

Abstract
Experimental models of sepsis in small and large animals and a variety of in vitro preparations have established several basic mechanisms that drive endothelial injury. This review is focused on what can be learned from the results of clinical studies of plasma biomarkers of endothelial injury and inflammation in patients with sepsis. There is excellent evidence that elevated plasma levels of several biomarkers of endothelial injury, including von Willebrand factor antigen (VWF), angiopoietin-2 (Ang-2), and soluble fms-like tyrosine kinase 1 (sFLT-1), and biomarkers of inflammation, especially interleukin-8 (IL-8) and soluble tumor necrosis factor receptor (sTNFr), identify sepsis patients with a higher mortality. There are also some data that elevated levels of endothelial biomarkers can identify which patients with non-pulmonary sepsis will develop acute respiratory distress syndrome (ARDS). If ARDS patients are divided among those with indirect versus direct lung injury, then there is an association of elevated levels of endothelial biomarkers in indirect injury and markers of inflammation and alveolar epithelial injury in patients with direct lung injury. New research suggests that the combination of biologic and clinical markers may make it possible to segregate patients with ARDS into hypo- versus hyper-inflammatory phenotypes that may have implications for therapeutic responses to fluid therapy. Taken together, the studies reviewed here support a primary role of the microcirculation in the pathogenesis and prognosis of ARDS after sepsis. Biological differences identified by molecular patterns could explain heterogeneity of treatment effects that are not explained by clinical factors alone.
Keywords: direct and indirect lung injury, acute lung injury (ALI)
Sepsis and acute respiratory distress syndrome (ARDS) are heterogeneous syndromes associated with high mortality rates and substantial healthcare costs in critically ill patients.1–4 Sepsis is defined as life-threatening organ dysfunction due to a dysregulated host response to infection.5 The clinical manifestations of severe sepsis syndrome involve organ dysfunction including encephalopathy, acute kidney injury, coagulopathy, and acute lung injury (ALI) or ARDS.
The initial site of injury in ARDS after sepsis or other etiologies of indirect lung injury is the injured lung microcirculation that leads to increased permeability pulmonary edema resulting in bilateral alveolar infiltrates on the chest radiograph and arterial hypoxemia.6,7 Considerable progress has been achieved by studying ARDS using broad clinical and physiologic criteria as illustrated in the Berlin Criteria for ARDS and the approach taken by the NHLBI ARDS network in testing therapies by the broad definition of bilateral pulmonary infiltrates and a Pa02/Fi02 < 300 mmHg. However, the failure of pharmacologic therapies in ARDS highlights the importance of recognizing the heterogeneity of ARDS and finding approaches to classify ARDS patients in categories or phenotypes that might be responsive to specific therapeutic approaches that may not be effective in all ARDS patients. Thus, in keeping with the goal of personalized medicine, there is a high priority for matching the optimal treatment to the appropriate patients by identifying meaningful subtypes within a heterogeneous population of critically ill patients.8 For targeted therapies, “splitting” study populations into endotypes on the basis of differences in biology—by biomarker concentrations, gene expression profile, or genotype—may enhance our ability to identify effective treatments.
Several clinical disorders can precipitate ARDS, including pulmonary or non-pulmonary sepsis, pneumonia, and aspiration of gastric contents. While there are several important clinical and demographic risk factors for both sepsis9 and ARDS,10–12 this review will focus on the biological mechanisms involving the microcirculation in the development of ALI after sepsis in ARDS patients. We will discuss recently published biomarker studies to highlight what is known about sepsis-induced endothelial injury in humans and then describe the relationship of endothelial injury in sepsis to ARDS with an emphasis on human studies.
Identifying cases of sepsis for clinical and research purposes is challenging and the approach has changed over time.13 In this article, we discuss studies that have used various definitions of sepsis contemporary to the date of their publication. Similarly, we will use the terms ALI and ARDS interchangeably and discuss studies that identified this outcome by a variety of evolving criteria.6
Endothelial injury from human sepsis
The endothelial lining is in continuous contact with circulating cells and soluble proteins (Fig. 1). The importance of the vascular endothelium in the pathophysiology of sepsis is well established and is regulated by pathways that control vascular permeability, coagulation, and systemic inflammation (Table 1).14–16 Severe sepsis causes an upregulation of several pro-inflammatory adhesion molecules, release of pro-inflammatory mediators including cytokines and lipid products, and is accompanied by procoagulant factors, activated neutrophils, platelets, and monocytes, all of which disrupt the barrier function of the microcirculation and also result in impaired tissue perfusion and oxygenation.16,17 Figure 1 highlights some of these pathways of injury. We describe the findings from three recent publications of prognostic biomarker studies in large cohorts of patients with sepsis to frame what is currently known about the importance of the vascular endothelium in predicting clinical outcomes after sepsis. In the next section, we will discuss the role of endothelial injury in the pathogenesis and prognosis of ARDS after sepsis.
Fig. 1.

The pathways by which sepsis induces injury to the endothelium. Sepsis upregulates the expression of selectins on the endothelium (P- and E-selectins), to which activated leukocytes (both neutrophils and monocytes) and platelet aggregates can adhere, and induce an increase in endothelial permeability. The potential role of neutrophil extracellular traps (NETS) and histone release is also included as well as olfactomedin 4, lipocalin 2, and CD 24, and bacterial permeability increasing protein, products primarily of neutrophils. Some circulating factors in the plasma are both biomarkers of injury and also enhance the injury, including Ang-2 and VEGF. In addition, the diagram shows circulating factors that enhance inflammation such as IL-8 and IL-6, sTNFr-2. Markers of endothelial injury also include vWF and sFLT-1, the circulating VEGF receptor. Also noted in the diagram are components of the activated protein C complex including protein C, protein S, factor V, and thrombomodulin because sepsis deranges the normal function of activated protein C leading to a pro-coagulant environment.
Table 1.
Selected clinical studies of endothelial and inflammatory biomarkers in sepsis.
| Biomarker | References |
|---|---|
| Vascular permeability | |
| Angiopoietin-2 (Ang-2) | 19,53–56 |
| von Willebrand Factor (vWF) | 19,29,43,45,57 |
| Vascular endothelial growth factor (VEGF) | 19,50,58,59 |
| Soluble FMS-like tyrosine kinase-1 (sFlt-1) | 19,60 |
| Coagulation | |
| Thrombomodulin (TM) | 19,61 |
| Plasminogen activator inhibitor-1 (PAI-1) | 60,62–64 |
| P-selectin | 65 |
| E-selectin | 15,64,65 |
| Protein C | 63,64,66,67 |
| Inflammation | |
| Interleukin-6 (IL-6) | 20,56,68,69 |
| Interleukin-8 (IL-8) | 23,56,69,70 |
| Soluble tumor necrosis factor receptor-1 (sTNFR-1) | 23,56 |
Two recently published studies leveraged clinical data and biospecimens from the Protocolized Care for Early Septic Shock (ProCESS) trial18 to test the prognostic value of biomarkers representing various domains of sepsis pathophysiology. The ProCESS trial was a multi-center randomized control trial (RCT) that enrolled patients with sepsis early in the course of illness, while they were still in the Emergency Department. The size of the trial and the study design targeting intervention for early sepsis lends strength to the two biology studies described below.
In 2017, Hou et al. reported results from a large biomarker study of 1341 individuals enrolled in the ProCESS trial split into derivation and validation subgroups.19 The authors measured baseline plasma levels of several proteins associated with endothelial cell permeability and hemostasis and tested the associations with mortality. The baseline values of several markers of endothelial injury were higher in patients who died compared to those who survived, including angiopoietin-2 (Ang-2), soluble fms-like tyrosine kinase 1 (sFLT-1; the soluble vascular endothelial growth factor [VEGF] receptor), thrombomodulin (TM), and von Willebrand factor (vWF). The authors analyzed the prognostic value of a panel of multiple biomarkers to identify patients with septic shock who had an increased risk of death. Lactate is a biomarker indicating impaired oxygen delivery to tissues and is used in clinical practice to identify high-risk patients with sepsis and to guide fluid resuscitation. Sequential Organ Failure Assessment (SOFA) score predicts mortality among patients with suspected infections and is commonly used as a marker of severity of illness in clinical research.5 Interestingly, sFLT-1 had a point estimate for mortality discrimination (derivation area under the curve [AUC] = 0.74; validation = 0.70), similar to lactate (AUC = 0.74) and SOFA score (AUC = 0.73). Models testing if sFLT-1 added prognostic value to multivariate models incorporating lactate or SOFA score were not reported. The plasma levels of the biomarkers were not affected by the resuscitation strategies tested in the ProCESS trial. The resuscitation treatment protocols may not have altered mortality because they did not have a direct effect on altering the fundamental pathways of endothelial injury, as reflected in part by the levels of the circulating plasma biomarkers. Because several of the markers were associated with higher mortality, the results support the importance of endothelial injury in the pathogenesis of poor clinical outcomes in sepsis and suggest that measuring sFLT-1 in patients with sepsis may help identify those patients at the highest risk for death. However, based on the data in this study, it is not clear if sFLT-1 would add prognostic value beyond lactate and SOFA score.
Findings from a separate biomarker study conducted in 628 of the participants enrolled in the ProCESS trial were reported by Kellum et al. in 2017.20 In this study, the authors made serial measurements (baseline, 6, 24, and 72 hours after initiation of study treatment) of plasma and urine biomarkers representative of four pathophysiologic domains: inflammation, coagulation, oxidative stress, and tissue hypoxia. For each domain, a primary marker and up to two secondary markers were measured as follows: inflammation: interleukin (IL)-6 (primary), tumor necrosis factor (TNF), and IL-10; coagulation: thrombin-antithrombin complex (TAT) (primary) and D-dimers; oxidative stress: urine isoprostane; and tissue hypoxia: lactate. The authors tested the association of the biomarkers with mortality and also used linear mixed effects models to determine if the trajectory of the biomarker measurement differed by treatment protocol. As in the study by Hou et al.,19 all of the biomarkers were higher at baseline among those who died compared to survivors. Despite these associations, none of the biomarkers alone, or models with combinations of biomarkers, discriminated well for mortality. Although lactate was the best-performing biomarker, when added to a parsimonious regression model using clinical variables, it did improve the area under the receiver operating characteristic (ROC) curve for predicting death among patients with sepsis. Similar to the findings reported by Hou et al., the biomarker profiles over time were not significantly different among the three resuscitation treatment arms of the ProCESS trial. The authors analyzed treatment effect on mortality among quartiles of the baseline biomarker measurements. Contrary to their hypothesis, the authors found an interaction between treatment effect and baseline IL-6 and IL-10 and mortality, suggesting that protocol-based resuscitation benefited patients with less-inflammation. In other words, among individuals with lower baseline inflammatory biomarkers, the group that received more intravenous fluids, had a lower mortality rate than individuals who received less intravenous fluids. A similar interaction was found in a secondary analysis of data from patients with ARDS enrolled in the Fluid and Catheter Treatment Trial (FACCT).21 Famous et al. found that patients with a subphenotype characterized by lower inflammatory markers had a differential response to fluid therapy. Mortality in this group with less inflammation was 26% with fluid-conservative strategy versus 18% with fluid-liberal strategy.22 Taken together, these findings from ProCESS and FACCT suggest that defining a biologic profile using markers of inflammation early in the course of critical illness could guide clinical decision-making. While the mechanisms underpinning these observations are not clear, both studies suggest that a more liberal fluid resuscitation strategy may benefit critically ill patients with less inflammation.
Taken together, the findings from these two biomarker studies of patients enrolled in the ProCESS trial indicate that patients with sepsis who develop more severe disturbances in any of the domains studied—inflammation, coagulation, endothelial permeability, oxidative stress, and tissue hypoxia—have a worse prognosis and higher mortality rates. Thus, these two recent clinical studies confirm experimental data that have identified the severity of endothelial injury and inflammation as major mechanisms driving the severity of organ injury. However, these studies find that the levels of plasma biomarkers did not add substantial prognostic value beyond models that incorporate clinical risk factors and plasma lactate.
Mikacenic et al. recently published results from a study with a large derivation and internal and external validation cohorts describing a two-biomarker model that identifies patients at low risk for death and organ dysfunction after sepsis.23 The authors studied patients admitted to the ICU in academic institutions enrolled in prospective cohort studies of patients with systemic inflammatory response syndrome (SIRS), most of whom had sepsis. The derivation and internal validation cohort enrolled participants between 2006 and 2010 and obtained plasma samples within 24 h of ICU admission. The external validation cohort enrolled participants between 1999 and 2010 and obtained plasma samples within 48 h of ICU admission. The investigators tested the value of a panel of biomarkers of inflammation, microvascular injury, and apoptosis along with a panel of clinical predictors and developed a model to predict mortality. Specifically, the authors measured several plasma biomarkers including interleukin-8 (IL-8) and soluble tumor necrosis factor receptor 1 (sTNFR-1) within 24 h of ICU admission in the derivation and internal validation cohort. The authors analyzed the data using least absolute shrinkage and selection operator (LASSO) regression, a method that identifies a subset of predictors and enhances prediction accuracy. The LASSO analysis method eliminated clinical predictors from the model and the final model included only the two plasma biomarkers: IL-8 and sTNFR-1. Low levels of these biomarkers had a high negative predictive value for 28-day mortality and persistent organ dysfunction. There were similar results when the cohorts were restricted to patients meeting criteria for Sepsis-2 and Sepsis-3.24 This two-biomarker model did not include clinical variables but performed similarly to APACHE III (a multivariable measure of severity of critical illness) and was superior to the day 0 SOFA score in predicting 28-day mortality. The authors also measured plasma concentrations of Ang-1, Ang-2, IL-6, soluble vascular cell adhesion molecule-1, granulocyte-colony stimulating factor, and soluble Fas. The authors tested models that included all of these biomarkers, as well as clinical variables in the model development and testing process, but ultimately found that a simple model including only IL-8 and sTNFR-1 performed best. The study was limited by a very low mortality rate of 9–13% but does establish the value of potentially using levels of two biomarkers for a negative predictive value of 28-day mortality and persistent organ dysfunction.
The strengths of these three recently published studies include large sample sizes of heterogenous populations of critically ill patients. The studies by Hou et al.19 and Mikacenic et al.23 used validation cohorts to test the clinical prediction models they developed. The secondary studies of the ProCESS trial data are strengthened by the enrollment of patients in the Emergency Department, early in the course of sepsis, limiting confounding by treatment interventions and strengthening the value of these biomarkers for the purposes of cohort enrichment and triage decisions.25 The Emergency Department-based studies stand in contrast to the Mikacenic et al. study which measured plasma biomarkers obtained up to 24 h and 48 h after ICU admission in both the internal and external cohorts. Another limitation of the Mikacenic et al. study is the enrollment of patients from nearly 20 years ago (1999), raising some concerns of confounding of clinical outcomes by temporal trends in sepsis treatment, including changes in clinical practice spurred by the landmark study of Early Goal Directed Therapy by Rivers et al.26 and the Surviving Sepsis Campaign. Another important difference in the Mikacenic et al. study is the application of the model to identify patients with a low risk of mortality. A model that identifies patients at low risk of death could be useful in clinical triage decisions if the biomarkers are measured very early in the course of illness using a point of care assay with immediately available results. Lactate and sFLT-1 seem to be the most promising early biomarkers that predict mortality after sepsis. Whether measurements of IL-8 and sTNFR-1 discriminate well for mortality after sepsis remains to be further validated. To answer this question, these biomarkers could be tested in a study design that enrolls patients with contemporary standard of care treatment for sepsis with plasma samples obtained earlier in the course of illness. However, the association of lower plasma levels of IL-8 and sTNFR-1 with survival reported by Mikacenic et al. and the interaction between treatment effect and baseline IL-6 an IL-10 on mortality reported by Kellum et al. raise the possibility that treatment effects in sepsis could differ among patients with higher and lower inflammatory profiles. Findings from secondary analyses of large RCTs identified a hyperinflammatory subphenotype of ARDS patients, characterized in part by differences in plasma levels of IL-6, IL-8, and sTNFR-1. In these studies, there was a differential response to treatment with protocols of positive end-expiratory pressure (PEEP) and fluid balance strategies.22,27 Future studies could test differential treatment effects among patients with sepsis who present with different molecular phenotypes providing that point-of-care measurements are available for measurement of key biomarkers.
Endothelial injury from sepsis and the development of ARDS
These three recent studies of mortality in patients with sepsis evaluated the prognostic value of biomarkers from several pathophysiologic domains related to the microvascular circulation: inflammation, vascular permeability, coagulation, oxidative stress, and tissue hypoxia. In particular, markers of inflammation (IL-6, IL-8, IL-10, and sTNFR-1), vascular permeability (sFLT-1), and tissue hypoxia (lactate) showed promise in identifying the risk of death and possibly a differential response to fluid resuscitation therapy. In addition to conferring a high risk of death, sepsis is associated with a variety of organ dysfunctions, including acute kidney injury, cardiovascular failure (shock), coagulation abnormalities including thrombocytopenia, altered mental status, and also lung injury and ARDS. We will now turn our attention to the relationship between the vascular endothelium and ARDS after sepsis.
ARDS is a heterogeneous syndrome characterized by impaired oxygenation and bilateral infiltrates on chest radiograph.28 A variety of clinical risk factors have been associated with the development of ARDS and fall into two broad categories: direct and indirect lung injury (Fig. 2). Common causes of direct lung injury are pneumonia and aspiration of gastric contents. Sepsis is the most common cause of indirect lung injury. Together, pneumonia, aspiration, and sepsis account for 85% of ARDS cases enrolled in recent clinical trials.28 Because there appear to be different biological profiles between patients with direct and indirect mechanisms of lung injury, these classifications are useful for research purposes.29,30 However, in clinical practice these entities are not always distinct. A patient with lobar pneumonia, a direct lung injury, may subsequently develop sepsis. In this clinical presentation, pulmonary infection first causes local injury of the lung epithelium and lung endothelium, the injury progresses to generalized systemic illness manifesting as sepsis, followed by more diffuse, bilateral lung endothelial injury resulting in respiratory failure and ARDS, and often other organ injury (Fig. 3). The early, exudative phase of ARDS is characterized by damage to the alveolar epithelium and endothelium leading to the accumulation of protein-rich edema fluid within the pulmonary interstitium and alveoli. When evaluating studies of pathogenesis and prognosis in ARDS, we find it useful to refer to a conceptual framework that distinguishes endothelial from epithelial lung injury and direct from indirect causes of lung injury. In this article, we distinguish what is known about the role of the vascular endothelium in ARDS caused by sepsis, a form of indirect lung injury, from other etiologies of ARDS. In clinical research, the distinction between pulmonary and non-pulmonary sepsis can be a useful extension of the direct and indirect lung injury framework discussed above.
Fig. 2.
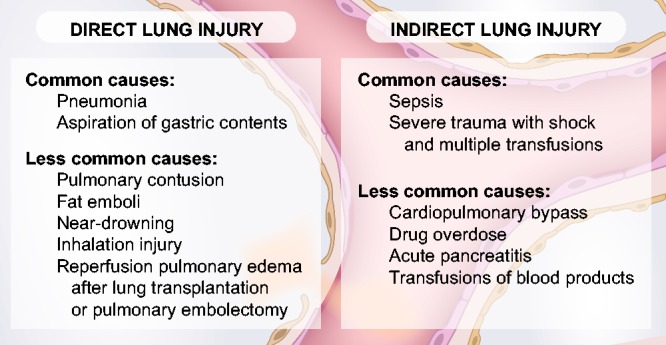
The common and less common causes of ARDS sepsis segregated into direct and indirect causes, including sepsis.
Fig. 3.

Three chest radiographs showing progression of lobar pneumonia to ARDS in a 55-year-old man. (a) The patient presented with lobar pneumonia (blood culture positive for S. pneumoniae) with consolidation of the right lower and middle lobes. (b) Radiograph now shows that the consolidation has progressed to involve more of the air spaces in the right lung and the patient has required endotracheal intubation (see turquoise arrows) because of progressive arterial hypoxemia, tachypnea with a respiratory rate of 40 per minute, and a high work of breathing. (c) Patient’s chest radiograph now shows bilateral pulmonary infiltrates consistent with ARDS. The Pa02/Fi02 is 120 mmHg. A central venous catheter has also been inserted (yellow arrow) through the right internal jugular vein to infuse vasopressor treatment with norepinephrine as the patient has developed shock unresponsive to 3 L intravenous Ringer’s lactate. In addition to ARDS, the patient has acute kidney injury from sepsis with oliguria and a serum creatinine that has risen to 3.1 mg/dL.
In vitro studies, animal models, and translational research have elucidated important mechanisms involving alterations in the permeability of vascular endothelium and interactions between the endothelium and the innate immune system in the pathogenesis of ARDS after sepsis. Translational studies identified a key role of circulating Ang-2 in pulmonary vascular leak and disruption of the endothelial architecture in sepsis.31 In animal models, extracellular histones have been implicated in sepsis pathophysiology and organ dysfunction in sepsis.32 Xu et al. found that mice challenged with a sub-lethal dose of histones showed characteristics of mice with sepsis, including neutrophil accumulation in the alveolar microvessels, vacuolated endothelial cells, and platelet- and fibrin-rich microthrombi in pulmonary circulation. Alterations in vascular permeability in ARDS pathogenesis after sepsis usually require the interaction of neutrophils and platelets with the microvasculature.7,17 Histopathology and in vivo microscopy studies support the key role of the interaction of platelets and neutrophils in mouse models of ALI after LPS administration and cecal ligation and puncture-induced sepsis. These mechanisms include the formation of neutrophil extracellular traps (NETS) and neutrophil platelet aggregation in the pulmonary microcirculation.33,34
Most of the evidence supporting the importance of the innate immune system in sepsis-related ARDS derives from animal models and translational studies using human-based samples in animal and in vitro experiments to assess changes in vascular permeability.35–39 These mechanisms of lung injury are challenging to study in humans because they require more complex measurements to detect cell–cell interactions such as flow cytometry and lung intravital microscopy.40,41 Recent gene-expression studies in human participants suggest that several neutrophil-related pathways may be involved in the early pathogenesis of sepsis-related ARDS.42 In a case control study, gene expression study of blood samples obtained within 24 h of ICU admission comparing 29 patients with sepsis and ARDS to 28 patients with sepsis alone, there was early differential gene expression of mediators related to the initial neutrophil response to infection: olfactomedin 4, lipocalin 2, CD24, and bactericidal/permeability increasing protein.42 These gene expression differences were robust to adjustment for several potential confounders, including mechanisms for direct lung injury, pneumonia, and aspiration.
When summarizing the literature describing the pathogenesis of ARDS after sepsis in humans, it is important to bear in mind that many studies do not distinguish subpopulations of patients with ARDS after sepsis from ARDS from other causes of lung injury. Additional heterogeneity arises from study designs that measure biomarkers at different time points in the course of illness. Furthermore, the standard of care treatment for both ARDS and sepsis has changed over time and may alter the pattern of biomarker expression and the risk of developing ARDS after sepsis.
Circulating vWF is predominantly released by vascular endothelial cells and to a lesser extent by megakaryocytes and platelets. vWF can regulate vascular permeability and has been measured in several studies of patients at risk for ARDS, some of whom have sepsis. Results from a prospective cohort study of vWF in non-pulmonary sepsis were published in 1990 by Rubin et al.43 Plasma vWF was measured in 45 patients with non-pulmonary sepsis without evidence of lung injury. In this cohort, 15 individuals (33%) subsequently developed lung injury. Among those who developed lung injury, plasma vWF was significantly higher than in patients who progressed to develop ARDS. Notably, there was a significantly higher mortality rate in the group that developed lung injury (93% vs. 30%). The authors concluded that in patients with non-pulmonary sepsis at risk for lung injury, elevated plasma vWf is a useful, early biomarker of endothelial injury and it has both predictive and prognostic value. In a later study, published in 1995, Moss et al. measured vWF at 6-h intervals over 48 h in a prospective cohort study of 96 individuals at risk for ARDS and 30 of these participants had sepsis.44 Plasma for these measurements was obtained within 6 h of developing an ARDS risk factor. Of the 30 patients with sepsis, ten developed ARDS. The study did not consider non-pulmonary sepsis separately from pulmonary sepsis. Among individuals with sepsis, the authors found no difference in vWF levels between those who developed ARDS and those who did not. Reconciling these findings with those reported by Rubin et al. requires consideration of several key differences in study design. The later study included both pulmonary and non-pulmonary sepsis. This is perhaps the most important difference in study design as there has been more recent work showing the pathogenesis and prognosis of ARDS differs between direct and indirect mechanisms of lung injury.12,29 The earlier study excluded patients with any evidence of lung injury at enrollment while the later study only excluded patients who met full diagnostic criteria for ARDS at enrollment. There were also differences between the two studies in the ascertainment of the outcome, ARDS.
In a more recent study published in 2013, Agrawal et al. studied 167 patients at risk for ARDS, including 46 (28%) with sepsis.45 The authors measured vWF along with several other biomarkers of endothelial permeability and inflammation: Ang2, Ang2: Ang1, IL-8, and receptor for advanced glycation end products (sRAGE). Similar to the finding in the Moss et al. study, in this study, baseline vWF was not different between those who progressed to develop ARDS and those who did not. Similarly, sRAGE did not differ between those who developed ARDS and those who did not. Baseline Ang2, Ang2: Ang1, and IL-8 were higher among those who later developed ARDS (n = 19 [11%]) compared to those who did not. In univariate logistic regression analyses, higher Ang-2 and IL-8 levels were associated with higher risk of ARDS. This association between Ang-2 and ARDS did not differ between those with sepsis and those without sepsis. In other words, the association between baseline Ang-2 levels in this cohort and ARDS was independent of sepsis. In contrast, the association between IL-8 and ARDS was attenuated by adjustment for sepsis, suggesting that in this study, sepsis was a confounder and both contributed to the increased levels of IL-8 as well as the risk of ARDS. When evaluating these findings in the context of what is known about the role of endothelial injury in ARDS after sepsis, there are several important considerations. This study enrolled patients with a variety of risk factors for ARDS. Furthermore, the investigators did not distinguish between patients with pulmonary and non-pulmonary sepsis. The severity of illness of patients enrolled in this study was substantially lower than in the Rubin et al. study. In the Agrawal et al. study there was a 25% mortality rate and 11% incidence of ARDS, compared to 51% mortality rate and 33% incidence of lung injury in the Rubin et al. study. The three aforementioned studies indicate that markers of endothelial injury and inflammation—including vWF, Ang-2, and IL-8—perform differently in the varied cohorts of patients at risk for ARDS. Thus, it seems critical to interpret the associations between the biomarkers and the development of ARDS in the context of the specific risk factor for lung injury.
The biology and prognosis of ARDS differs between direct and indirect mechanisms of lung injury12 and the disparate findings of the association of vWF and ARDS in the studies discussed above can be attributed to different study bases: (1) patients with non-pulmonary sepsis; (2) patients with a sepsis from both pulmonary and non-pulmonary sources; and (3) patients with a variety of risk factors for ARDS, with 28% of participants having sepsis. In a case control study of 100 patients with severe sepsis comparing individuals with ARDS to those without ARDS, Ware et al. evaluated a panel of 11 biomarkers reflecting different aspects of the pathophysiology of ARDS.30 The authors described a multivariate model incorporating a panel of five biomarkers—SP-D, RAGE, CC-16, IL-8, and IL-6—with an area under the ROC curve of 0.75 (95% confidence interval [CI] = 0.7–0.84) for diagnosis of ARDS. Of note, in this study, Ang-2 did not differ significantly between those with ARDS and those without ARDS. The authors found that among study participants with severe sepsis, biomarkers of lung epithelial injury and inflammation were the most useful for discriminating patients with ARDS from those without ARDS. This finding is somewhat surprising because sepsis is generally considered a risk factor for indirect lung injury that would be expected to act through biological pathways involving the endothelium. A limitation of this study is that there was no distinction between pulmonary and non-pulmonary sources of severe sepsis. The authors did not report the incidence of pneumonia in the cases and controls, nor does the article indicate if the models were adjusted for the source of infection. It is possible that the association between ARDS and the biomarker profile of epithelial injury reflects a difference in the pathophysiology between pulmonary and non-pulmonary sepsis and ARDS.
A study published by Calfee et al. in 2015 builds on this distinction between direct and indirect lung injury. The authors measured biomarkers of endothelial injury (Ang-2, vWF), alveolar epithelial injury (SPD, sRAGE), and inflammation (IL-6, IL-8) in two populations: one prospective single-center cohort study and one large multicenter RCT.29 All study participants with indirect lung injury had non-pulmonary sepsis. The authors tested whether each biomarker differed between individuals with direct vs. indirect lung injury. They found that indirect lung injury, caused by non-pulmonary sepsis, was characterized by a molecular phenotype consistent with more severe endothelial injury and less severe alveolar epithelial injury. The opposite pattern was identified in patients with direct lung injury. In keeping with the finding from Rubin et al.,43 in the multicenter study cohort, vWF was higher among participants with non-pulmonary sepsis compared to those with direct lung injury. These associations were robust to adjustments for severity of illness. Compared to individuals with ARDS from pneumonia or aspiration, those with ARDS from non-pulmonary sepsis had higher levels of Ang-2 on study enrollment. While IL-6 and IL-8 were not different between participants with indirect vs. direct lung injury in the single center study of 100 patients, in the multicenter study that included 853 patients IL-6 and IL-8 were higher among those with indirect lung injury. Taken together with the findings from the earlier studies of vWF, Ang-2, and IL-8, the results suggest that endothelial permeability and inflammation play a key role in the pathophysiology of ARDS after non-pulmonary sepsis, a specific etiology of indirect lung injury. However, in more heterogeneous populations of patients at risk for ARDS the associations between these biomarkers and the subsequent development of ARDS are less consistent.
Prognosis of patients with ARDS after sepsis
A study published in 2004 evaluated the prognostic value of vWF in patients with ARDS.46 vWF was measured in 559 patients with ARDS enrolled in the National Heart, Lung, and Blood Institute ARDS Network RCT of low tidal volume ventilation, the ARMA trial.46,47 All individuals enrolled in this study had established ARDS and 141 participants (25%) had sepsis. Pulmonary sepsis was not distinguished from non-pulmonary sepsis. In this cohort of patients with ARDS, baseline vWF levels were similar in patients with and without sepsis and were significantly higher in non-survivors compared to survivors. vWF levels did not differ by treatment arm. The authors concluded that the degree of endothelial activation and injury was strongly associated with outcomes in ARDS, regardless of the presence or absence of sepsis, and was not modulated by a protective ventilatory strategy.
A similar study was conducted in a cohort of patients enrolled in different ARDS Network RCT testing a two fluid management treatment protocols and the use of pulmonary artery catheters in patients with ARDS.21 Calfee et al. measured day 0 and day 3 plasma vWF and Ang-2 in patients enrolled in the FACTT trial and tested the association of these biomarkers with mortality.48 There were 24% of the 917 individuals in this biomarker substudy of FACTT had sepsis. The authors distinguished patients with infection-related lung injury from those with non-infection causes. However, they did not distinguish between pneumonia and sepsis as the cause of ARDS in the infection-related lung injury group. Similar to the findings from the biomarker studies in ARMA trial discussed above, the authors found that vWF was significantly associated with mortality in both participants with and without infection-related lung injury and that vWF levels were not affected by fluid therapy. In other words, in patients with ARDS, vWF predicted mortality, but this association appeared to act independently of infection or sepsis. The authors also measured Ang-2 in this cohort of patients enrolled in the FACTT study. Unlike vWF, the association between Ang-2 and mortality differed between individuals with infection and those without infection. Among participants without infection, baseline Ang-2 levels were associated with higher morality (odds ratio [OR] = 2.43 per 1-log increase in Ang-2, P < 0.001). Among individuals with infection (pneumonia or sepsis), baseline Ang-2 was not different between survivors and those who died. Among those with infection, patients whose plasma Ang-2 levels increased from day 0 to day 3, there was more than double the odds of death compared with patients whose Ang-2 levels declined over the same period of time. This difference was statistically significant. Taken together, these findings show that among patients with ARDS, Ang-2 has differential prognostic value for mortality depending on the presence or absence of infection. The authors also found that Ang-2 was highest among patients with sepsis. Among participants with sepsis, a fluid conservative strategy was associated with a 15% greater decline in Ang-2 levels from day 0 to day 3 compared with fluid-liberal therapy in patients with infection-related ALI. This difference was statistically significant. These findings suggest that fluid conservative therapy preferentially lowers plasma Ang-2 levels over time and thus may be beneficial in part by decreasing endothelial inflammation and are supported by earlier experimental studies showing a pro-inflammatory response of the endothelium to high vascular pressures.49
In a study of 259 children with ARDS, Zinter et al. measured vWF, Ang-2, and VEGF on study day 1 and day 3 of study enrollment and tested the association of these biomarkers with mortality.50 There was a 15% mortality rate in this cohort. Sepsis was the ARDS risk factor in 22% of patients and sepsis was the most common of indirect lung injury risk factor (n = 56 individuals with sepsis, 57% of 97 participants with indirect lung injury). Analyses were stratified by prior hematopoietic cellular transplantation (HCT), given the severe ARDS phenotype of this subgroup. The authors did not report findings for models testing the association of the Ang-2, VEGF, or vWF and mortality adjusted for direct or indirect mechanisms of lung injury, or for patients with sepsis after ARDS. Compared to patients with direct lung injury, patients with indirect lung injury had higher plasma Ang-2 on day 1 but not on day 3. In logistic regression models adjusted for age, sex, race, and PaO2:FiO2 ratio, day 1 and day 3 Ang-2 levels were associated with mortality (OR = 3.7, 95% CI = 1.1–11.5, P = 0.027 and OR = 10.2, 95% CI = 2.2–46.5, P = 0.003, for each log10 increase in Ang-2 on day1 and day 3, respectively). These associations were independent of vWF and VEGF levels. Nearly half of the cohort (45%) had a rising Ang-2 levels between days 1 and 3; this was strongly associated with mortality, particularly among children with HCT. Children with indirect lung injury had similar vWF levels compared with direct lung injury patients. In the aforementioned studies, the association of vWF with mortality was compared between adult participants with and without infection. In this study, the authors distinguished indirect and direct causes of lung injury. Importantly, sepsis was the most common cause on indirect lung injury in this cohort and pneumonia was the most common cause of direct lung injury. It is therefore difficult to compare some of the reported findings of Ang-2 and vWF as prognostic biomarkers in sepsis after ARDS, the focus of this review. In contrast to the studies discussed earlier, although children who died had higher vWF on day 1 compared to survivors, the association between vWF and mortality in univariate logistic regression models was not significant. The authors attribute this to three outliers of high vWF measurements among children who survived. VEGF was not measured in the previously discussed ARDS Network prognostic biomarker studies. In this cohort of pediatric patients with ARDS, patients with indirect lung had similar VEGF levels compared to those with direct lung injury. Plasma VEGF was not associated with mortality in pediatric ARDS.
Thombomodulin (TM) is an activator of protein C and a biomarker for endothelial injury. A different substudy of 449 individuals enrolled in the FACTT trial tested the association between soluble thrombomodulin (sTM) and mortality among patients with ARDS.51 In this cohort, 22% of patients had sepsis. The authors also tested the association between TM gene polymorphisms and levels of sTM at baseline to assess whether genetic heterogeneity contributed to differential TM gene expression in patients with ARDS. Plasma sTM levels were higher among patients with non-pulmonary sepsis compared to those without. Plasma sTM was higher in non-survivors than survivors at baseline and on day 3. In analyses adjusted for non-pulmonary sepsis, among other variables, each log increase in sTM at baseline and at day 3 more than doubled the odds of death, OR = 2.4 (95% CI = 1.5–3.8, P < 0.001) and OR = 2.8 (95% CI = 1.7–4.7, P < 0.001), respectively. The addition of sTM to the APACHEIII scale significantly improved discrimination for mortality from 77% to 80% (P < 0.03). The authors found no differences in sTM levels by single nucleotide polymorphism (SNP) genotype. They concluded that the lack of association between the sTM levels and genetic variants indicated that the increased levels of sTM among those who died reflected the severity of endothelial damage rather than genetic heterogeneity. In patients with ARDS, sTM was associated with death. Although sTM levels were highest among participants with non-pulmonary sepsis, the association between sTM and death was independent of non-pulmonary sepsis.
Conclusions
Based on the existing biomarker data from clinical studies, what can be concluded about the role of the vascular endothelium in the pathogenesis and prognosis of ARDS after sepsis? Taken together, the studies reviewed here support a primary role of the microcirculation in the pathogenesis and prognosis of ARDS after sepsis. Although preclinical studies demonstrate the importance of pulmonary endothelial injury in ARDS, clinical studies are limited by the lack of a specific pulmonary endothelial marker. The interactions among the innate immune system and the regulation of vascular permeability and alveolar epithelial injury are key drivers of ARDS after sepsis. Furthermore, when considering the results from the clinical studies described here, a few important patterns emerge. The timing of biomarker measurements, the information gained from serial biomarker measurements, and the general distinction between direct and indirect lung injury affect the interpretation of the study results. Additionally, among patients with sepsis, distinguishing pulmonary from non-pulmonary sources of infection changes the performance of some biomarkers of endothelial injury. A summary of the findings of studies of ARDS after sepsis, organized by biomarkers of interest, is presented in Table 2.
Table 2.
Summary of endothelial and inflammatory biomarker studies in ARDS after sepsis.
| Biomarker | Outcome | Study population | Key findings |
|---|---|---|---|
| Vascular permeability | |||
| Ang-2 | ARDS | Adults with a variety of risk factors for ARDS, including sepsis | Higher Ang-2 was associated with ARDS, independent of sepsis45 |
| Mortality | ARDS related to infection (pneumonia or sepsis) | Rising Ang-2 levels were associated with death48 | |
| Mortality | Children with ARDS from a variety of causes, including sepsis | Rising Ang-2 levels were associated with death. Patients with sepsis not analyzed separately from other causes of indirect lung injury50 | |
| ARDS | Case control study of adults with sepsis, ARDS vs. no ARDS. No distinction between non-pulmonary and pulmonary source | No difference in Ang-2 levels between those with and without ARDS30 | |
| vWF | ARDS | Non-pulmonary sepsis | Higher vWF was associated with ARDS29,43 |
| Mortality | ARDS from a variety of causes, including sepsis | Higher vWF was associated with death, independent of sepsis46,48 | |
| ARDS | Sepsis, no distinction between non-pulmonary and pulmonary source | vWF was not associated with ARDS45,57 | |
| Mortality | Children with ARDS from a variety of causes, including sepsis | vWF was not associated with death. Patients with sepsis not analyzed separately from other causes of indirect lung injury50 | |
| Coagulation | |||
| TM | Mortality | ARDS from a variety of causes, including non-pulmonary sepsis | Higher sTM was associated with death, independent of non-pulmonary sepsis51 |
| Inflammation | |||
| IL-6 | ARDS | Case control study of adults with sepsis, ARDS vs. No ARDS. No distinction between non-pulmonary and pulmonary source | IL-6 was higher among ARDS cases compared to controls30 |
| IL-8 | ARDS | Adults with a variety of risk factors for ARDS, including sepsis | Higher IL-8 was associated with ARDS, independent of vasopressor use45 |
| ARDS | Case control study of adults with sepsis, ARDS vs. no ARDS. No distinction between non-pulmonary and pulmonary source | IL-8 was higher among ARDS cases compared to controls30 | |
Unless otherwise specified, all clinical studies listed above are cohort studies of adult patients.
Ang-2, Angiopoietin-2; vWF, von Willebrand Factor; TM, thrombomodulin; IL-6, Interleukin-6; IL-8, Interleukin-8; ARDS, acute respiratory distress syndrome.
How can these data inform future study design? The heterogeneity of results in the biomarker studies above seems to be in part driven by heterogeneous patient populations. Taken together, these findings suggest that future biomarker studies should differentiate patients with sepsis as the primary risk factor for ARDS from other causes of lung injury. Further differentiation between pulmonary and non-pulmonary sepsis may allow researchers to better understand specific biologic pathways involved in ARDS after sepsis. This could be accomplished through a study design with a well-defined patient population, or using adjusted or stratified data analyses of more heterogeneous cohorts. These distinctions may provide valuable insights into the biological differences among subgroups of patients. It follows that biological differences identified by distinct molecular patterns could explain heterogeneity of treatment effects that are not explained by clinical factors alone.52
Acknowledgements
The authors thank Diana Lim for help with designing and formatting the figures in this manuscript.
Conflict of interest
The author(s) declare that there is no conflict of interest.
Funding
CMH was supported by National Heart, Lung, and Blood Institute (NHLBI) Career Development Grant, NHLBI K23HL-133495. MAM was supported by HL51856, HL110969, and HL10871301.
References
- 1.Liu V, Escobar GJ, Greene JD, et al. Hospital deaths in patients with sepsis from 2 independent cohorts. JAMA 2014; 312: 90–92. [DOI] [PubMed] [Google Scholar]
- 2.Bellani G, Laffey JG, Pham T, et al. Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA 2016; 315: 788–800. [DOI] [PubMed] [Google Scholar]
- 3.Torio C MB. National Inpatient Hospital Costs: The Most Expensive Conditions by Payer, 2013. HCUP Statistical Brief #204. Rockville, MD: Agency for Healthcare Research and Quality, 2016. Available at: https://www.hcup-us.ahrq.gov/reports/statbriefs/sb204-Most-Expensive-Hospital-Conditions.jsp [accessed 25 Aug 2017].
- 4.Bice T, Cox CE, Carson SS. Cost and health care utilization in ARDS–different from other critical illness? Semin Respir Crit Care Med 2013; 34: 529–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seymour CW, Liu VX, Iwashyna TJ, et al. Assessment of Clinical Criteria for Sepsis: For the Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016; 315: 762–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ranieri VM, Rubenfeld GD, Thompson BT, et al. Acute respiratory distress syndrome: the Berlin Definition. JAMA 2012; 307: 2526–2533. [DOI] [PubMed] [Google Scholar]
- 7.Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. J Clin Invest 2012; 122: 2731–2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prescott HC, Calfee CS, Thompson BT, et al. Toward smarter lumping and smarter splitting: rethinking strategies for sepsis and acute respiratory distress syndrome clinical trial design. Am J Respir Crit Care Med 2016; 194: 147–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gotts JE, Matthay MA. Sepsis: pathophysiology and clinical management. BMJ 2016; 353: i1585. [DOI] [PubMed] [Google Scholar]
- 10.Moss M, Bucher B, Moore FA, et al. The role of chronic alcohol abuse in the development of acute respiratory distress syndrome in adults. JAMA 1996; 275: 50–54. [PubMed] [Google Scholar]
- 11.Calfee CS, Matthay MA, Kangelaris KN, et al. Cigarette smoke exposure and the acute respiratory distress syndrome. Crit Care Med 2015; 43: 1790–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luo L, Shaver CM, Zhao Z, et al. Clinical predictors of hospital mortality differ between direct and indirect ARDS. Chest 2017; 151: 755–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seymour CW, Coopersmith CM, Deutschman CS, et al. Application of a framework to assess the usefulness of alternative sepsis criteria. Crit Care Med 2016; 44: e122–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shapiro NI, Schuetz P, Yano K, et al. The association of endothelial cell signaling, severity of illness, and organ dysfunction in sepsis. Crit Care 2010; 14: R182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Skibsted S, Jones AE, Puskarich MA, et al. Biomarkers of endothelial cell activation in early sepsis. Shock 2013; 39: 427–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aird WC. The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood 2003; 101: 3765–3777. [DOI] [PubMed] [Google Scholar]
- 17.Evans CE, Zhao YY. Impact of thrombosis on pulmonary endothelial injury and repair following sepsis. Am J Physiol Lung Cell Mol Physiol 2017; 312: L441–L451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yealy DM, Kellum JA, Huang DT, et al. A randomized trial of protocol-based care for early septic shock. N Engl J Med 2014; 370: 1683–1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hou PC, Filbin MR, Wang H, et al. Endothelial permeability and hemostasis in septic shock: results from the ProCESS Trial. Chest 2017; 152: 22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kellum JA, Pike F, Yealy DM, et al. Relationship between alternative resuscitation strategies, host response and injury biomarkers, and outcome in septic shock: analysis of the Protocol-based Care for Early Septic Shock study. Crit Care Med 2017; 45: 438–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wiedemann HP, Wheeler AP, Bernard GR, et al. Comparison of two fluid-management strategies in acute lung injury. N Engl J Med 2006; 354: 2564–2575. [DOI] [PubMed] [Google Scholar]
- 22.Famous KR, Delucchi K, Ware LB, et al. Acute respiratory distress syndrome subphenotypes respond differently to randomized fluid management strategy. Am J Respir Crit Care Med 2017; 195: 331–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mikacenic C, Price BL, Harju-Baker S, et al. A two biomarker model predicts mortality in the critically ill with sepsis. Am J Respir Crit Care Med 2017; 196: 1004–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abraham E. New definitions for sepsis and septic shock: continuing evolution but with much still to be done. JAMA 2016; 315: 757–759. [DOI] [PubMed] [Google Scholar]
- 25.Ware LB. Biomarkers in critical illness: new insights and challenges for the future. Am J Respir Crit Care Med 2017; 196: 944–945. [DOI] [PubMed] [Google Scholar]
- 26.Rivers E, Nguyen B, Havstad S, et al. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med 2001; 345: 1368–1377. [DOI] [PubMed] [Google Scholar]
- 27.Calfee CS, Delucchi K, Parsons PE, et al. Subphenotypes in acute respiratory distress syndrome: latent class analysis of data from two randomised controlled trials. Lancet Respir Med 2014; 2: 611–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thompson BT, Chambers RC, Liu KD. Acute respiratory distress syndrome. N Engl J Med 2017; 377: 562–572. [DOI] [PubMed] [Google Scholar]
- 29.Calfee CS, Janz DR, Bernard GR, et al. Distinct molecular phenotypes of direct vs indirect ARDS in single-center and multicenter studies. Chest 2015; 147: 1539–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ware LB, Koyama T, Zhao Z, et al. Biomarkers of lung epithelial injury and inflammation distinguish severe sepsis patients with acute respiratory distress syndrome. Crit Care 2013; 17: R253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Parikh SM, Mammoto T, Schultz A, et al. Excess circulating angiopoietin-2 may contribute to pulmonary vascular leak in sepsis in humans. PLoS Med 2006; 3: e46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu J, Zhang X, Pelayo R, et al. Extracellular histones are major mediators of death in sepsis. Nat Med 2009; 15: 1318–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seeley EJ, Matthay MA, Wolters PJ. Inflection points in sepsis biology: from local defense to systemic organ injury. Am J Physiol Lung Cell Mol Physiol 2012; 303: L355–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Asaduzzaman M, Lavasani S, Rahman M, et al. Platelets support pulmonary recruitment of neutrophils in abdominal sepsis. Crit Care Med 2009; 37: 1389–1396. [DOI] [PubMed] [Google Scholar]
- 35.Yadav H, Kor DJ. Platelets in the pathogenesis of acute respiratory distress syndrome. Am J Physiol Lung Cell Mol Physiol 2015; 309: L915–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Clark SR, Ma AC, Tavener SA, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med 2007; 13: 463–469. [DOI] [PubMed] [Google Scholar]
- 37.Fox ED, Heffernan DS, Cioffi WG, et al. Neutrophils from critically ill septic patients mediate profound loss of endothelial barrier integrity. Crit Care 2013; 17: R226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fein AM, Grant MM, Niederman MS, et al. Neutrophil-endothelial cell interaction in critical illness. Chest 1991; 99: 1456–1462. [DOI] [PubMed] [Google Scholar]
- 39.Schmidt EP, Yang Y, Janssen WJ, et al. The pulmonary endothelial glycocalyx regulates neutrophil adhesion and lung injury during experimental sepsis. Nat Med 2012; 18: 1217–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ortiz-Munoz G, Mallavia B, Bins A, et al. Aspirin-triggered 15-epi-lipoxin A4 regulates neutrophil-platelet aggregation and attenuates acute lung injury in mice. Blood 2014; 124: 2625–2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kiefmann R, Heckel K, Schenkat S, et al. Platelet-endothelial cell interaction in pulmonary micro-circulation: the role of PARS. Thromb Haemost 2004; 91: 761–770. [DOI] [PubMed] [Google Scholar]
- 42.Kangelaris KN, Prakash A, Liu KD, et al. Increased expression of neutrophil-related genes in patients with early sepsis-induced ARDS. Am J Physiol Lung Cell Mol Physiol 2015; 308: L1102–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rubin DB, Wiener-Kronish JP, Murray JF, et al. Elevated von Willebrand factor antigen is an early plasma predictor of acute lung injury in nonpulmonary sepsis syndrome. J Clin Invest 1990; 86: 474–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moss M, Ackerson L, Gillespie MK, et al. von Willebrand factor antigen levels are not predictive for the adult respiratory distress syndrome. Am J Respir Crit Care Med 1995; 151: 15–20. [DOI] [PubMed] [Google Scholar]
- 45.Agrawal A, Matthay MA, Kangelaris KN, et al. Plasma angiopoietin-2 predicts the onset of acute lung injury in critically ill patients. Am J Respir Crit Care Med 2013; 187: 736–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ware LB, Eisner MD, Thompson BT, et al. Significance of von Willebrand factor in septic and nonseptic patients with acute lung injury. Am J Respir Crit Care Med 2004; 170: 766–772. [DOI] [PubMed] [Google Scholar]
- 47.Brower RG, Matthay MA, et al. Acute Respiratory Distress Syndrome Network. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med 2000; 342: 1301–1308. [DOI] [PubMed] [Google Scholar]
- 48.Calfee CS, Gallagher D, Abbott J, et al. Plasma angiopoietin-2 in clinical acute lung injury: prognostic and pathogenetic significance. Crit Care Med 2012; 40: 1731–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kuebler WM, Ying X, Singh B, et al. Pressure is proinflammatory in lung venular capillaries. J Clin Invest 1999; 104: 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zinter MS, Spicer A, Orwoll BO, et al. Plasma angiopoietin-2 outperforms other markers of endothelial injury in prognosticating pediatric ARDS mortality. Am J Physiol Lung Cell Mol Physiol 2016; 310: L224–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sapru A, Calfee CS, Liu KD, et al. Plasma soluble thrombomodulin levels are associated with mortality in the acute respiratory distress syndrome. Intensive Care Med 2015; 41: 470–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Iwashyna TJ, Burke JF, Sussman JB, et al. Implications of heterogeneity of treatment effect for reporting and analysis of randomized trials in critical care. Am J Respir Crit Care Med 2015; 192: 1045–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kumpers P, van Meurs M, David S, et al. Time course of angiopoietin-2 release during experimental human endotoxemia and sepsis. Crit Care 2009; 13: R64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fisher J, Douglas JJ, Linder A, et al. Elevated plasma angiopoietin-2 levels are associated with fluid overload, organ dysfunction, and mortality in human septic shock. Crit Care Med 2016; 44: 2018–2027. [DOI] [PubMed] [Google Scholar]
- 55.David S, Mukherjee A, Ghosh CC, et al. Angiopoietin-2 may contribute to multiple organ dysfunction and death in sepsis*. Crit Care Med 2012; 40: 3034–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mikacenic C, Hahn WO, Price BL, et al. Biomarkers of endothelial activation are associated with poor outcome in critical illness. PLoS One 2015; 10: e0141251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Moss M, Gillespie MK, Ackerson L, et al. Endothelial cell activity varies in patients at risk for the adult respiratory distress syndrome. Crit Care Med 1996; 24: 1782–1786. [DOI] [PubMed] [Google Scholar]
- 58.Zhang R-Y, Liu Y-Y, Qu H-P, et al. The angiogenic factors and their soluble receptors in sepsis: friend, foe, orboth? Crit Care 2013; 17: 446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yang KY, Liu KT, Chen YC, et al. Plasma soluble vascular endothelial growth factor receptor-1 levels predict outcomes of pneumonia-related septic shock patients: a prospective observational study. Crit Care 2011; 15: R11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Skibsted S, Jones AE, Puskarich MA, et al. Biomarkers of endothelial cell activation in early sepsis. Shock 2013; 39: 427–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Katayama S, Nunomiya S, Koyama K, et al. Markers of acute kidney injury in patients with sepsis: the role of soluble thrombomodulin. Crit Care 2017; 21: 229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hoshino K, Kitamura T, Nakamura Y, et al. Usefulness of plasminogen activator inhibitor-1 as a predictive marker of mortality in sepsis. J Intensive Care 2017; 5: 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Iba T, Kidokoro A, Fukunaga M, et al. Association between the severity of sepsis and the changes in hemostatic molecular markers and vascular endothelial damage markers. Shock 2005; 23: 25–29. [DOI] [PubMed] [Google Scholar]
- 64.Trzeciak S, Jones AE, Shapiro NI, et al. A prospective multicenter cohort study of the association between global tissue hypoxia and coagulation abnormalities during early sepsis resuscitation. Crit Care Med 2010; 38: 1092–1100. [DOI] [PubMed] [Google Scholar]
- 65.Vassiliou AG, Mastora Z, Orfanos SE, et al. Elevated biomarkers of endothelial dysfunction/activation at ICU admission are associated with sepsis development. Cytokine 2014; 69: 240–247. [DOI] [PubMed] [Google Scholar]
- 66.Shaw AD, Vail GM, Haney DJ, et al. Severe protein C deficiency is associated with organ dysfunction in patients with severe sepsis. J Crit Care 2011; 26: 539–545. [DOI] [PubMed] [Google Scholar]
- 67.Ostrowski SR, Haase N, Muller RB, et al. Association between biomarkers of endothelial injury and hypocoagulability in patients with severe sepsis: a prospective study. Crit Care 2015; 19: 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gomez HG, Gonzalez SM, Londono JM, et al. Immunological characterization of compensatory anti-inflammatory response syndrome in patients with severe sepsis: a longitudinal study*. Crit Care Med 2014; 42: 771–780. [DOI] [PubMed] [Google Scholar]
- 69.Rondina MT, Carlisle M, Fraughton T, et al. Platelet-monocyte aggregate formation and mortality risk in older patients with severe sepsis and septic shock. J Gerontol A Biol Sci Med Sci 2015; 70: 225–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Calfee CS, Thompson BT, Parsons PE, et al. Plasma interleukin-8 is not an effective risk stratification tool for adults with vasopressor-dependent septic shock. Crit Care Med 2010; 38: 1436–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]