

Abstract
A series of natural products-based phenyl sulfone derivative and their property-based analogues were investigated as potential growth inhibitors of Trypanosoma brucei. Trypanosoma brucei is a kinetoplastid protozoan parasite that causes trypanosomiasis. In this work, we found that nopol- and quinoline-based phenyl sulfone derivative were the most active and selective for T. brucei, and they were not reactive towards the active thiol of T. brucei’s cysteine protease rhodesain. A thiol reactive variant of the quinoline-based phenyl sulfone was subsequently investigated and found to be a moderate inhibitor of rhodesain. The quinoline-based compound that is not reactive towards rhodesain can serve a template for phenotypic-based lead discovery while its thiol-active congener can serve as template for structure-based investigation of new antitrypanosomal agents.
Keywords: Trypanosoma brucei, Cysteine protease, Natural products, Quinoline
Graphical Abstract

Human African Trypanosomiasis (HAT), one of the neglected tropical diseases (NTDs), caused by protozoan Trypanosoma brucei is a declining public health problem on the African continent due to a gradual decrease in the number of reported cases in the past few years. It is most prevalent, at the moment, in the Democratic Republic of the Congo. Historically, the lack of adequate and rapid diagnostic tools as well as lack of effective, safe, and accessible medicines to treat HAT resulted in the death of hundreds of thousands of people. Despite the decrease in reported case, the lack of good network of primary healthcare facilities in most rural and remote places on the continent as well as the possibility of continuous transmission of the parasite from animal reservoirs to humans, make the disease a continuous threat to millions of people.1–4 Discovery and development of effective oral drugs remains a key objective in combating the disease. In this regard, a promising drug candidate, nitroimidazole fexinidazole, is in the approval stages for the treatment of human African trypanosomiasis. It would be the first approved oral medicine to treat human African trypanosomiasis in several decades. Fexinidazole is also being investigated as a potential treatment for Chagas Disease.5,6 Despite these recent gains, the drug development pipeline for HAT is sparse and there is need for continued investment and investigation into new chemical entities that can be developed as treatments and/or as prophylactic agents against the disease. Many plant-derived natural products have been reported as antiprotozoal agents. See review by Schmidt and colleagues.7 In addition, natural products have been widely explored in anti-infective drug discovery. Most anti-infective agents are natural products-based/inspired.8 However, due to the complexity and scarcity of most active agents, follow-up studies are usually difficult and rarely pursued in NTDs drug discovery.
The compounds described in this work were synthesized as outlined in Schemes 1 and 2. For compounds 5 to 25, allyl phenyl sulfone (1) was reacted with bromine to obtain the 1,2-dibromide (2), in good yield (93%). This was followed by dehalogenation of the vicinal dibromide with sodium carbonate in diethyl ether to obtain (E)-((3-bromoprop-1-en-1-yl)sulfonyl)benzene (3). Compounds 4a–c were obtained via etherification reaction between the appropriate 4-hydroxyphenylacetic acids and 3 in ethanol, using potassium hydroxide and sodium iodide. Compounds 4a–c were then used to synthesize the corresponding amides (5–10) and esters (11–25) using CDI or DCC and DMAP as coupling reagents.9–13 Detailed synthesis and compound characterization data are provided as supporting information.
Scheme 1.
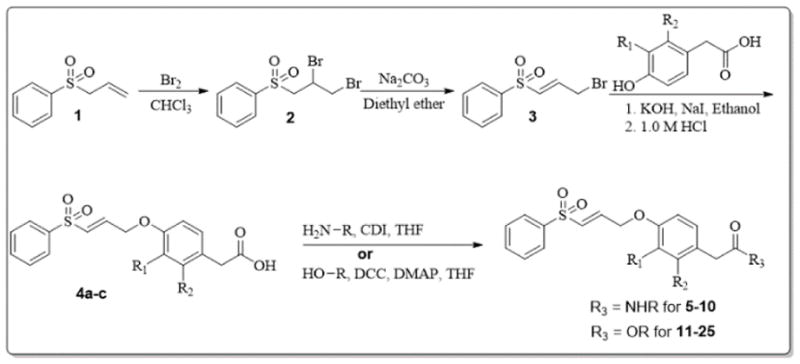
Synthesis of target compounds 5–25.
Scheme 2.
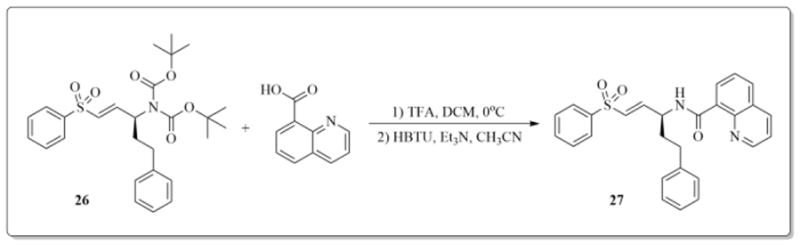
Synthesis of target compound 27.
The compounds were subsequently tested for their ability to inhibit the growth of T. brucei in vitro.14 The parasites were exposed to the compounds for 48 hours. Most of the compounds displayed selective but moderate growth inhibitory activity against T. brucei when compared with mammalian cells (Hep G2).15 Compounds derived from 8-aminoquinoline (9), (1R)-nopol (15, 24), 6-bromo-2-naphthol (16), (+) fenchol (21) and 4-benzylphenol (23) were the most active (Table 1). The 8-aminoquinoline-based compound (9), being the most selective, was evaluated for in vivo antitrypanosomal activity. Two groups of T. brucei (STIB795)-infected mice were treated for 4 consecutive days intraperitoneally with 50 mg/kg/day and 100 mg/kg/day of 9, respectively.16 The infected mice were positive for parasites 24 hours posttreatment, suggesting that compound 9 lack in vivo efficacy. Several generations of aminoquinoline-based compounds have found clinical use in the treatment of malaria but not in the treatment of trypanosomiasis.17 This is perhaps due to the unique mechanism of action of aminoquinolines in plasmodium-infected cells. However, there are increasing reports of quinoline-based growth inhibitors of trypanosomes, although, the mechanism of action of the quinoline-based compounds have not been deciphered.18–21
Table 1.
The antitrypanosomal activities of compounds 5–27.

| |||||
|---|---|---|---|---|---|
| R1 | R2 | R3 | T. brucei IC50 | Hep G2 IC50 | |
| 5 | H | H |

|
11.72 ± 0.83 | >20 |
| 6 | H | H |

|
10.77 ± 0.31 | >20 |
| 7 | H | H |

|
>20 | >20 |
| 8 | H | H |

|
>20 | >20 |
| 9 | H | H |

|
0.76 ± 0.11 | >80 |
| 10 | H | H |

|
5.45 ± 0.20 | >20 |
| 11 | H | H |

|
7.16 ± 0.42 | >20 |
| 12 | H | H |

|
5.62 ± 0.65 | >20 |
| 13 | H | H |

|
>20 | >20 |
| 14 | H | H |

|
>20 | >20 |
| 15 | H | H |

|
2.01 ± 0.12 | >80 |
| 16 | H | H |

|
2.18 ± 0.25 | >80 |
| 17 | H | H |

|
6.04 ± 0.03 | >20 |
| 18 | H | H |

|
>20 | >20 |
| 19 | H | H |

|
>20 | >20 |
| 20 | H | H |

|
5.63 ± 0.61 | >20 |
| 21 | H | H |

|
4.04 ± 0.01 | 11.9 ± 1.03 |
| 22 | H | H |

|
>20 | >20 |
| 23 | H | H |

|
1.47 ± 0.40 | >80 |
| 24 | Cl | H |
|
3.04 ± 0.07 | >80 |
| 25 | H | F |
|
7.03 ± 0.17 | >20 |
| 27 |

|
5.97 ± 0.12 | >80 | ||
| Suramin | 0.004 ± 0.001 | n/a | |||
| Podophyllotoxin | n/a | 0.008 ± 0.0003 | |||
The presence of the vinyl sulfone moiety in 5–25 suggests that they are potential covalent inhibitors of trypanosoma cysteine proteases. Compounds 5–25 were then tested for inhibitory activity against the major cathepsin L protease in T. brucei, rhodesain.
Rhodesain is a validated drug target and it is known to be essential for the survival and infectivity of the parasite. Its role in the ability of the parasite to proliferate has been extensively investigated.22–24 None of the compounds displayed noteworthy inhibition of the protease at 20 μM. The inactivity of the compounds may be because of the proximity of the vinylic Michael acceptor to the phenoxide oxygen in 5–25. It is also possible that the compounds are just unable to adopt favorable orientation at the active site of the protease. Nevertheless, a quinoline-based thiol reactive structural variant of 9 was synthesized and tested for protease inhibition, and for trypanocidal activity. Compound 27 was synthesized from boc-protected (E)-5-phenyl-1-(phenylsulfonyl)pent-1-en-3-amine (26). Compound 26 was a generous gift from Prof. J Love (University of British Columbia), and it was reported by Kiemele and co-workers in 2016.25 Compound 27 was able to completely inactivate rhodesain at 20 μM after 1 hour of incubation with estimated IC50 value of 800 nM, and a Kinact/Ki value of 99 M−1s−1 (Figure 1).26
Figure 1.
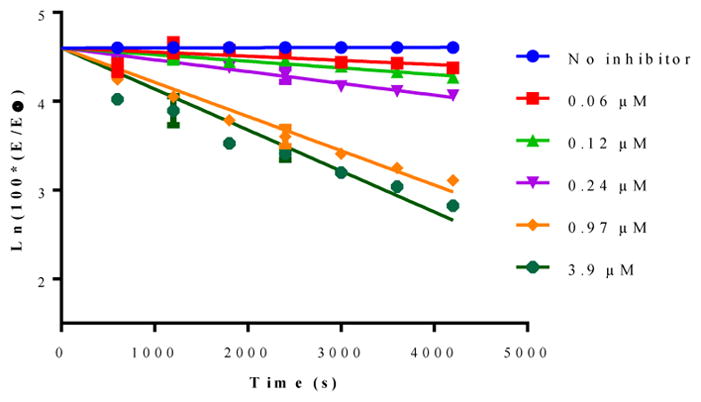
Pseudo-first order inhibition plots for compound 27.
It has a moderate antitrypanosomal activity with an IC50 value of 5.97 uM. Compound 27 was also tested for inhibitory activity against human cathepsin L, but it was inactive (from 0.1 μM to 125 μM).26 Crystallographic investigation of rhodesain-inhibitor (27) complex has been attempted but it has not been successful. However, it is still being pursued. In order to understand the interactions responsible for the inhibitory activity of 27 on rhodesain, template docking was used.27 Compound 27 was docked in the previously reported crystal structure of rhodesain using the bound ligand (D1R) as template.28 Three of the top five docking poses suggests that the vinyl group is in the vicinity of active thiol (Cys25), while the homophenyl moiety occupies the P1 site, and the quinoline moiety occupies the P2 site (Figure 2a and 2b). The phenyl sulfone moiety is predicted to have steric interactions with Gln19 and His162 while the quinolyl motif have steric interactions with Met68. Quite noticeable is the empty P3 site.
Figure 2.

a. The superposition of docked compound 27 (Yellow) and D1R (Blue) at the active site of rhodesain; b. The interaction plot of compound 27 with active site residues.
In conclusion, a series of phenyl sulfone natural products-based compounds were synthesized and evaluated as potential antitrypanosomal agents. Quinoline- and nopol-based compounds, 9 and 15, were the most active and selective. The quinoline-based compound (9) can serve a template for phenotypic-based lead discovery and the thiol-active compound (27) may serve as template for structure-based investigation of new covalent antitrypanosomal agents.
Supplementary Material
Acknowledgments
This work was carried out in part by resources made available by the US National Institutes of Health (SC2GM109782, SC3GM122629, and G12MD007581). JC and SW were supported by NSF-RISE (HRD-1547836). We thank Prof. Jennifer Love (University of British Columbia) for the gift of compound 26.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.WHO. Third WHO Report on Neglected Tropical Diseases. World Health Organization; Geneva: 2015. Investing to Overcome the Global Impact of Neglected Tropical Diseases. [Google Scholar]
- 2.De Kyvon MALC, Maakaroun-Vermesse Z, Lanotte P, Priotto G, Perez-Simarro P, Guennoc AM, De Toffol B, Paris L, Bernard L, Goudeau A, Chandenier J, Desoubeaux G. Emerg Infect Dis. 2016;22:935–937. doi: 10.3201/eid2205.160133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mukadi P, Lejon V, Barbé B, Gillet P, Nyembo C, Lukuka A, Likwela J, Lumbala C, Mbaruku J, Vander Veken W, Mumba D, Lutumba P, Muyembe JJ, Jacobs J. PLOS ONE. 2016;11:e0146450. doi: 10.1371/journal.pone.0146450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lumbala C, Simarro PP, Cecchi G, Paone M, Franco JR, Kande Betu Ku Mesu V, Makabuza J, Diarra A, Chansy S, Priotto G, Mattioli RC, Jannin JG. Int J Health Geogr. 2015:14. doi: 10.1186/s12942-015-0013-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nagle A, Khare S, Kumar A, Supek F, Buchynskyy A, Mathison C, Chennamaneni N, Pendem N, Buckner F, Gelb M, Molteni V. Chem Rev. 2014;114:11332–11333. doi: 10.1021/cr500365f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaiser M, Bray MA, Cal M, Bourdin TB, Torreele E, Brun R. Antimicrob Agents Chemother. 2011;55:5602. doi: 10.1128/AAC.00246-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schmidt TJ, Khalid SA, Romanha AJ, Alves TMA, Biavatti MW, Brun R, Da Costa FBa, de Castro SL, Ferreira VF, de Lacerda MVG, Lago JHG, Leon LL, Lopes NP, Das Neves Amorin RC, Niehues M, Ogungbe IV, et al. Curr Med Chem. 2012;19:2128–2175. doi: 10.2174/092986712800229023. [DOI] [PubMed] [Google Scholar]
- 8.a Newman DJ, Cragg GM, Snader KM. J Nat Prod. 2003;66:1022–1037. doi: 10.1021/np030096l. [DOI] [PubMed] [Google Scholar]; b Newman DJ, Cragg GM. J Nat Prod. 2007;70:461–477. doi: 10.1021/np068054v. [DOI] [PubMed] [Google Scholar]; c Newman DJ, Cragg GM. J Nat Prod. 2012;75:311–335. doi: 10.1021/np200906s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zindel J, de Meijere A. Synthesis. 1994;3:190–194. [Google Scholar]
- 10.Ishizaki M, Yamada M, Watanabe SI, Hoshino O, Nishitani K, Hayashida M, Tanaka A, Hara H. Tetrahedron. 2004;60:7973–7981. [Google Scholar]
- 11.Neises B, Steglich W. Angew Chem Int Ed. 1978;17:522–524. [Google Scholar]
- 12.Kamiński K, Rapacz A, Łuszczki JJ, Latacz G, Obniska J, Kieć-Kononowicz K, Filipek B. Bioorgan Med Chem. 2015;23:2548–2561. doi: 10.1016/j.bmc.2015.03.038. [DOI] [PubMed] [Google Scholar]
- 13.Lafrance D, Bowles P, Leeman K, Rafka R. Org Lett. 2011;13:2322–2325. doi: 10.1021/ol200575c. [DOI] [PubMed] [Google Scholar]
- 14.Trypanosoma brucei assay: The growth inhibitory activity of the compounds was evaluated using the Alamar blue assay. Bloodstream forms of T. brucei brucei (strain 427) cultured in HMI-9-medium supplemented with 10% FBS, 10% Serum plus (SAFC), 0.05 mM bathocuproinesulfonate, 1.5 mM L-cysteine, 1 mM hypoxanthine, 0.2 mM β-mercaptoethanol, 0.16 mM thymidine, 1 mM pyruvate, and 0.0125% Tween 80 were dispensed into sterile 96-well plates at 5 X 103 cells/well, and treated with compounds for 48 hours. The compounds were prepared in DMSO and were tested in triplicates with a total assay volume of 100 μL. Next, Alamar blue (20 μL) was added and the plate was incubated at 37°C for 4 hours. Immediately following incubation, fluorescence signals were read (λex 530 nm, λem 590nm). IC50 values were determined by testing compounds in a dose range of 0.3–50 μM. Suramin was used as positive control.
- 15.Cytotoxicity Assay: Human hepatocarcinoma cell line (Hep G2, CRL-11997™) was used for cytotoxicity studies. The cells were grown in complete medium (DMEM: F12 containing L-glutamine and sodium bicarbonate, 10% FBS, 1% penicillin/streptomycin, 0.0125% Tween 80) incubated at 37°C in a 5% CO2 environment. Once 80–90% confluent, the cells were washed with phosphate-buffered saline (PBS), treated with 0.25% (w/v) of trypsin/EDTA, counted and suspended in fresh complete media. Into 96-well plates, 198 μL of 5 × 105 cells/mL were seeded and incubated for about 24 hours. Cells were treated with the compounds prepared in DMSO for 72 hours. After 72 hours, the old cell medium is removed and fresh DMEM:F12 medium containing MTT (5 mg/mL in PBS) was added to the cells, and incubated for 1.5 hour. The MTT-containing medium was gently removed and replaced with DMSO (200 μL/well). Lyzed cells were then repeatedly resuspended in DMSO using multichannel pipette in order to allow the formazan crystals to dissolve. Plates were incubated for 10 minutes and absorbance were measured at 550 nm. All compounds were tested in triplicates. SDS (10%) was used as assay positive control and podophyllotoxin was used as cytotoxicity control.
- 16.In vivo assay: T. b. brucei STIB795-Luc model was used for in vivo studies. The studies were conducted at the Swiss Tropical and Public Health Institute (Basel, Switzerland). They were approved by the veterinary office of the Canton Basel-Stadt, Switzerland under license number 2813. Four female NMRI mice were used per experimental group. A control group and two treatment groups (50 mg/kg/day and 100 mg/kg/day). Each mouse was inoculated intraperitoneally (ip) with 1 × 104 bloodstream forms of STIB795-Luc. Treatments were administered ip in water (plus 10% DMSO) 3 days post-infection. All mice were monitored for parasitemia by live imaging. Moribund mice were euthanized after detection of parasitemia.
- 17.Parhizgar AR, Tahghighi A. Iran J Med Sci. 2017;42:115. [PMC free article] [PubMed] [Google Scholar]
- 18.Ramírez–Prada J, Robledo SM, Vélez ID, del Pilar Crespo M, Quiroga J, Abonia R, Montoya A, Svetaz L, Zacchino S, Insuasty B. Eur J Med Chem. 2017;131:237–254. doi: 10.1016/j.ejmech.2017.03.016. [DOI] [PubMed] [Google Scholar]
- 19.Nefertiti A, Batista M, Da Silva P, Batista D, Da Silva C, Peres R, Torres-Santos E, Cunha-Junior E, Holt E, Boykin DW, Brun R. Antimicrob Agents Ch. 2017 doi: 10.1128/AAC.01936-17. AAC-01936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carvalho L, Martínez-García M, Pérez-Victoria I, Manzano JI, Yardley V, Gamarro F, Pérez-Victoria JM. Antimicrob Agents Ch. 2015;59:6151–6160. doi: 10.1128/AAC.00879-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Di Pietro O, Vicente-García E, Taylor MC, Berenguer D, Viayna E, Lanzoni A, Sola I, Sayago H, Riera C, Fisa R, Clos MV. Eur J Med Chem. 2015;105:120–137. doi: 10.1016/j.ejmech.2015.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.a Scory S, Caffrey CR, Stierhof YD, Ruppel A, Steverding D. Exp Parasitol. 1999;91:327–333. doi: 10.1006/expr.1998.4381. [DOI] [PubMed] [Google Scholar]; b Troeberg L, Morty RE, Pike RN, Lonsdale-Eccles JD, Palmer JT, McKerrow JH, Coetzer TH. Exp Parasitol. 1999;91:349–355. doi: 10.1006/expr.1998.4386. [DOI] [PubMed] [Google Scholar]; c Steverding D, Sexton DW, Wang X, Gehrke SS, Wagner GK, Caffrey CR. Int J Parasitol. 2012;42:481–488. doi: 10.1016/j.ijpara.2012.03.009. [DOI] [PubMed] [Google Scholar]
- 23.Ettari R, Tamborini L, Angelo IC, Micale N, Pinto A, De Micheli C, Conti P. J Med Chem. 2013;56:5637. doi: 10.1021/jm301424d. [DOI] [PubMed] [Google Scholar]
- 24.Mott B, Ferreira R, Simeonov A, Jadhav A, Ang K, Leister W, Shen M, Silveira J, Doyle P, Arkin M, McKerrow J, Inglese J, Austin C, Thomas C, Shoichet B, Maloney D. J Med Chem. 2010;53:52. doi: 10.1021/jm901069a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kiemele ER, Wathier M, Bichler P, Love JA. Total synthesis of K777: Successful application of transition-metal-catalyzed alkyne hydrothiolation toward the modular synthesis of a potent cysteine protease inhibitor. Org lett. 2016;18:492–495. doi: 10.1021/acs.orglett.5b03535. [DOI] [PubMed] [Google Scholar]
- 26.a. T. brucei cathepsin L (rhodesain) was expressed in Pichia pastoris. The inhibition assays were carried out in 50 mM sodium acetate (assay buffer) pH 5.5, containing 2 mM DTT and 0.01% Triton X-100. The enzyme (5 μL, final concentration of 0.8 nM) and test compound (5 μL, final concentration of 20 μM) mixtures were pre-incubated for 1 hour, followed by the addition of 100 μL of the substrate, Z-Phe-Arg-AMC (10 μM), in sodium acetate buffer pH 5.5. Fluorescence (RFU/sec) resulting from proteolytic cleavage of the substrate was monitored at 25 °C (λex 355 nm and λem 460 nm) on a PolarStar Omega plate reader (BMG LABTECH, Germany). E-64 (10 μM) was used as positive control (100% inhibition). For time dependent inhibition studies, rhodesain-compound assay mixture includes 30 μL of compound 27 in DMSO, 30 μL of rhodesain in assay buffer, and 180 μL of assay buffer. Aliquots (10 μL) of rhodesain-27 mixture were assayed every 10 minutes using Z-Phe-Arg-AMC (10 μM) as substrate. Fluorescence (RFU/sec) resulting from proteolytic cleavage of the substrate was monitored for 7 minutes as described above. Compound 27 was assayed at the following concentrations: 0, 0.06, 0.12, 0.24, 0.97, and 3.9 μM., b. Cathepsin L (CatL) from human liver (Millipore Sigma) was used for protease selectivity studies. Human CatL (0.58 nM) was assayed in 400 mM sodium acetate (100 μL) pH 5.5, containing 8 mM DTT, 4 mM EDTA, and 0.01% Triton X-100. Human CatL and 27 were pre-incubated for 1 hour in assay buffer, followed by the addition of 100 μL of the substrate, Z-Phe-Arg-AMC (20 μM), in sodium acetate buffer and the plate was read as described above.
- 27.Molecular structure of compound 27 was built using SPARTAN ’10 for Windows. Its geometries were optimized using the MMFF 94 force field. The docking simulations were carried out using the MolDock docking algorithm of the Molegro Virtual Docker in the template docking mode. The X-ray crystal structure (PDB ID: 2P7U) of rhodesain was used for the docking calculation. The bound inhibitor in 2P7U was used as template molecule. A docking sphere (15 Å radius) was placed on the binding site (X = −9.38; Y = 1.66; Z = 10.38) in order to allow different orientations of the ligand to be searched. The docking algorithm was set at maximum iterations of 1500 with a simplex evolution population size of 50, and a minimum of 50 runs. The 2D representations of rhodesain-27 complex was prepared using MOE 2016.
- 28.Kerr ID, Lee JH, Farady CJ, Marion R, Rickert M, Sajid M, Pandey KC, Caffrey CR, Legac J, Hansell E, McKerrow JH. J Bio Chem. 2009;284:25697–25703. doi: 10.1074/jbc.M109.014340. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.