

Abstract
Recent evidence highlights a role for sex and hormonal status in regulating cellular responses to ischemic brain injury and neurodegeneration. A key pathological event in ischemic brain injury is the opening of a mitochondrial permeability transition pore (MPT) induced by excitotoxic calcium levels, which can trigger irreversible damage to mitochondria accompanied by the release of proapoptotic factors. However, sex differences in brain MPT modulation have not yet been explored. Here, we show that mitochondria isolated from female mouse forebrain have a lower calcium threshold for MPT than male mitochondria, and that this sex difference depends on the MPT regulator cyclophilin D (CypD). We also demonstrate that an estrogen receptor beta (ERβ) antagonist inhibits MPT and knockout of ERβ decreases the sensitivity of mitochondria to the CypD inhibitor, cyclosporine A. These results suggest a functional relationship between ERβ and CypD in modulating brain MPT. Moreover, co-immunoprecipitation studies identify several ERβ binding partners in mitochondria. Among these, we investigate the mitochondrial ATPase as a putative site of MPT regulation by ERβ. We find that previously described interaction between the oligomycin sensitivity-conferring subunit of ATPase (OSCP) and CypD is decreased by ERβ knockout, suggesting that ERβ modulates MPT by regulating CypD interaction with OSCP. Functionally, in primary neurons and hippocampal slice cultures, modulation of ERβ has protective effects against glutamate toxicity and oxygen glucose deprivation, respectively. Taken together, these results reveal a novel pathway of brain MPT regulation by ERβ that could contribute to sex differences in ischemic brain injury and neurodegeneration.
Keywords: Estrogen receptor, calcium, permeability transition, cyclophilin D, mitochondria
1. Introduction
There is a growing body of evidence describing sex differences in ischemic brain injury. Many factors are thought to contribute to increased overall stroke risk, severity and mortality rate in women compared to men, including longer average life expectancy, sex-specific comorbidities, post-menopausal hormonal changes, and hormonal replacement therapies [1, 2]. Furthermore, recent studies have highlighted cellular mechanisms, which differ between males and females, that may contribute to sex differences in stroke incidence and outcome [3].
One important cellular phenomenon in ischemic brain injury is mitochondrial permeability transition (MPT) [4, 5]. MPT is the opening of a non-selective pore in the inner mitochondrial membrane that results in unregulated flow of solutes and influx of water into the mitochondrial matrix. MPT results in swelling of mitochondria, outer membrane rupture, and release of proapoptotic factors [6]. A key trigger for MPT pore opening is a high level of calcium in the mitochondrial matrix. Calcium enters mitochondria through the mitochondrial calcium uniporter (MCU) using mitochondrial membrane potential as a driving force [7]. A moderate calcium influx in mitochondria is critical for activation of matrix dehydrogenases of the TCA cycle that stimulates oxidative metabolism [8]. However, in ischemia reperfusion, when neurons are flooded with calcium due to the opening of plasma membrane channels and release from internal calcium stores [9, 10], mitochondria are overloaded with calcium and MPT occurs [11]. However, it is important to note that, alternatively to irreversible and damaging MPT, reversible and transient MPT can lead to opening of a low-conductance pore that may serve as a protective calcium extrusion mechanism [12].
The precise composition and mechanisms of regulation of the MPT pore remain to be fully elucidated, although several candidate structural and modulatory proteins have been identified (reviewed in [13, 14]). It is widely accepted that a key MPT modulatory protein is cyclophilin D (CypD), a matrix protein, which upon binding calcium translocates to the mitochondrial inner membrane to promote MPT [15, 16]. In the absence of CypD [17, 18] or under its pharmacological inhibition [19, 20], MPT can still occur, but the calcium threshold for its activation is much higher. Mitochondrial calcium retention capacity is typically used in animal models as a quantitative assay for MPT, and the presence of a CypD-dependent MPT has also been verified in adult human brain mitochondria [21].
Although sex differences in MPT have been observed in heart [22, 23], the molecular mechanisms underlying these differences are unknown. Furthermore, whether males and females differ in brain MPT has not been explored. Sex hormones are clear potential candidates for being modulators of mitochondrial function. It is known that estrogens affect intracellular calcium dynamics [24, 25] and mitochondrial function [26], in both receptor dependent and independent manners [27]. Two estrogen receptors, alpha (ERα) and beta (ERβ), were originally identified as nuclear receptors [28, 29]. More recently both receptors have also been shown to localize to mitochondria [30], although mitochondrial localizations has been more thoroughly characterized for ERβ [31, 32]. Both male and female mouse brain contain ERβ in many regions, and some areas have more ERβ-positive cells in females than in males [33], which may contribute to sex differences in MPT.
Despite the known effects of estrogen on mitochondrial function and the finding of ERs in mitochondria, until now the role of estrogen receptors in sex differences in brain MPT regulation has not been explored. In this study, we investigated sex differences in calcium-induced MPT in mouse brain mitochondria, the mechanisms of ERβ and CypD MPT modulation, and the potential involvement of such mechanisms in in vitro models of ischemia reperfusion injury.
2. Materials and Methods
2.1 Animals
CypDKO mice in the C57Bl/6J background have been described previously [17]. ERβKO mice in the C57Bl/6J background were from The Jackson Laboratories (strain B6.129P2-Esr2tm1Unc/J). CypDKO and ERβKO were crossed to obtain ERβ/CypD double KO animals. Heterozygote males and females were bred to obtain homozygote double KO animals. Adult animals (130 days of age) were used to isolate brain mitochondria for the calcium capacity and membrane potential measurements, embryonic cortical neuronal cultures were prepared from day 18 embryos, and postnatal day 5 animals were used for the organotypic hippocampal slice cultures.
All experiments were approved by the Institutional Animal Care and Use Committee of the Weill Medical College of Cornell University and carried out in compliance with the National Institute of Health guidelines for the care and use of laboratory animals.
2.2 Calcium Capacity and Membrane Potential Measurements
Isolation and purification of mouse forebrain mitochondria was performed as previously described [34, 35]. Mitochondria (0.1 mg protein/mL) were suspended in 125 mM KCl, 20 mM HEPES, 1 mM MgCl2 (pH 7.2), 2 mM KH2PO4, 25 μM EGTA, 0.2 mM ATP, 1 μM rotenone and 5 mM succinate. Fura-FF (0.2 μM, Life Technologies) was used to measure calcium uptake following repeated pulses of calcium (25 μM) every 150 seconds, until mitochondria were no longer able to take up calcium. Measurements were made at 37°C in an F-7000 spectrofluorimeter (Hitachi) using 340/380 nm excitation and 510 nm emission. Mitochondrial calcium capacity was calculated as previously described [35] by plotting the fluorescence values measured 75 seconds after each calcium addition. The inflection point in the curve obtained by plotting such values represented the time at which MPT is initiated. To correlate the fluorescence values to calcium concentration, calcium standards were added to the same buffer to create a calibration curve. Mitochondrial calcium capacity measurements were performed in absence or presence of the cyclophilin D inhibitor cyclosporine A (CsA, Sigma), the ERβ inhibitor 4-[2-Phenyl-5,7-bis(trifluoromethyl)pyrazolo[1,5-a]pyrimidin-3-yl]phenol (PHTPP, Tocris Bioscience) or the ERα inhibitor 1,3-Bis(4-hydroxyphenyl)-4-methyl-5-[4-(2-piperidinylethoxy)phenol]-1H-pyrazole dihydrochloride (MPP, Tocris Bioscience), added prior to calcium additions.
Safranin O (2 μM, Sigma) was used to measure membrane potential using 495nM excitation and 586nm emission, in the conditions described above. Following sequential calcium boluses added to 0.2 mg mitochondria, antimycin A (1 μg/mL) was added to inhibit the respiratory chain and induce maximal depolarization. The percent of maximal depolarization for each calcium addition was plotted in order to determine the amount of calcium required to induce 50% of the maximal achievable depolarization (Δψm50). To test the ability of mitochondria to generate Δψm, sequential additions of the uncoupler SF6847 (2 nM each, Malonoben, Tocris Bioscience) were made, and the decrease in Δψm after each addition was plotted.
2.3 Western Blot Analyses
Brain mitochondria samples were prepared in Laemmli buffer with β-mercaptoethanol, then separated by SDS-PAGE on 12% Tris-acrylamide/bis-acrylamide gels and transferred to polyvinylidene fluoride membranes (BioRad). Membranes were probed overnight with the following primary antibodies: anti-Tim23 (BD Transduction Laboratories), anti-mitochondrial calcium uniporter (MCU, Sigma), anti-voltage dependent anion channel (VDAC, Abcam), anti-mitochondrial calcium uptake 1 (MICU1, Abcam), and anti-cyclophilin D (CypD Mitoscience). Membranes were incubated with horseradish peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch) and detected via chemiluminescence.
2.4 Primary Cortical Neuronal Culture and Glutamate Toxicity
Primary cortical neuron cultures were prepared from embryonic day 18 mouse embryos as described in [36], and cultured for 14 days on polyornithine and laminin coated 96-well plates. Neurons were exposed to 100 μM glutamate for 24 hours, after 1 hour of pretreatment with or without 10 μM 17β-estradiol or vehicle (ethanol). 17β-estradiol was maintained in the culture for the duration of glutamate exposure. MTT was added to culture medium at a final concentration of 0.5 mg/ml. The plates were incubated for 3 hours, and then cells were solubilized in dimethyl sulphoxide. Absorbance was measured at 595 nm.
2.5 Hippocampal Slice Culture Preparation and Oxygen Glucose Deprivation
Hippocampal slice cultures (coronal slices, 350 μm thick) were prepared as previously described [37, 38], and cultured on Millicel CM membrane inserts (Millipore). After 14 days in culture, slices were imaged for three consecutive days after staining with propidium iodide (PI) in the culture medium (5 μg/mL). On the first day, baseline PI images were captured (Flbasal) in a transmission fluorescence microscope with a 2× objective, and then OGD was performed. Briefly, slices were washed in OGD buffer (125 mM NaCl, 5 mM KCl, 1.2 mM Na2PO4, 26 mM NaHCO3, 1.8 mM CaCl2, 0.9 mM MgCl2, 10 mM HEPES, pH 7.4), and then incubated in OGD buffer without glucose in an anoxic gas chamber for one hour, and then returned to normal culture medium. On the second day, post-OGD PI images were captured (FlOGD). Then, slices were treated with 1 mM N-Methyl-D-aspartic acid (NMDA) for 24 hours. On the third day, post-NMDA PI images were captured, representing the maximum death in each slice (Flmax). The same imaging settings were used at all time points, and images from the CA1 hippocampal region were used to calculate the proportion of OGD-induced death using the formula (FlOGD − Flbasal)/(Flmax − Flbasal) × 100%.
2.6 Cell Culture, Transfection, and FLAG Co-immunoprecipitation
COS-7 cells were cultured in high-glucose Dulbecco’s modified Eagle’s medium (DMEM, Life Technologies) supplemented with 5% fetal bovine serum (Atlanta Biologicals) and 1% antibiotic-antimycotic (Life Technologies). Cells were transfected with the ERβ-Flag construct (Addgene #35562) using the Lonza Nucleofector device according to the manufacturer’s instructions. Enriched mitochondrial fractions were prepared from transfected cells. Briefly, cells were harvested and homogenized in mannitol-sucrose buffer (225 mM mannitol, 75 mM sucrose, 5 mM HEPES, 1 mM EGTA, 1 mg/mL fatty-acid free BSA, pH 7.4), and centrifuged at 4°C at 800 rcf for 5 minutes. Supernatants were centrifuged at 10,000 rcf for 10 minutes, and pellets washed in BSA-free mannitol-sucrose buffer and then resuspended in mannitol-sucrose buffer. Protein concentrations were quantified using a BCA assay kit (Pierce).
Co-Immunoprecipitation (Co-IP) experiments were performed on mitochondrial fractions using a co-Immunoprecipitation kit (Pierce). Briefly, FLAG antibody (Sigma F1804) or normal mouse IgGs (ThermoFisher) were covalently coupled to IP resins. Mitochondria (0.5 mg) were resuspended in Co-IP lysis/wash buffer and incubated with the resins overnight. Proteins were eluted in elution buffer and eluates neutralized with Tris-HCl pH 8.5. Western blot using FLAG antibody was performed to confirm the presence of ERβ-Flag in the Co-IP eluates.
2.7 Protein extraction, digestion and Tandem Mass Tag (TMT) labeling
Proteins were denatured in 200 mM triethylammoniumbicarbonate, 7M urea, 2M thiourea and 0.2% SDS, pH 8.5 for 1 hour. Each sample (2 μg) was reduced with 11.9 mM tris(2-carboxyethyl) phosphine for 1 hr at 37°C, alkylated with 20 mM iodoacetamide for 1 hr in the dark and quenched with 20 mM dithiothreitol (DTT). The proteins were precipitated with acetone and frozen overnight, reconstituted in 90 μL of 100 mM triethylammoniumbicarbonate and digested with 0.2 μg trypsin for 18 h at 37 °C. The TMT 6-plex labels were reconstituted with 45 μL of anhydrous ACN and added to each of the digest samples for 1 hour at room temperature. The peptides from the 6 samples were mixed with each tag: 126-tag (mock IP #1), 127-tag (ERβ-FLAG IP #1), 128-tag (ERβ-FLAG + 17βE #1) 129-tag (mock IP #2), 130-tag (ERβ-FLAG IP #2), and 131-tag (ERβ-FLAG + 17βE #2). The six samples were pooled, evaporated to dryness and subjected to cation exchange chromatography fractionation using a PolyLC strong cationexchange cartridge (PolyLC Inc. Columbia, MD). The pooled peptides were then reconstituted with 3 mL of loading buffer (10 mM potassium phosphate pH 3.0, 25% ACN), and the pH adjusted to 3.0 with formic acid. After conditioning of the strong-cation exchange (SCX) cartridge (PolySULFOETHYL A, 10 mm id × 14 mm, particle size 12 μm, pore size 300 Å), the sample was loaded and washed with an additional 2 mL of loading buffer. The peptides were eluted in three isocratic steps by 1 mL of loading buffer containing 50 mM, 150 mM and 500 mM KCl for each step. Desalting of SCX fractions was carried out using solid phase extraction (SPE) on Sep-Pak® Cartridges (Waters, Milford, MA) and the eluted peptides were evaporated to dryness.
2.8 Nano-scale reverse phase chromatography and tandem mass spectrometry (nanoLC-MS/MS)
Each fraction was reconstituted in 15 μL 0.5% FA, and nanoLC-ESI-MS/MS analysis was carried out using an Orbitrap Elite (Thermo-Fisher Scientific, San Jose, CA) mass spectrometer equipped with a “CorConneX” nano ion source device (CorSolutions LLC, Ithaca, NY). The Orbitrap was interfaced with a Dionex UltiMate3000RSLCnano system (Thermo, Sunnyvale, CA). Each sample was injected onto a PepMap 100 C-18 RP nano trap column (5 μm, 100 μm × 20 mm, Thermo) with nanoViper Fittings at 20 μL/min flow rate and then separated on a PepMap C-18 RP nano column (3 μm, 75μm × 15 cm), and eluted in a 120 min gradient of 5% to 38% acetonitrile (ACN) in 0.1% formic acid at 300 nL/min., followed by a 5-min ramping to 95% ACN-0.1% FA and a 5-min hold at 95% ACN-0.1% FA. The Orbitrap Elite was operated in positive ion mode (nano spray voltage 1.6 kV, source temperature at 250 °C). The instrument was operated in data-dependent acquisition (DDA) mode using FT mass analyzer for one survey MS scan for selecting top 15 precursor ions followed by data-dependent HCD-MS/MS scans on the precursor peptides with multiple charged ions above a threshold ion count of 8000 with normalized collision energy of 37%. MS survey scans at a resolution of 60,000 (fwhm at m/z 400), for the mass range of m/z 375-1600, and MS/MS scans at 15,000 resolution for the mass range, m/z 100-2000. Dynamic exclusion parameters were set at repeat count 1, an exclusion list size of 500, 50s exclusion duration with ±10 ppm exclusion mass width. The activation time was 0.1 ms for HCD analysis. All data were acquired under Xcalibur 2.2 operation software (Thermo-Fisher Scientific).
All MS and MS/MS raw spectra from TMT experiments were processed using Proteome Discoverer 1.4 (PD1.4, Thermo) and the spectra used for subsequent database search using Mascot Daemon (version 2.3.02, Matrix Science, Boston, MA). The human RefSeq sequence plus Chlorocebus_aethiops_plus_flander_virus database containing 37,469 sequence entries was downloaded on August 28, 2013 from NCBInr and used for database searches. The default search settings used for 6-plex TMT quantitative processing and protein identification in Mascot server were: two mis-cleavages for full trypsin with fixed carbamidomethyl modification of cysteine, fixed 6-plex TMT modifications on lysine and N-terminal amines and variable modifications of methionine oxidation, and deamidation on asparagines/glutamine residues. The peptide mass tolerance and fragment mass tolerance values were 15 ppm and 100 mDa. The significant scores at 95% confidence interval for the peptides defined by a Mascot probability analysis (www.matrixscience.com/help/scoring_help.html#PBM) greater than “identity” were used as filters along with a p value of < 0.05 (expectation value). The resulting peptides are considered to be confidently-identified peptides with at least 2 unique peptides per protein. Intensities of the reporter ions from TMT tags upon fragmentation were used for quantification of peptides.
2.9 OSCP Co-Immunoprecipitation
Enriched mitochondrial fractions from HEK293 cells or purified brain mitochondria were prepared as described above. The OSCP antibody (Santa Cruz #sc-365162) or normal mouse IgGs (ThermoFisher) were coupled to the resin provided in the co-immunoprecipitation kit (Pierce) and co-IP of mitochondrial preparations (0.5 mg) was performed overnight in the following buffer: 50mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, 0.1% NP-40, pH 7.4 [39]. Proteins were eluted from the resin using the provided buffer and samples subjected to Western blot, as described above.
2.10 Statistical analyses
Data are presented as mean values and standard error of the mean. For experiments in isolated mitochondria, differences between two groups were analyzed using Student’s t-test with a 95% confidence interval. When multiple groups were compared, data were analyzed by ANOVA followed by Bonferroni post-hoc correction. In both cases, data were analyzed using GraphPad Prism software.
3. Results
3.1 Sex difference in brain mitochondria calcium capacity depends on CypD
To investigate sex differences in calcium-induced MPT we used mitochondria freshly isolated from adult mouse forebrain (130 days of age). Mitochondria were prepared via centrifugation of forebrain homogenates in a Percoll gradient. Calcium capacity, defined as the maximal amount of calcium that mitochondria could take up before undergoing MPT, was measured with a fluorimetric approach. Mitochondria were resuspended in buffer containing respiratory substrates and the ratiometric calcium probe Fura-FF, and sequential calcium boluses (25 μM each) were added to the cuvette. Mitochondrial calcium uptake was indicated by a deflection in the fluorescence ratio trace. The calcium concentration was increased until uptake was no longer observed. The calcium concentration at which MPT is initiated (MPT threshold) was determined as described in the methods. Representative traces of calcium uptake in male and female brain mitochondria are shown in figure 1A. The quantification of MPT threshold demonstrated that mitochondrial calcium capacity was significantly lower in females compared to males (p = 0.012, n = 8, Fig. 1B).
Figure 1. Sex difference in brain mitochondrial calcium capacity.
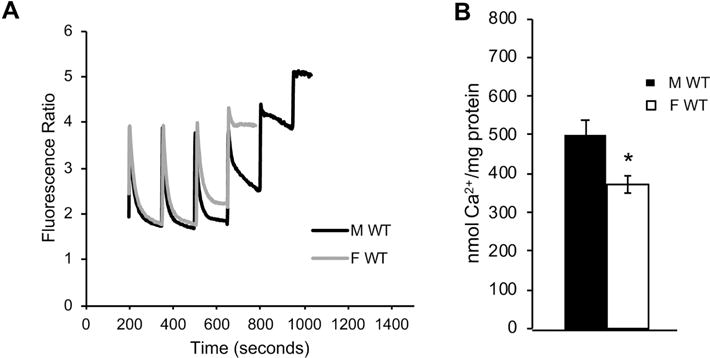
A) Representative fluorimetric traces of calcium uptake in mitochondria purified from male (black line) and female (gray line) adult (130 days of age) mouse forebrain. B) Quantification of brain mitochondrial calcium capacity in wild type males and females. n = 8 animals per group; * p<0.05.
In order to understand the mechanism underlying the sex difference in brain mitochondrial calcium capacity, we investigated the involvement of CypD. The CypD inhibitor CsA (1 μM) added to the cuvette immediately prior to the start of the calcium boluses, increased calcium capacity in both male and female brain mitochondria (by 91 ± 16 % and 62% ± 6, respectively, n = 4, p = 0.001 and p = 0.0085, Fig. 2A,B), and abolished the sex difference (p = 0.72). Importantly, we further established the role of CypD by studying brain mitochondrial calcium capacity in males and females with genetic ablation of CypD (CypDKO). CypDKO increased calcium capacity in both male and female brain mitochondria and abolished the sex difference (n = 8, p = 0.93, Fig. 2C,D). Taken together, these pharmacological and genetic results indicate that CypD is required for the sex difference in mouse brain mitochondrial calcium capacity.
Figure 2. The sex difference in brain mitochondrial calcium capacity is abolished by pharmacological inhibition or genetic ablation of CypD.
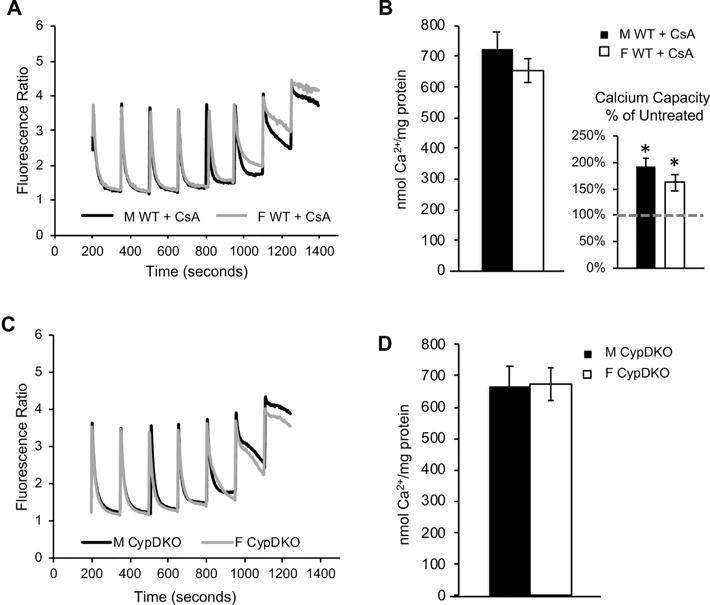
A) Representative fluorimetric traces of calcium uptake in the presence of CsA (1 μM) in mitochondria purified from male (black line) and female (gray line) adult mouse forebrain. B) Quantification of calcium capacity in male and female brain mitochondria in the presence of CsA. Calcium capacity in CsA treated mitochondria shown as percentage of vehicle treated calcium capacity (dashed line) in the figure inset. * p<0.05 vs. untreated; n = 4 animals per group. C) Representative fluorimetric traces of calcium uptake in mitochondria purified from male (black line) and female (gray line) adult CypDKO mouse forebrain. D) Quantification of brain mitochondrial calcium capacity in CypDKO mice. n = 8 animals per group.
3.2 ERβ modulates brain MPT in a CypD-dependent manner
Because of the sex difference in brain mitochondrial calcium capacity and the localization of ERs in brain mitochondria, we examined the possibility that selective antagonists of ERβ or ERα modulate brain mitochondrial calcium capacity. Mitochondria were treated with the ERβ inhibitor PHTPP (5 μM) prior to calcium additions. Calcium capacity was markedly increased by PHTPP treatment to approximately the same extent in both male (by 62% ± 3, or 198 ± 10 nmol Ca2+/mg protein, p = 0.004, n = 3, Fig. 3A) and female (by 57% ± 15, or 168 ± 38 nmol Ca2+/mg protein, p = 0.005, n = 3) brain mitochondria relative to untreated mitochondria. This finding is particularly interesting as molecules that increase the calcium threshold for MPT are under investigation as therapeutic agents to prevent apoptosis induction [40–43]. Furthermore, because PHTPP affected calcium capacity within minutes in purified mitochondria, any transcriptional effects of the drug could virtually be excluded, suggesting that ERβ in mitochondria acts as an MPT regulator. PHTPP is highly selective for ERβ, with minimal effects on ERα [44]. However, to exclude an effect of ERα on mitochondrial calcium capacity, we used the ERα antagonist MPP, which had no significant effects on calcium capacity in male or female mitochondria (respectively, p = 0.51 and 0.15, n = 4, Fig. 3B), suggesting that ERα is not involved.
Figure 3. Inhibition of ERβ, but not ERα, increases brain mitochondrial calcium capacity.
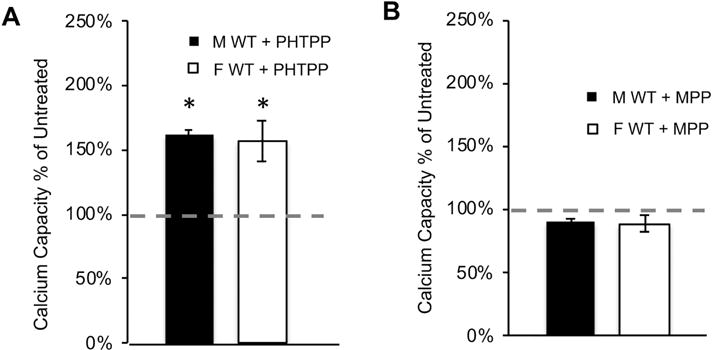
A) Quantification of calcium capacity in wild type mitochondria treated with the ERβ antagonist PHTPP (5 μM). Data are represented as percent of calcium capacity of respective vehicle-treated mitochondria (dashed line). * p<0.05 vs. untreated; n = 3 animals per group. B) Quantification of mitochondrial calcium capacity of wild type mitochondria treated with ERα antagonist MPP (5 μM). Data are represented as percent of calcium capacity of respective vehicle-treated mitochondria. n = 4 animals per group.
To further examine the role of ERβ in brain MPT and verify the specificity of the effects of PHTPP, we utilized ERβKO mice. Similar to wild type mice, ERβKO brain mitochondria calcium capacity was lower in females than in males (p = 0.004, n = 8, Fig. 4A), suggesting that ERβ alone is not sufficient to determine the sex difference. Nevertheless, addition of PHTPP to ERβKO mitochondria failed to increase calcium capacity in males and females (respectively, p = 0.83 and p = 0.51, n = 3, Fig. 4B), indicating that the increase of calcium capacity by PHTPP observed in wild type mice (Fig. 3A) requires ERβ, and is not the result of an off-target effect of the drug. Unlike the strong effect of CsA on wild type mitochondria (Fig. 2B inset), CsA modestly increased calcium capacity in both male and female ERβKO mitochondria (Fig. 4C), an effect that did not reach statistical significance (respectively, 38% ± 6, p = 0.24 and 24% ± 6, p = 0.23, n = 6).
Figure 4. ERβKO brain mitochondria have decreased sensitivity to CsA.
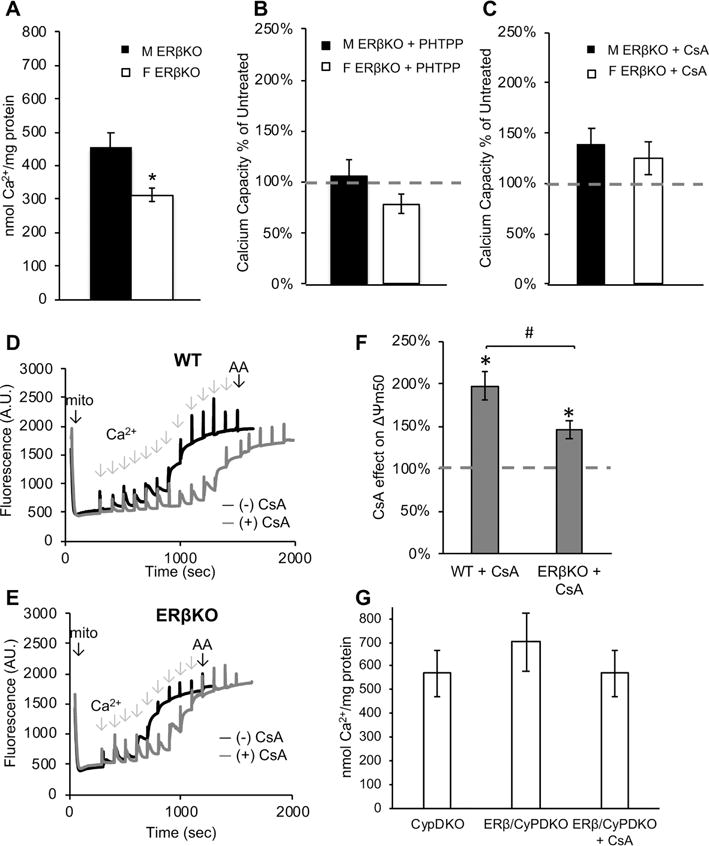
A) Quantification of calcium capacity in male and female ERβKO brain mitochondria. * p<0.05; n = 8 animals per group. B) Quantification of mitochondrial calcium capacity of ERβKO mitochondria treated with PHTPP (5 μM). Data are represented as percent of calcium capacity of respective vehicle-treated mitochondria (dashed line). n = 3 animals per group. C) Quantification of mitochondrial calcium capacity of ERβKO mitochondria treated with CsA (1 μM). Data are represented as percent of calcium capacity of respective vehicle-treated mitochondria. n = 6 animals per group. D) Representative trace of Δψm in wild type (WT) brain mitochondria with (gray line) and without (black line) CsA (1 μM). Sequential calcium additions and final addition of antimycin A (AA) are indicated with arrows. E) Representative trace of Δψm in ERβKO brain mitochondria with (gray line) and without (black line) CsA treatment. F) Quantification of half-maximum Δψm in the presence of CsA, expressed as percentage of vehicle-treated, in wild type and ERβKO mitochondria. * p<0.05 vs. vehicle-treated; # p<0.05; n = 6 animals per group. G) Quantification of female brain mitochondrial calcium capacity in CypDKO and ERβ/CypDKO, and ERβ/CypDKO treated with CsA. n = 3 animals per group.
To further explore the functional interaction between ERβ and CypD in handling of mitochondrial calcium, since mitochondria expend membrane potential to take up calcium, we compared the mitochondrial depolarization in response to calcium additions in wild type and ERβKO mice, in the presence or absence of CsA. Using Safranin O to assess changes in mitochondrial membrane potential (Δψm) [45], we measured mitochondrial depolarization following sequential bolus calcium additions (25 μM each), equivalent to those used for Fura-FF measurements. Each calcium addition followed by mitochondrial uptake causes a decrease of Δψm, ultimately resulting in maximal loss of Δψm when mitochondria undergo MPT. Example traces of Safranin O fluorescence upon sequential calcium additions for wild type and ERβKO mitochondria are shown in figure 4D and 4E, respectively. In wild type mitochondria, CsA increased the amount of calcium required to induce 50% of the maximal achievable depolarization by 100% (Δψm50, Fig. 4F). However, in ERβKO mitochondria CsA only increased by 50% the amount of calcium required for Δψm50 (p = 0.02, n = 6). Lastly, we tested brain mitochondrial calcium capacity in ERβ/CypD double KO mice compared to CypDKO to assess whether the absence of ERβ modifies the effect of CypDKO. Calcium capacity was not significantly different in the two genotypes (p = 0.44, n= 3, Fig. 4G).
Taken together, these data indicate that there is a functional relationship between ERβ and CypD in modulating calcium-induced MPT in brain, and since ERβKO did not affect calcium capacity in CypDKO mitochondria, we concluded that ERβ was functionally upstream of CypD in regulating MPT.
3.3 ERβKO does not affect mitochondrial calcium-related protein expression or Δψm
ERβ was first identified as a nuclear receptor that regulates the expression of an estrogen-responsive transcriptional program [28, 46]. However, here we show that the ERβ antagonist PHTPP affects mitochondrial calcium capacity within minutes, suggesting fast-acting, non-transcriptional mechanisms by which ERβ modulates MPT. To exclude that changes in calcium capacity were caused by altered levels of the proteins involved in mitochondrial calcium handling we compared protein expression in wild type and ERβKO brain mitochondria. The levels of calcium handling proteins were assessed by Western blot of mitochondrial extracts. The intensities of the immunoreactive bands for the mitochondrial calcium uniporter (MCU), voltage-dependent anion channel (VDAC), mitochondrial calcium uptake 1 (MICU1), and CypD, using the integral inner membrane protein Tim23 as a loading reference, were unchanged in ERβKO mitochondria (n = 3-6, Fig. 5A,B). Furthermore, no differences were observed in the levels of these proteins between sexes (Fig. 5A,B).
Figure 5. Calcium-related protein expression and bioenergetic capacity of brain mitochondria are not affected by ERβKO.
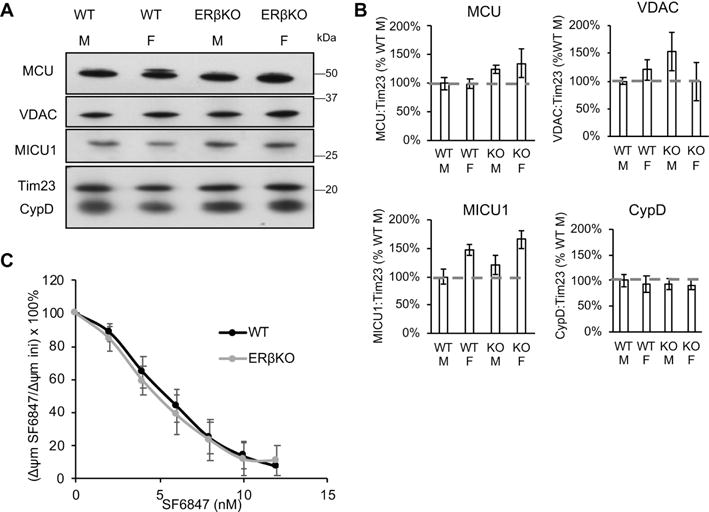
A) Western blot of MCU, VDAC, MICU1, CypD, and Tim23 as mitochondrial protein loading control in wild type (WT) and ERβKO male and female mouse brain mitochondria. B) Quantification of Western blot band immunoreactivity for each protein as a ratio to Tim23 mitochondrial loading control, and expressed as a percentage WT male. n = 3-6 animals per group. C) Depolarization of wild type and ERβKO mitochondria in response to sequential additions (2 nM each) of uncoupler SF6847. Data are calculated as the percentage of the initial membrane potential. n = 4 animals per group.
Since mitochondrial calcium buffering ability is closely tied to Δψm [47], we determined whether wild type and ERβKO brain mitochondria differed in the ability to generate Δψm. To this end, we added sequential boluses (2 nM each) of the uncoupler SF6847 to purified brain mitochondria, and assessed mitochondrial depolarization upon each addition. Upon each addition of the uncoupler, mitochondria attempt to maintain their Δψm. Therefore, the uncoupler concentration response curve can be taken as a measure of respiratory chain competence. Results indicated that wild type and ERβKO mitochondria did not differ in their ability to generate Δψm (n = 4, Fig. 5C), because the concentration response curve upon SF6847 treatment was virtually identical in the two genotypes. Together, these data excluded that the modulation of brain MPT by ERβ was due to effects on the expression of key mitochondrial calcium proteins or changes in Δψm.
3.4 ERβKO attenuates glutamate toxicity in cortical neurons and OGD-induced death in hippocampal slice cultures
Since calcium-induced MPT plays a role in ischemic brain injury [4, 5] and sex differences in ischemia-reperfusion injury have been extensively reported (Recently reviewed in [2, 3]), we hypothesized that modulation of the ERβ-CypD axis of MPT could be involved. To address this hypothesis, we compared the effects of ERβKO in two different cultured models that are relevant to ischemia-reperfusion injury. First, we exposed primary mouse embryonic (E18) cortical neuronal cultures to glutamate (100 μM) for 24 hours, with or without pre-treatment with 17β-estradiol (17βE, 10 μM), which was maintained in the medium during glutamate exposure. Cell viability measured by MTT assay showed a significant protective effect of 17βE in wild type neurons relative to untreated (p < 0.0001, n = 10-18, by two-way ANOVA with Bonferroni correction, Fig. 6A). Furthermore, the genetic ablation in ERβKO neurons alone was protective (p < 0.039, n = 10-18, by two-way ANOVA with Bonferroni correction), and 17βE did not confer additional protection, suggesting that the protective effect of 17βE was dependent on ERβ.
Figure 6. ERβKO is protective in ischemic injury models.
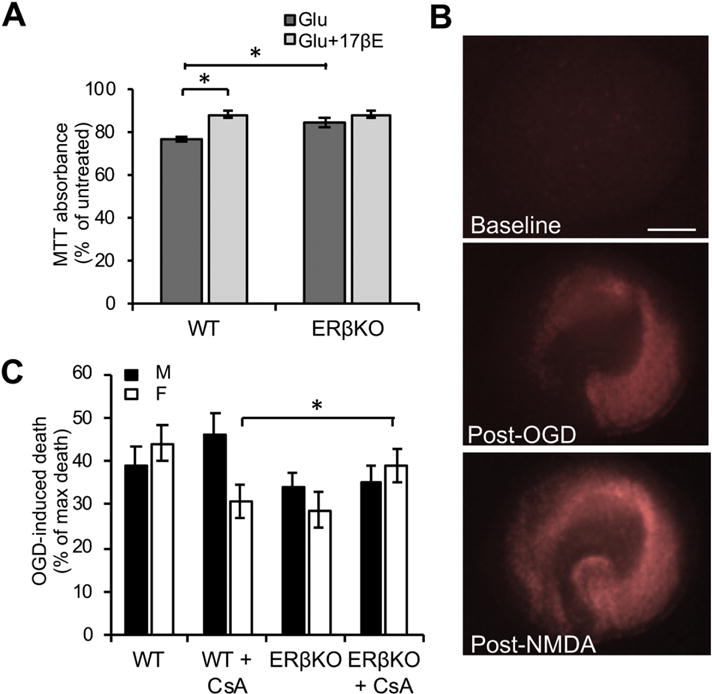
A) Quantification of MTT assay absorbance values in wild type (WT) or ERβKO primary cortical neuronal cultures treated with glutamate (100 μM), with or without 17βE. Data are expressed as percentage of untreated cultures. * p < 0.05 by two-way ANOVA with Bonferroni post-hoc analysis; n = 10-18 independent wells. B) Representative images of hippocampal slice cultures stained with propidium iodide (PI) (indicating cell death) at baseline, 24 hours post-oxygen glucose deprivation (OGD), and 24 hours post-treatment with 1 mM NMDA. C) Quantification of the percentage of PI fluorescence 24 hours post-OGD over the percentage of PI fluorescence post-NMDA treatment, with subtraction of fluorescence prior to any treatment. * p < 0.05 by two-way ANOVA with Bonferroni post-hoc analysis; n = 22-41 slices per group over 8 experiments.
Second, organotypic hippocampal slice cultures were prepared from postnatal day 5 wild type or ERβKO littermates and exposed to anoxic and glucose-free buffer (oxygen glucose deprivation, OGD). Slices were treated with propidium iodide (PI) to label dead cells, and images were captured before and 24 hours after OGD exposure. After capturing images of PI fluorescence, the same slices were treated with NMDA (1 mM) to induce maximal cell death. A representative set of images of hippocampal slices subjected to this protocol is shown in Figure 6B. There was a significant interaction effect of genotype and sex (F (1,115) = 5.25, p = 0.024, by two-way ANOVA, Fig. 6C). In slices from male mice, genetic ablation of ERβ did not affect cell death induced by OGD, but in slices from females, ERβKO had less OGD-induced cell death, when compared to wild type (p = 0.03, n = 22-41, by two-way ANOVA with Bonferroni correction). Taken together, results in these two models suggest that the mechanisms underlying the pathological responses to ischemic injury differ in males and females, and that ERβ is a key player in these differences.
3.5 ERβ interacts with mitochondrial proteins in an estrogen-dependent manner
To begin investigating the mechanism of ERβ modulation on MPT we designed a cell-based system to identify ERβ binding partners in mitochondria using an unbiased approach based on co-immunoprecipitation (co-IP) and protein mass spectrometry (MS). Since the specificity of commercially available antibodies against ERβ is controversial [48], we used a recombinant FLAG-tagged human ERβ DNA construct to express the full-length, 59 kDa protein in cultured cells. Western blot using anti-FLAG antibody allowed for both the detection and the IP of the expressed protein. The recombinant ERβ-FLAG protein was detected in whole cell homogenates, as well as in the cytosolic and enriched mitochondrial fractions, confirming that the protein localizes to multiple cellular compartments, including mitochondria (Fig. 7A). To address the role of the ERβ ligand estrogen in regulating its interactions with mitochondrial proteins, mitochondria extracted from cells transfected with ERβ-FLAG were incubated with estrogen (17βE, 10 μM) or vehicle (ethanol) for 10 minutes and throughout all steps of protein co-IP. This concentration of 17βE was used because it improved neuronal viability upon glutamate excitotoxicity, and therefore we inferred that it could also affect potential ERβ protein-protein interactions. A FLAG antibody covalently bound to resins was used to co-IP proteins. As negative control, cells mock transfected with empty plasmid were used. Finally, proteins were eluted and subjected to MS. Two independent experiments were conducted and only proteins that were detected and identified with high confidence (95%) in both experiments were considered. Among these, we selected only proteins known to reside in mitochondria and were >2 fold enriched in ERβ expressing cells treated with 17βE relative to mock, and which were not enriched in cells treated with vehicle.
Figure 7. ERβKO decreases the interaction between OSCP and CypD in brain mitochondria.
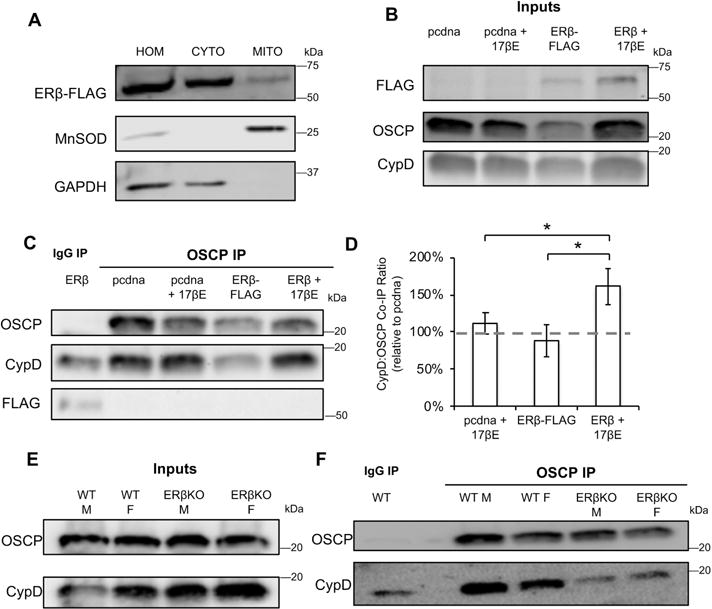
A) Western blot of total homogenate (HOM), cytosolic (CYTO) and mitochondrial (MITO) fractions of cells transfected with ERβ-FLAG. Manganese SOD and GAPDH are used as mitochondrial and cytosolic markers, respectively. B) Western blot of FLAG, OSCP and CypD in enriched mitochondrial fractions from cells expressing empty vector (pcdna) or ERβ-FLAG, with and without 17βE treatment. C) Western blot of co-IP eluate from IgG IP or OSCP co-IP. D) Quantification of the ratio of CypD:OSCP in co-IP eluates. Data are expressed relative to the CypD: OSCP ratio in pcdna transfected cells. * p < 0.05 by one-way ANOVA with Bonferroni post-hoc analysis; n = 7 independent biological replicates. E) Western blot of OSCP and CypD in purified brain mitochondria from wild type (WT) and ERβKO males and females. F) Western blot of co-IP eluate from IgG IP or OSCP co-IP.
MS of proteins co-immunoprecipitated with ERβ identified a relatively small number of peptides that fulfilled the enrichment criteria described above (Table 1). One of them was the trifunctional protein b (HADHB), an enzyme that catalyzes three steps of β-oxidation and interacts with complex I of the respiratory chain in the inner mitochondrial membrane. Calmodulin, hsp10, and several of the mitochondrial ribosome protein components were also identified. In addition, the co-IP revealed subunit c of the F0 ATPase and subunit α of the F1 ATPase as ERβ binding partners, which was particularly intriguing because ATPase was previously identified as an ERβ binding partner in rat brain [49]. Furthermore, both the β subunit [50] and the c ring [51]of the ATPase were found to be structural components of the MPT pore, and the ATPase complex was found to be a binding partner of CypD [52, 53].
Table 1.
Peptides identified by mass spectrometry of proteins eluted after Co-IP with ERβ-FLAG. Numbers indicate the ratio of the abundance of the peptides enriched in FLAG Co-IP in either the presence or the absence of 17βE, normalized by the peptide abundance in the Co-IP eluate from mock transfected cells.
| FLAG Co-IP + 17βE / mock | FLAG Co-IP / mock | |
|---|---|---|
| HADHB (mito trifunctional protein) | 3.9 | 0.9 |
| Calmodulin 2 | 3.1 | 0.8 |
| Hsp10 | 2.5 | 0.9 |
| Ribsomal protein L34 | 3.3 | 1.1 |
| Ribosomal protein L30 | 6.9 | 1.1 |
| Ribosomal protein L35 | 5.2 | 0.9 |
| Ribsomal protein L13 | 7.4 | 0.9 |
| Ribosomal protein S10 | 5.1 | 0.8 |
| ATPase subunit α (F1) | 2.6 | 0.9 |
| ATPase subunit c (F0) | 3.3 | 0.9 |
Since it was shown that CypD binds the ATPase complex at the level of the oligomycin sensitivity-conferring protein (OSCP) in the lateral stalk of the complex [39, 52], we investigated whether ERβ-FLAG could bind directly to OSCP. We transfected cells with ERβ-FLAG or empty vector (pcdna) and treated them with either vehicle or 10 μM 17βE for 24 hours. The presence of ERβ-FLAG in the mitochondrial fractions was confirmed by Western blot (Fig. 7B). Samples were subjected to co-IP using an antibody against OSCP or an IgG control and eluates were probed for OSCP, FLAG and CypD. OSCP was successfully pulled down in all samples, but we did not detect ERβ-FLAG interacting with OSCP (Fig. 7C). This result was in agreement with the ERβ-FLAG co-IP MS data, as OSCP was not identified. Although a small amount of CypD was found in the IgG co-IP, indicating non-specific binding, it was clearly enriched in the OSCP co-IP, confirming previous observations [39, 52]. Notably, addition of 17ßE to ERβ-FLAG transfected cells resulted in an increase in the amount of CypD which co-immunoprecipitated with OSCP relative to pcdna-transfected or vehicle-treated cells (Fig. 7D).
Since there was no direct interaction between ERβ and OSCP, we tested the hypothesis that ERβ could modulate the interaction between OSCP and CypD. We performed co-IP of OSCP in purified brain mitochondria from wild type and ERβKO male and female mice. Western blot indicated that expression of OSCP and CypD did not differ among sexes and genotypes (Fig. 7E). Interestingly, co-IP of OSCP in these samples revealed that the interaction between OSCP and CypD was greatly reduced in male and female ERβKO mitochondria, to levels comparable to the non-specific IgG control (Fig. 7F), confirming the hypothesis that ERβ affects OSCP-CypD binding.
4.Discussion
We have identified a novel pathway for MPT modulation in brain that can contribute to sex differences in ischemic brain injury. We first demonstrated a sex difference in brain mitochondrial calcium capacity whereby female brain mitochondria undergo MPT at a lower calcium concentration than male mitochondria. Decreased mitochondrial calcium capacity reflects an increased propensity for calcium-dependent MPT pore opening. The consequences of increased MPT sensitivity depend on the properties of the pore opening. MPT could be an irreversible permeabilization event, resulting in mitochondrial swelling and release of solutes, including calcium and pro-apoptotic factors [6, 21], or it could be a reversible, limited permeabilization event that can facilitate calcium release from the mitochondrial matrix and prevent calcium overload [12]. Previous studies in rat heart show that female mitochondria are more resistant to calcium-induced swelling following a single calcium bolus than male mitochondria [22] and have improved recovery from calcium-induced depolarization [23]. Furthermore, in heart mitochondrial calcium overload is a key cellular event following ischemia reperfusion, which profoundly affects the outcome of ischemia [14]. Taken together, this evidence suggests that sex differences in calcium-induced MPT could be similar to the differences observed in heart ischemia [54]. Little is known about the mechanisms of sex differences in ischemic brain injury and its potential links to MPT. However, because of the increased risk and worse outcomes of stroke in females, particularly in early menopause [1], it is possible that sex hormones and their receptors play a role in influencing stroke by modulating brain MPT. Therefore, it could be hypothesized that decreased calcium capacity in female brain mitochondria worsens ischemic brain injury. On the other hand, a protective reversible opening of a low-conductance pore permeable to calcium cannot be ruled out, since our experimental paradigm with consecutive boluses of calcium is designed to force brain mitochondria into irreversible pore opening. Future experiments will address this possibility.
We used pharmacological and genetic approaches to begin to address the mechanisms underlying sex differences in brain MPT. We found that both inhibition and deletion of CypD eliminated the sex differences in mitochondrial calcium capacity, suggesting that CypD is a necessary component of the mechanism underlying sex differences. We also demonstrated that the ERβ antagonist PHTPP increased mitochondrial calcium capacity. These findings were particularly intriguing, because they suggested a functional link between ERβ and CypD. The effect of PHTPP in increasing brain mitochondrial calcium capacity was more modest than the effect of CsA. We hypothesize that the reason for this difference could be that the effect of ERβ inhibition occurs at the level of MPT regulation upstream of CypD, via modulation of protein-protein interactions. The effect of ERβ inhibition on MPT can therefore be less pronounced than blocking CypD itself pharmacologically or genetically.
Since MPT inhibition has been explored as a therapeutic target [55], but its use has been traditionally challenging due to off target effects of CypD inhibition [56], we deemed that targeting ERβ could have applications in modulating brain mitochondrial calcium overload in pathological conditions, such as ischemia. This led us to further investigate the role of ERβ in MPT.
We found that ERβ modulated sensitivity to CsA, as the inhibitory effect of CsA on MPT in brain mitochondria was attenuated by ERβKO, in both males and females. These data further confirmed the functional relationship between ERβ and CypD in modulating calcium capacity. While PHTPP increased calcium capacity, ERβKO did not. The difference between pharmacological inhibition and genetic ablation could be explained if binding of the inhibited receptor to a yet unknown target induces conformational changes and downstream effects that cannot occur in the absence of the receptor.
Similar to our results in brain mitochondria, it was previously shown that male and female cardiac mitochondria also contain similar amounts of CypD, VDAC, and ATPase [22], highlighting a role for protein-protein interaction, rather than protein expression levels, as the putative mechanism causing a blunted CsA effect in ERβKO brain mitochondria. Therefore, using co-IP of mitochondrial proteins with a tagged ERβ followed by unbiased MS, we searched for candidate ERβ interactors in mitochondria. Interestingly, one of the identified proteins, HADHB, was previously shown to bind ERβ [57]. Although the involvement of HADHB in MPT is still unknown, its location and interactions with inner membrane components and ERβ, as well as its effects on cardiolipin [58] make it an intriguing candidate for MPT regulation. The list also included Calmodulin [59] and hsp10 [60], which have been implicated in MPT regulation, although the mechanisms are unknown, and further studies will be needed to determine how these proteins affect MPT in brain. Several of the mitochondrial ribosome protein components were also identified. The strong enrichment of the interaction of these proteins with ERβ by estrogen is surprising and the functional meaning unclear, especially in relationship to MPT. However, it is possible that ERβ in mitochondria has other functions, which remain to be elucidated.
The co-IP followed by MS also revealed subunit c of the F0 ATPase and subunit α of the F1 ATPase as ERβ binding partners. This was a particularly interesting result for three reasons. First, ATPase was previously identified as an ERβ binding partner in rat brain [49]. Second, both dimers of the β subunit [50] and the c ring [51] of ATPase were proposed to be structural components of the MPT pore. Third, ATPase was found to be a binding partner of CypD at the OSCP subunit of the complex [39, 52, 53]. Notably, co-IP studies using OSCP antibody in cultured cells did not detect a physical interaction between ERβ and OSCP, but showed an interaction between OSCP and CypD, which was enhanced by the addition of 17βE to ERβ-FLAG transfected cells. Furthermore, in brain mitochondria, ERβKO decreased the binding of CypD to OSCP. Therefore, modulation of the interaction between OSCP and CypD could be a mechanism of MPT regulation by ERβ in brain mitochondria.
To explore whether ERβ plays a role in modulating sex differences in the outcome of ischemic injury, we used cortical neuronal culture and organotypic hippocampal slice culture systems. We first demonstrated in embryonic cortical neurons that both 17βE and ERβKO are protective against glutamate toxicity. Previous reports have shown that female-derived slice cultures were less susceptible to OGD injury than males [61], however, we found the same baseline susceptibility to hippocampal cell death induced by OGD in male and female slices. However, we found that female-derived slices were protected by ERβKO, while male-derived cultures were not. The results in both primary neuronal cultures as well as organotypic hippocampal slices support the hypothesis that ERβ plays a role in modulating sex differences in ischemic injury. They also contribute to an increasing body of evidence on the role of ERβ in neuronal vulnerability, since it was shown that ERβKO is protective under oxidative stress in primary neuronal culture models [62].
The observation that both 17βE and ERβKO can be neuroprotective may appear puzzling. However, since 17βE modulates the affinity of CypD for OSCP, we propose that the conformation of estrogen-bound ERβ is different than unbound. It is unknown whether changes in the interaction between CypD and OSCP have positive or negative effects on MPT. Similarly, it is unknown if modulation of the interaction by ERβKO is a positive or negative regulator of MPT. Therefore, it is plausible that, while estrogen is clearly a positive regulator of ERβ transcriptional functions, it serves as a negative regulator of cell toxicity by modulating the molecular machinery involved in membrane permeabilization to calcium.
In conclusion, this work identifies new molecular players involved in brain MPT regulation and mitochondrial calcium capacity. Furthermore, the findings reported here shed new light on the mechanisms of sex differences in MPT, a key pathogenic event in ischemic brain injury. Importantly, the modulation of brain MPT may also be pathologically relevant to other sex biased brain degenerative conditions, in which mitochondrial calcium handling is altered, such as amyotrophic lateral sclerosis [34, 35], Alzheimer’s disease [63] and Parkinson’s disease [64]. Taken together, these findings suggest that the function of ERβ in modulating MPT could become a therapeutic target.
Highlights.
Female brain mitochondria are more susceptible to permeability transition (MPT)
ERβ regulates calcium-induced MPT in brain mitochondria
ERβ affects the interaction of cyclophilin D with ATPase MPT components
ERβ modulates ischemic/excitotoxic injury in cell and slice culture models
Acknowledgments
Funding
This work was funded by NIH/NINDS pre-doctoral Fellowship F31NS090715 and by R01NS095692 and by NIH SIG grant 1S10RR025449-01 support for the Orbitrap mass spectrometer.
Abbreviations
- MPT
mitochondrial permeability transition
- ERβ
estrogen receptor beta
- CypD
cyclophilin D
- CsA
cyclosporine A
- MCU
mitochondrial calcium uniporter
- OSCP
oligomycin sensitivity-conferring protein
- Δψm
mitochondrial membrane potential
- VDAC
voltage dependent anion channel
- MICU1
mitochondrial calcium uptake 1
- MS
mass spectrometry
- Co-IP
co-immunoprecipitation
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.M. Writing Group. Lloyd-Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, De Simone G, Ferguson TB, Ford E, Furie K, Gillespie C, Go A, Greenlund K, Haase N, Hailpern S, Ho PM, Howard V, Kissela B, Kittner S, Lackland D, Lisabeth L, Marelli A, McDermott MM, Meigs J, Mozaffarian D, Mussolino M, Nichol G, Roger VL, Rosamond W, Sacco R, Sorlie P, Roger VL, Thom T, Wasserthiel-Smoller S, Wong ND, Wylie-Rosett J, C. American Heart Association Statistics, S. Stroke Statistics Heart disease and stroke statistics–2010 update: a report from the American Heart Association. Circulation. 2010;121:e46–e215. doi: 10.1161/CIRCULATIONAHA.109.192667. [DOI] [PubMed] [Google Scholar]
- 2.Girijala RL, Sohrabji F, Bush RL. Sex differences in stroke: Review of current knowledge and evidence. Vasc Med. 2017;22:135–145. doi: 10.1177/1358863X16668263. [DOI] [PubMed] [Google Scholar]
- 3.Spychala MS, Honarpisheh P, McCullough LD. Sex differences in neuroinflammation and neuroprotection in ischemic stroke. J Neurosci Res. 2017;95:462–471. doi: 10.1002/jnr.23962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Racay P, Tatarkova Z, Chomova M, Hatok J, Kaplan P, Dobrota D. Mitochondrial calcium transport and mitochondrial dysfunction after global brain ischemia in rat hippocampus. Neurochem Res. 2009;34:1469–1478. doi: 10.1007/s11064-009-9934-7. [DOI] [PubMed] [Google Scholar]
- 5.Starkov AA, Chinopoulos C, Fiskum G. Mitochondrial calcium and oxidative stress as mediators of ischemic brain injury. Cell Calcium. 2004;36:257–264. doi: 10.1016/j.ceca.2004.02.012. [DOI] [PubMed] [Google Scholar]
- 6.Crompton M, Virji S, Doyle V, Johnson N, Ward JM. The mitochondrial permeability transition pore. Biochem Soc Symp. 1999;66:167–179. doi: 10.1042/bss0660167. [DOI] [PubMed] [Google Scholar]
- 7.De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011;476:336–340. doi: 10.1038/nature10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McCormack JG, Halestrap AP, Denton RM. Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol Rev. 1990;70:391–425. doi: 10.1152/physrev.1990.70.2.391. [DOI] [PubMed] [Google Scholar]
- 9.Tymianski M, Charlton MP, Carlen PL, Tator CH. Source specificity of early calcium neurotoxicity in cultured embryonic spinal neurons. J Neurosci. 1993;13:2085–2104. doi: 10.1523/JNEUROSCI.13-05-02085.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peng TI, Greenamyre JT. Privileged access to mitochondria of calcium influx through N-methyl-D-aspartate receptors. Mol Pharmacol. 1998;53:974–980. [PubMed] [Google Scholar]
- 11.Gouriou Y, Demaurex N, Bijlenga P, De Marchi U. Mitochondrial calcium handling during ischemia-induced cell death in neurons. Biochimie. 2011;93:2060–2067. doi: 10.1016/j.biochi.2011.08.001. [DOI] [PubMed] [Google Scholar]
- 12.Bernardi P, von Stockum S. The permeability transition pore as a Ca(2+) release channel: new answers to an old question. Cell Calcium. 2012;52:22–27. doi: 10.1016/j.ceca.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Giorgio V, Guo L, Bassot C, Petronilli V, Bernardi P. Calcium and regulation of the mitochondrial permeability transition. Cell Calcium. 2017 doi: 10.1016/j.ceca.2017.05.004. [DOI] [PubMed] [Google Scholar]
- 14.Halestrap AP, Richardson AP. The mitochondrial permeability transition: a current perspective on its identity and role in ischaemia/reperfusion injury. J Mol Cell Cardiol. 2015;78:129–141. doi: 10.1016/j.yjmcc.2014.08.018. [DOI] [PubMed] [Google Scholar]
- 15.Bernardi P, Broekemeier KM, Pfeiffer DR. Recent progress on regulation of the mitochondrial permeability transition pore; a cyclosporin-sensitive pore in the inner mitochondrial membrane. J Bioenerg Biomembr. 1994;26:509–517. doi: 10.1007/BF00762735. [DOI] [PubMed] [Google Scholar]
- 16.Connern CP, Halestrap AP. Recruitment of mitochondrial cyclophilin to the mitochondrial inner membrane under conditions of oxidative stress that enhance the opening of a calcium-sensitive non-specific channel. Biochem J. 1994;302(Pt 2):321–324. doi: 10.1042/bj3020321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, Hetz C, Danial NN, Moskowitz MA, Korsmeyer SJ. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci U S A. 2005;102:12005–12010. doi: 10.1073/pnas.0505294102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Basso E, Fante L, Fowlkes J, Petronilli V, Forte MA, Bernardi P. Properties of the permeability transition pore in mitochondria devoid of Cyclophilin D. J Biol Chem. 2005;280:18558–18561. doi: 10.1074/jbc.C500089200. [DOI] [PubMed] [Google Scholar]
- 19.Broekemeier KM, Dempsey ME, Pfeiffer DR. Cyclosporin A is a potent inhibitor of the inner membrane permeability transition in liver mitochondria. J Biol Chem. 1989;264:7826–7830. [PubMed] [Google Scholar]
- 20.Szabo I, Zoratti M. The giant channel of the inner mitochondrial membrane is inhibited by cyclosporin A. J Biol Chem. 1991;266:3376–3379. [PubMed] [Google Scholar]
- 21.Hansson MJ, Morota S, Chen L, Matsuyama N, Suzuki Y, Nakajima S, Tanoue T, Omi A, Shibasaki F, Shimazu M, Ikeda Y, Uchino H, Elmer E. Cyclophilin D-sensitive mitochondrial permeability transition in adult human brain and liver mitochondria. J Neurotrauma. 2011;28:143–153. doi: 10.1089/neu.2010.1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Milerova M, Drahota Z, Chytilova A, Tauchmannova K, Houstek J, Ostadal B. Sex difference in the sensitivity of cardiac mitochondrial permeability transition pore to calcium load. Mol Cell Biochem. 2016;412:147–154. doi: 10.1007/s11010-015-2619-4. [DOI] [PubMed] [Google Scholar]
- 23.Arieli Y, Gursahani H, Eaton MM, Hernandez LA, Schaefer S. Gender modulation of Ca(2+) uptake in cardiac mitochondria. J Mol Cell Cardiol. 2004;37:507–513. doi: 10.1016/j.yjmcc.2004.04.023. [DOI] [PubMed] [Google Scholar]
- 24.Zhao L, Brinton RD. Estrogen receptor alpha and beta differentially regulate intracellular Ca(2+) dynamics leading to ERK phosphorylation and estrogen neuroprotection in hippocampal neurons. Brain Res. 2007;1172:48–59. doi: 10.1016/j.brainres.2007.06.092. [DOI] [PubMed] [Google Scholar]
- 25.Zhang L, Blackman BE, Schonemann MD, Zogovic-Kapsalis T, Pan X, Tagliaferri M, Harris HA, Cohen I, Pera RA, Mellon SH, Weiner RI, Leitman DC. Estrogen receptor beta-selective agonists stimulate calcium oscillations in human and mouse embryonic stem cell-derived neurons. PLoS One. 2010;5:e11791. doi: 10.1371/journal.pone.0011791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nilsen J, Chen S, Irwin RW, Iwamoto S, Brinton RD. Estrogen protects neuronal cells from amyloid beta-induced apoptosis via regulation of mitochondrial proteins and function. BMC Neurosci. 2006;7:74. doi: 10.1186/1471-2202-7-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Simpkins JW, Yi KD, Yang SH, Dykens JA. Mitochondrial mechanisms of estrogen neuroprotection. Biochim Biophys Acta. 2010;1800:1113–1120. doi: 10.1016/j.bbagen.2009.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tremblay GB, Tremblay A, Copeland NG, Gilbert DJ, Jenkins NA, Labrie F, Giguere V. Cloning, chromosomal localization, and functional analysis of the murine estrogen receptor beta. Mol Endocrinol. 1997;11:353–365. doi: 10.1210/mend.11.3.9902. [DOI] [PubMed] [Google Scholar]
- 29.Greene GL, Gilna P, Waterfield M, Baker A, Hort Y, Shine J. Sequence and expression of human estrogen receptor complementary DNA. Science. 1986;231:1150–1154. doi: 10.1126/science.3753802. [DOI] [PubMed] [Google Scholar]
- 30.Chen JQ, Delannoy M, Cooke C, Yager JD. Mitochondrial localization of ERalpha and ERbeta in human MCF7 cells. Am J Physiol Endocrinol Metab. 2004;286:E1011–1022. doi: 10.1152/ajpendo.00508.2003. [DOI] [PubMed] [Google Scholar]
- 31.Yang SH, Liu R, Perez EJ, Wen Y, Stevens SM, Jr, Valencia T, Brun-Zinkernagel AM, Prokai L, Will Y, Dykens J, Koulen P, Simpkins JW. Mitochondrial localization of estrogen receptor beta. Proc Natl Acad Sci U S A. 2004;101:4130–4135. doi: 10.1073/pnas.0306948101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Milner TA, Ayoola K, Drake CT, Herrick SP, Tabori NE, McEwen BS, Warrier S, Alves SE. Ultrastructural localization of estrogen receptor beta immunoreactivity in the rat hippocampal formation. J Comp Neurol. 2005;491:81–95. doi: 10.1002/cne.20724. [DOI] [PubMed] [Google Scholar]
- 33.Milner TA, Thompson LI, Wang G, Kievits JA, Martin E, Zhou P, McEwen BS, Pfaff DW, Waters EM. Distribution of estrogen receptor beta containing cells in the brains of bacterial artificial chromosome transgenic mice. Brain Res. 2010;1351:74–96. doi: 10.1016/j.brainres.2010.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Damiano M, Starkov AA, Petri S, Kipiani K, Kiaei M, Mattiazzi M, Flint Beal M, Manfredi G. Neural mitochondrial Ca2+ capacity impairment precedes the onset of motor symptoms in G93A Cu/Zn-superoxide dismutase mutant mice. J Neurochem. 2006;96:1349–1361. doi: 10.1111/j.1471-4159.2006.03619.x. [DOI] [PubMed] [Google Scholar]
- 35.Kim HJ, Magrane J, Starkov AA, Manfredi G. The mitochondrial calcium regulator cyclophilin D is an essential component of oestrogen-mediated neuroprotection in amyotrophic lateral sclerosis. Brain. 2012;135:2865–2874. doi: 10.1093/brain/aws208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim HJ, Magrane J. Isolation and culture of neurons and astrocytes from the mouse brain cortex. Methods Mol Biol. 2011;793:63–75. doi: 10.1007/978-1-61779-328-8_4. [DOI] [PubMed] [Google Scholar]
- 37.Gahwiler BH, Thompson SM, Muller D. Preparation and maintenance of organotypic slice cultures of CNS tissue. Curr Protoc Neurosci. doi: 10.1002/0471142301.ns0611s09. Chapter 6 (2001) Unit 6 11. [DOI] [PubMed] [Google Scholar]
- 38.Zhou P, Qian L, D’Aurelio M, Cho S, Wang G, Manfredi G, Pickel V, Iadecola C. Prohibitin reduces mitochondrial free radical production and protects brain cells from different injury modalities. J Neurosci. 2012;32:583–592. doi: 10.1523/JNEUROSCI.2849-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Beck SJ, Guo L, Phensy A, Tian J, Wang L, Tandon N, Gauba E, Lu L, Pascual JM, Kroener S, Du H. Deregulation of mitochondrial F1FO-ATP synthase via OSCP in Alzheimer’s disease. Nat Commun. 2016;7:11483. doi: 10.1038/ncomms11483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Argaud L, Gateau-Roesch O, Muntean D, Chalabreysse L, Loufouat J, Robert D, Ovize M. Specific inhibition of the mitochondrial permeability transition prevents lethal reperfusion injury. J Mol Cell Cardiol. 2005;38:367–374. doi: 10.1016/j.yjmcc.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 41.Le Lamer S, Paradis S, Rahmouni H, Chaimbault C, Michaud M, Culcasi M, Afxantidis J, Latreille M, Berna P, Berdeaux A, Pietri S, Morin D, Donazzolo Y, Abitbol JL, Pruss RM, Schaller S. Translation of TRO40303 from myocardial infarction models to demonstration of safety and tolerance in a randomized Phase I trial. J Transl Med. 2014;12:38. doi: 10.1186/1479-5876-12-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Warne J, Pryce G, Hill JM, Shi X, Lenneras F, Puentes F, Kip M, Hilditch L, Walker P, Simone MI, Chan AW, Towers GJ, Coker AR, Duchen MR, Szabadkai G, Baker D, Selwood DL. Selective Inhibition of the Mitochondrial Permeability Transition Pore Protects against Neurodegeneration in Experimental Multiple Sclerosis. J Biol Chem. 2016;291:4356–4373. doi: 10.1074/jbc.M115.700385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Martin LJ, Fancelli D, Wong M, Niedzwiecki M, Ballarini M, Plyte S, Chang Q. GNX-4728, a novel small molecule drug inhibitor of mitochondrial permeability transition, is therapeutic in a mouse model of amyotrophic lateral sclerosis. Front Cell Neurosci. 2014;8:433. doi: 10.3389/fncel.2014.00433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Compton DR, Sheng S, Carlson KE, Rebacz NA, Lee IY, Katzenellenbogen BS, Katzenellenbogen JA. Pyrazolo[1,5-a]pyrimidines: estrogen receptor ligands possessing estrogen receptor beta antagonist activity. J Med Chem. 2004;47:5872–5893. doi: 10.1021/jm049631k. [DOI] [PubMed] [Google Scholar]
- 45.Akerman KE, Wikstrom MK. Safranine as a probe of the mitochondrial membrane potential. FEBS Lett. 1976;68:191–197. doi: 10.1016/0014-5793(76)80434-6. [DOI] [PubMed] [Google Scholar]
- 46.Mosselman S, Polman J, Dijkema R. ER beta: identification and characterization of a novel human estrogen receptor. FEBS Lett. 1996;392:49–53. doi: 10.1016/0014-5793(96)00782-x. [DOI] [PubMed] [Google Scholar]
- 47.Nicholls DG, Crompton M. Mitochondrial calcium transport. FEBS Lett. 1980;111:261–268. doi: 10.1016/0014-5793(80)80806-4. [DOI] [PubMed] [Google Scholar]
- 48.Snyder MA, Smejkalova T, Forlano PM, Woolley CS. Multiple ERbeta antisera label in ERbeta knockout and null mouse tissues. J Neurosci Methods. 2010;188:226–234. doi: 10.1016/j.jneumeth.2010.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alvarez-Delgado C, Mendoza-Rodriguez CA, Picazo O, Cerbon M. Different expression of alpha and beta mitochondrial estrogen receptors in the aging rat brain: interaction with respiratory complex V. Exp Gerontol. 2010;45:580–585. doi: 10.1016/j.exger.2010.01.015. [DOI] [PubMed] [Google Scholar]
- 50.Giorgio V, von Stockum S, Antoniel M, Fabbro A, Fogolari F, Forte M, Glick GD, Petronilli V, Zoratti M, Szabo I, Lippe G, Bernardi P. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc Natl Acad Sci U S A. 2013;110:5887–5892. doi: 10.1073/pnas.1217823110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alavian KN, Beutner G, Lazrove E, Sacchetti S, Park HA, Licznerski P, Li H, Nabili P, Hockensmith K, Graham M, Porter GA, Jr, Jonas EA. An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc Natl Acad Sci U S A. 2014;111:10580–10585. doi: 10.1073/pnas.1401591111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Giorgio V, Bisetto E, Soriano ME, Dabbeni-Sala F, Basso E, Petronilli V, Forte MA, Bernardi P, Lippe G. Cyclophilin D modulates mitochondrial F0F1-ATP synthase by interacting with the lateral stalk of the complex. J Biol Chem. 2009;284:33982–33988. doi: 10.1074/jbc.M109.020115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chinopoulos C, Konrad C, Kiss G, Metelkin E, Torocsik B, Zhang SF, Starkov AA. Modulation of F0F1-ATP synthase activity by cyclophilin D regulates matrix adenine nucleotide levels. FEBS J. 2011;278:1112–1125. doi: 10.1111/j.1742-4658.2011.08026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ostadal B, Ostadal P. Sex-based differences in cardiac ischaemic injury and protection: therapeutic implications. Br J Pharmacol. 2014;171:541–554. doi: 10.1111/bph.12270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sileikyte J, Forte M. Shutting down the pore: The search for small molecule inhibitors of the mitochondrial permeability transition. Biochim Biophys Acta. 2016;1857:1197–1202. doi: 10.1016/j.bbabio.2016.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Laupacis A, Keown PA, Ulan RA, McKenzie N, Stiller CR. Cyclosporin A: a powerful immunosuppressant. Can Med Assoc J. 1982;126:1041–1046. [PMC free article] [PubMed] [Google Scholar]
- 57.Zhou Z, Zhou J, Du Y. Estrogen receptor beta interacts and colocalizes with HADHB in mitochondria. Biochem Biophys Res Commun. 2012;427:305–308. doi: 10.1016/j.bbrc.2012.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Taylor WA, Mejia EM, Mitchell RW, Choy PC, Sparagna GC, Hatch GM. Human trifunctional protein alpha links cardiolipin remodeling to beta-oxidation. PLoS One. 2012;7:e48628. doi: 10.1371/journal.pone.0048628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Toledo FD, Perez LM, Basiglio CL, Ochoa JE, Sanchez Pozzi EJ, Roma MG. The Ca(2)(+)-calmodulin-Ca(2)(+)/calmodulin-dependent protein kinase II signaling pathway is involved in oxidative stress-induced mitochondrial permeability transition and apoptosis in isolated rat hepatocytes. Arch Toxicol. 2014;88:1695–1709. doi: 10.1007/s00204-014-1219-5. [DOI] [PubMed] [Google Scholar]
- 60.He L, Lemasters JJ. Heat shock suppresses the permeability transition in rat liver mitochondria. J Biol Chem. 2003;278:16755–16760. doi: 10.1074/jbc.M300153200. [DOI] [PubMed] [Google Scholar]
- 61.Li H, Pin S, Zeng Z, Wang MM, Andreasson KA, McCullough LD. Sex differences in cell death. Ann Neurol. 2005;58:317–321. doi: 10.1002/ana.20538. [DOI] [PubMed] [Google Scholar]
- 62.Yang SH, Sarkar SN, Liu R, Perez EJ, Wang X, Wen Y, Yan LJ, Simpkins JW. Estrogen receptor beta as a mitochondrial vulnerability factor. J Biol Chem. 2009;284:9540–9548. doi: 10.1074/jbc.M808246200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Supnet C, Bezprozvanny I. The dysregulation of intracellular calcium in Alzheimer disease. Cell Calcium. 2010;47:183–189. doi: 10.1016/j.ceca.2009.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Martin LJ, Semenkow S, Hanaford A, Wong M. Mitochondrial permeability transition pore regulates Parkinson’s disease development in mutant alpha-synuclein transgenic mice. Neurobiol Aging. 2014;35:1132–1152. doi: 10.1016/j.neurobiolaging.2013.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]