

ABSTRACT
Tissue factor (TF) is the main initiator of coagulation and procoagulant phospholipids (PPL) are key components in promoting coagulation activity in blood. Both TF and PPL may be presented on the surface of extracellular vesicles (EVs), thus contributing to their procoagulant activity. These EVs may constitute a substantial part of pathological hypercoagulability that is responsible for triggering a higher risk of thrombosis in certain patients. The aim of this study was to describe a model system for the isolation of EVs required for investigating their effect on coagulation. Differential ultracentrifugation (DUC) with and without a single washing step was used to isolate and evaluate the procoagulant capacity of EVs from healthy volunteers through analysis of thrombin generation and PPL activity. Ultracentrifugation at 20,000 × g and 100,000 × g resulted in pellets containing larger vesicles and smaller vesicles, respectively. Isolation yield of particle concentration was assessed by nanoparticle tracking analysis. Immunoelectron microscopy and western blotting revealed vesicles positive for the commonly used EV-marker CD9. Plasma proteins and lipoproteins were co-isolated with the EVs; however, application of a washing step clearly diminished the amount of contaminants. The isolated EVs were capable of enhancing thrombin generation, mainly due to PPL predominantly present in pellets from 20,000 × g centrifugation, and correlated with the activity measured by a PPL activity assay. Thus, DUC was proficient for the isolation of EVs with minimal contamination from plasma proteins and lipoproteins, and the setup can be used to study EV-associated procoagulant activity. This may be useful in determining the procoagulant activity of EVs in patients at potentially increased risk of developing thrombosis, e.g. cancer patients.
Abbreviations: TF: Tissue factor; PL: Phospholipids; EVs: Extracellular vesicles; FXa: Activated coagulation factor X; TGA: Thrombin generation assay; PPL: Procoagulant phospholipids; DUC: Differential ultracentrifugation; NTA: Nanoparticle tracking analysis; TEM: Transmission electron microscopy; SPP: Standard pool plasma; CTI: Corn trypsin inhibitor; 20K: 20,000 × g; 100K: 100,000 × g; FVIII: Coagulation factor VIII
KEYWORDS: Electron microscopy, extracellular vesicles, isolation, microvesicles, thrombin generation, tissue factor, procoagulant phospholipids
Introduction
The coagulation processes in haemostasis are mainly initiated by tissue factor (TF). An important cofactor in haemostasis, besides calcium, is anionic phospholipids (PL); this procoagulant factor is necessary for the formation of the important coagulation complexes binding to this surface. Membrane-associated TF, and anionic PL, mainly phosphatidylserine, are embedded in the membrane of extracellular vesicles (EVs) circulating in the blood [1,2]. EVs are commonly divided into smaller vesicles, mainly exosomes, and larger vesicles, mainly microvesicles, dependent on size, biogenesis, and phenotypic origin [3,4]. It is probably mostly microvesicles that carry TF and phosphatidylserine and therefore, they are possibly the most procoagulant of the EVs [5,6]. EVs are elevated in the blood of patients in a variety of diseases, such as cancer, and may play a significant role in the development of venous thromboembolism [7–9].
Isolating exosomes and microvesicles can be achieved through various techniques, including size exclusion chromatography, immunoaffinity precipitation, and ultracentrifugation, however, each technique has both advantages and limitations [10]. There are different available methods to study the procoagulant activity of EVs. Hisada et al. describes two assays to measure microvesicle TF-dependent generation of activated coagulation factor X (FXa) [11]. Another approach is to evaluate the overall procoagulant effect of EVs by using a modified version of the thrombin generation assay (TGA) described by Hemker et al. [12]. This has been tested in patients with thrombosis or cancer [13,14]. Marchetti and co-workers [15] reported evidence of procoagulant phospholipid (PPL) activity in thrombocythemia patients using a combination of TGA and a PPL activity assay. However, the methods available to study the procoagulant effect of EVs retain certain limitations. Thus, the combination of assays with increased sensitivity towards TF and phosphatidylserine and quantitative and phenotypical analyses of EVs may convey a more accurate profile of procoagulant EV activity. This may, in a clinical perspective, help clarify the role of EVs in coagulation in identifying certain procoagulant patient groups or individual patients at risk of developing venous thromboembolism.
Therefore, the aim of this study was to compare the procoagulant effect of EVs of different sizes isolated from healthy donors using differential ultracentrifugation (DUC) and to examine whether a post isolation washing step is imperative for the preparation of an EV sample devoid of lipoproteins and plasma remnants. DUC was chosen as it is a commonly used isolation method for the preparation of EVs. The isolated EVs were quantitated and validated for specific phenotypical features using nanoparticle tracking analysis (NTA), western blot, and transmission electron microscopy (TEM) with immunogold labelling. The procoagulant effect of the isolated EVs was investigated by TGA and a PPL activity assay.
Materials and methods
Sample collection and EV isolation
To examine the procoagulant effect of EVs, blood was collected from 12 healthy fasting volunteers in the morning. Six of these 12 donors also delivered samples to test the effect of contact activation inhibition by corn trypsin inhibitor (CTI). All volunteers had given informed consent and the study was conducted in accordance with the Declaration of Helsinki. Blood samples were collected from the antecubital vein with tourniquet and a 21-gauge needle in 6 ml 3.2% (0.105 M) citrated tubes (BD Vacutainer, BD Biosciences, San Jose, CA, USA). The blood samples were left 15 min at room temperature after collection, before platelet-free plasma was extracted by a double centrifugation at 2,500 × g for 15 min according to international recommendations [10,16], leaving 1 cm plasma both above the buffy coat and the pellet in the subsequent centrifugation step. Platelet-free-plasma was obtained within one hour from blood collection. Standard pooled plasma (SPP) was collected once from one donor in 6 ml standard citrate tubes and once from another donor in 4.5 ml specialized corn trypsin inhibitor (CTI) tubes (final concentration of CTI in blood: 50 µg/ml CTI, Haematologic Technologies, Essex Junction, VT, USA) annotated SPP and SPP+CTI, respectively. SPP and SPP+CTI were extracted by double-centrifugation at 2,500 × g for 15 min similarly to the procedure described above. Larger EVs were pelleted from 1 ml plasma at 20,000 × g (20K) for 30 min at 4°C. The supernatant was further centrifuged at 100,000 × g (100K) for 60 min at 4°C to pellet smaller EVs. Pellets resuspended in SPP+CTI were subjected to a washing step after the initial centrifugation step by resuspension in 1 ml phosphate-buffered saline (PBS) and re-centrifugation at the same g-force. For direct comparison of washed and non-washed pellets on thrombin generation an additional portion of blood for SPP was obtained from one of the initial SPP donors. Followed by the initial preparation of platelet-free-plasma, this portion of SPP was eventually deprived of EVs using a 100,000 × g centrifugation for 60 min at 4°C. EV isolation was performed in an Avanti J-30i ultracentrifuge with a JA-30.50 rotor, k-factor 280 (Beckman Coulter, Brea, CA, USA). All pellets, washed and unwashed, were finally resuspended in either 200 µl SPP or SPP+CTI (i.e. these suspensions had a 5-fold higher concentration of EVs) for TGA and PPL activity assay or PBS for NTA, TEM, western blot, and protein assay. All plasma samples and EV suspensions were stored at −80°C until analysis.
Nanoparticle tracking analysis
Particle concentration and size determination was measured using NTA and performed on a LM10-HS system (Malvern Instruments Ltd, Malvern, UK) equipped with a 405 nm laser and a Luca-DL EMCCD camera (Andor Technology, Belfast, UK). The analysis was performed to confirm isolation of particles in the EV size range. Silica Microspheres (0.1 µm, Polysciences, Hirchberg, Germany) were used as standard to establish software settings. For analysis, samples were diluted in PBS to obtain an average of 20–80 particles per frame and a total of 5 × 30 s videos were captured for each sample. The specific settings used were camera level 10, detection threshold 2, and blur 9 × 9 were employed. The addition of 0.03% Tween 20 (Bio-Rad, Copenhagen, Denmark) would prevent EVs in the suspensions from aggregating [17] and this way amend the outcome of the NTA, therefore, this was also evaluated. Data was processed by the Nanosight NTA software version 3.0 (Malvern Instruments Ltd, Malvern, UK) to determine particle concentration and size.
Transmission electron microscopy and immunogold labelling
TEM was used for the structural characterization of isolated EVs. TEM was performed according to the protocol adopted by Vogel et al. [17]. Procedurally, five microliters of EV suspension was mounted on a carbon coated, glow discharged 300-mesh nickel grid (SPI Supplies, Chester, PA, USA) for 30 s, then stained with 1 to 3 drops of 1% (w/v) phosphotungstic acid (Ted Pella, Caspilor AB, Lindingö, Sweden), pH 7.0, for 15 s and blotted dry for a few seconds on filter paper. EV marker CD9 was used for phenotypic characterization of vesicles. For immunogold labelling, samples were positioned for 30 s on an identical grid as described above and washed 3 × 5 min in PBS. Subsequently, the grids were blocked for 5 min in one drop 0.5% ovalbumin (Sigma-Aldrich, St. Louis, MO, USA) in PBS. After blockage, the grids were incubated in one drop of primary anti-CD9 antibody (clone M-L13, BD Pharmingen, San Jose, CA, USA) 1:50 in 0.5% ovalbumin in PBS for 30 min at 37°C. After incubation, grids were washed 3 × 5 min in PBS and incubated with secondary goat anti-mouse antibody conjugated to 10 nm colloidal gold (British BioCell, Cardiff, UK) 1:25 in 0.5% ovalbumin in PBS for 30 min at 37°C. Grids were subsequently washed with 3 drops of PBS for 5 min, before incubation on 3 drops of 1% cold fish gelatin (Sigma-Aldrich, St. Louis, MO, USA) in PBS for 10 min each. The grids were finally washed 3 × 5 min in PBS, before negative staining with 1 drop of 1% (w/v) phosphotungstic acid at pH 7.0 and blotted dry for a few seconds. Images were obtained with a transmission electron microscope (JEM 1010, JEOL, Eching, Germany) operated at 60 keV coupled to an electron-sensitive CCD camera (KeenView, Olympus, Center Valley, PA, USA). For size determination of visible EVs, a grid-size replica (2,160 lines/mm) and the ImageJ 1.50r software (NIH, Bethesda, MD, USA) was used.
Western blotting
Western blotting was used to validate the isolated EVs for EV-associated proteins, and was conducted using the common EV marker CD9 and coagulation protein TF. Additionally, lipoprotein contamination was determined by examining apolipoprotein B levels in the pellets. Pellet-material subjected to SDS-PAGE consisted of a single EV suspension pool (equal volume from all donors). Proteins were separated in MiniProtean TGX 4–15% gels (Bio-Rad, Copenhagen, Denmark) under non-reducing conditions. EV suspensions were loaded in equal amounts, i.e. 10 µl of each EV suspension pool were diluted 1:1 with Laemmli sample buffer (Bio-Rad, Copenhagen, Denmark) and 20 µl were loaded into each designated lane in order to enable relative comparisons between samples on the same gel in the following analysis of markers. The proteins were then transferred to Amersham Hybond 0.45 PVDF blotting membranes (GE Healthcare, Broendby, Denmark) and blocked for 1 h in 5% (w/v) skim milk in Tris-Glycine buffer. Primary antibodies were mouse monoclonal anti-human CD9 (clone M-L13, BD Pharmingen, San Jose, CA, USA), monoclonal mouse anti-CD142 (clone HTF-1, BD Pharmingen, San Jose, CA, USA), or monoclonal mouse apolipoprotein B (clone F2C9, Thermo Scientific, Waltham, MA, USA). All primary antibodies were diluted 1:1000 in 5% (w/v) skim milk in Tris-Glycine buffer and incubated with the membranes overnight at 4°C. Secondary labelling was performed with horseradish peroxidase-conjugated polyclonal goat anti-mouse antibodies (Dako, Glostrup, Denmark, 1:30,000 dilution, 2 h at room temperature). Antibody positive controls were: platelet lysate for CD9, diluted plasma for apolipoprotein B, and plasma from LPS-stimulated blood for TF. Samples were developed with ECL Prime Western Blotting detection reagent (GE Healthcare, Broendby, Denmark) and detected on a PXi 4 system (Syngene, Cambridge, UK) with the GeneSys software 1.5.4.0 (Syngene, Cambridge, UK). Band intensity was determined with the ImageJ 1.50r software (NIH, Bethesda, MD, USA) to compute relative differences in marker expression between the different pellet pools.
Protein and factor VIIIa quantitation
In order to determine the extent of co-isolation of larger coagulation factors, the activity of coagulation factor VIII (FVIII) in the EV suspensions dissolved in PBS was determined by performing a modified activated partial thromboplastin time test (HemosIL and SynthASil, Instrumentation Laboratory, München, Germany), using FVIII-deficient plasma and optimized for low concentrations of FVIII, on an ACL TOP 500 CTS system (Instrumentation Laboratory, München, Germany). Total protein content of the pellet suspensions was analysed with the Pierce BCA Protein Assay Kit (Thermo Scientific, Waltham, MA, USA) on a Fluostar Optima (BMG Labtech, Ortenberg, Germany) at wavelength 562 nm. Measurement of haemoglobin, leucocytes, and platelets were performed on a Sysmex XN-9000 Automated Hematology System (Sysmex Corporation, Kobe, Japan).
Thrombin generation assay
The thrombin generation in SPP (n = 12) or SPP+CTI (n = 6) spiked with EVs was measured using the calibrated automated thrombogram as described by Hemker et al. [12] by mixing 80 µl of each sample with 20 µl trigger reagent and 20 µl buffer containing a fluorogenic substrate and CaCl2 (FluCa kit, Thrombinoscope BV, Maastricht, The Netherlands). Thrombin generation was triggered with one of three different reagents, i.e. MP (4 µM PL), PRP (1 pM TF), and PPPlow (1 pM TF and 4 µM PL, Thrombinoscope BV, Maastricht, The Netherlands), or without addition of supplementary PL or TF (no trigger reagent). Each sample was calibrated against calibrator containing a fixed amount of thrombin-α2-macroglobulin complex (Thrombin Calibrator, Thrombinoscope BV, Maastricht, The Netherlands) diluted in the same sample. To determine the TF-associated thrombin generation when using no trigger, thrombin generation was assessed in SPP spiked with the isolated EVs in the presence and absence of anti-CD142 (clone HTF-1). Additionally, thrombin generation in washed compared to unwashed pellets was determined using no trigger and the three different reagents in three of the donors. The reaction was monitored on a fully automated and computer-controlled Fluoroscan Ascent (Thermo Scientific, Waltham, MA, USA) with a 390/460 filter set using the Thrombinoscope software 5.0 (Thrombinoscope BV, Maastricht, The Netherlands).
Procoagulant phospholipid activity assay
PPL activity in SPP spiked with the unwashed EVs was analysed with the FXa-based clotting STA-Procoag PPL kit (Stago, Asnières, France). The test measures a clotting time solely dependent on PPL in the sample, i.e. the more PPL activity the shorter clotting time. EV suspension, 25 µl, diluted 1:1 in Owren-Koller buffer (Stago, Asnières, France) were mixed with 25 µl PL-free plasma and triggered with 100 µl 0.001 IU FXa and 0.025 M CaCl2. The clotting time was measured on an automated STA-Compact (Stago, Asnieres, France) in agreement with the manufacturer’s protocol.
Statistical analysis
Data are expressed as mean ± standard deviation. Parametric (Student’s t-test) or nonparametric (Mann-Whitney U test) was used to compare differences in particle size, particle quantity, and coagulation activity of the different pellet types, including washed/unwashed and ±CTI. Paired samples t-test was used to detect differences between paired groups, e.g. comparison of washed and unwashed pellets and effect of washing. Pearson correlation coefficient was used to identify associations between coagulation assays and NTA measurements. All statistical analyses were performed using IMB SPSS Statistics 23 (SPSS, Chicago, IL, USA) and Graph Pad Prism 6 (GraphPad Software, La Jolla, CA, USA).
Results
Donor characteristics
The donors were all healthy with a mean age of 35 years (range 23–63) and comprised 6 males and 6 females. None of the donors were on pain medication prior to sample collection. One female used progestogen-only pills. All donors had a complete haematology investigation, including erythrocytes, haemoglobin, leucocytes, and platelets. All values were within normal reference ranges.
EV isolation
When compared to the original plasma particle concentration, the amount of particles isolated in the unwashed pellets was approximately 5–8% and for the washed pellets less than 1% (Table 1). On average, there were significantly more particles in the 100K pellets compared to the 20K pellets in unwashed pellets (42%, P < 0.05, Figure 1(a)) and the mean size was 13 nm smaller (11%, P < 0.05, Figure 1(b)). Introduction of a single washing step, resulted in reduced particle concentrations in the pellets to almost 5% of the equivalent unwashed pellet (P < 0.01). While comparing the individual donors, the mean particle size in the 20K pellets was 32 nm larger (P < 0.01) than their subsequent 100K pellets in washed pellets. Furthermore, the washed 20K pellets were generally larger than their corresponding unwashed 20K pellets (31%, P < 0.05, Table 1). A substantial overlap in distribution of particle size was observed between pellets, although the 20K pellets contained approximately 50% more particles larger than 200 nm independent of whether washing was applied or not. Moreover, it was clear that washing increased the fractions of particles above 100 nm in size, especially in the 20K pellets (Figure 1(c)). The addition of 0.03% Tween 20 to eliminate eventual aggregation of the isolated EV pellets, resulted in similar NTA measurements compared to no detergent (data not shown). The particles were analysed by electron microscopy to confirm the presence of EVs and to detect possible EV subpopulations. TEM revealed structures that resembled EVs of various sizes (Figure 2(a,b)). Immunogold labelling identified several EVs of different sizes that were positive for CD9 in both washed and unwashed pellets. For the washed 100K pellets, the relative intensity extrapolated from the western blotting band revealed a reduction of almost 50% in CD9 expression, whereas the CD9 expression in the 20K pellets remained unchanged (Figure 2(f)). Additionally, for the unwashed samples, a faint TF (CD142, clone HTF-1) band was detected in both 20K and 100K pellets (3-fold higher in the 20K pellets), however, washing resulted in the loss of the TF bands.
Table 1.
The main findings from NTA.
| Original plasma | 20K pellets | P-value | 100K pellets | P-value | |
|---|---|---|---|---|---|
| Unwashed pellets (n = 12) | |||||
| Particle concentration (part./ml) | 1,66E+12 ± 1,28E+12 | 9,23E+10 ± 4,37E+10 | <0.01 | 1,31E+11 ± 8,85E+10 | <0.01 |
| Mean particle size (nm) | 85 ± 13 | 119 ± 14 | <0.001 | 106 ± 11 | <0.001 |
| Washed pellets (n = 6) | |||||
| Particle concentration (part./ml) | 1,45E+12 ± 9,34E+11 | 5,73E+09 ± 1,24E+09 | <0.05 | 8,31E+09 ± 2,97E+09 | <0.05 |
| Mean particle size (nm) | 82 ± 6 | 158 ± 11 | <0.0001 | 126 ± 12 | <0.001 |
Significantly less particles were present in the pellets compared to the original donor plasma, but these had a larger mean particle size. A series of 5 × 30 s videos were taken for each sample. P values are representing the findings from the pellets compared against the original donor plasma. The videos were captured at camera level 10 and the particles tracked at detection threshold 2 with 9 × 9 blur.
Figure 1.
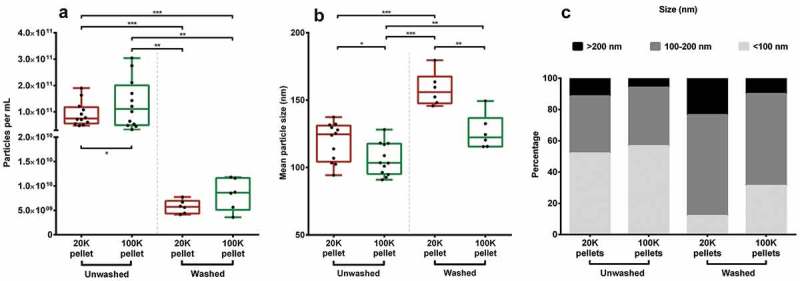
NTA performed on pellets. (A) Concentration of particles and (B) size determination of particles in each pellet group. *P < 0.05. **P < 0.01. ***P < 0.001. (C) Size distribution of the different pellets divided into three fractions, i.e. <100 nm, 100–200 nm, and >200 nm. EV suspensions were diluted to obtain a particle count per frame within the recommended limits. Capture duration was 5 × 30 s at camera level 10 and the videos were analysed at detection threshold 2 with 9 × 9 blur.
Figure 2.
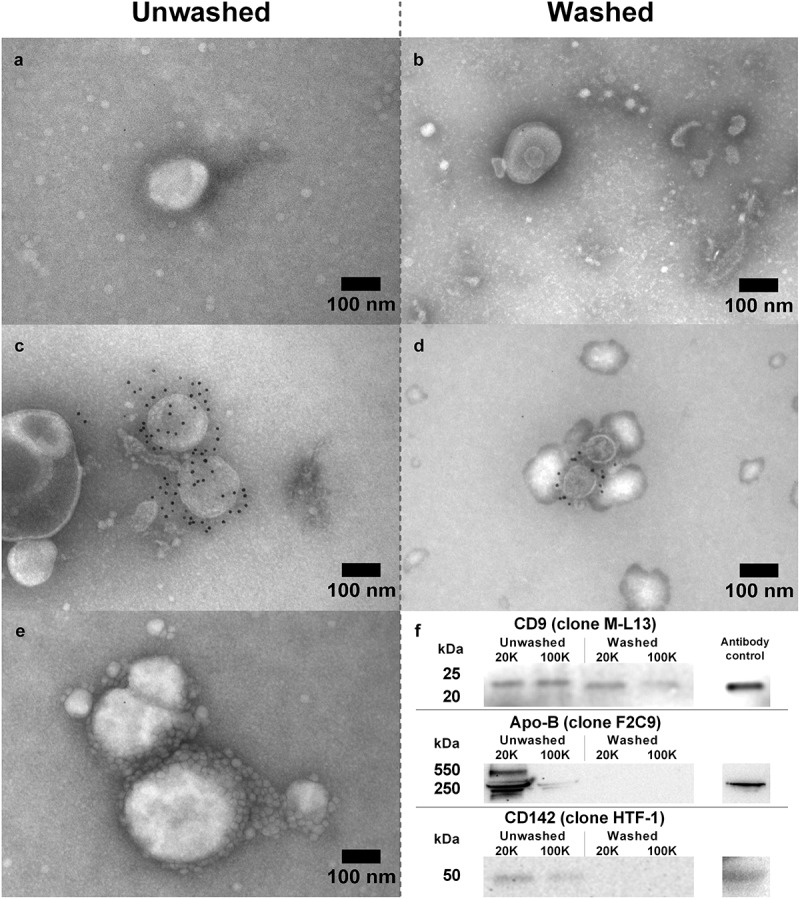
TEM and western blotting was applied to verify the presence of EV subpopulations in both washed and unwashed pellets. TEM showing EV-characteristics, i.e. shape and size in (A) unwashed and (B) washed pellets. Immunogold labelling with anti-CD9 (clone M-L13) bound to EVs in unwashed (C) and washed (D) pellets confirming presence of CD9-positive subpopulations. (E) Small (<50 nm) vesicular structures adhering to or residing in proximity to larger vesicles. (F) Western blot with anti-CD9, anti-CD142 (clone HTF-1), and anti-apolipoprotein B (clone F2C9) on a pellet pool for each pellet type showing the presence of EV marker CD9 and the removal of apolipoprotein B after washing in PBS.
The images from TEM revealed that a substantial amount of small particles (<50 nm) are co-isolated in the unwashed pellets, but this co-isolation was considerably reduced when pellets were washed (Figure 2(a-c)). In some cases, predominantly in unwashed pellets, it appeared that these smaller particle-shaped structures adhered to or resided in proximity to larger particles (Figure 2(e)). Apolipoproteins-B100 and -B48 were present in unwashed pellets with approximately 90% of the apolipoproteins being present in the 20K pellet pool and 10% in the 100K pellet pool (Figure 2(f)). After washing, apolipoprotein B bands were not present in either pellet type. The protein content of the pellets pools was quantitated to determine the extent of co-isolated plasma proteins (potentially including coagulation factors). After introduction of a single washing step, the protein content in 20K pellets was reduced from 2158.1 ± 68.3 µg/ml to 50.1 ± 0.7 µg/ml and the 100K pellets from 1276.9 ± 36.9 µg/ml to 146.9 ± 2.3 µg/ml, i.e. a reduction in protein content of 88–98% in the two pellet types. Moreover, FVIII measurement did not reveal any activity in either pellet type.
EVs effect on coagulation
To demonstrate the potential role of EVs in coagulation, the isolated EVs resuspended in SPP were measured using TGA without or in the presence of three different trigger reagents (Figure 3). EVs “spiked” into SPP and analysed without trigger reagent resulted in thrombograms depicting an overall increase in thrombin generation compared to non-spiked SPP, especially for the 20K pellets but with a considerable inter-individual variation (Figure 3). Using the MP reagent, containing only PL, this variation diminished. Inhibition of TF activity with anti-CD142 (clone HTF-1) resulted in little or no changes to the thrombin generation profile, thus indicating that TF is not the determining factor when using no trigger reagents (Supplementary Figure 1). When the pellets were resuspended in SPP+CTI, little or no thrombin was generated when using no trigger or the MP reagent. When thrombin generation was triggered with the PRP reagent (TF but no PL), both 20K and 100K pellets demonstrated an increased thrombin generation in SPP and SPP+CTI, with the 20K pellet exhibiting the most profound thrombin generation effect. When the PPPlow-reagent was used to trigger coagulation, the overall thrombin generation was higher with the 100K pellets compared to the 20K pellets (Figure 3(g)). This observation was consistent for EVs from all healthy volunteers, however, this was not apparent when SPP+CTI were used (Figure 3 (h).
Figure 3.
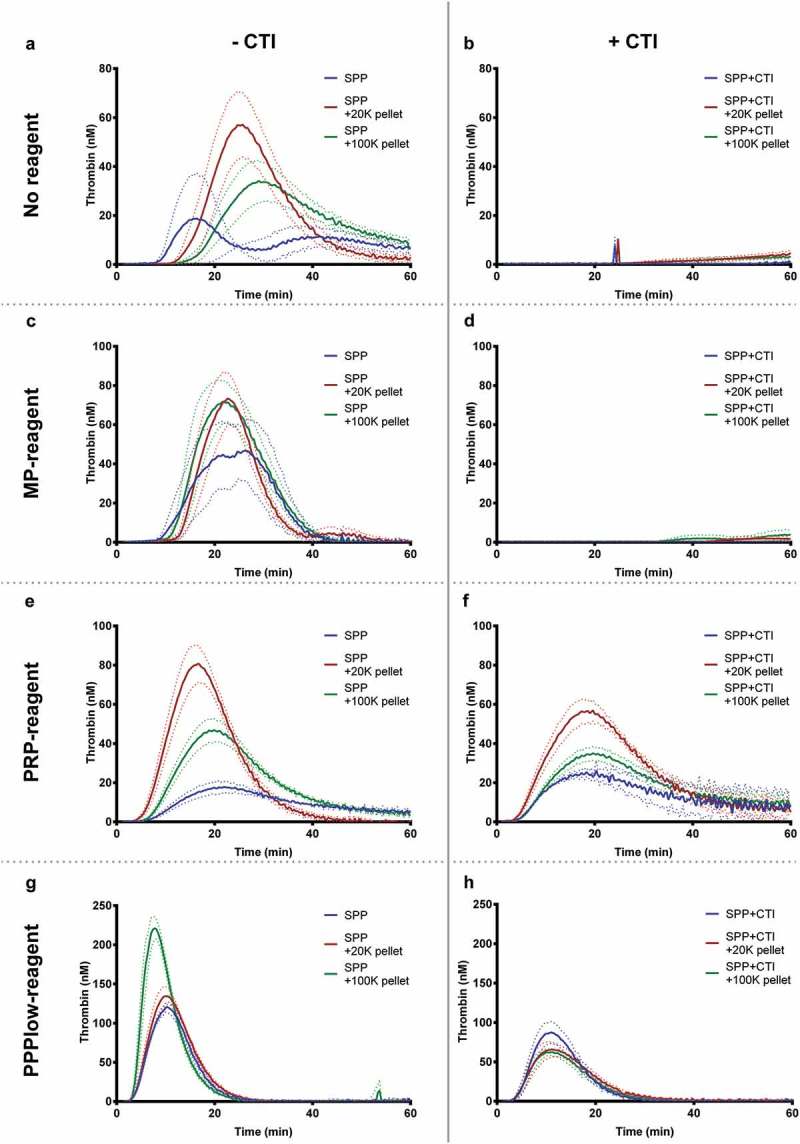
Thrombograms (mean ± SEM) depicting the effect on thrombin generation of EV-containing pellets “spiked” into SPP with (n = 6) or without (n = 12) collected with CTI. PBS was added to the standard pool plasma that was not spiked with EVs (control). Thrombograms were generated with different trigger either MP, PRP, or PPPlow reagent or without addition of trigger reagents (depicted vertically). The effect of EVs on thrombin generation seems to mainly be driven by PPL. The use of CTI is depicted in the horizontally.
In order to directly compare the effect of a washing step on coagulation, thrombin generation was performed on samples treated with or without washing. The ratio between the various measures of thrombin generation on washed and unwashed pellets are shown in (Table 2). In general, the calculated ratios were close to 1.0 (i.e. no effect of washing), although, there was a tendency of a minor decrease in thrombin generation from the washed pellets. This effect, however, was not significant and inconsistent between the different donors.
Table 2.
The effect of a single washing step on thrombin generation using no trigger reagent, MP, PRP, or PPPlow.
| ETP (nM*min) | Peak thrombin (nM) | Lag time (min) | Time-to-peak (min) | |
|---|---|---|---|---|
| No reagent | ||||
| 20K pellets | 1.08 ± 0.07 | 0.83 ± 0.17 | 1.14 ± 0.10 | 1.14 ± 0.10 |
| 100K pellets | 0.88 ± 0.14 | 0.71 ± 0.36 | 1.25 ± 0.22 | 1.24 ± 0.26 |
| MP reagent | ||||
| 20K pellets | 1.03 ± 0.07 | 0.97 ± 0.07 | 1.05 ± 0.12 | 1.05 ± 0.10 |
| 100K pellets | 0.95 ± 0.11 | 1.00 ± 0.20 | 1.17 ± 0.12 | 1.13 ± 0.12 |
| PRP reagent | ||||
| 20K pellets | 1.21 ± 0.17 | 1.00 ± 0.21 | 1.03 ± 0.04 | 1.06 ± 0.06 |
| 100K pellets | 1.18 ± 0.07 | 0.71 ± 0.12 | 1.08 ± 0.10 | 1.20 ± 0.15 |
| PPPlow reagent | ||||
| 20K pellets | 1.14 ± 0.31 | 1.06 ± 0.27 | 1.06 ± 0.02* | 1.02 ± 0.04 |
| 100K pellets | 1.21 ± 0.14 | 1.23 ± 0.23 | 0.97 ± 0.07 | 0.97 ± 0.05 |
Parameters ETP, peak thrombin, lag time, and time-to-peak are presented as a mean ± SD of a ratio (washed/unwashed) calculated for both 20K and 100K pellets from each donor. No significant differences are observed between the procoagulant effect of washed and unwashed pellets, except for lag time in 20K pellets when using PPPlow reagent with a slight increase in washed pellets. *P < 0.05.
PPL in the pellets shortened the PPL clotting time with the most pronounced effect among 20K pellets (Figure 4(a)). The 20K pellets reduced the clotting time by almost 44% (P < 0.001), whereas the 100K pellets only resulted in an 18% reduction (P < 0.01) on average. The PPL activity correlated strongly with thrombin generation measured with PRP-reagent (containing only TF); Figure 4 (b) depicts the correlation between peak of thrombin generation and PPL clotting time. This correlation disappeared when PPL clotting time was correlated to thrombin generation measured with PPPlow-reagent (containing both TF and PL). There appears to be a correlation between the mean particle size and PPL activity in the 20K pellets, where a larger mean particle size is equivalent to a shorter PPL clotting time, i.e. increased PPL activity (Figure 4 (c), although not significant (P = 0.092). This trend was only present when the pellets were washed. The main findings in TGA and PPL activity assay are summarized in (Table 3).
Figure 4.
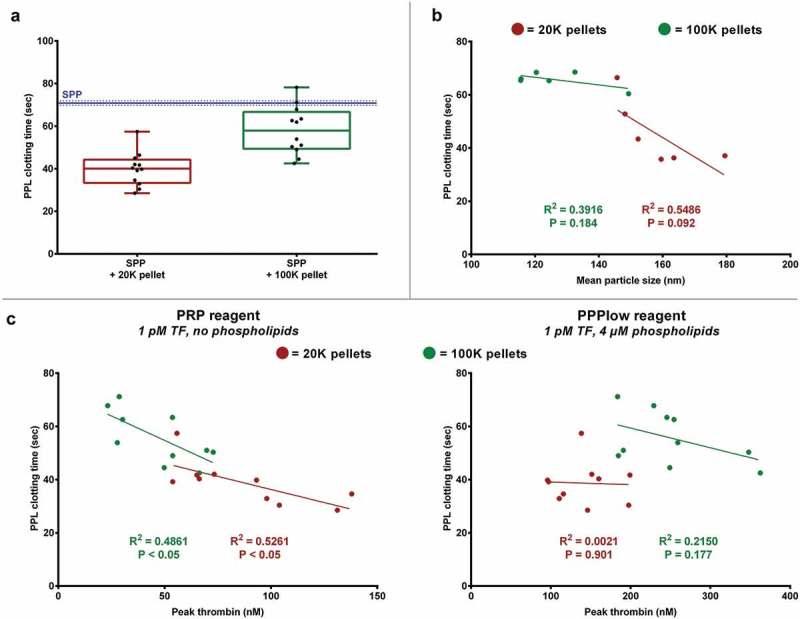
EV-associated PPL activity measured by PPL activity assay. (A) EVs reduce the PPL clotting time of SPP, mainly those in 20K pellets, thus increasing the PPL activity. The blue and dotted lines represents the reference PPL clotting time (mean ± SD, n = 12) of SPP without addition of EVs. (B) The 20K pellets showed tendency towards larger particles correlating with increased PPL activity, although this was not significant. (C) A significant correlation between PPL clotting time and thrombin generation using PRP reagent (containing no PL) was observed, but this disappeared when using PPPlow reagent (containing PL). *P < 0.001.
Table 3.
The main findings from thrombin generation and analysis of PPL when “spiked” into SPP with (n = 12) or without (n = 6) CTI.
| Thrombin generation assay |
−CTI |
+CTI |
||||
|---|---|---|---|---|---|---|
| SPP | +20K | +100K | SPP | +20K | +100K | |
| No reagent | ||||||
| ETP (nM*min) | 570 ± 218 | 1021 ± 477 | 884 ± 399 | |||
| Peak thrombin (nM) | 17 ± 1 | 81 ± 39 | 46 ± 22 | |||
| Lag time (min) | 28.9 ± 3.4 | 19.4 ± 4.2 | 24.5 ± 7.7 | |||
| Time-to-peak (min) | 41.0 ± 3.6 | 28.7 ± 5.4 | 35.8 ± 9.3 | |||
| Velocity index (nM/min) | 0.8 ± 0.7 | 2.2 ± 1.0 | 4.3 ± 3.0 | |||
| MP reagent | ||||||
| ETP (nM*min) | 888 ± 15 | 1184 ± 264 | 1192 ± 241 | |||
| Peak thrombin (nM) | 81 ± 1 | 105 ± 27 | 118 ± 43 | |||
| Lag time (min) | 16.3 ± 0.2 | 18.5 ± 3.6 | 17.2 ± 2.8 | |||
| Time-to-peak (min) | 23.0 ± 0.2 | 25.4 ± 4.1 | 23.5 ± 3.4 | |||
| Velocity index (nM/min) | 12.2 ± 0.1 | 14.1 ± 7.6 | 20.7 ± 13.2 | |||
| PRP reagent | ||||||
| ETP (nM*min) | 543 ± 34 | 1292 ± 209 | 1071 ± 263 | 794 ± 283 | 1426 ± 186 | 1055 ± 355 |
| Peak thrombin (nM) | 18 ± 3 | 88 ± 29 | 48 ± 18 | 25 ± 2 | 57 ± 13 | 35 ± 8 |
| Lag time (min) | 8.2 ± 0.1 | 7.3 ± 0.8 | 8.3 ± 0.8 | 5.5 ± 0.2 | 5.8 ± 0.5 | 6.3 ± 0.2 |
| Time-to-peak (min) | 22.3 ± 0.9 | 17.5 ± 1.9 | 20.5 ± 1.6 | 18.8 ± 1.5 | 19.4 ± 2.0 | 20.0 ± 1.0 |
| Velocity index (nM/min) | 1.3 ± 0.3 | 9.1 ± 4.3 | 4.1 ± 1.9 | 1.8 ± 0.0 | 4.4 ± 1.4 | 2.6 ± 0.6 |
| PPPlow reagent | ||||||
| ETP (nM*min) | 1219 ± 76 | 1415 ± 202 | 1969 ± 289 | 1009 ± 317 | 864 ± 239 | 857 ± 111 |
| Peak thrombin (nM) | 120 ± 7 | 141 ± 36 | 251 ± 59 | 90 ± 33 | 70 ± 26 | 69 ± 11 |
| Lag time (min) | 4.2 ± 0.1 | 4.5 ± 0.4 | 4.2 ± 0.5 | 4.9 ± 0.6 | 4.7 ± 0.5 | 4.7 ± 0.7 |
| Time-to-peak (min) | 10.3 ± 0.2 | 10.7 ± 1.3 | 7.9 ± 1.2 | 11.1 ± 1.2 | 12.1 ± 1.4 | 11.5 ± 1.7 |
| Velocity index (nM/min) | 19.7 ± 0.8 | 27.0 ± 11.6 | 76.8 ± 40.2 | 15.2 ± 6.8 | 10.2 ± 5.8 | 11.0 ± 3.5 |
| PPL activity assay | ||||||
| PPL clotting time (sec) | 70.9 ± 0.8 | 39.9 ± 7.5 | 58.0 ± 10.7 | 68.1 ± 0.4 | 45.3 ± 11.1 | 65.7 ± 2.7 |
Parameters ETP, peak thrombin, lag time, time-to-peak, and velocity index are listed as mean ± SD. Data from TGA with no trigger reagent or using the MP reagent are not available due to very little to no thrombin generated.
Examining the activity in the supernatants after centrifugation and removal of first 20K and subsequently 100K supernatants showed a markedly reduced thrombin generation compared to the original plasma (Figure 5(a)), and this observation was consistent and independent of trigger reagent used. PPL activity decreased in a similar way, i.e. increased PPL clotting times, in both the 20K and 100K supernatants (Figure 5 (b)).
Figure 5.
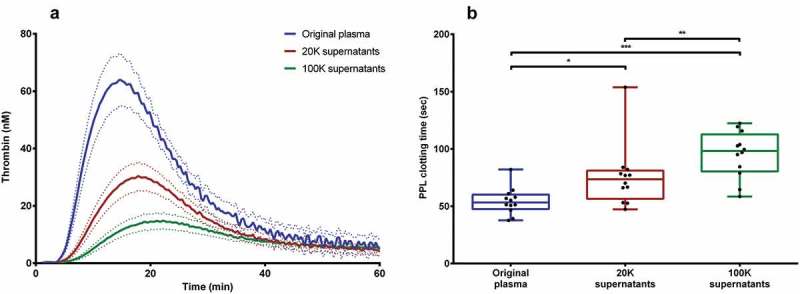
Loss of procoagulant activity from plasma from healthy donors after DUC and EV removal. (A) Thrombin generation (using PRP reagent) decreased in the donor plasma as result of EV-removal during DUC. Dotted lines represent mean ± SEM. (B) PPL activity in the donor plasma was reduced significantly after DUC and EV-removal. *P < 0.05; **P < 0.01; ***P < 0.001.
Discussion
The aim of this study was to establish a method to isolate plasma EVs for the determination of procoagulant activity and to examine whether a single washing step is essential for the removal of lipoproteins and plasma remnants for clinical studies. Using DUC, we isolated EVs of various sizes and demonstrated that whilst a single washing step was beneficial for the removal of excess proteins and lipids, there is a concurrent loss of some EVs and a minimal effect on coagulation activity. The TGA and PPL activity assay results of the 5-fold up-concentrated EVs, clearly indicating that the procoagulant effect is mostly driven by PPL activity from the larger EVs. Thus, this is a model system to investigate procoagulant EVs potentially present in patients at increased risk of thrombosis.
EVs are involved in coagulation processes, and it has been suggested that they may have a thrombogenic effect in various diseases. Several methods have been used for EV isolation, but each method has its advantages and disadvantages. We have tested DUC, the most commonly used isolation technique [18], as it is a simple approach and has a rather high throughput. Furthermore, it has the advantage that EVs are concentrated in a pellet suitable for undiluted resuspension in SPP for further coagulation analysis. A disadvantage of DUC is that non-EV components with similar dimensions and densities are also isolated with the EVs and this technique often results in co-isolation of plasma proteins [19]. Nevertheless, DUC appears to be a well-suited isolation method for the investigation of the procoagulant activity of EVs. Ideally, we expect to have microvesicles in the 20K pellets and smaller vesicles, mainly exosomes, in the 100K pellets. However, our results show that both pellet types contained particles of various sizes; yet with almost 50% more vesicles >200 nm in the 20K pellets compared to the 100K pellets (Figure 1 (c)). DUC has a limited capability to separate EVs by size [20] explaining the size-overlap we observed between 20K and 100K pellets in unwashed pellets. Washing of pellets in PBS after the isolation procedure resulted in an overall increase in mean particle size, a better size-separation between pellet types, and an overall decrease in particle concentration for both 20K and 100K pellets. This, however, results in the loss of some CD9+ EVs in the 100K pellets. In addition, we discern that washing of the pellets resulted in an increase in particles >100 nm and a reduction in the particles <100 nm in both 20K pellets and 100K pellets. Since the concentration of the smaller particles was significantly reduced, the increase in mean particle size could probably be partly attributed to the removal of lipoproteins, but possibly also aggregates of proteins or other plasma material (Figure 1 (c)), as it is well known that lipoproteins interfere with the particle quantitation by NTA [10,21]. This observation was in accordance with the reduced protein content shown by protein quantitation, and also the western blot findings, where we confirmed that the presence of apolipoprotein B, a marker for both lipoprotein fractions LDL, IDL, VLDL, and chylomicrons, disappeared upon washing. Through TEM we observed that small particles (Figure 2 (e)) were found in close proximity to larger vesicle-like structures, mainly in the unwashed pellets, and the size of these was accordant with that of lipoproteins. Ultracentrifugation has been reported to instigate aggregation of platelet vesicles [22], but this may also be the case for lipoproteins and other vesicle-like structures. Addition of low concentrations of Tween 20 in the EV suspension has previously been utilized to prevent EV aggregation [17]. Nevertheless, we found that addition of Tween 20 to the samples did not change particle size and concentration measured by NTA. The introduction of a washing step seemed advantageous because the protein content was reduced by more than 88%, which likely eliminates the risk of coagulation factors (not associated to vesicles) in plasma contaminating the pellets. FVIII, a relatively high molecular weight protein, displayed no activity in our pellet material indicating that the thrombin generation was not influenced by this coagulation protein and probably other coagulation factors. Western blotting and electron microscopy evidently showed that CD9+ EVs were present in the pellets, but also that the CD9 expression in the 100K pellets was reduced after washing. Additionally, the minute TF expression found in unwashed pellets was not detectable after washing, indicating a possible loss of some procoagulant TF+ EVs. Nevertheless, no significant loss of functionality in regard to coagulation was observed when the effect of washing was tested with TGA (Table 2). Therefore, it seems beneficial to include a washing process in the EV isolation step to diminish plasma contaminants. The washing procedure adopted to isolate EVs in the present study is similar to recent research regarding EV-isolation through ultracentrifugation [20,23,24]. Although we observed a moderate overlap in EV size distribution in between the two pellet types, we find DUC suitable to isolate EVs of various sizes for further analysis of procoagulant activity. Others recommend using DUC combined with a density gradient [25,26], which may improve the size-separation of EVs, i.e. microvesicles vs. exosomes, and consequently be a beneficial addition to the isolation step. However, this was not done in the present study because we anticipate that the different types of gradient medium could influence the procoagulant capacity EVs and the subsequent coagulation analysis.
The procoagulant activity of the EVs, i.e. the pellets, was examined with TGA using various trigger reagents in the presence and/or absence of TF and PL. TF primarily promotes a faster initiation of thrombin generation i.e. shorter lag time, whereas PPL primarily have an effect on the amount of thrombin generated (peak and ETP) [27]. Using no trigger reagent, where only isolated EVs and the coagulation factors in SPP can trigger coagulation, a small but variable amount of thrombin was generated (Figure 3 (a)). If only PL was added, i.e. MP reagent, the thrombin generated were almost the same in original SPP and SPP spiked with EVs indicating that the differences in (Figure 3 (a)) are mainly driven by PPL on the isolated EVs. Inhibition with anti-CD142 did not reduce thrombin generation, thus suggesting that this reaction is mainly initiated by contact activation and, furthermore, that healthy donors do not contain any or very little TF+ EVs. Supporting this observation, no thrombin generation was seen when SPP+CTI was used (Figure 3(d)), as documented by Boknäs and colleagues [27]. When TF was used as trigger, i.e. PRP reagent, a significant difference was observed between pure SPP and SPP spiked with EVs (Figure 3 (e)). This difference disappeared for SPP containing 20K pellets when PL also was added via the trigger reagent, i.e. PPPlow reagent (Figure 3 (g)), i.e. in accordance with the presence of PPL in the pellets. However, with both TF and PL present in the trigger reagent, SPP containing 100K pellets actually had a higher thrombin generation than pure SPP and SPP containing 20K pellets. This was unexpected but most of the donors showed this difference, although the differences between 20K and 100K pellets differed substantially. Since this effect was only apparent when TF as well as PL were added, it indicates that the 100K pellet may contain some other factors that increase the coagulation activity. Measuring PPL with the PPL activity assay demonstrated that 20K pellets had a higher PPL content than the 100K pellets. This is in agreement with the correlation between the TGA findings using PRP-reagent and PPL activity assay (Figure 4 (c)), however, when PL is supplied by the reagent (PPPlow-reagent) the correlation between thrombin generation and PPL activity disappears. Furthermore, we observed a tendency towards a higher PPL activity in the larger particles (Figure 4 (b)). The removal of 20K pellets and 100K pellets revealed a reduced activity of PPL in the subsequent supernatants (Figure 5), similar to findings by Marchetti et al. [15]. Our findings indicated that EVs from healthy persons carry no or a very low amounts of TF, but contain some PPL which is also in agreement with the findings of others [28,29].
Unfortunately, in the present study SPP and SPP+CTI were drawn from two different donors, and therefore a comparison of procoagulant activity of EVs between the two SPP types should be evaluated with caution. While CTI inhibit contact activation it may also have an impact on other coagulation factors [30]. Consequently, it may not be optimal to use plasma with CTI added, and as an alternative, the activity of TF can be demonstrated by the addition of anti-CD142 antibodies. It may be advantageous to use SPP deprived of EVs, e.g. by ultracentrifugation, which also was used in the experiment examining the effect of the washing step on thrombin generation (Table 2), thus increasing the assay sensitivity. Nevertheless, in the present study a procoagulant effect of EVs from different donors was demonstrated when spiked into SPP not deprived of EVs and was not much different when ultracentrifuged SPP was used. In this study, we have used non-pathological samples having a low TF-activity [28,29]. However, to determine whether the method described in the present study can be used to identify patients at increased risk of thrombosis warrants further investigation.
In summary, we have successfully isolated EVs of different sizes from plasma by DUC and confirm the presence of an EV population positive for the commonly used EV marker CD9. The inclusion of a washing step after the isolation of EVs appears to be beneficial for application in EV characterization analysis and coagulation analysis. A combination of TGA, using different reagents, and a PPL activity assay has the potential to demonstrate the presence (or not) of TF and PPL in the EV suspensions which is useful for the characterization of pathological samples, e.g. from cancer patients.
Funding Statement
This work was supported by the Danish Council for Independent Research (Grant number 4183-00268) and the Obel Family Foundation (Grant number 26145)
Acknowledgements
We thank the Department of Clinical Immunology at Aalborg University Hospital for the use of their ultracentrifuge for the isolation of EVs from plasma. This work was supported by the Danish Council for Independent Research (Grant number 4183-00268) and the Obel Family Foundation (Grant number 26145).
Disclosure statement
The authors report no conflicts of interest.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Mackman N, Tilley RE, Key NS.. Role of the extrinsic pathway of blood coagulation in hemostasis and thrombosis. Arterioscler Thromb Vasc Biol. 2007;27(8):1687–12. [DOI] [PubMed] [Google Scholar]
- [2].Owens AP, Mackman N. Microparticles in hemostasis and thrombosis. Circ Res. 2011;108:1284–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Van Der Pol E, Hoekstra AG, Sturk A, et al. Optical and non-optical methods for detection and characterization of microparticles and exosomes. J Thromb Haemost. 2010;8(12):2596–2607. [DOI] [PubMed] [Google Scholar]
- [4].Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013;200(4):373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].György B, Szabó TG, Pásztói M, et al. Membrane vesicles, current state-of-the-art: emerging role of extracellular vesicles. Cell Mol Life Sci. 2011;68(16):2667–2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Giesen PL, Rauch U, Bohrmann B, et al. Blood-borne tissue factor: another view of thrombosis. Proc Natl Acad Sci U S A. 1999;96:2311–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Zwicker JI, Liebman HA, Neuberg D, et al. Tumor-derived tissue factor-bearing microparticles are associated with venous thromboembolic events in malignancy. Clin Cancer Res. 2009;15(22):6830–6840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Tesselaar MET, Romijn FPHTM, Van Der Linden IK, et al. Microparticle-associated tissue factor activity: a link between cancer and thrombosis? J Thromb Haemost. 2007;5(3):520–527. [DOI] [PubMed] [Google Scholar]
- [9].Davila M, Amirkhosravi A, Coll E, et al. Tissue factor-bearing microparticles derived from tumor cells : impact on coagulation activation. J Thromb Haemost. 2008;6:1517–1524. [DOI] [PubMed] [Google Scholar]
- [10].Witwer KW, Buzás EI, Bemis LT, et al. Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. J Extracell Vesicles. 2013;2:20360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hisada Y, Alexander W, Kasthuri R, et al. Measurement of microparticle tissue factor activity in clinical samples: a summary of two tissue factor-dependent FXa generation assays. Thromb Res Elsevier Ltd. 2016;139:90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hemker HC, Giesen P, AlDieri R, et al. The calibrated automated thrombogram (CAT): a universal routine test for hyper- and hypocoagulability. Pathophysiol Haemost Thromb. 2002;32(5–6):249–253. [DOI] [PubMed] [Google Scholar]
- [13].Bidot L, Jy W, Bidot C, et al. Microparticle-mediated thrombin generation assay: increased activity in patients with recurrent thrombosis. J Thromb Haemost. 2008;6(6):913–919. [DOI] [PubMed] [Google Scholar]
- [14].Gheldof D, Hardij J, Cecchet F, et al. Thrombin generation assay and transmission electron microscopy: a useful combination to study tissue factor-bearing microvesicles. J Extracell Vesicles. 2013;2:19728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Marchetti M, Tartari CJ, Russo L, et al. Phospholipid-dependent procoagulant activity is highly expressed by circulating microparticles in patients with essential thrombocythemia. Am J Hematol. 2014;89(1):68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lacroix R, Judicone C, Mooberry M, et al. Standardization of pre-analytical variables in plasma microparticle microparticle determination: results of the International Society on Thrombosis and Haemostasis SSC Collaborative workshop. J Thromb Haemost. 2013;150(2):137–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Vogel R, Coumans FAW, Maltesen RG, et al. A standardized method to determine the concentration of extracellular vesicles using tunable resistive pulse sensing. J Extracell Vesicles. 2016;5:31242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Gardiner C, Di VD, Sahoo S, et al. Techniques used for the isolation and characterization of extracellular vesicles: results of a worldwide survey. J Extracell Vesicles. 2016;5:32945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Baranyai T, Herczeg K, Onódi Z, et al. Isolation of exosomes from blood plasma: qualitative and quantitative comparison of ultracentrifugation and size exclusion chromatography methods. PLoS One. 2015;10(12):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Théry C, Amigorena S, Raposo G, et al. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol. 2006;Chapter 3 Unit 322: 1–29. [DOI] [PubMed] [Google Scholar]
- [21].Mørk M, Handberg A, Pedersen S, et al. Prospects and limitations of antibody-mediated clearing of lipoproteins from blood plasma prior to nanoparticle tracking analysis of extracellular vesicles. J Extracell Vesicles. 2017;6(1):1308779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Yuana Y, Böing AN, Grootemaat AE, et al. Handling and storage of human body fluids for analysis of extracellular vesicles. J Extracell Vesicles. 2015;4:29260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kalra H, Adda CG, Liem M, et al. Comparative proteomics evaluation of plasma exosome isolation techniques and assessment of the stability of exosomes in normal human blood plasma. Proteomics. 2013;13(22):3354–3364. [DOI] [PubMed] [Google Scholar]
- [24].Caby M-P, Lankar D, Vincendeau-Scherrer C, et al. Exosomal-like vesicles are present in human blood plasma. Int Immunol. 2005;17(7):879–887. [DOI] [PubMed] [Google Scholar]
- [25].BJ Tauro, DW Greening, RA Mathias, et al. Comparison of ultracentrifugation, density gradient separation, and immunoaffinity capture methods for isolating human colon cancer cell line LIM1863-derived exosomes. Methods Elsevier Inc. 2012;56(2):293–304. [DOI] [PubMed] [Google Scholar]
- [26].Welton JL, Khanna S, Giles PJ, et al. Proteomics analysis of bladder cancer. Mol Cell Proteomics. 2010;9:1324–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Boknäs N, Faxälv L, Lindahl TL, et al. Contact activation: important to consider when measuring the contribution of tissue factor-bearing microparticles to thrombin generation using phospholipid-containing reagents. J Thromb Haemost. 2014;12(4):515–518. [DOI] [PubMed] [Google Scholar]
- [28].Butenas S, Bouchard BA, Brummel-Ziedins KE, et al. Tissue factor activity in whole blood. Blood. 2005;105(7):2764–2770. [DOI] [PubMed] [Google Scholar]
- [29].Santucci RA, Erlich J, Labriola J, et al. Measurement of tissue factor activity in whole blood. Thromb Haemost. 2000;83(3):445–454. [PubMed] [Google Scholar]
- [30].Hansson KM, Nielsen S, Elg M, et al. The effect of corn trypsin inhibitor and inhibiting antibodies for FXIa and FXIIa on coagulation of plasma and whole blood. J Thromb Haemost. 2014;12(10):1678–1686. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.