

Abstract
Despite the discovery more than 15 years ago that patients with hereditary pulmonary arterial hypertension (HPAH) inherit BMP type 2 receptor (BMPR2) mutations, it is still unclear how these mutations cause disease. In part, this is attributable to the rarity of HPAH and difficulty obtaining tissue samples from patients with early disease. However, in addition, limitations to the approaches used to study the effects of BMPR2 mutations on the pulmonary vasculature have restricted our ability to determine how individual mutations give rise to progressive pulmonary vascular pathology in HPAH. The importance of understanding the mechanisms by which BMPR2 mutations cause disease in patients with HPAH is underscored by evidence that there is reduced BMPR2 expression in patients with other, more common, non-hereditary form of PAH, and that restoration of BMPR2 expression reverses established disease in experimental models of pulmonary hypertension. In this paper, we focus on the effects on endothelial function. We discuss some of the controversies and challenges that have faced investigators exploring the role of BMPR2 mutations in HPAH, focusing specifically on the effects different BMPR2 mutation have on endothelial function, and whether there are qualitative differences between different BMPR2 mutations. We discuss evidence that BMPR2 signaling regulates a number of responses that may account for endothelial abnormalities in HPAH and summarize limitations of the models that are used to study these effects. Finally, we discuss evidence that BMPR2-dependent effects on endothelial metabolism provides a unifying explanation for the many of the BMPR2 mutation-dependent effects that have been described in patients with HPAH.
Keywords: BMPR2 mutations, pulmonary arterial hypertension, metabolism, pulmonary hypertension experimental, pulmonary endothelium
BMPR2 mutations and endothelial cell dysfunction in hereditary pulmonary arterial hypertension
Hereditary pulmonary arterial hypertension (HPAH) is a rare disorder that is characterized by progressive, obliterative remodeling of pre-capillary pulmonary arteries associated with increased pulmonary vascular resistance, ultimately leading to right heart failure and death.1 In 2000, two independent groups reported an association between heterozygous germline mutations in the bone morphogenetic protein (BMP) type 2 receptor gene, BMPR2, and cases of autosomal dominant inherited pulmonary arterial hypertension (PAH).2,3 It is now recognized that ∼80% of PAH patients with a family history and ∼20% of those with no known family history have germline mutations at the BMPR2 locus.4 Since this discovery, germline mutations have been identified in at least nine other genes in patients with BMPR2 mutation “negative” HPAH, most commonly ENDOGLIN and ALK1,5,6 many of which intersect with the BMP signaling pathway and are expressed at high levels in pulmonary endothelial cells (PECs).5 The significance of these findings is underscored by the observation that BMPR2 expression is also reduced in the lungs of patients with other, non-hereditary idiopathic or disease-associated forms of PAH,7 and in cultured PECs from patients with idiopathic PAH (IPAH).8 In addition, reduced levels of Bmpr2 have been described in a variety of experimental models of pulmonary hypertension (PH) that do not involve genetic manipulation of Bmpr2 signaling,9–11 and restoration of Bmpr2 expression or signaling prevents, and even reverses, established disease in different experimental models of PH.12–14 These findings suggest that defects in BMPR2 expression and/or signaling contribute to common pathophysiological processes in different forms of PAH. In addition, these findings suggest that the use of different strategies to enhance BMPR2 expression and/or function represent promising therapeutic approaches for the treatment of patients with PAH.15
One of the common features of the pulmonary vascular pathophysiology in PAH is that there is a hyper-proliferative phenotype involving diverse cells types including pulmonary vascular endothelium, smooth muscle, fibroblasts, and inflammatory cells.16,17 These changes lead to decreased right ventricular function and, eventually, right heart failure and death.17 While the primary cellular targets of disease in patients with IPAH and disease-associated PAH remain to be established, in HPAH there is good evidence that PECs may be the primary drivers of disease pathophysiology: PECs express high levels of BMPR2 in vivo and in vitro,7,18 and mice with conditional deletion of Bmpr2 restricted to the endothelium develop spontaneous PH.19
In this review, therefore, we will focus on the role of PEC dysfunction in mediating BMPR2 mutation-dependent effects in HPAH. We will discuss some of the controversies and challenges in this area, focusing specifically on mutation-dependent effects that different BMPR2 mutations may have on PEC function and susceptibility to HPAH, and whether there are quantitative and/or qualitative differences between the effects of different BMPR2 mutations in this disease. We will review BMPR2-dependent pathways that may account for abnormalities in PEC function in patients with HPAH, limitations of the models that are used to study these effects, and how BMPR2-dependent effects on metabolism may be the underlying mechanism mediating BMPR2-mutation associated PEC dysfunction in HPAH.
Are there genotype-phenotype relationships between BMPR2 mutations in human HPAH?
Since first described,2,3 more than 380 different genetic variants across the BMPR2 locus have been identified in patients with HPAH.5 These mutations occur throughout the BMPR2 open reading frame and include missense mutations (predicted to give rise to a single amino-acid exchanges), in-frame exon deletions associated with splice site mutations (predicted to delete exon segments), as well as non-sense and frameshift BMPR2 mutations (predicted to give rise to BMPR2 truncations and/or resulting in loss of expression of the mutant product as a result of activation of non-sense mediated messenger RNA [mRNA] decay [NMD]20). Given the diversity of BMPR2 mutations that have been described in patients with HPAH, and the fact that some of these mutant products, when expressed, may exert additional dominant negative effects on BMPR2 signaling, a number of investigators have sought to determine whether there are BMPR2 genotype–phenotype associations that are predictive of disease severity in HPAH. This question has been much more difficult to address and the issue is still debated.
A recent meta-analysis of eight cohorts of patients with idiopathic, hereditary, and anorexigen-associated PAH that were systematically tested for BMPR2 mutations lends support to the hypothesis that all BMPR2 mutations behave in the same way.4 The main finding of this study was that BMPR2 mutation-positive PAH patients present at a younger age and have more severe disease than patients without mutations. However, the same analysis did not reveal differences in disease severity between patients with missense BMPR2 mutations and patients with other BMPR2 mutations.4 These findings are consistent with an earlier study which also failed to show differences in disease severity between HPAH patients carrying missense, truncating, large rearrangement, or BMPR2 splice site mutations.21 However, two other studies have shown that patients with truncating mutations predicted to give rise to NMD have later onset and less severe disease than patients with BMPR2 mutations that are predicted to give rise to an expressed mutant product.22,23 These discrepancies may have resulted from differences in the classification of the BMPR2 mutations. The two papers showing positive associations between truncating mutations and improved outcomes, defined BMPR2 mutations either according to their NMD status, or by the presence of C-terminal mutations predicted to give rise to NMD.23,24 In contrast, the two papers that did not report an association between mutation types and disease severity, compared missense mutations with all other mutations.4,21 Since the latter includes in-frame deletions as well as truncating mutations that do not activate NMD, some of these patients may also express mutant products that may have dominant inhibitory effects on BMPR2 function. This problem is confounded by the observation that some truncating mutations, such as the BMPR2 W13X mutation, which is predicted to activate NMD and give rise to a null allele, actually escape NMD and expresses mutant receptor products.25 Moreover, while the ability of any mutation to induce NMD can be evaluated in different cell types (this is often performed in Epstein–Barr virus transformed lymphocytes for genetic studies in HPAH22,25), mRNA stability and decay varies between cell types under different conditions,26 so that the ability of a given BMPR2 mutation to activate NMD in a particular target cell in vivo may be impossible to determine (see the discussion below in relation to PH susceptibility of Bmpr2R899X/+ mice12). Given these challenges, it is unlikely we will be able to answer the question as to whether the severity of HPAH depends on the type of BMPR2 mutation from clinical studies alone.
An experimental approach to identify genotype–phenotype relationships in PH
To address this question, therefore, we have taken an alternative, experimental approach by evaluating the PH susceptibility of mice carrying two different germline Bmpr2 mutations.27 For this, we used two established mouse lines: Bmpr2ΔEx4-5/+ mice in which there is an out of frame deletion of the fourth and fifth exons of Bmpr2 (Bmpr2+/− mice),28 and Bmpr2ΔEx2/+ mice in which there is an in-frame deletion of the second exon of Bmpr2 (Fig. 1a).29 Germline BMPR2 mutations giving rise to out-of-frame deletions of exons 4 and 5, and in-frame deletions of exon 2, have been described in patients with HPAH.30 In previous studies, we showed that PECs from Bmpr2ΔEx2/+ mice express high levels of the Bmpr2ΔEx2 mutant allelic product, that this product is abnormally processed and retained in the endoplasmic reticulum (ER), and that chemical chaperones, which enhance maturation of misfolded proteins in the ER, restore Bmpr2ΔEx2 protein expression at the plasma membrane (PM) and enhance canonical Bmpr2-dependent Smad1/5 signaling in PECs.18 This is illustrated in Fig. 1b in which we use two different anti-Bmpr2 antibodies to demonstrate that the lower molecular weight band on Western blot of PEC lysates represents the Bmpr2ΔEx2 protein product lacking sequences encoded by exon 2 of Bmpr2. Moreover, we demonstrated that cultured lymphocytes from a HPAH patient with a splice site mutation in the second intron of BMPR2,30 also express a BMPR2ΔEx2 mutant receptor that is not properly trafficked to the PM.18 These findings indicate that Bmpr2ΔEx2/+ mice provide a genetic model of a HPAH BMPR2 mutations that escapes NMD and expresses a mutant receptor that may exert dominant negative effects on the remaining wild type (WT) BMPR2 allelic product. In contrast, Bmpr2+/− mice provide a genetic model of non-sense or frameshift HPAH BMPR2 mutations that are subject to NMD.
Fig. 1.

Characterization of the Bmpr2ΔEx2 mutant protein product in PECs from Bmpr2ΔEx2/+ mice. (a) Schematic representation of the Bmpr2 gene. Exons 1–3 encode the extracellular domain of Bmpr2; exon 4 encodes most of the transmembrane domain; exons 5–13 encode the intracellular kinase domain and C-terminal tail. In-frame deletion of Exon 2 in Bmpr2ΔEx2/+ mutant mice, and corresponding amino-acid sequences recognized by the anti-BMPR2 antibodies clone 18 and ASQ, are indicated. (b) Conditionally immortalized PECs (ciPECs) were isolated from WT control and Bmpr2ΔEx2/+ mice. These cells were generated, as previously described,18,37,88 by crossing WT or Bmpr2 mutant mice on a C57Bl/6 background with C57Bl/6 H-2Kb-tsA58 SV40 large T antigen transgenic mice (Charles River's “Immortomouse”). Primary PECs were isolated from macerated mouse lungs perfused with trypsin and selected using microvascular endothelial cell medium under permissive conditions (in the presence of Interferon-γ at 33℃). For experiments, cells were transferred to 37℃ without Interferon-γ for 3–5 days to inhibit SV40 large T antigen activity, enabling the cells to become quiescent and differentiate into polarized PECs.18,37,88 Western blot using mouse monoclonal Clone 18 (BD) and ASQ (in house) anti-BMPR2 antibodies in WT and Bmpr2ΔEx2/+ ciPEC lysates, as described.18 Both cell lines expressed a 150-kDa WT Bmpr2 product which is detected using both antibodies. Bmpr2ΔEx2/+ ciPECs also express a 130-kDa product that is detected with Clone 18, which recognizes a C-terminal peptide sequence that is preserved in both WT Bmpr2 and in the Bmpr2ΔEx2 mutant product, but not the ASQ antibody, which recognizes an amino-acid sequence encoded by exon 2. These western blots have not previously been published but similar studies have been published in Frump et al.18
We had already shown that Bmpr2ΔEx2/+ mice have increased susceptibility to PH in response to prolonged hypoxic exposure when compared with WT littermates.31 This contrasted with previous studies that did not show an increase in the PH response to prolonged hypoxia in Bmpr2+/− mice.32 However, mouse strains affect the severity of hypoxic PH responses,33 and the two Bmpr2 mutant lines were maintained on different backgrounds at the time these studies were performed. Both Bmpr2 mutant lines were, therefore, back-crossed at least ten generations onto pure C57BL/6 backgrounds so that we could perform a direct comparison of PH susceptibility between the two lines without the confounding influence of the background stain. We used two experimental models of PH: three weeks of exposure to 10% oxygen (normobaric hypoxia); and three weeks of exposure to 10% oxygen in combination with weekly injections of the non-specific vascular endothelial growth factor receptor (VEGFR) small molecule inhibitor, SU5416, as described.34 Consistent with our earlier studies,31 Bmpr2ΔEx2/+ mice developed slightly higher right ventricular systolic pressures (RVSP) compared to WT and Bmpr2+/− mice in response to hypoxia alone, but there was a further increase in RVSPs in Bmpr2ΔEx2/+ mice compared with Bmpr2+/− mice in response to SU5416 and hypoxia (Fig. 2a and b).27 This was associated with increased peripheral vessel muscularization, thickening of the resistance-sized pulmonary arterioles, and reduced luminal diameters in resistance level arterioles (Fig. 2c–e).27 These findings demonstrated for the first time that there is a genotype–phenotype relationship between the type of Bmpr2 mutation and PH susceptibility. While limited to experimental models of PH that do not recapitulate the structural changes seen in the pulmonary vasculature of patients with established PAH, these studies suggest that different BMPR2 mutations types could have distinct effects on PAH severity in HPAH. In support of this hypothesis, a recent study demonstrated that a mouse with the germline, heterozygous HPAH-associated C-terminal truncation mutations, Bmpr2 R899X, develops spontaneous PH by the age of six months.12 The authors also reported that the same Bmpr2+/− mice used in our studies did not develop spontaneous PH at the same age. However, they also provide evidence that the Bmpr2 R899X mutation activates NMD in pulmonary vascular smooth muscle cells (PVSMCs) from these mice, suggesting that Bmpr2R899X/+ mice should have the same PH phenotype as heterozygous null Bmpr2+/− mutant mice. The fact that Bmpr2R899X/+ mice appear to have greater susceptibility to PH than Bmpr2+/− mice, suggests that the Bmpr2 R899X mutation escapes NMD in vivo. Since mRNA stability and decay varies between cell types,26 it is possible that while the Bmpr2 R899X mutation is subject to NMD in cultured PVSMs, it escapes NMD and is expressed as a mutant protein product in other cell types in vivo. Since loss of Bmpr2 in endothelium is sufficient to promote spontaneous PH in mice,19 we would argue that this is likely to be in the pulmonary endothelium.
Fig. 2.

Differential susceptibility of Bmpr2+/− and Bmpr2ΔEx2/+ mutant mice to PH. (a, b) Right ventricular systolic pressure (RVSP) measurements in male WT, Bmpr2+/−, and Bmpr2ΔEx2/+ mutant mice all maintained on a pure C57Bl/6 background ( > 10 generations) after exposure to normoxia or 10% oxygen for three weeks (a) or treatment with the VEGFR2 antagonist, SU5416 (or vehicle) subcutaneously at 20 mg/kg weekly ± 10% oxygen for three weeks, as indicated (b). RVSP measurements were obtained from pressure traces on right heart catheterization with Millar Instruments PVR-1035 pressure/volume catheters in anesthetized mice accessed via the right jugular vein. Individual data points shown, with means ± SEM indicated. (c) Peripheral muscularization. Lung sections underwent two-color immunofluorescence staining for Von Willebrand factor and α-SMA. The percent circumference of vessels covered with smooth muscle cells was determined in 20 round or oval sections of 20–50-µM diameter inter-acinar vessels per mouse. (d) Vessel wall thickness (mean of two orthogonal outer diameter–inner vessel diameters expressed as the percentage of outer diameter), measured in 10 round or oval sections of 20–50-µM diameter intrapulmonary vessels. (e) The range of vessel wall thicknesses. Individual data points represent the percentage of total vessels measured in each group with vessel wall thicknesses within the indicated ranges. One-way ANOVA with Bonferroni correction for between-group comparisons: *P < 0.05; **P < 0.01; ***P < 0.005; #P < 0.0001. Bars indicate statistically significant between group differences. Figure modified from Figs. 2 and 3 in Frump et al. with permission from the publishers.27
Bmpr2 mutation-dependent effects on pulmonary endothelial cell signaling and function
Based on our earlier findings that increased susceptibility of Bmpr2ΔEx2/+ mice to hypoxic PH was associated with reduced endothelium-dependent vasodilation of resistance-sized intrapulmonary arteries,31 we compared levels of activating serine (S) 1177 and inhibitory threonine (T) 495 endothelial nitric oxide synthase (eNOS) phosphorylation in WT, Bmpr2+/−, and Bmpr2ΔEx2/+ mouse lungs. Under basal conditions, there were reduced levels of pS1177 eNOS in both Bmpr2+/− and Bmpr2ΔEx2/+ mouse lungs, associated with a marked reduction in pT495 eNOS in Bmpr2ΔEx2/+ but not Bmpr2+/− mouse lungs (Fig. 3a and b).27 These effects were preserved in Bmpr2ΔEx2/+ but not Bmpr2+/− mouse lungs after three weeks of exposure to SU5416 and hypoxia (Fig. 3c and d). Since persistent de-phosphorylation of T495 eNOS is associated with eNOS uncoupling,35 these findings may explain why Bmpr2ΔEx2/+ mice have reduced endothelium-dependent vasodilatation in pulmonary vasculature,31 and potentially why Bmpr2ΔEx2/+ mice have greater susceptibility to experimental PH.
Fig. 3.

Regulation of eNOS in mouse lungs from WT, Bmpr2+/−, and Bmpr2ΔEx2/+ mice. (a) Western blots of lung lysates from normoxic mice demonstrating phosphorylated S1177 and T495 and total eNOS, and β-actin. Molecular weight markers indicated. (b) Quantification of band densities corrected for total eNOS protein, as indicated. (c) Western blots of lung lysates from WT, Bmpr2+/−, and Bmpr2ΔEx2/+ mice treated with SU5416 and 10% oxygen for three weeks. (d) Quantification of band densities corrected for total eNOS protein, as indicated. Results expressed as mean ± SEM based on western blot data shown normalized to WT normoxic controls. One-way ANOVA with Bonferroni correction for between-group comparisons: *P < 0.05; **P < 0.01; #P < 0.005, as indicated. (a, b) Modified from Fig. 4 in Frump et al. with permission from the publishers.27 (c, d) Unpublished data.
We have also evaluated alterations in caveolar function and Src kinase signaling in PECs from the two Bmpr2 mutant lines. As part of our studies to determine the intracellular localization of the Bmpr2ΔEx2 mutant product in PECs,18 we evaluated the use of antibodies against caveolin-1 (Cav-1) and Cavin-1, integral components of caveolae,36 which can be used as PM markers in endothelial cells. To our surprise, while Cav-1 and Cavin-1 performed well as PM markers in WT PECs, they performed poorly in Bmpr2ΔEx2/+ and Bmpr2+/− PECs.37 Further analysis of Bmpr2+/− PECs showed that this resulted from increased caveolar endocytosis-associated increased phosphorylation of tyrosine (Y) 14 Cav-1 and reduced PEC barrier function.37 Since Src kinase activation regulates pY14 Cav-1-dependent caveolar endocytosis,38 we explored the role of Src kinase in mediating these effects. Consistent with previous reports demonstrating increase levels of activating Y416 phosphorylation of Src kinase in the lungs of patients with PAH,39,40 Bmpr2+/− PECs had increased levels of pY416 Src, and inhibition of Src kinase activity using small molecule Src family kinase inhibitors PP2 and SKI606, reduced levels of pY14 Cav-1, decreased caveolar endocytosis, and improved Bmpr2+/− PEC barrier function in vitro.37 However, while our preliminary studies suggested that there was also increased caveolar endocytosis in Bmpr2ΔEx2/+ PECs, unlike Bmpr2+/− PECs, this is not associated with increased pY416 Src in these cells (Fig. 4). While the mechanisms regulating Src kinase activation in Bmpr2+/− PECs remain to be established, these findings suggest that different mechanisms regulate increased caveolar endocytosis in Bmpr2+/− and Bmpr2ΔEx2/+ PECs, and that the two Bmpr2 mutations have distinct effects on Src kinase activity. Taken together, our discovery that there are differences in PEC function and signaling in the two Bmpr2 mouse mutants illustrates the importance of determining whether different categories of BMPR2 mutation have distinct effects on pulmonary vascular cell function. It remains to be determined whether these differences result from e differences in Bmpr2 signaling between the two germline Bmpr2 mutations or whether this is a qualitative effect resulting from specific pathologic properties of the Bmpr2ΔEx2 mutant allelic product when expressed in PECs.
Fig. 4.
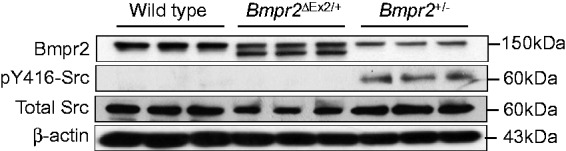
Activating Y416 phosphorylation of Src kinase in Bmpr2+/− but not Bmpr2ΔEx2/+ mouse PECs. ciPECs were isolated from WT control, Bmpr2+/−, and Bmpr2ΔEx2/+ mice. Western blot using Clone 18 anti-BMPR2 antibodies, pY416, and total Src kinase, and β-actin in WT, Bmpr2+/−, and Bmpr2ΔEx2/+ ciPEC lysates, as indicated. Reduced WT Bmpr2 expression seen in both Bmpr2+/− and Bmpr2ΔEx2/+ ciPECs compared to WT cells. Bmpr2ΔEx2/+ ciPECs also express a prominent Bmpr2ΔEx2 mutant product. pY416 Src kinase is only detected in Bmpr2+/− ciPECs. BMPR2 western blot was modified from Fig. 1 in Frump et al. with permission from the publishers.27 Corresponding pY416 and total Src kinase western blot studies have not previously been published.
Limitations of our current experimental models of BMPR2 mutation-associated associated PAH
While Bmpr2ΔEx2/+ mice develop moderately severe PH in response to hypoxia and SU5416 (Fig. 2b),27 unlike human PAH, careful analysis of vessel wall to luminal diameter ratios in pulmonary resistance vessels did not provide any evidence that this was associated with obstructive pulmonary vascular remodeling of these vessels (Fig. 2e).27 Furthermore, while changes in endothelial eNOS and Src kinase activation could account for some of the early events associated with the development of PAH in patients carrying BMPR2 mutations, they are unlikely to account for the obstructive vascular remodeling that occurs in patients with PAH. For example, while mice with eNOS deficiency develop mild PH associated with muscularization of peri-acinar pulmonary vessels, they do not develop severe obliterative pulmonary vascular remodeling.41,42 In addition, while Bmpr2+/− mice have increased susceptibility to PH in response to inflammatory challenges and serotonin treatment,43–45 there is no evidence that decreased barrier function contributes to the PH phenotype of these mice and no evidence that any of the germline heterozygous Bmpr2 mouse mutant lines that have been studied develop severe obliterative pulmonary vascular remodeling either spontaneously, with aging, or when exposed to models of experimental PH.12,27,31,32,43–45 In part, this may be because we do not yet know how to model the secondary hits (either genetic or environmental) that are required to induce PAH in BMPR2 mutation carriers. However, this may also be because mouse models of PH cannot recapitulate the severity of the obstructive pulmonary vascular remodeling which develops over many years in patients with PAH. Rats show much more robust proliferative and hemodynamic responses to a variety of different experimentally induced PH models than mice.46,47 For this reason, there was considerable interest in the development of genetic models of germline Bmpr2 mutations in rats. However, initial reports indicate that rats carrying two different truncating mutations at the Bmpr2 locus only develop mild spontaneous and hypoxia-induced PH without evidence of severe obliterative pulmonary vascular remodeling.48,49 An alternative approach has been to study the effects of conditionally deleting Bmpr2 expression in different cell types. For example, a subset of mice with conditional deletion of Bmpr2 in endothelial cells develop spontaneous PH associated with decreased endothelial barrier function, perivascular inflammation, and proliferative vascular remodeling that mimic some of the features of human PAH.19,50–52 However, pulmonary vascular remodeling is relatively mild and patchy. In addition, there is no evidence that complete loss of BMPR2 (loss of heterozygosity) occurs in vascular lesions from HPAH patients carrying heterozygous BMPR2 mutations,53 suggesting that complete loss of Bmpr2 expression, while providing insight into the function of Bmpr2 in the pulmonary vasculature, may not model the effects that germline BMPR2 mutations have on the pulmonary vasculature in patients with HPAH. The same maybe said of the transgenic mouse models that have been developed to over-express two different HPAH-associated BMPR2 mutations in different cell types.54–60 While over-expression of mutant receptors in these mice provides insight into the role of BMPR2 in regulating the pulmonary vasculature, these transgenic mice may not model the distinct effects that the same heterozygous germline mutations have on pulmonary vascular function in patients with HPAH.
BMPR2-dependent cellular and molecular pathways promoting PEC dysfunction
Given the limitations of our current in vivo models of BMPR2 mutation-associated HPAH, we have become dependent on results of studies, largely based on cell culture models, that recapitulate some of the observed or inferred features of established human PAH, without being able to test the functional significance of these changes directly using in vivo genetic models that recapitulate the human disease. Based largely on the analysis of a rat model of PH in which rats exposed to hypoxia (or unilateral pneumonectomy) and treated with SU5416 develop severe PH, with marked and progressive obstructive pulmonary vascular remodeling, it has become a widely held view in the PH research community that one of the early initiating events in any form of PAH, including BMPR2 mutation-associated HPAH, is the induction of widespread PEC apoptosis.61–64 This hypothesis goes on to suggest that widespread loss of endothelial cell integrity is followed by uncontrolled proliferative expansion of surviving pulmonary endothelium, giving rise to the obliterative neo-intimal proliferation as well as secondary smooth muscle cell proliferation and perivascular inflammation seen in this rat model and patients with established PAH.65 In support of this hypothesis, a number of groups have shown that BMPR2 deficiency in PECs reduces cell viability by increasing susceptibility to apoptosis.8,12,66,67 The majority of these studies based their conclusion on the effects of siRNA knockdown of BMPR2 in PECs. In some cases, these findings were validated in PECs isolated from IPAH patients (without known BMPR2 mutations),8,66,67 and in one case using blood outgrowth endothelial progenitor cells from patients with HPAH and known BMPR2 mutations.12 However, it is notable that none of these studies explored whether there was a genotype-dependent response in PECs from HPAH patients carrying different types of BMPR2 mutation and there is, as yet, no evidence that these effects are sufficient to account for the obliterative vasculopathy seen in patients with HPAH.
A number of downstream mechanisms have been shown to mediate these BMPR2-dependent effects on PEC survival, ranging from abnormalities in BRCA1-dependent DNA repair67 to activation of canonical Wnt signaling associated with reduced Apelin secretion.8 Other studies have identified BMPR2-dependent effects on Slug and HMGA1-dependent endothelium to mesenchyme transition (EndoMT)68 on the expression of chemokines and inflammatory cell growth factors by PECs50–52,69 and on increased production of mitochondrial reactive oxygen species by PECs.52,56 However, these studies also based their findings largely on siRNA knockdown or over-expression of HPAH-associated BMPR2 mutant constructs in PECs. While some studies were validated in IPAH patient-derived PECs52,69 or in vivo using mice with conditional deletion of Bmpr2 in endothelial cells,51,52 limitations of these models means that we cannot determine the functional significance of these BMPR2-dependent effects on obliterative vascular remodeling that is characteristic of HPAH.
Is metabolic reprogramming a common mediator of BMPR2 mutation effects in PECs?
An investigator reviewing the large number of cell signaling events that may promote PAH disease susceptibility and PEC dysfunction in mice or humans carrying heterozygous germline BMPR2 mutations might naturally ask whether there is a common mechanism mediating these effects or whether all of these effects could result from interference with the highly pleotropic actions of a single receptor. While there is no definitive answer to this question, we believe that BMPR2-dependent defects in cellular metabolism described in HPAH patients and in cultured PECs with reduced BMPR2 expression and/or signaling, could account for diverse BMPR2 mutation-dependent effects in PECs.
A number of studies have shown that there are abnormalities in cellular metabolism, including evidence of reduced mitochondrial oxidative phosphorylation and increased glycolytic flux in the right ventricle, pulmonary vasculature, and in skeletal muscle of patients with PAH and in a variety of experimental models of PH (reviewed in Sutendra and Michelakis70). Moreover, treatment with drugs that increase mitochondrial glucose oxidation, such as the pyruvate dehydrogenase kinase inhibitor, dichloroacetic acid, which leads to redirection of glucose metabolism away from lactate production and towards mitochondrial oxidative metabolism,71 also prevents PH and proliferative vascular remodeling in rat models of monocrotaline (MCT)-induced PH, and in the Fawn hood rat, a model of spontaneous PH.72–74 This led to the hypothesis that the proliferative vasculopathy in PAH results from metabolic reprogramming, reminiscent of the Warburg effect in cancer. In the classical Warburg effect, high levels of metabolic intermediates of glycolysis resulting from increased glycolytic flux, drive the anabolic reactions that are required for rapid cellular growth.75 As an example of this, there is evidence of increased glycolysis and reduced mitochondrial oxidative phosphorylation in isolated PECs from patients with IPAH, and in vivo, PET imaging data indicate that there is increased uptake of 18F-fluoro-deoxy glucose, indicative of increased glucose flux in lungs and right ventricles of patients with IPAH.76,77 HIF1α, which is a driver of the metabolic switch from mitochondrial glucose oxidation to glycolysis in cancer,75 may also drive metabolic reprogramming in IPAH PECs.78
RNA profiling and analysis of metabolic intermediates in PECs over-expressing PAH-associated BMPR2 mutations indicate that BMPR2 signaling abnormalities regulate widespread changes in cellular metabolism.79 These studies demonstrated not only increased glycolysis and reduced TCA cycle activity, but also upregulation of the pentose phosphate shunt, along with increased activity of pathways involved in glutamine and aspartate metabolism in PECs over-expressing BMPR2 mutant products. In addition, mitochondrial substrate use is switched from glucose to glutamine in these PECs, and the hyper-proliferative phenotype of PECs over-expressing HPAH-associated BMPR2 mutations is dependent on glutamine levels in the cell culture media.80 This effect is also seen in human PAH, in HIV-associated PH in monkeys, and in MCT-induced PH in rats, and inhibition of glutaminolysis using glutaminase inhibitors also prevents proliferative vascular remodeling in rat MCT PH.80,81 These metabolic changes are likely not only to affect cellular bioenergetics and growth, but may also influence diverse cellular functions and signaling responses. This may be of particular importance in endothelial cells which have relatively low mitochondrial content and depend largely of glycolysis rather than mitochondrial glucose oxidation to generate ATP.82 This suggests that the principal effects of alterations in mitochondrial metabolism in PECs is not to influence cellular bioenergetics, but rather to modify other cellular effects of mitochondria on PEC function. These include: (1) mitochondrial de-polarization associated with reduced mitochondrial oxidation which increases sensitivity to apoptosis;83 (2) increased calcium release resulting from mitochondrial depolarization which affects calcium-dependent changes in cell function (contraction, proliferation, and migration), and calcium-dependent signaling responses;83 (3) alterations in the cellular redox state resulting from reduced mitochondrial oxidative phosphorylation and shunting through the pentose phosphate pathway, both of which influence physiologic reactive oxygen species (ROS) signaling and ROS-dependent cellular pathophysiology;84,85 and (4) changes in the levels of metabolic intermediates which are altered by changes in the metabolic flux. These changes can have widespread effects on cell signaling and gene transcription through modifications in protein phosphorylation (which requires ATP) and acetylation (which requires acetyl-co-A and is modified by NAD-dependent deacetylases, Sirtuins, which are regulated by mitochondrial NAD/NADH levels), and gene regulation through histone acetylation (which requires acetyl-co-A) and methylation (histone demethylases are regulated by succinate/α-ketoglutarate levels).86,87
Taken together, these observations suggest that BMPR2-associated metabolic reprogramming could explain many of the observed effects of BMPR2 mutations on PEC function. However, it remains to be seen whether these changes actually account for the observed effects of BMPR2 signaling defects on PEC survival, inflammation, and oxidative stress, or whether these are sufficient to account for the marked obliterative vascular remodeling seen in BMPR2 mutation carriers with established HPAH. Furthermore, we are unaware of any published data in which the effects of HPAH-associated, heterozygous germline BMPR2 mutations have been evaluated on PEC metabolism. This is notable not only because of our findings that different types of BMPR2 mutation may have distinct effects of PEC function (as discussed above), but also because distinct effects on PEC metabolism have been described from BMPR2 siRNA knockdown studies, in which both basal aerobic glycolysis and mitochondrial glucose oxidation have been shown to be increased in PECs with reduced BMPR2 expression.52 This discrepancy between the effects of BMPR2 siRNA knockdown and BMPR2 mutation over-expression on PEC metabolism, while not unexpected given the differences between the model systems used, does suggest that there may be quantitative and/or qualitative differences in PEC metabolism depending on the degree and/or manner by which BMPR2 signaling is modified. This also suggests that different types of BMPR2 mutation may have distinct effects on PEC metabolism.
Summary
In this review, we have focused on the effects that heritable germ line mutations at the BMPR2 locus have on PEC function in patients with HPAH. We have discussed both clinical and experimental evidence for genotype–phenotype relationships between BMPR2 mutations and disease severity in HPAH, and how different mutations may have distinct effects on endothelial cell function. We reviewed evidence suggesting that BMPR2 signaling defects regulate a number of different cellular responses that may account for endothelial abnormalities in HPAH, but also discuss limitations of the in vitro and in vivo model systems that have been used to study these effects in the context of clinically meaningful endpoints. We also summarized published data indicating that there are widespread BMPR2-dependent effects on endothelial metabolism that could provide a unifying explanation for diverse BMPR2 mutation-dependent effects described in patients with HPAH. Future studies will need to focus on the development of more robust in vitro and in vivo experimental models that will enable us to explore the effects that different types of germline BMPR2 mutations have on endothelial cell function, whether these can all be attributed to effects on endothelial cell metabolism, and ultimately whether these effects are sufficient to account for the severe, obliterative pulmonary vascular remodeling that typically occurs in HPAH patients with established pulmonary vascular disease.
Acknowledgments
The authors thank the organizing committee and the American Thoracic Society for the opportunity to present this work to the 2017 Grover Conference. The authors also thank Josh Fessel for his helpful suggestions and comments in writing this review and James West for his assistance with performing mouse right heart catheterizations.
2017 Grover Conference Series
This review article is part of the 2017 Grover Conference Series. The American Thoracic Society and the conference organizing committee gratefully acknowledge the educational grants provided for the support of this conference by Actelion Pharmaceuticals US, Inc., Gilead Sciences, Inc., and United Therapeutics Corporation. Additionally, the American Thoracic Society is grateful for the support of the Grover Conference by the American Heart Association, the Cardiovascular Medical Research and Education Fund, the National Institutes of Health.
Conflict of interest
The author(s) declare that there is no conflict of interest.
Funding
Work from the de Caestecker laboratory described in this presentation was supported by R01 HL093057 and AHA 16GRNT31310011. AP was recipient of an AHA Pre-Doctoral Research Training Fellowship during 2012–2014 while working in the de Caestecker lab.
References
- 1.Runo JR, Loyd JE. Primary pulmonary hypertension. Lancet 2003; 361: 1533–1544. [DOI] [PubMed] [Google Scholar]
- 2.Deng Z, Morse JH, Slager SL, et al. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet 2000; 67: 737–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lane KB, Machado RD, et al. International PPH Consortium. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nat Genet 2000; 26: 81–84. [DOI] [PubMed] [Google Scholar]
- 4.Evans JD, Girerd B, Montani D, et al. BMPR2 mutations and survival in pulmonary arterial hypertension: an individual participant data meta-analysis. Lancet Respir Med 2016; 4: 129–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Machado RD, Southgate L, Eichstaedt CA, et al. Pulmonary arterial hypertension: a current perspective on established and emerging molecular genetic defects. Hum Mutat 2015; 36: 1113–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garcia-Rivas G, Jerjes-Sanchez C, Rodriguez D, et al. A systematic review of genetic mutations in pulmonary arterial hypertension. BMC Med Genet 2017; 18: 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Atkinson C, Stewart S, Upton PD, et al. Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation 2002; 105: 1672–1678. [DOI] [PubMed] [Google Scholar]
- 8.Alastalo TP, Li M, Perez Vde J, et al. Disruption of PPARgamma/beta-catenin-mediated regulation of apelin impairs BMP-induced mouse and human pulmonary arterial EC survival. J Clin Invest 2011; 121: 3735–3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morty RE, Nejman B, Kwapiszewska G, et al. Dysregulated bone morphogenetic protein signaling in monocrotaline-induced pulmonary arterial hypertension. Arterioscler Thromb Vasc Biol 2007; 27: 1072–1078. [DOI] [PubMed] [Google Scholar]
- 10.Takahashi H, Goto N, Kojima Y, et al. Downregulation of type II bone morphogenetic protein receptor in hypoxic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 2006; 290: L450–458. [DOI] [PubMed] [Google Scholar]
- 11.Long L, Crosby A, Yang X, et al. Altered bone morphogenetic protein and transforming growth factor-beta signaling in rat models of pulmonary hypertension: potential for activin receptor-like kinase-5 inhibition in prevention and progression of disease. Circulation 2009; 119: 566–576. [DOI] [PubMed] [Google Scholar]
- 12.Long L, Ormiston ML, Yang X, et al. Selective enhancement of endothelial BMPR-II with BMP9 reverses pulmonary arterial hypertension. Nat Med 2015; 21: 777–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Long L, Yang X, Southwood M, et al. Chloroquine prevents progression of experimental pulmonary hypertension via inhibition of autophagy and lysosomal bone morphogenetic protein type II receptor degradation. Circ Res 2013; 112: 1159–1170. [DOI] [PubMed] [Google Scholar]
- 14.Spiekerkoetter E, Tian X, Cai J, et al. FK506 activates BMPR2, rescues endothelial dysfunction, and reverses pulmonary hypertension. J Clin Invest 2013; 123: 3600–3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Orriols M, Gomez-Puerto MC, Ten Dijke P. BMP type II receptor as a therapeutic target in pulmonary arterial hypertension. Cell Mol Life Sci 2017; 74: 2979–2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boucherat O, Vitry G, Trinh I, et al. The cancer theory of pulmonary arterial hypertension. Pulm Circ 2017; 7: 285–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Voelkel NF, Gomez-Arroyo J, Abbate A, et al. Pathobiology of pulmonary arterial hypertension and right ventricular failure. Eur Respir J 2012; 40: 1555–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frump AL, Lowery JW, Hamid R, et al. Abnormal trafficking of endogenously expressed BMPR2 mutant allelic products in patients with heritable pulmonary arterial hypertension. PLoS One 2013; 8: e80319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hong KH, Lee YJ, Lee E, et al. Genetic ablation of the BMPR2 gene in pulmonary endothelium is sufficient to predispose to pulmonary arterial hypertension. Circulation 2008; 118: 722–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khajavi M, Inoue K, Lupski JR. Nonsense-mediated mRNA decay modulates clinical outcome of genetic disease. Eur J Hum Genet 2006; 14: 1074–1081. [DOI] [PubMed] [Google Scholar]
- 21.Girerd B, Montani D, Eyries M, et al. Absence of influence of gender and BMPR2 mutation type on clinical phenotypes of pulmonary arterial hypertension. Respir Res 2010; 11: 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Austin ED, Phillips JA, Cogan JD, et al. Truncating and missense BMPR2 mutations differentially affect the severity of heritable pulmonary arterial hypertension. Respir Res 2009; 10: 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Girerd B, Coulet F, Jais X, et al. Characteristics of pulmonary arterial hypertension in affected carriers of a mutation located in the cytoplasmic tail of bone morphogenetic protein receptor type 2. Chest 2015; 147: 1385–1394. [DOI] [PubMed] [Google Scholar]
- 24.Austin ED, Hamid R, Ahmad F. Somatic mutations in pulmonary arterial hypertension: primary or secondary events?. Am J Respir Crit Care Med 2010; 182: 1094–1096. [DOI] [PubMed] [Google Scholar]
- 25.Hamid R, Hedges LK, Austin E, et al. Transcripts from a novel BMPR2 termination mutation escape nonsense mediated decay by downstream translation re-initiation: implications for treating pulmonary hypertension. Clin Genet 2010; 77: 280–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perez-Ortin JE, Alepuz P, Chavez S, et al. Eukaryotic mRNA decay: methodologies, pathways, and links to other stages of gene expression. J Mol Biol 2013; 425: 3750–3775. [DOI] [PubMed] [Google Scholar]
- 27.Frump AL, Datta A, Ghose S, et al. Genotype-phenotype effects of Bmpr2 mutations on disease severity in mouse models of pulmonary hypertension. Pulm Circ 2016; 6: 597–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beppu H, Kawabata M, Hamamoto T, et al. BMP type II receptor is required for gastrulation and early development of mouse embryos. Dev Biol 2000; 221: 249–258. [DOI] [PubMed] [Google Scholar]
- 29.Delot EC, Bahamonde ME, Zhao M, et al. BMP signaling is required for septation of the outflow tract of the mammalian heart. Development 2003; 130: 209–220. [DOI] [PubMed] [Google Scholar]
- 30.Cogan JD, Pauciulo MW, Batchman AP, et al. High frequency of BMPR2 exonic deletions/duplications in familial pulmonary arterial hypertension. Am J Respir Crit Care Med 2006; 174: 590–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frank DB, Lowery J, Anderson L, et al. Increased susceptibility to hypoxic pulmonary hypertension in Bmpr2 mutant mice is associated with endothelial dysfunction in the pulmonary vasculature. Am J Physiol Lung Cell Mol Physiol 2008; 294: L98–109. [DOI] [PubMed] [Google Scholar]
- 32.Beppu H, Ichinose F, Kawai N, et al. BMPR-II heterozygous mice have mild pulmonary hypertension and an impaired pulmonary vascular remodeling response to prolonged hypoxia. Am J Physiol Lung Cell Mol Physiol 2004; 287: L1241–1247. [DOI] [PubMed] [Google Scholar]
- 33.Tada Y, Laudi S, Harral J, et al. Murine pulmonary response to chronic hypoxia is strain specific. Exp Lung Res 2008; 34: 313–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ciuclan L, Bonneau O, Hussey M, et al. A novel murine model of severe pulmonary arterial hypertension. Am J Respir Crit Care Med 2011; 184: 1171–1182. [DOI] [PubMed] [Google Scholar]
- 35.Lin MI, Fulton D, Babbitt R, et al. Phosphorylation of threonine 497 in endothelial nitric-oxide synthase coordinates the coupling of L-arginine metabolism to efficient nitric oxide production. J Biol Chem 2003; 278: 44719–44726. [DOI] [PubMed] [Google Scholar]
- 36.Parat MO. The biology of caveolae: achievements and perspectives. Int Rev Cell Mol Biol 2009; 273: 117–162. [DOI] [PubMed] [Google Scholar]
- 37.Prewitt AR, Ghose S, Frump AL, et al. Heterozygous null bone morphogenetic protein receptor type 2 mutations promote SRC kinase-dependent caveolar trafficking defects and endothelial dysfunction in pulmonary arterial hypertension. J Biol Chem 2015; 290: 960–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hu G, Minshall RD. Regulation of transendothelial permeability by Src kinase. Microvasc Res 2009; 77: 21–25. [DOI] [PubMed] [Google Scholar]
- 39.Courboulin A, Paulin R, Giguere NJ, et al. Role for miR-204 in human pulmonary arterial hypertension. J Exp Med 2011; 208: 535–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Paulin R, Meloche J, Jacob MH, et al. Dehydroepiandrosterone inhibits the Src/STAT3 constitutive activation in pulmonary arterial hypertension. Am J Physiol Heart Circ Physiol 2011; 301: H1798–1809. [DOI] [PubMed] [Google Scholar]
- 41.Fagan KA, Fouty BW, Tyler RC, et al. The pulmonary circulation of homozygous or heterozygous eNOS-null mice is hyperresponsive to mild hypoxia. J Clin Invest 1999; 103: 291–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Quinlan TR, Li D, Laubach VE, et al. eNOS-deficient mice show reduced pulmonary vascular proliferation and remodeling to chronic hypoxia. Am J Physiol Lung Cell Mol Physiol 2000; 279: L641–650. [DOI] [PubMed] [Google Scholar]
- 43.Long L, MacLean MR, Jeffery TK, et al. Serotonin increases susceptibility to pulmonary hypertension in BMPR2-deficient mice. Circ Res 2006; 98: 818–827. [DOI] [PubMed] [Google Scholar]
- 44.Song Y, Coleman L, Shi J, et al. Inflammation, endothelial injury, and persistent pulmonary hypertension in heterozygous BMPR2-mutant mice. Am J Physiol Heart Circ Physiol 2008; 295: H677–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Song Y, Jones JE, Beppu H, et al. Increased susceptibility to pulmonary hypertension in heterozygous BMPR2-mutant mice. Circulation 2005; 112: 553–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gomez-Arroyo J, Saleem SJ, Mizuno S, et al. A brief overview of mouse models of pulmonary arterial hypertension: problems and prospects. Am J Physiol Lung Cell Mol Physiol 2012; 302: L977–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Colvin KL, Yeager ME. Animal models of pulmonary hypertension: matching disease mechanisms to etiology of the human disease. J Pulm Respir Med 2014; 4: 198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hautefort A, Rucker-Martin C, Capuano V, et al. Heterozygote Bmpr2 mutatiuon in the rat: a new strategy to understand PAH pathobiology. Am J Respir Crit Care Med 2016; 193: A3876. [Google Scholar]
- 49.Ranchoux B, Antigny F, Rucker-Martin C, et al. Endothelial-to-mesenchymal transition in pulmonary hypertension. Circulation 2015; 131: 1006–1018. [DOI] [PubMed] [Google Scholar]
- 50.Burton VJ, Ciuclan LI, Holmes AM, et al. Bone morphogenetic protein receptor II regulates pulmonary artery endothelial cell barrier function. Blood 2011; 117: 333–341. [DOI] [PubMed] [Google Scholar]
- 51.Burton VJ, Holmes AM, Ciuclan LI, et al. Attenuation of leukocyte recruitment via CXCR1/2 inhibition stops the progression of PAH in mice with genetic ablation of endothelial BMPR-II. Blood 2011; 118: 4750–4758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Diebold I, Hennigs JK, Miyagawa K, et al. BMPR2 preserves mitochondrial function and DNA during reoxygenation to promote endothelial cell survival and reverse pulmonary hypertension. Cell Metab 2015; 21: 596–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Machado RD, James V, Southwood M, et al. Investigation of second genetic hits at the BMPR2 locus as a modulator of disease progression in familial pulmonary arterial hypertension. Circulation 2005; 111: 607–613. [DOI] [PubMed] [Google Scholar]
- 54.Bryant AJ, Robinson LJ, Moore CS, et al. Expression of mutant bone morphogenetic protein receptor II worsens pulmonary hypertension secondary to pulmonary fibrosis. Pulm Circ 2015; 5: 681–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fessel JP, Chen X, Frump A, et al. Interaction between bone morphogenetic protein receptor type 2 and estrogenic compounds in pulmonary arterial hypertension. Pulm Circ 2013; 3: 564–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fessel JP, Flynn CR, Robinson LJ, et al. Hyperoxia synergizes with mutant bone morphogenic protein receptor 2 to cause metabolic stress, oxidant injury, and pulmonary hypertension. Am J Respir Cell Mol Biol 2013; 49: 778–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Majka S, Hagen M, Blackwell T, et al. Physiologic and molecular consequences of endothelial Bmpr2 mutation. Respir Res 2011; 12: 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Trammell AW, Talati M, Blackwell TR, et al. Pulmonary vascular effect of insulin in a rodent model of pulmonary arterial hypertension. Pulm Circ 2017; 7: 624–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.West J, Fagan K, Steudel W, et al. Pulmonary hypertension in transgenic mice expressing a dominant-negative BMPRII gene in smooth muscle. Circ Res 2004; 94: 1109–1114. [DOI] [PubMed] [Google Scholar]
- 60.West J, Harral J, Lane K, et al. Mice expressing BMPR2R899X transgene in smooth muscle develop pulmonary vascular lesions. Am J Physiol Lung Cell Mol Physiol 2008; 295: L744–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Taraseviciene-Stewart L, Kasahara Y, Alger L, et al. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J 2001; 15: 427–438. [DOI] [PubMed] [Google Scholar]
- 62.Abe K, Toba M, Alzoubi A, et al. Formation of plexiform lesions in experimental severe pulmonary arterial hypertension. Circulation 2010; 121: 2747–2754. [DOI] [PubMed] [Google Scholar]
- 63.Toba M, Alzoubi A, O'Neill KD, et al. Temporal hemodynamic and histological progression in Sugen5416/hypoxia/normoxia-exposed pulmonary arterial hypertensive rats. Am J Physiol Heart Circ Physiol 2014; 306: H243–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Happe CM, de Raaf MA, Rol N, et al. Pneumonectomy combined with SU5416 induces severe pulmonary hypertension in rats. Am J Physiol Lung Cell Mol Physiol 2016; 310: L1088–1097. [DOI] [PubMed] [Google Scholar]
- 65.Voelkel NF, Cool C, Taraceviene-Stewart L, et al. Janus face of vascular endothelial growth factor: the obligatory survival factor for lung vascular endothelium controls precapillary artery remodeling in severe pulmonary hypertension. Crit Care Med 2002; 30: S251–256. [DOI] [PubMed] [Google Scholar]
- 66.Teichert-Kuliszewska K, Kutryk MJ, Kuliszewski MA, et al. Bone morphogenetic protein receptor-2 signaling promotes pulmonary arterial endothelial cell survival: implications for loss-of-function mutations in the pathogenesis of pulmonary hypertension. Circ Res 2006; 98: 209–217. [DOI] [PubMed] [Google Scholar]
- 67.Li M, Vattulainen S, Aho J, et al. Loss of bone morphogenetic protein receptor 2 is associated with abnormal DNA repair in pulmonary arterial hypertension. Am J Respir Cell Mol Biol 2014; 50: 1118–1128. [DOI] [PubMed] [Google Scholar]
- 68.Hopper RK, Moonen JR, Diebold I, et al. In pulmonary arterial hypertension, reduced BMPR2 promotes endothelial-to-mesenchymal transition via HMGA1 and its target slug. Circulation 2016; 133: 1783–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sawada H, Saito T, Nickel NP, et al. Reduced BMPR2 expression induces GM-CSF translation and macrophage recruitment in humans and mice to exacerbate pulmonary hypertension. J Exp Med 2014; 211: 263–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sutendra G, Michelakis ED. The metabolic basis of pulmonary arterial hypertension. Cell Metab 2014; 19: 558–573. [DOI] [PubMed] [Google Scholar]
- 71.Kankotia S, Stacpoole PW. Dichloroacetate and cancer: new home for an orphan drug?. Biochim Biophys Acta 2014; 1846: 617–629. [DOI] [PubMed] [Google Scholar]
- 72.McMurtry MS, Bonnet S, Wu X, et al. Dichloroacetate prevents and reverses pulmonary hypertension by inducing pulmonary artery smooth muscle cell apoptosis. Circ Res 2004; 95: 830–840. [DOI] [PubMed] [Google Scholar]
- 73.Li B, Yan J, Shen Y, et al. Dichloroacetate prevents but not reverses the formation of neointimal lesions in a rat model of severe pulmonary arterial hypertension. Mol Med Rep 2014; 10: 2144–2152. [DOI] [PubMed] [Google Scholar]
- 74.Bonnet S, Michelakis ED, Porter CJ, et al. An abnormal mitochondrial-hypoxia inducible factor-1alpha-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: similarities to human pulmonary arterial hypertension. Circulation 2006; 113: 2630–2641. [DOI] [PubMed] [Google Scholar]
- 75.Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol 2011; 27: 441–464. [DOI] [PubMed] [Google Scholar]
- 76.Xu W, Koeck T, Lara AR, et al. Alterations of cellular bioenergetics in pulmonary artery endothelial cells. Proc Natl Acad Sci U S A 2007; 104: 1342–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lundgrin EL, Park MM, Sharp J, et al. Fasting 2-deoxy-2-[18F]fluoro-D-glucose positron emission tomography to detect metabolic changes in pulmonary arterial hypertension hearts over 1 year. Ann Am Thorac Soc 2013; 10: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fijalkowska I, Xu W, Comhair SA, et al. Hypoxia inducible-factor1alpha regulates the metabolic shift of pulmonary hypertensive endothelial cells. Am J Pathol 2010; 176: 1130–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fessel JP, Hamid R, Wittmann BM, et al. Metabolomic analysis of bone morphogenetic protein receptor type 2 mutations in human pulmonary endothelium reveals widespread metabolic reprogramming. Pulm Circ 2012; 2: 201–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Egnatchik RA, Brittain EL, Shah AT, et al. Dysfunctional BMPR2 signaling drives an abnormal endothelial requirement for glutamine in pulmonary arterial hypertension. Pulm Circ 2017; 7: 186–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bertero T, Oldham WM, Cottrill KA, et al. Vascular stiffness mechanoactivates YAP/TAZ-dependent glutaminolysis to drive pulmonary hypertension. J Clin Invest 2016; 126: 3313–3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Eelen G, de Zeeuw P, Simons M, et al. Endothelial cell metabolism in normal and diseased vasculature. Circ Res 2015; 116: 1231–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Marcu R, Zheng Y, Hawkins BJ. Mitochondria and Angiogenesis. Adv Exp Med Biol 2017; 982: 371–406. [DOI] [PubMed] [Google Scholar]
- 84.Kanaan GN, Harper ME. Cellular redox dysfunction in the development of cardiovascular diseases. Biochim Biophys Acta 2017; 1861: 2822–2829. [DOI] [PubMed] [Google Scholar]
- 85.D'Alessandro A, El Kasmi KC, Plecita-Hlavata L, et al. Hallmarks of pulmonary hypertension: mesenchymal and inflammatory cell metabolic reprogramming. Antioxid Redox Signal 2018; 28: 230–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Matilainen O, Quiros PM, Auwerx J. Mitochondria and epigenetics - crosstalk in homeostasis and stress. Trends Cell Biol 2017; 27: 453–463. [DOI] [PubMed] [Google Scholar]
- 87.Stram AR, Payne RM. Post-translational modifications in mitochondria: protein signaling in the powerhouse. Cell Mol Life Sci 2016; 73: 4063–4073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Anderson L, Lowery JW, Frank DB, et al. Bmp2 and Bmp4 exert opposing effects in hypoxic pulmonary hypertension. Am J Physiol Regul Integr Comp Physiol 2010; 298: R833–842. [DOI] [PMC free article] [PubMed] [Google Scholar]