

Abstract
Small ubiquitin-like modifier (SUMO) modification is an important post-translational modification (PTM) that mediates signal transduction primarily through modulating protein-protein interactions. Similar to ubiquitin modification, SUMOylation is directed by a sequential enzyme cascade including E1-activating enzyme (SAE1/SAE2), E2-conjugation enzyme (Ubc9), and E3-ligase (i.e., PIAS family, RanBP2, and Pc2). However, different from ubiquitination, an E3 ligase is non-essential for the reaction but does provide precision and efficacy for SUMO conjugation. Proteins modified by SUMOylation can be identified by in vivo assay via immunoprecipitation with substrate-specific antibodies and immunoblotting with SUMO-specific antibodies. However, the demonstration of protein SUMO E3 ligase activity requires in vitro reconstitution of SUMOylation assays using purified enzymes, substrate, and SUMO proteins. Since in the in vitro reactions, usually SAE1/SAE2 and Ubc9, alone are sufficient for SUMO conjugation, enhancement of SUMOylation by a putative E3 ligase is not always easy to detect. Here, we describe a modified in vitro SUMOylation protocol that consistently identifies SUMO modification using an in vitro reconstituted system. A step-by-step protocol to purify catalytically active K-bZIP, a viral SUMO-2/3 E3 ligase, is also presented. The SUMOylation activities of the purified K-bZIP are shown on p53, a well-known target of SUMO. This protocol can not only be employed for elucidating novel SUMO E3 ligases, but also for revealing their SUMO paralog specificity.
Keywords: Molecular Biology, Issue 131, In vitro SUMOylation, K-bZIP, SUMO E3 ligase, p53, Post-translational modification, Ubc9
Introduction
SUMO modification was initially identified as a reversible post-translational modification (PTM) that regulates protein stability1. In addition to direct conjugation, SUMO can also be attached to a protein through non-covalent interaction by SUMO interaction motifs (SIMs)2. Similar to the binding of tyrosyl-phosphorylation by molecules harboring Src homology 2 (SH2) or phosphotyrosine binding (PTB) domains3,4, SUMO modification provides an additional interaction platform for selective recruitment of SIM-containing effector proteins5,6. In addition to regulation of signal transduction, SUMOylation of transcriptional factors and chromatin remodeling molecules serve to modulate gene expression7,8. Studies of genome-wide SUMOylation patterns have revealed that SUMO modification is associated with either positive9,10 or negative10,11,12 transcription regulation, in part due to the paralogue specificity of SUMOylation.
There are three major SUMO isoforms for protein conjugating present in mammalian cells; these include SUMO-1, and the highly homologous SUMO-2 and SUMO-3 (referred to as SUMO-2/3)13. SUMO is usually conjugated to the lysine (K) residue within the ψKxD/E consensus motif in the target protein. The SIM in SUMO E3 ligases is responsible for the paralogue specificity14. In contrast to ubiquitination pathways containing hundreds of E3 ligases, there have only been a few SUMO E3 ligases identified15. SUMO E3 ligases are defined by properties including their ability to (i) bind Ubc9, (ii) bind SUMO moiety via a SIM domain, and (iii) enhance SUMO transfer from Ubc9 onto substrate. E3 ligase is not absolutely required for SUMO conjugation16, but provides substrate and SUMO-paralogue specificity. Since usually only a small fraction of the total substrate protein is SUMO-modified, detection of SUMOylated proteins in vivo is always a challenge. Therefore, accurate and reproducible assays are needed in order to elucidate the precise function of SUMO modification.
The in vitro SUMOylation assay, which evaluates the ability of purified Ubc9 to catalyze SUMOylation of substrate proteins, is a well-accepted assay for studying SUMO modification17. However, SUMO E3 ligase activity in most cases cannot be detected or can only be detected with mutated SUMO ligase with high SUMO E3 ligase activity and low substrate specificity by the standard protocol because of the presence of an abundant amount of Ubc918. The overall success of this assay largely depends upon the careful titration of purified Ubc9. The in vitro SUMOylation assay described here is modified from a standard SUMOylation protocol (see Table of Materials). The observation of increased global SUMO modification during Kaposi's sarcoma-associated herpesvirus (KSHV) reactivation prompted us to identify the viral SUMO E3 ligase that may be responsible for the up-regulation of SUMOylation. Following a small-scale screening of the interacting proteins of viral SUMO E3 ligase K-bZIP, p53 was identified as a novel substrate. In this protocol, we show in detail in insect cells the steps involved in the expression and purification of wild-type K-bZIP, a viral SUMO E3 ligase with SUMO-2/3 specificity. The ability of the purified K-bZIP to enhance SUMOylation of p53, a well-known SUMO substrate, is evaluated in the presence of reduced amounts of E1 and E2 enzymes. Using this modified SUMOylation assay, researchers can reliably define the abilities of SUMO E3 ligase to SUMOylate novel or known substrates, which is a primary step in the study of protein SUMOylation. Moreover, this method helps identify novel SUMO E3 ligases with low ligase activity but high substrate specificity.
Protocol
1. Preparation of Baculovirus Expression Constructs
- Purify SUMO E3 ligase K-bZIP by cloning the cDNA of K-bZIP19 into a dual expression baculovirus vector (see Table of Materials) with an N-terminal epitope-tag. We have had success using an octapeptide tag (indicated throughout the protocol as vector-tag-K-bZIP). NOTE: The DNA template for K-bZIP polymerase chain reaction (PCR) cloning is cDNA reverse transcribed from RNA isolated from TREx F3H3-K-Rta BCBL-1 cells following doxycycline treatment20.
- Separate and extract the digested DNA following electrophoresis on a 0.8% agarose gel22. Ligate the insert (K-bZIP) and vector by T4 DNA ligase at 16 °C for 30 min according to manufacturer's instructions (see Table of Materials).
- Add 5 µL of ligation DNA to 50 µL competent E. coli cells "A" (see Table of Materials) in a 1.5 mL tube. Put on ice for 30 min. Then, incubate the 1.5 mL tube in a 42 °C water bath for 45 s and immediately put the tube on ice for 3 min.
- Add 600 µL S.O.C. medium (0.5% (w/v) yeast extract, 2% (w/v) tryptone, 10 mM NaCl, 2.5 mM KCl, 20 mM MgSO4, and 4% (w/v) glucose) into the tube and incubate at 37 °C for 1 h. Then, centrifuge the tube at room temperature for 3 min at 600 x g.
- Select 1 colony to inoculate in 5 mL LB medium containing 100 µg/mL ampicillin and culture at 37 °C for 16 h with 250 rpm shaking. Then, extract the plasmid, vector-tag-K-bZIP, by a plasmid extraction kit according to the manufacturer's instructions (see Table of Materials).
- Generate the recombinant bacmid harboring the tag-K-bZIP by transformation of the vector-tag-K-bZIP into competent E. coli cells "B" (see Table of Materials).
- Mix 0.2 µg vector-tag-K-bZIP plasmid and 100 µL competent E. coli cells "B" in a 1.5 mL tube and put on ice for 30 min. Incubate the tube in 42 °C water bath for 45 s and immediately put the tube on ice for 3 min.
- Add 900 µL of S.O.C. medium into the tube and incubate at 37 °C for 4 h with rotation at 50 rpm. Next, take 10 µL of the medium, add an additional 50 µL of S.O.C medium to spread on LB agar plates containing 50 µg/mL kanamycin, 7 µg/mL gentamicin, 10 µg/mL tetracycline, 100 µg/mL galactosidase substrate, and 40 µg/mL isopropyl β-D-1-thiogalactopyranoside (IPTG), and incubate the plates for 48 h at 37 °C.
- Inoculate one white colony in 5 mL LB medium containing 50 µg/mL kanamycin, 7 µg/mL gentamicin, and 10 µg/mL tetracycline. Incubate at 37 °C with 250 rpm shaking for 16 h.
- Isolate the recombinant bacmid DNA harboring the tag-K-bZIP, quantify, and re-suspend at a concentration of 1 µg/µL25.
- Generate recombinant baculoviruses expressing tag-K-bZIP by transfecting 1 µg of recombinant bacmid DNA containing tag-K-bZIP into Sf9 cells in one well of a 6-well plate.
- Seed 2 x 106 Sf9 cells (see Table of Materials) in 2 mL Grace's Insect Medium supplied with 10% fetal bovine serum (FBS) and 1% gentamicin in each well of a 6 well plate, then incubate at 27 °C for 1 h.
- Maintain a log phase culture of Sf9 cells in Grace's Insect Medium supplied with 10% FBS, 1% gentamicin, and 1% detergent C (see Table of Materials) at 27 °C in orbital suspension at 140 rpm. Count cells using a hemocytometer and trypan blue staining; cell viability should exceed 95%. Maintain cells using a subcultivation ratio of 1:3 every 2-3 days (minimal density ~0.5 x 106 cells/mL).
- Add 2 µL bacmid DNA (1 µg/µL) and 98 µL reduced serum media (see Table of Materials) into a 2 mL tube. Then, add 4 µL transfection reagent (see Table of Materials) to the tube and mix well. Incubate at room temperature for 15 min.
- Add this transfection mixture (104 µL) into each well, and incubate at 27 °C for 12-16 h.
- Remove exhausted media with gentle aspiration, and replace with 2 mL of fresh Grace's Insect Medium supplied with 10% FBS and 1% gentamicin. The Sf9 cells adhere loosely to the plate surface and will not be disturbed with gentle aspiration.
- Collect recombinant baculovirus after transfection and amplify baculovirus to obtain high-titer stocks (P1) for further experiments.
- 72 h after changing medium, scrape the Sf9 cells using a sterile cell lifter, collect the supernatant from each individual well in a 1.5 mL tube and vortex for 10 s.
- Centrifuge at 4 °C for 3 min at 150 x g. Collect supernatant as P0 baculovirus and aliquot in 1.5 mL tubes (1 mL baculovirus in each tube). Store at -80 °C.
- Seed 1 x 107 Sf9 cells in 10 mL Grace's Insect Medium supplied with 10% FBS and 1% gentamicin in a 10 cm Petri dish and incubate at 27 °C for 1 h.
- For amplification of recombinant baculoviruses expressing tag-K-bZIP, add 0.5 mL P0 baculovirus to the seeded Sf9 cells and incubate at 27 °C for 48 h.
- Collect supernatant as P1 baculovirus in a 15 mL tube and store at 4 °C for up to 1 month.
2. Purification of K-bZIP
Maintain a log phase culture of Sf9 cells in Grace's Insect Medium supplied with 10% FBS, 1% gentamicin, and 1% detergent C at 27 °C in orbital suspension at 140 rpm as described in step 1.3.2.
On the day of transduction, add 1 mL of baculovirus-containing supernatant (P1) into 250 mL of Sf9 cells with a density of 2 x 106 cells/mL.
Incubate Sf9 cells for 72 h at 27 °C on an orbital shaker with 140 rpm shaking, then collect by centrifugation at 2,740 x g for 15 min at 4 °C.
Remove supernatant and lyse cell pellets in 10 mL lysis buffer (20 mM HEPES pH 7.9, 0.5 M NaCl, 1% detergent A (see Table of Materials), 2% glycerol, protease inhibitor cocktail) and rotate at 50 rpm using a suspension mixer at 4 °C for 30 min.
Centrifuge cell lysate at 15,000 x g for 15 min to collect the supernatant.
To purify tag-K-bZIP, incubate the cell lysates (10 mL) with 50 µL of antibody-tagged magnetic beads in 14 mL polypropylene tubes, and rotate at 50 rpm for 3 h at 4 °C using a suspension mixer. Following protein capture, pellet the beads at 800 x g centrifugation for 30 s at 4 °C. Remove most of the supernatant, re-suspend the beads in the residual volume of about 1 mL, and transfer to a 1.5 mL tube for washing.
Wash the captured protein by placing the tube containing the beads on a magnetic stand, and remove the supernatant once the magnetic beads have fully adhered to the side of the tube.
- Wash the beads with lysis buffer four times.
- Add 1 mL lysis buffer into 1.5 mL tubes, and invert the tubes 10 times. Place the 1.5 mL tube on a magnetic stand and remove the supernatant as described in 2.7.
- Repeat the previous step another 3 times.
- Wash the beads with phosphate buffered saline (PBS) once.
- Add 1 mL PBS (137 mM NaCl, 2.7 mM KCl, 8 mM Na2HPO4, 1.5 mM KH2PO4) into 1.5 mL tubes, and invert the tubes 10 times.
- Put the 1.5 mL tube on a magnetic stand and then remove the supernatant.
Elute tag-K-bZIP protein from the antibody-tagged magnetic beads by adding 100 µL of 150 µg/mL octapeptide diluted in PBS into a 1.5 mL tube. Rotate the tube for 10 min at 50 rpm by suspension mixer at room temperature. Then, place the tube back on the magnetic stand and collect the K-bZIP-containing supernatant in a clean 1.5 mL tube.
Analyze 1-5 µL purified tag-K-bZIP protein by SDS-PAGE followed by Coomassie blue staining26. Load 2 µg, 1 µg, and 0.5 µg samples of bovine serum albumin (BSA) on the same gel and use it for quantification with an image processing program, like ImageJ. Estimate the K-bZIP concentration by comparison to the BSA standards.
The purified K-bZIP is now ready to be used in each SUMOylation assay. Dilute the K-bZIP in PBS to a final concentration of 100 ng/µL and store in small aliquots at -80 °C.
3. SUMOylation Assay
Add 3 µL of 100 ng/µL of purified tag-K-bZIP into the master mix of each in vitro SUMOylation reaction. Components in master mix contain 1 µL protein buffer, 1x SUMOylation buffer, 0.5 µL p53 protein (0.5 mg/mL), 25 nM of E1 activating enzyme (stock conc.: 1 µM), 50 nM of E2 conjugating enzyme (stock conc.: 10 µM), and 250 nM each of the SUMO peptides (SUMO-1, SUMO-2, or SUMO-3; stock conc.: 5 µM) to a final volume of 17 µL.
Mix the contents gently and incubate the reaction at 30 °C for 3 h. Stop the reaction by adding 20 µL of 2x SDS-PAGE loading buffer (100 mM Tris-HCL pH 6.8, 4% sodium dodecyl sulfate, 0.2% (w/v) bromophenol blue, 20% (v/v) glycerol, and 200 mM β-mercaptoethanol) and denature the samples at 95 °C for 5 min.
- Separate samples on SDS-PAGE, transfer the separated proteins onto PVDF membrane by using a semi-dry electrophoretic transfer apparatus, and analyze by Western blot using anti-p53 antibody.
- Set up the gel apparatus as in step 2.11, and load 20 µL of sample into gel wells.
- Run the 10% gel in a constant current at 80 V for about 120 min (until the dye band reaches the bottom of the gel). After completion of electrophoresis, turn off the power supply.
- Disconnect the gel apparatus and gel cassettes to take out the gel, and float the gel into semi-dry transfer buffer (48 mM Tris-HCl, 39 mM glycine, 20% methanol) for 5 min.
- Take another container and soak the PVDF membrane in methanol for 1 min. Then, take PVDF out of methanol and put in semi-dry transfer buffer. Gently agitate the membrane for 5 min.
- Remove the safety cover of the semi-dry electrophoretic apparatus.
- Pre-wet filter paper, and prepare a gel sandwich on the bottom platinum anode as follows: filter paper, PVDF membrane, gel, and filter paper.
- Secure cathode plate and safety cover, then run blot at 15 V constant current for 90 min. Turn off the power supply, disconnect semi-dry apparatus, and take out the PVDF membrane.
- Block PVDF membrane with blocking buffer (5% non-fat milk in TBST buffer (137 mM NaCl2, 20 mM Tris-HCl, 0.1% detergent B (see Table of Materials), pH 7.6)) at room temperature for 1 h with 30 rpm shaking by orbital shaker.
- Hybridize the PVDF membrane with anti-p53 antibody diluted (1:1,000) in blocking buffer for 12-16 h at 4 °C with 30 rpm shaking using a suspension mixer.
- Take out the PVDF membrane into a container and put in TBST. Rinse PVDF membrane with TBST twice more.
- Soak the PVDF membrane in TBST for 30 min with 45 rpm shaking by orbital shaker.
- Hybridize the PVDF membrane with anti-rabbit antibody conjugated with horseradish peroxidase (HRP) diluted (1:4,000) in blocking buffer for 1 h at room temperature with 30 rpm shaking using a suspension mixer.
- Wash the PVDF membrane with TBST 3 times as described in step 3.4.10.
- Soak the PVDF membrane in TBST for 30 min with 45 rpm shaking using a suspension mixer. Remove TBST and add PBS to preserve the PVDF membrane at 4 °C for up to 12 h.
- Mix the enhanced chemiluminescent substrate (ECL substrate) reagent 1 and 2 (1:1) (see Table of Materials). Remove the PVDF membrane from the PBS, blot briefly with a paper towel or light-duty wiper to absorb excess moisture, and immediately add 400 µL of ECL reagent to the surface of each membrane for 3-5 min. Remove excess ECL reagent by briefly blotting, but do not allow the membrane to completely dry.
- Expose the blot using a luminescence imaging system or autoradiography film27.
Representative Results
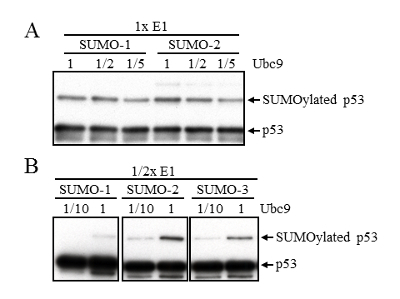
According to the information provided by the manufacturer, the standard amount of E1 and E2 enzyme in the SUMOylation assay is 50 nM and 500 nM, respectively. The minimal amount of E2 conjugating enzyme Ubc9 that is able to SUMOylate p53 was first determined by an in vitro SUMOylation assay. As low as one-fifth of the amount of Ubc9 used in the standard in vitro SUMOylation assay protocol was able to efficiently SUMOylate p53 (Figure 1A). Therefore, half of the amount of E1 enzyme in combination with one-tenth of the amount of Ubc9 used in the standard protocol was used for another in vitro SUMOylation assay. A significant reduction of SUMOylation efficiency was observed as compared with standard protocol (Figure 1B).
Thus, the ability of SUMO E3 ligase K-bZIP to enhance p53 SUMOylation was determined using these modified in vitro SUMOylation conditions. Tagged K-bZIP purified from Sf9 cells (Figure 2A) was employed in a SUMOylation reaction containing lower amounts of E1 and E2 Ubc9 enzymes. Under these modified conditions, K-bZIP could efficiently catalyze the SUMOylation of p53 (Figure 2B). Immunoblotting with anti-p53 antibody confirmed the similar total amounts of p53 in each reaction, and also detected SUMO modified p53.
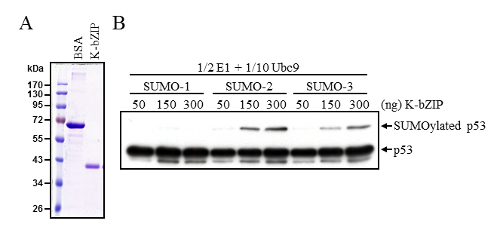
Figure 1: Determination of the minimal amount of SUMO enzymes used in the in vitro SUMOylation assay. (A) p53 SUMOylation was evaluated in an in vitro SUMOylation assay that included one half and one fifth the amount of Ubc9 enzyme used in standard protocol. After 3 hours of in vitro SUMOylation reaction, SUMOylated p53 enzymes were examined by Western blot with anti-p53 antibody. (B) In vitro SUMOylation of p53 was further analyzed by using half the amount of E1 enzyme in combination with one-tenth the amount of Ubc9 used in the standard protocol. A Western blot was performed as described as in (A). Please click here to view a larger version of this figure.
Figure 2: Enhancement of p53 SUMOylation by SUMO E3 ligase K-bZIP. (A) Purified baculovirus-expressed K-bZIP protein, analyzed by SDS-PAGE followed with Coomassie blue staining. 1 µg BSA and 10 µL purified K-bZIP were loading for comparative the protein quantity. (B) K-bZIP enhanced p53 SUMOylation was evaluated by using an in vitro SUMOylation reaction with half the amount of E1 enzyme and one tenth the amount of Ubc9 recommend in the standard protocol. SUMOylated p53 were investigated by Western blot as described in Figure 1A. Please click here to view a larger version of this figure.
Discussion
The in vitro SUMOylation protocol described here is routinely used to establish the SUMOylation status of identified Ubc9 substrates. The major limitation using the standard protocol to study the SUMOylation function of SUMO E3 ligase is the abundance of SUMO E1 activating and E2 conjugating enzymes Ubc9 that maximize the SUMO conjugation in in vitro systems. Considering this challenge, we believe that titration of the amount of SUMO E1 and E2 enzymes to the level that one can barely detect their activity in SUMOylation is the only way available to determine SUMO E3 ligase catalytic activities using this in vitro SUMOylation system. Following this hypothesis, we successfully elucidated the E3 ligase activity of K-bZIP and identified it as a novel SUMO E3 ligase with specificity towards SUMO-2/3.
The critical step within the protocol is the incorporation of lower amounts of E1 and E2. Using these lower amounts, as shown in Figure 2, novel SUMO E3 ligases with low ligase activity may be able to be identified with preservation of their specificity towards substrate and SUMO paralogues. A major limitation of the technique is the requirement for a high-quality antibody recognizing the SUMO substrate, since only a very small proportion of the substrate can be SUMO modified.
Though many proteins show the potential to increase SUMOylation after overexpression in vivo, defining a SUMO E3 ligase must be demonstrated by a reconstituted in vitro SUMOylation system using purified E1, E2, and E3 enzymes. As opposed to the hundreds and perhaps thousands of ubiquitin E3 ligases identified, until now only a few SUMO E3 ligases have been found. This may be due in part to the use of the traditional standard in vitro SUMOylation assay which maximizes SUMO conjugation efficiency and consequently hinders the SUMOylation function of potential SUMO E3 ligases. The present protocol describes a simple and consistent assay to probe SUMOylation enhanced by a SUMO E3 ligase. The described method is essential for the identification and characterization of novel SUMO E3 ligases.
Disclosures
The authors disclose no potential conflicts of interest.
Acknowledgments
This work was supported by grants from the Ministry of Science and Technology (MOST, 105-2320-B-010-007-MY3 to PCC), from the National Health Research Institute (NHRI-EX105-10215BC to PCC), from the Ministry of Science and Technology (MOST 105-2314-B-400-019 to HJK) and from the National Health Research Institute (NHRI MG-105-SP-10, NHRI MG-106-SP-10 to HJK). This work was also supported partly with National Yang-Ming University on manuscript publication to PCC. The funders had no role in study design, data collection and analysis, decision to publish, or reparation of the manuscript.
References
- Bies J, Markus J, Wolff L. Covalent attachment of the SUMO-1 protein to the negative regulatory domain of the c-Myb transcription factor modifies its stability and transactivation capacity. J Biol Chem. 2002;277(11):8999–9009. doi: 10.1074/jbc.M110453200. [DOI] [PubMed] [Google Scholar]
- Lin DY, et al. Role of SUMO-interacting motif in Daxx SUMO modification, subnuclear localization, and repression of sumoylated transcription factors. Mol Cell. 2006;24(3):341–354. doi: 10.1016/j.molcel.2006.10.019. [DOI] [PubMed] [Google Scholar]
- Welsh M, Jamalpour M, Zang G, Akerblom B. The role of the Src Homology-2 domain containing protein B (SHB) in beta cells. J Mol Endocrinol. 2016;56(1):21–31. doi: 10.1530/JME-15-0228. [DOI] [PubMed] [Google Scholar]
- Huang WQ, et al. Structure, function, and pathogenesis of SHP2 in developmental disorders and tumorigenesis. Curr Cancer Drug Targets. 2014;14(6):567–588. doi: 10.2174/1568009614666140717105001. [DOI] [PubMed] [Google Scholar]
- Perry JJ, Tainer JA, Boddy MN. A SIM-ultaneous role for SUMO and ubiquitin. Trends Biochem Sci. 2008;33(5):201–208. doi: 10.1016/j.tibs.2008.02.001. [DOI] [PubMed] [Google Scholar]
- Sun H, Leverson JD, Hunter T. Conserved function of RNF4 family proteins in eukaryotes: targeting a ubiquitin ligase to SUMOylated proteins. EMBO J. 2007;26(18):4102–4112. doi: 10.1038/sj.emboj.7601839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang J, Shi Y, Valin A, Xuan Y, Gill G. Direct binding of CoREST1 to SUMO-2/3 contributes to gene-specific repression by the LSD1/CoREST1/HDAC complex. Mol Cell. 2009;34(2):145–154. doi: 10.1016/j.molcel.2009.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyst MJ, Stancheva I. A role for SUMO modification in transcriptional repression and activation. Biochem Soc Trans. 2007;35:1389–1392. doi: 10.1042/BST0351389. Pt 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HW, et al. Chromatin modification by SUMO-1 stimulates the promoters of translation machinery genes. Nucleic Acids Res. 2012;40(20):10172–10186. doi: 10.1093/nar/gks819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosonina E, Duncan SM, Manley JL. SUMO functions in constitutive transcription and during activation of inducible genes in yeast. Genes Dev. 2010;24(12):1242–1252. doi: 10.1101/gad.1917910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang PC, et al. The chromatin modification by SUMO-2/3 but not SUMO-1 prevents the epigenetic activation of key immune-related genes during Kaposi's sarcoma associated herpesvirus reactivation. BMC Genomics. 2013;14:824. doi: 10.1186/1471-2164-14-824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neyret-Kahn H, et al. Sumoylation at chromatin governs coordinated repression of a transcriptional program essential for cell growth and proliferation. Genome Res. 2013;23(10):1563–1579. doi: 10.1101/gr.154872.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatham MH, et al. Polymeric chains of SUMO-2 and SUMO-3 are conjugated to protein substrates by SAE1/SAE2 and Ubc9. J Biol Chem. 2001;276(38):35368–35374. doi: 10.1074/jbc.M104214200. [DOI] [PubMed] [Google Scholar]
- Mattoscio D, Segre CV, Chiocca S. Viral manipulation of cellular protein conjugation pathways: The SUMO lesson. World J Virol. 2013;2(2):79–90. doi: 10.5501/wjv.v2.i2.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gareau JR, Lima CD. The SUMO pathway: emerging mechanisms that shape specificity, conjugation and recognition. Nat Rev Mol Cell Biol. 2010;11(12):861–871. doi: 10.1038/nrm3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernier-Villamor V, Sampson DA, Matunis MJ, Lima CD. Structural basis for E2-mediated SUMO conjugation revealed by a complex between ubiquitin-conjugating enzyme Ubc9 and RanGAP1. Cell. 2002;108(3):345–356. doi: 10.1016/s0092-8674(02)00630-x. [DOI] [PubMed] [Google Scholar]
- Knipscheer P, et al. Ubc9 sumoylation regulates SUMO target discrimination. Mol Cell. 2008;31(3):371–382. doi: 10.1016/j.molcel.2008.05.022. [DOI] [PubMed] [Google Scholar]
- Vethantham V, Manley JL. In vitro sumoylation of recombinant proteins and subsequent purification for use in enzymatic assays. Cold Spring Harb Protoc. 2009;2009(1):5121. doi: 10.1101/pdb.prot5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SF, Robinson DR, Miller G, Kung HJ. Kaposi's sarcoma-associated herpesvirus encodes a bZIP protein with homology to BZLF1 of Epstein-Barr virus. J Virol. 1999;73(3):1909–1917. doi: 10.1128/jvi.73.3.1909-1917.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WS, Hsu HW, Campbell M, Cheng CY, Chang PC. K-bZIP Mediated SUMO-2/3 Specific Modification on the KSHV Genome Negatively Regulates Lytic Gene Expression and Viral Reactivation. PLoS Pathog. 2015;11(7):1005051. doi: 10.1371/journal.ppat.1005051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor CD, Metcalf E, Wrighton CJ, Harris TJ, Saunders JR. RsrII--a novel restriction endonuclease with a heptanucleotide recognition site. Nucleic Acids Res. 1984;12(17):6701–6708. doi: 10.1093/nar/12.17.6701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helling RB, Goodman HM, Boyer HW. Analysis of endonuclease R-EcoRI fragments of DNA from lambdoid bacteriophages and other viruses by agarose-gel electrophoresis. J Virol. 1974;14(5):1235–1244. doi: 10.1128/jvi.14.5.1235-1244.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sezonov G, Joseleau-Petit D, D'Ari R. Escherichia coli physiology in Luria-Bertani broth. J Bacteriol. 2007;189(23):8746–8749. doi: 10.1128/JB.01368-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingold EB. Bacterial transformation. Methods Mol Biol. 1985;2:237–240. doi: 10.1385/0-89603-064-4:237. [DOI] [PubMed] [Google Scholar]
- Brulois KF, et al. Construction and manipulation of a new Kaposi's sarcoma-associated herpesvirus bacterial artificial chromosome clone. J Virol. 2012;86(18):9708–9720. doi: 10.1128/JVI.01019-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227(5259):680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Ahmed S, Holt M, Riedel D, Jahn R. Small-scale isolation of synaptic vesicles from mammalian brain. Nat Protoc. 2013;8(5):998–1009. doi: 10.1038/nprot.2013.053. [DOI] [PubMed] [Google Scholar]