in κ-OR affinity to the phenyl ester 10 (32-fold loss46 vs. 47-fold loss,37 respectively). Other acetate replacements that are not H-bond donating can be safely introduced, as in thioacetate 28 (3–25-fold loss in affinity, 1.7-fold loss in potency).47,53
A series of unsaturated esters revealed an interesting feature of the SalA chemotype. While α,β-unsaturated esters, exemplified by 29, showed depressed affinity (6.2-fold worse binding), β,γ-unsaturated esters were more successful, as in 30 (3.4-fold worse binding) and 31 (2.1-fold worse binding). Aided by computational study, the authors determined that additional spacing enabled the alkene to occupy a small hydrophobic subpocket formed by Asp138, Tyr139, and Met142.48 However, since no consensus binding model has been reached, this conclusion is not definitive.
The types of analogs that most consistently improve affinities and potencies are alkoxyl methyl ethers, as in 32–34. These compounds mimic SalA with a modified oxidation state: the carbonyl of the acetate is replaced with a C—O single bond. In this class, MOM SalB (32) binds 1.8–4-fold more efficiently, and is 4.5–7.7-fold more potent than SalA.40,44,54,55 Six other alkoxyl methyl ethers have been reported with stronger affinities or potencies than SalA, including EOM SalB (33, 13–63-fold more potent).40,54 In general, restricting rotation of these compounds as in tetrahydropyran 34 does not significantly aid binding values (1.7-fold worse affinity than SalA in one report, 1.2-fold better affinity in another).40,54 One study explored additional C2 substitution, as in 35. This compound demonstrates a 3.6-fold loss in affinity relative to SalA, and therefore approximately a 10-fold loss relative to the parent MOM SalB.55 Other, larger substituents (see SI) were even worse, but the study did not extensively sample polar functionality along this vector.
A second H-bond acceptor as in malonate 36 seems beneficial—this compound demonstrates a 3-fold improvement in affinity relative to SalA. However, the trend does not translate to potency, as 36 is 27-fold less potent than SalA.49 Other malonic esters and different spacing of the carbonyls are detrimental (see SI). Lastly, restricting bond rotation of the acetate as in spiro-lactone 37 results in only a 2.9-fold loss in potency,43 a somewhat surprising result given the loss in activity demonstrated by 2-epi-1.
With active doses as low as 200 µg in humans,56 improvement of SalA potency is not necessarily the most important goal in a medicinal chemistry campaign. Instead, improvement of its metabolic lifetime, brain residence time and removal of its hallucinogenic effects take priority. In this vein, several of the above compounds stand out. N-methylacetamide 24 is more metabolically stable than SalA in vitro, both with human and rat liver microsomes, with or without the P450 co-factor NADPH.51 An in vivo assay with 24 demonstrated a 6-fold increase over SalA in the duration of its prodepressant-like effects. Mesyl SalB (26) also has longer-lived in vivo effects compared to the parent SalA: 26 increases the latency of tail-withdrawal in a hot water tail-withdrawal assay for 60 minutes, while SalA shows an impact only to 15 minutes.57
MOM SalB (32), EOM SalB (33), and THP Sal B (34) are all demonstrated to have improved metabolism. MOM SalB (32) was observed to have antinociceptive and hypothermic effects for 120 and 90 minutes, respectively, in contrast to SalA’s 20 minute antinociceptive effect.58 EOM SalB (33) was shown to be more metabolically stable than SalA in vivo with [11C]-labeled versions of both compounds. SalA was metabolized such that 50% of the observed radioactivity was from metabolites after 5 minutes in female baboons, while 33 took ~30 minutes to reach 50% metabolism.27 33 was not observed to have a longer brain residence time than SalA when both were administered intravenously (both cleared to half their maximum brain concentration in under ten minutes). However, when administered intraperitoneally, the brain concentration of 33 after 65 minutes was nearly triple that of SalA.27 These data indicate that the slower route of administration was more effective for the more metabolically stable compound, but that its increased stability did not translate to increased brain residence time. In another test, THP SalB (34) showed an antinociceptive effect in a hot water tail-withdrawal test for 45 minutes, in comparison to SalA’s 30 minute performance.59 EOM SalB and THP SalB were also observed to attenuate drug seeking behavior (specifically, cocaine) in vivo.60
Cinnamic ester 14 (termed “PR-38”), a mixed µ-OR and κ-OR (and cannabinoid) agonist is orally bioavailable, and preliminary results indicate a lack of CNS-associated locomotive side-effects.61 This compound and malonate 36 (termed “PR-37”) were both observed to have antipruritic effects in vivo. PR-37’s effects were counteracted by a κ-OR selective antagonist, while PR-38’s antipruritis was blocked by a µ-OR selective antagonist.62 The restricted spiro-lactone 37 is more stable when exposed to rat liver microsomes wi or without the co-factor NADPH (after 2.5 hours, about twice as much material remains vs. SalA), but the authors did not observe a longer-lived effect in vivo.43
Assays to reliably measure G protein bias have appeared more recently, and represent a minority set among assays used to evaluate SalA analogs. Structure–activity relationships to explain G protein bias have not been described for the SalA chemotype, and in general the differences between the two types of active states are not well understood. Thiocyanate RB-64 (8) demonstrates bias in favor of G protein signaling over βarrestin2 recruitment. As mentioned above, RB-64 was initially thought to be a covalent binder of κ-OR in vitro, but subsequent unpublished results indicate it may not be irreversibly binding in vivo63
Analogs at C4
In contrast to the acetate group at C2, selective cleavage of the methyl ester at C4 requires more forcing conditions that consistently epimerize C8 and require separation of these diastereomers. The C4 region of salvinorin may fit into a small pocket of the κ-OR bound by Trp-287 and Tyr-320,32 depending on binding orientation. Accordingly, analogs that are bulkier at this position bind less well. Conversion of the SalA (1) methyl ester to larger alkyl esters like propyl ester 38 results in a total loss of affinity (Ki > 1000 nM).64 Alternatively, changing the functionality to other related functional groups like a carboxylic acid (39)30,64,65 or an aldehyde (40)65 also causes a complete loss of affinity. The carboxylate anion may introduce unfavorable electron repulsion, and the aldehyde may introduce unfavorable H-bond as the hydrate. The corresponding alcohol 41 has produced conflicting reports. One report found its affinity reduced 87-fold versus SalA, but its potency non-existent to 10 µM—the profile of an antagonist.38 Another report found it to bind to κ-OR 380-fold worse than SalA, and to produce a partial response (68%) at 10 µM.33 In contrast, Ref. 65 determined this alcohol to bind the κ-OR only 33-fold worse than SalA, and produce a full activity response with an EC50 only 4.6-fold higher than SalA. To the best of our knowledge, this disparity has not been resolved. While bulkier alkyl esters almost uniformly cause complete loss of binding, introduction of additional polar functionality can rescue some of that lost affinity. MOM ether 42, while comparable in size to the inactive propyl ester 38, retains some affinity (77-fold drop vs SalA) and activity (13-fold drop vs SalA).64
Replacing the methyl ester with an H-bond donor as in amide 43 results in a significant loss of affinity (540-fold). The loss in activity in this compound is much harsher: 43 only elicits a 71% response at 10 µM.33 This partial agonism may mirror that of H-bond donor alcohol 41, if the two different reported activities of 41 can be reconciled. Dimethylamide 44, without an H-bond donor, does not show any evidence of binding.33 Removing the carbonyl in favor of ethers (45)65 or amines (46, 47)33 results in a complete loss of affinity. Use of alcohol 41 to prepare esters with alternative regiochemistry as in acetate 48 predictably results in total loss of binding (12 compounds evaluated, see SI). The lone exception, cyclopropyl ester 49, has 170-fold worse binding to κ-OR and 80-fold lower activity, compared to SalA.65 Interestingly, increased steric bulk with amino acids did not uniformly remove all affinity: alanine derivative 50 only loses affinity by a factor of 21, and it loses activity by a factor of 10. Serine derivative 51, in contrast, was completely inactive.64
Scaffold Analogs
Deep-seated changes to the SalA scaffold are harder to come by. A few changes to the A and C rings have been reported. Removal of the C1 ketone in the A ring (52) causes a modest loss (3–6.2-fold) in potency and affinity (4.5-fold), whereas introduction of an H-bond donor as in alcohol 53 results in a steeper loss in affinity (280-fold).38,66 In fact, alcohol 53 was reported in one publication as an antagonist of κ-OR.66 Protecting this alcohol did recover some activity, as in mesylate 54 (60-fold less active than SalA).66 Fragmentation and more freedom of rotation in the A ring resulted in 56–140-fold losses in activity, as exemplified by acetal 55 (56-fold less active, see SI for others).43 Modification of the geometry of the A ring by introducing unsaturation (56, 57) also harmed activity at κ-OR. Acetate 56 was 14-fold less active than SalA, but benzoate 57 (termed “kurkinorin”) was reported to be completely inactive at κ-OR (EC50 > 10 µM) while improving µ-OR activity.42 In comparison to the saturated benzoate 10 (Fig. 2), this is a dramatic change in activity, and improvement in selectivity (see µ-OR section below).
The C-ring lactone of SalA has been the target of even less derivatization. In general, alteration of the lactone results in a modest loss in affinity or potency: tetrahydropyran derivative 58 has 1.5-fold less affinity for κ-OR, and 4.8-fold lower activity; lactol 59 has 15-fold lower affinity, and 1.7-fold lower activity; and dihydropyran 60 has 1.5-fold worse affinity, and 14-fold worse activity.38 Fragmentation of this ring, as in 61 (480-fold loss in affinity) generally results in steeper drops in affinity.50
A variety of chemical conditions cause epimerization of the C8 carbon (see Table 1). These analogs are often tested when they arise, and are in general significantly less active than their 8-nat counterparts. The majority of them show no affinity (Ki > 1 µM) at κ-OR. A few select examples show improvements against their counterpart, but none are within an order of magnitude of SalA’s affinity or activity (see SI).
Table 1.
Conditions in which C8 epimerization have been observed
| Conditions: | Reference: |
|---|---|
| NH3, MeOH, 0 °C | 37 |
| Na2CO3, MeOH, 0 °C | 37 |
| NaHCO3, DMPU or DMF, 150 °C | 38 |
| K2CO3, MeOH, 0 °C | 44 |
| Alyklamide oxime, EDCI, HOBt, CH2Cl2, 23 °C; | 50, 67, 68 |
| PhMe, 110 °C | |
| CCl4, PPh3, PhH, 80 °C | 53 |
| NaH, DMF, −40 °C | 55 |
| LiI, pyridine, 120 °C | 64, 65 |
| RSO2NH2, AcOH, 95 °C | 67 |
| RB(OH)2, Pd2(dba)3, SPhos, K3PO4, PhMe, 100 °C | 69 |
| LiOH | 70 |
| NaOH, MeOH/H2O; 1M HCl; Ac2O, pyridine | 71 |
| neat, 248 °C | 72 |
Analogs at the Furan
The furan ring in SalA has been another desirable target for alteration, in part because of the well-known metabolic lability of furans.73 The role of the furan—H-bonding, pi-stacking, or hydrophobic interactions—has not yet been fully elucidated, but its importance is clear: complete removal of substitution at C12, as in methylene analog 62, results in an 1800-fold loss in affinity.50 Alternating the regiochemistry of the furan (63), which could disrupt or alter a hydrogen-bonding interaction, results in only a 1.2-fold loss in affinity and a 3.5-fold loss in activity relative to the parent SalA.74 Hydrogenation of the furan ring (64), in contrast to the reduced and fragmented analog 61 (Fig. 4), results in only a moderate loss in affinity (7–39-fold) and activity (2.7-fold).38,50 One diastereomer (13R) retains more affinity (1.9-fold loss vs. SalA) than the mixture demonstrated.50 Installing the furan with the opposite stereochemistry at C12, as in analog 12-epi-1, causes a moderate loss in affinity (2.3–16-fold loss). In two reports, this compound demonstrates partial agonist behavior (67–73% maximum response) for G protein signaling68,75; a third report observes a full response (109% vs. (−)-U50,488) in the same test.74 Interestingly, 12-epi-1 demonstrated G protein signaling without effecting receptor internalization, but only when acting at the human κ-OR. At the mouse κ-OR, 12-epi-1 was internalization biased.76
Figure 4.

Selected examples of core modifications to SalA
One of the most effective strategies for selective alteration of the furan ring has been oxidative cleavage. The resulting carboxylic acid 75 has been used to prepare the majority of furan replacement analogs (as opposed to differently substituted furans, like 79–87). The aryl ketones prepared from this process in general experience a moderate loss in affinity and activity: furyl ketones 65 and 66 show an 11-fold and 20-fold loss in affinity, respectively.74 Activity was only tested for the 2-furyl regioisomer 65, but a 53-fold loss was observed. Thiophene analog 67 was more competent (4.2-fold less affinity, 16-fold less activity).74 Phenyl ketone 68 has 9.5-fold less affinity and is 57-fold less active. A series of substituted benzene rings lose at least an order of magnitude of affinity relative to SalA (see SI), with the lone exception being the meta-carboxamide analog 69 (5.4-fold less affinity, 18-fold less activity).74 Reduction of the ketone spacer as in alcohol 70 results in a compound with a slightly improved profile than the ketone (8-activity) fold less affinity, 17-fold less activity).68 Other secondary alcohols of this sort have not yet been reported.
The oxidative cleavage product has also been used to prepare a series of amides, esters, and ethers. A series of 22 secondary amides, exemplified by methyl amide 7168 or phenyl amide 72,77 were found to have a Ki above 1 µM. Tertiary amides, as in piperidine 73, did not demonstrate a similar drop (73 has 19-fold less affinity and 120-fold less activity than SalA).77 Other compounds which remove the H-bond donor of the secondary amides, like methyl ester 74, also recover affinity at κ-OR (74 has 62-fold less affinity and 170-fold less activity than SalA).68 Carboxylic acid 75, presumably bound as the negatively charged carboxylate, demonstrates an even better profile (22-fold worse affinity, 80-fold worse activity) than the series of esters.68 Methyl ether 76 has 200-fold less affinity than SalA; other related ethers and esters demonstrate a similar profile.68
The furan ring has also been selectively altered by Diels–Alder reaction with alkynes to prepare oxanorbornadiene derivatives like 77 (see SI). These compounds could be converted to the corresponding substituted benzene analog, as in 78. In general, these substituted benzene rings demonstrate a moderate loss in affinity (78 shows a 39-fold loss in affinity). Surprisingly, the more sterically demanding oxanorbornadienes show a better profile than their benzene counterparts: 77 only loses 8-fold affinity vs. SalA.78
Selective substitution of the furan ring of SalA has been achieved via the bromination product 79. This compound itself displays a similar profile to SalA (1.2–1.6-fold worse affinity, 1.1–1.3-fold loss in activity),50,68,79 and has been used as a coupling partner to further substitute the furan ring. Phenyl substitution (80) results in a 2.5–43-fold loss in activity69,79; more substituted benzene rings, regardless of electronics, were worse (see SI). The lone exception, the ortho-fluoro analog, was roughly equipotent (1.2 ± 0.1 nM vs. 1.3 ± 0.4 nM) to the unadorned benzene ring.79 Polar substituents result in steep drops in potency, for example nitrile 81 is 310-fold less potent than SalA.79 Small nonpolar substituents (like cyclopropyl analog 82) result in compounds with better profiles than polar functionality, but they still experience moderate drops in activity—cyclopropyl analog 82 is 33-fold less active than SalA.79 Introduction of a trifluoromethyl group to C2 as in analog 83 results in a 1000-fold drop in potency, but an alternative regioisomer, compound 84, is an order of magnitude more potent than the C2 analog (100-fold less active than SalA).79 To the best of our knowledge, this is the only compound yet prepared to probe C5 substitution. Vinyl-substituted compound 85 demonstrates 2.2–32-fold lower potency,68,79 whereas the alkyne analog 86 is more active than the parent SalA (1.6-fold more potent).79 Any additional substitution to the alkyne results in a steep drop in potency (6 compounds, of which 87 is the most potent with an 870-fold drop in EC50).79
Recent Synthetic Analogs
SalA’s intriguing activity profile has made it a desirable target for total synthesis: four syntheses from three groups have been reported.80–83 These routes, while creative and aesthetically pleasing, are suboptimal for analog generation due to their length, throughput, and linearity. None of the original syntheses report activity data for any analogs. In 2017, our lab reported that deletion of the C20 methyl group allowed for a much shorter synthesis of the SalA scaffold and higher material throughput thanks to diminished C8 epimerization. The 20-nor-SalA target (20-nor-1) proved to be similarly active to SalA (6.8-fold drop in both affinity and potency).84 Its synthesis was divergent and enabled rapid analog development: benzene and thiophene analogs 90–91 had not been described previously in the parent system, but were available in 2 steps from intermediate 89. These simpler arene replacements were marginally more active than the previously described aryl ketones (66 & 68, iffinity Fig. 5). Phenyl analog 90 was 3.3-fold lower affinity than SalA, and experiences an 18-fold drop in potency. The 12-epi series, including 12-epi-20-nor-1, 12-epi-90, and 12-epi-91 have similar profiles (4.8–13-fold loss in affinity, 17-, 26-, or 93-fold loss in potency).84 This route also enabled the creation of more deep-seated changes to the scaffold. In particular, analog 92 featured a cyclohexanone in place of the C ring lactone; this change completely eliminated C8 epimerization and retained activity (1.3-fold less potent than U69,593, a benchmark agonist which is typically 4–10-fold less potent than SalA).85
Figure 5.

Selected examples of furan-modified analogs of SalA
An analog synthesis based on the 2006 Evans total synthesis reported SalA-inspired compound 94, which was 110-fold less potent than SalA.86 It is difficult to infer what causes this potency decline. C19,20 deletion or inclusion of a B ring hydrogen-bond donor may be offset by the beneficial MOM group at C2 (see 32, Fig. 2), whereas the methyl ether (see 45, Fig. 3) may exacerbate potency loss.
Figure 3.

Selected examples of C4-modified analogs of SalA
Analogs with µ-OR Activity
Early into SalA analog development, C2 aryl ester analogs were determined to increase affinity at µ-OR yet decrease κ-OR affinity and potency.46 This was first observed in phenyl ester 10, later termed “herkinorin.” Herkinorin has a Ki at µ-OR of 12 ± 1 nM, 7.5-fold lower than its affinity at κ-OR,46 and has been observed to avoid µ-OR receptor internalization and other βarrestin2 signaling pathways.87 The improved µ-OR profile extends to the series of substituted benzene rings reported (see SI), of which para-bromide 95 has the highest affinity for µ-OR (10 ± 1 nM) and the best selectivity (74-fold in favor of µ-OR over κ-OR).46,47 Other aryl rings like thiophene 11 or pyridine 13 display similar properties: both favor µ-OR binding by a factor of 26, thiophene 11 with a Ki at µ-OR of 10 ± 2 nM46,47 and pyridine 13 with a Ki at µ-OR of 73 ± 2 nM.37
In contrast to the deleterious nature of hydrogen-bond donating amides at C2 in κ-OR activity, phenyl amide analog 96 (termed “herkamide”) demonstrates strong activity at µ-OR: in one report its Ki was shown to be 3.1 ± 0.4 nM (selective over κ-OR by a factor of 2500),47 in another report its EC50 was determined to be 3.0 ± 0.6 nM (selective over κ-OR by a factor of 3300 and more potent than morphine at µ-OR in the same assay).42 Sulfonic esters like 27 were not found to be comparable to their ester counterparts in µ-OR activity (27 did not demonstrate any binding).46 PR-38 (14), a vinylogous aryl ester , demonstrates both µ-OR and κ-OR activity (see Fig. 2 above). As mentioned above (Fig. 4), unsaturation between C2 and C3, as in benzoate 57, changes the conformation of the compound such that κ-OR activity is completely lost, while µ-OR activity is improved.42 This compound, and the related series of substituted benzene esters (see SI), nearly completely reversed the natural selectivity of SalA. Unfortunately, 57 gained activity at δ-OR, and the series of related compounds tested did not improve upon its µ-OR/δ-OR selectivity.
Conclusions
The salvinorin A scaffold represents a promising, yet underdeveloped chemotype for modulation of opioid receptors, especially κ-OR.88 Several points of ambiguity remain. First, no consensus binding model has been settled: two recent papers describe opposite binding poses of SalA at the same site.17,84 Second, little pharmacology besides agonism has been observed and no predictive model developed. Third, unlike the morphinan class, SalA analogs lack predictable, discernable SAR trends for antagonism, partial agonism or biased agonism, although there is proof-of-principle for each. Finally, we could find no in vivo studies that identify all major metabolic degradation products of SalA (besides SalB); its short brain residence time may result from active efflux, or may point to an unknown metabolite. Regardless, application to chronic pain treatment requires that the short brain residence time of SalA be radically increased. Nevertheless, the inherent selectivity of SalA for κ-OR merely raise the profile of these challenges, heightening the reward, if not mitigating the risk. A bright spot on the horizon may be the two recent syntheses of the SalA scaffold, which will allow more diverse and more deep-seated scaffold modifications than the simpler peripheral changes already reported.84,86 Three important advances may be enabled by these chemical syntheses. First, covalent analogs of SalA may elucidate the binding site, pose and contacts within active17 and inactive89 receptor conformations. Second, diverse pharmacology besides mere agonism may become available to the SalA chemotype; biased agonism may be possible by taking structural lessons from other ligands. Third, the possibility of photochemical modulation of SalA binding presents the opportunity for temporal control of κ-OR signaling and all the lessons it may bring.90 To be sure, the future of the SalA scaffold is bright, illuminated largely by innovations in chemical synthesis and close ties with advanced pharmacology.
Supplementary Material
Figure 6.

Synthetic analogs of SalA
Figure 7.

Selected examples of µ-OR active analogs of SalA
Acknowledgments
We are grateful to the NIH (R35 GM122606) for financial support. We thank Professors Laura M. Bohn and Dale L. Boger for helpful conversations.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nahin RL. J. Pain. 2015;16:769–780. doi: 10.1016/j.jpain.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.U.S. National Institutes of Health. [Accessed December 2017];Pathways to Prevention Workshop: The Role of Opioids in the Treatment of Chronic Pain. 2014 Sep 29–30; https://prevention.nih.gov/docs/programs/p2p/ODPPainPanelStatementFinal_10-02-14.pdf.
- 3.Fine PG. Pain Med. 2011;12:996–1004. doi: 10.1111/j.1526-4637.2011.01187.x. [DOI] [PubMed] [Google Scholar]
- 4.U.S. Centers for Disease Control and Prevention. [Accessed December 2017];Vital Signs: Changes in Opioid Prescribing in the United States, 2006–2015. https://www.cdc.gov/mmwr/volumes/66/wr/mm6626a4.htm Published July 7, 2017.
- 5.U.S. Centers for Disease Control and Prevention. [Accessed December 2017];Annual Surveillance Reports of Drug-Related Risks and Outcomes — United States, 2017. https://www.cdc.gov/drugoverdose/pdf/pubs/2017-cdc-drug-surveillance-report.pdf Published August 31, 2017.
- 6.U.S. Centers for Disease Control and Prevention. [Accessed January 2018];U.S. Prescribing Rate Maps. https://www.cdc.gov/drugoverdose/maps/rxrate-maps.html Updated July 31, 2017.
- 7.Katz J, Goodnough A. The Opioid Crisis Is Getting Worse, Particularly for Black Americans. [Accessed December 2017];New York Times. https://www.nytimes.com/interactive/2017/12/22/upshot/opioid-deaths-are-spreading-rapidly-into-black-america.html Published December 22, 2017.
- 8.Cunningham CW, Rothman RB, Prisinzano TE. Pharmacol. Rev. 2011;63:316–347. doi: 10.1124/pr.110.003244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Merlin MD. Economic Botany. 2003;57:295–323. [Google Scholar]
- 10.Brownstein MJ. Proc. Natl. Acad. Sci. USA. 1993;90:5391–5393. doi: 10.1073/pnas.90.12.5391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Corbett AD, Henderson G, McKnight AT, Paterson SJ. Brit. J. Pharmacol. 2006;147:S153–S162. doi: 10.1038/sj.bjp.0706435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pert CB, Snyder SH. Science. 1973;179:1011–1014. doi: 10.1126/science.179.4077.1011. [DOI] [PubMed] [Google Scholar]
- 13.Pasternak GW, Pan Y-X. Pharmacol. Rev. 2013;65:1257–1317. doi: 10.1124/pr.112.007138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Al-Hasani R, Bruchas MR. Anesthesiology. 2011;115:1363–1381. doi: 10.1097/ALN.0b013e318238bba6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stein C. Annu. Rev. Med. 2016;67:433–451. doi: 10.1146/annurev-med-062613-093100. [DOI] [PubMed] [Google Scholar]
- 16.Rankovic Z, Brust TF, Bohn LM. Bioorg. Med. Chem. Lett. 2016;26:241–250. doi: 10.1016/j.bmcl.2015.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Che T, Majumdar S, Zaidi SA, Ondachi P, McCorvy JD, Wang S, Mosier PD, Uprety R, Vardy E, Krumm BE, Han GW, Lee M-Y, Pardon E, Steyaert J, Huang X-P, Strachan RT, Tribo AR, Pasternak GW, Carroll FI, Stevens RC, Cherezov V, Katritch V, Wacker D, Roth BL. Cell. 2018;172:1–13. doi: 10.1016/j.cell.2017.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reiter E, Ahn S, Shukla AK, Lefkowitz RJ. Annu. Rev. Pharmacol. Toxicol. 2012;52:179–197. doi: 10.1146/annurev.pharmtox.010909.105800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pradhan AA, Smith ML, Kieffer BL, Evans CJ. Brit. J. Pharmacol. 2012;167:960–969. doi: 10.1111/j.1476-5381.2012.02075.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Manglik A, Lin H, Aryal DK, McCorvy JD, Dengler D, Corder G, Levit A, Kling RC, Bernat V, Hübner H, Huang X-P, Sassano MF, Giguère PM, Löber S, Duan D, Scherrer G, Kobilka BK, Gmeiner P, Roth BL, Shoichet BK. Nature. 2016;537:185–190. doi: 10.1038/nature19112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carlezon WA, Jr, Krystal AD. Depression and Anxiety. 2016;33:895–906. doi: 10.1002/da.22500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ortega A, Blount JF, Manchand PS. J. Chem. Soc. Perkin Trans. I. 1982:2505–2508. [Google Scholar]
- 23.Roth BL, Baner K, Westkaemper R, Siebert D, Rice KC, Steinberg S, Ernsberger P, Rothman RB. Proc. Natl. Acad. Sci. USA. 2002;99:11934–11939. doi: 10.1073/pnas.182234399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hooker JM, Xu Y, Schiffer W, Shea C, Carter P, Fowler JS. NeuroImage. 2008;41:1044–1050. doi: 10.1016/j.neuroimage.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schmidt MS, Prisinzano TE, Tidgewell K, Harding W, Butelman ER, Kreek MJ, Murry DJ. J. Chromatog B. 2005;818:221–225. doi: 10.1016/j.jchromb.2004.12.041. [DOI] [PubMed] [Google Scholar]
- 26.Kutrzeba LM, Karamyan VT, Speth RC, Williamson JS, Zjawiony JK. Pharm. Biol. 2009;47:1078–1084. [Google Scholar]
- 27.Hooker JM, Munro TA, Béguin C, Alexoff D, Shea C, Xu Y, Cohen B. Neuropharmacology. 2009;57:386–391. doi: 10.1016/j.neuropharm.2009.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schmidt MD, Schmidt MS, Butelman ER, Harding WW, Tidgewell K, Murry DJ, Kreek MJ, Prisinzano TE. Synapse. 2005;58:208–210. doi: 10.1002/syn.20191. [DOI] [PubMed] [Google Scholar]
- 29.Tsujikawa K, Kuwayama K, Miyaguchi H, Kanamori T, Iwata YT, Inoue H. Xenobiotica. 2009;39:391–398. doi: 10.1080/00498250902769967. [DOI] [PubMed] [Google Scholar]
- 30.Munro TA, Duncan KK, Staples RJ, Xu W, Liu-Chen L-Y, Béguin C, Carlezon WA, Jr, Cohen BM. Beil J. Org. Chem. 2007;3:1. doi: 10.1186/1860-5397-3-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Prisinzano TE, Rothman RB. Chem. Rev. 2008;108:1732–1743. doi: 10.1021/cr0782269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vardy E, Mosier PD, Frankowski KJ, Wu H, Katritch V, Westkaemper RB, Aubé J, Stevens RC, Roth BL. J. Biol. Chem. 2013;288:34470–34483. doi: 10.1074/jbc.M113.515668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Béguin C, Richards MR, Li J-G, Wang Y, Xu W, Liu-Chen L-Y, Carlezon WA, Jr, Cohen BM. Bioorg Med. Chem. Lett. 2006;16:4679–4685. doi: 10.1016/j.bmcl.2006.05.093. [DOI] [PubMed] [Google Scholar]
- 34.Tidgewell K, Harding WW, Schmidt M, Holden KG, Murry DJ, Prisinzano TE. Bioorg. Med. Chem. Lett. 2004;14:5099–5102. doi: 10.1016/j.bmcl.2004.07.081. [DOI] [PubMed] [Google Scholar]
- 35.Béguin C, Richards MR, Wang Y, Chen Y, Liu-Chen L-Y, Ma Z, Lee DYW, Carlezon WA, Jr, Cohen BM. Bioorg. Med. Chem. Lett. 2005;15:2761–2765. doi: 10.1016/j.bmcl.2005.03.113. [DOI] [PubMed] [Google Scholar]
- 36.Chavkin C, Sud S, Jin W, Stewart J, Zjawiony JK, Siebert DJ, Toth BA, Hufeisen SJ, Roth BL. J. Pharmacol. Exp Ther. 2004;308:1197–1203. doi: 10.1124/jpet.103.059394. [DOI] [PubMed] [Google Scholar]
- 37.Harding WW, Tidgewell K, Byrd N, Cobb H, Dersch CM, Butelman ER, Rothman RB, Prisinzano TE. J. Med. Chem. 2005;48:4765–4771. doi: 10.1021/jm048963m. [DOI] [PubMed] [Google Scholar]
- 38.Munro TA, Rizzacasa MA, Roth BL, Toth BA, Yan F. J. Med. Chem. 2005;48:345–348. doi: 10.1021/jm049438q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee DYW, Yang L, Xu W, Deng G, Guo L, Liu-Chen L-Y. Bioorg. Med. Chem. Lett. 2010;20:5749–5752. doi: 10.1016/j.bmcl.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Prevatt-Smith KM, Lovell KM, Simpson DS, Day VW, Douglas JT, Bosch P, Dersch CM, Rothman RB, Kivell B, Prisinzano TE. Med. Chem. Commun. 2011;2:1217–1222. doi: 10.1039/C1MD00192B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harding WW, Schmidt M, Tidgewell K, Kannan P, Holden KG, Gilmour B, Navarro H, Rothman RB, Prisinzano TE. J. Nat. Prod. 2006;69:107–112. doi: 10.1021/np050398i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Crowley RS, Riley AP, Sherwood AM, Groer CE, Shivaperumal N, Biscaia M, Paton K, Schneider S, Provasi D, Kivell BM, Filizola M, Prisinzano TE. J. Med. Chem. 2016;59:11027–11038. doi: 10.1021/acs.jmedchem.6b01235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sherwood AM, Crowley RS, Paton KF, Biggerstaff A, Neuenswander B, Day VW, Kivell BM, Prisinzano TE. J. Med. Chem. 2017;60:3866–3878. doi: 10.1021/acs.jmedchem.7b00148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee DYW, Karnati VVR, He M, Liu-Chen L-Y, Kondaveti L, Ma Z, Wang Y, Chen Y, Beguin C, Carlezon WA, Jr, Cohen B. Bioorg. Med. Chem. Lett. 2005;15:3744–3747. doi: 10.1016/j.bmcl.2005.05.048. [DOI] [PubMed] [Google Scholar]
- 45.Yan F, Bikbulatov RV, Mocanu V, Dicheva N, Parker CE, Wetsel WC, Mosier PD, Westkaemper RB, Allen JA, Zjawiony JK, Roth BL. Biochemistry. 2009;48:6898–6908. doi: 10.1021/bi900605n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tidgewell K, Harding WW, Lozama A, Cobb H, Shah K, Kannan P, Dersch CM, Parrish D, Deschamps JR, Rothman RB, Prisinzano TE. J. Nat. Prod. 2006;69:914–918. doi: 10.1021/np060094b. [DOI] [PubMed] [Google Scholar]
- 47.Tidgewell K, Groer CE, Harding WW, Lozama A, Schmidt M, Marquam A, Hiemstra J, Partilla JS, Dersch CM, Rothman RB, Bohn LM, Prisinzano TE. Med. Chem. 2008;51:2421–2431. doi: 10.1021/jm701162g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Polepally PR, Huben K, Vardy E, Setola V, Mosier PD, Roth BL, Zjawiony JK. Eur. J. Med. Chem. 2014;85:818–829. doi: 10.1016/j.ejmech.2014.07.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Polepally PR, White K, Vardy E, Roth BL, Ferreira D, Zjawiony JK. Bioorg. Med. Chem. Lett. 2013;23:2860–2862. doi: 10.1016/j.bmcl.2013.03.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Simpson DS, Katavic PL, Lozama A, Harding WW, Parrish D, Descham ps JR, Dersch CM, Partilla JS, Rothman RB, Navarro H, Prisinzano TE. J. Med. Chem. 2007;50:3596–3603. doi: 10.1021/jm070393d. [DOI] [PubMed] [Google Scholar]
- 51.Béguin C, Potter DN, DiNieri JA, Munro TA, Richards MR, Paine TA, Berry L, Zhao Z, Roth BL, Xu W, Liu-Chen L-Y, Carlezon WA, Jr, Cohen BM. J. Pharmacol. Exp. Ther. 2008;324:188–195. doi: 10.1124/jpet.107.129023. [DOI] [PubMed] [Google Scholar]
- 52.Fichna J, Lewellyn K, Yan F, Roth BL, Zjawiony JK. Bioorg. Med. Chem. Lett. 2011;21:160–163. doi: 10.1016/j.bmcl.2010.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bikbulatov RV, Yan F, Roth BL, Zjawiony JK. Bioorg. Med. Chem. Lett. 2007;17:2229–2232. doi: 10.1016/j.bmcl.2007.01.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Munro TA, Duncan KK, Xu W, Wang Y, Liu-Chen L-Y, Carlezon WA, Jr, Cohen BM, Béguin C. Bioorg. Med. Chem. 2008;16:1279–1286. doi: 10.1016/j.bmc.2007.10.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee DYW, Deng G, Ma Z, Xu W, Yang L, Liu J, Dai R, Liu-Chen L-Y. Bioorg. Med. Chem. Lett. 2015;25:4689–4692. doi: 10.1016/j.bmcl.2015.06.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Siebert DJ. J. Ethnopharmacol. 1994;43:53–56. doi: 10.1016/0378-8741(94)90116-3. [DOI] [PubMed] [Google Scholar]
- 57.Simonson B, Morani AS, Ewald AWM, Walker L, Kumar N, Simpson D, Miller JH, Prisinzano TE, Kivell BM. Brit. J. Pharmacol. 2015;172:515–531. doi: 10.1111/bph.12692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang Y, Chen Y, Xu W, Lee DYW, Ma Z, Rawls SM, Cowan A, Liu-Chen L-Y. J. Pharmacol. Exp. Ther. 2008;324:1073–1083. doi: 10.1124/jpet.107.132142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Paton KF, Kumar N, Crowley RS, Harper JL, Prisinzano TE, Kivell BM. Eur. J. Pain. 2017;21:1039–1050. doi: 10.1002/ejp.1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ewald AWM, Bosch PJ, Culverhouse A, Crowley RS, Neuenswander B, Prisinzano TE, Kivell BM. Psychopharmacology. 2017;234:2499–2514. doi: 10.1007/s00213-017-4637-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Salaga M, Polepally PR, Sobczak M, Grzywacz D, Kamysz W, Sibaev A, Storr M, Do Rego JC, Zjawiony JK, Fichna J. J. Pharmacol. Exp. Ther. 2014;350:69–78. doi: 10.1124/jpet.114.214239. [DOI] [PubMed] [Google Scholar]
- 62.Salaga M, Polepally PR, Zielinska M, Marynowski M, Fabisiak A, Murawska N, Sobczak K, Sacharczuk M, Do Rego JC, Roth BL, Zjawiony JK, Fichna J. Brit. J. Pharmacol. 2015;172:4331–4341. doi: 10.1111/bph.13212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.White KL, Robinson JE, Zhu H, DiBerto JF, Polepally PR, Zjawiony JK, Nichols DE, Malanga CJ, Roth BL. J. Pharmacol. Exp. Ther. 2015;352:98–109. doi: 10.1124/jpet.114.216820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee DYW, He M, Kondaveti L, Liu-Chen L-Y, Ma Z, Wang Y, Chen Y, Li J-G, Beguin C, Carlezon WA, Jr, Cohen B. Bioorg. Med. Chem. Lett. 2005;15:4169–4173. doi: 10.1016/j.bmcl.2005.06.092. [DOI] [PubMed] [Google Scholar]
- 65.Lee DYW, He M, Liu-Chen L-Y, Wang Y, Li J-G, Xu W, Ma Z, Carlezon WA, Jr, Cohen B. Bioorg. Med. Chem. Lett. 2006;16:5498–5502. doi: 10.1016/j.bmcl.2006.08.051. [DOI] [PubMed] [Google Scholar]
- 66.Holden KG, Tidgewell K, Marquam A, Rothman RB, Navarro H, Prisinzano TE. Bioorg. Med. Chem. Lett. 2007;17:6111–6115. doi: 10.1016/j.bmcl.2007.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Harding WW, Schmidt M, Tidgewell K, Kannan P, Holden KG, Dersch CM, Rothman RB, Prisinzano TE. Bioorg. Med. Chem. Lett. 2006;16:3170–3174. doi: 10.1016/j.bmcl.2006.03.062. [DOI] [PubMed] [Google Scholar]
- 68.Béguin C, Duncan KK, Munro TA, Ho DM, Xu W, Liu-Chen L-Y, Carlezon WA, Jr, Cohen BM. Bioorg. Med. Chem. 2009;17:1370–1380. doi: 10.1016/j.bmc.2008.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Riley AP, Day VW, Navarro HA, Prisinzano TE. Org. Lett. 2013;15:5936–5939. doi: 10.1021/ol4027528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Paudel MK, Shirota O, Sasaki-Tabata K, Tanaka H, Sekita S, Morimoto S. J. Nat. Prod. 2013;76:1654–1660. doi: 10.1021/np400358n. [DOI] [PubMed] [Google Scholar]
- 71.Yang L, Xu W, Chen F, Liu-Chen L-Y, Ma Z, Lee DYW. Bioorg. Med. Chem. Lett. 2009;19:1301–1304. doi: 10.1016/j.bmcl.2009.01.078. [DOI] [PubMed] [Google Scholar]
- 72.Ma Z, Deng G, Lee DYW. Tet. Lett. 2010;51:5207–5209. doi: 10.1016/j.tetlet.2010.07.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.St Jean DJ, Jr, Fotsch C. J. Med. Chem. 2012;55:6002–6020. doi: 10.1021/jm300343m. [DOI] [PubMed] [Google Scholar]
- 74.Lovell KM, Vasiljevik T, Araya JJ, Lozama A, Prevatt-Smith KM, Day VW, Dersch CM, Rothman RB, Butelman ER, Kreek MJ, Prisinzano TE. Bioorg. Med. Chem. 2012;20:3100–3110. doi: 10.1016/j.bmc.2012.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Béguin C, Potuzak J, Xu W, Liu-Chen L-Y, Streicher JM, Groer CE, Bohn LM, Carlezon WA, Jr, Cohen BM. Bioorg. Med. Chem. Lett. 2012;22:1023–1026. doi: 10.1016/j.bmcl.2011.11.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.DiMattio KM, Ehlert FJ, Liu-Chen L-Y. Eur. J. Pharmacol. 2015;761:235–244. doi: 10.1016/j.ejphar.2015.05.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Simpson DS, Lovell KM, Lozama A, Han N, Day VW, Dersch CM, Rothman RB, Prisinzano TE. Org. Biomol. Chem. 2009;7:3748–3756. doi: 10.1039/b905148a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lozama A, Cunningham CW, Caspers MJ, Douglas JT, Dersch CM, Rothman RB, Prisinzano TE. J. Nat. Prod. 2011;74:718–726. doi: 10.1021/np1007872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Riley AP, Groer CE, Young D, Ewald AW, Kivell BM, Prisinzano TE. J. Med. Chem. 2014;57:10464–10475. doi: 10.1021/jm501521d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Scheerer JR, Lawrence JF, Wang GC, Evans DA. J. Am. Chem. Soc. 2007;129:8968–8969. doi: 10.1021/ja073590a. [DOI] [PubMed] [Google Scholar]
- 81.Nozawa M, Suka Y, Hoshi T, Suzuki T, Hagiwara H. Org. Lett. 2008;10:1365–1368. doi: 10.1021/ol800101v. [DOI] [PubMed] [Google Scholar]
- 82.Hagiwara H, Suka Y, Nojima T, Hoshi T, Suzuki T. Tetrahedron. 2009;65:4820–4825. [Google Scholar]
- 83.Line NJ, Burns AC, Butler SC, Casbohm J, Forsyth CJ. Chem. - Eur. J. 2016;22:17983–17986. doi: 10.1002/chem.201604853. [DOI] [PubMed] [Google Scholar]
- 84.Roach JJ, Sasano Y, Schmid CL, Zaidi S, Katritch V, Stevens RC, Bohn LM, Shenvi RA. ACS Cent. ScI. 2017;3:1329–1336. doi: 10.1021/acscentsci.7b00488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hirasawa S, Cho M, Brust TF, Roach JJ, Bohn LM, Shenvi RA. ChemRxiv. 2018 doi: 10.26434/chemrxiv.5728286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sherwood AM, Williamson SE, Crowley RS, Abbott LM, Day VW, Prisinzano TE. Org Lett. 2017;19:5414–5417. doi: 10.1021/acs.orglett.7b02684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Groer CE, Tidgewell K, Moyer RA, Harding WW, Rothman RB, Prisinzano TE, Bohn LM. Mol. Pharmacol. 2007;71:549–557. doi: 10.1124/mol.106.028258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cruz A, Domingos S, Gallardo E, Martinho A. Phytochemistry. 2017;137:9–14. doi: 10.1016/j.phytochem.2017.02.001. [DOI] [PubMed] [Google Scholar]
- 89.Wu H, Wacker D, Mileni M, Katritch V, Han GW, Vardy E, Liu W, Thompson AA, Huang X-P, Carroll FI, Mascarella SW, Westkaemper RB, Mosier PD, Roth BL, Cherezov V, Stevens RC. Nature. 2012;485:327–332. doi: 10.1038/nature10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Broichhagen J, Frank JA, Trauner D. Acc. Chem. Res. 2015;48:1947–1960. doi: 10.1021/acs.accounts.5b00129. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.