

Abstract
DNA damage induces specific signaling and repair responses in the cell, which is critical for protection of genome integrity. Laser microirradiation became a valuable experimental tool to investigate the DNA damage response (DDR) in vivo. It allows real-time high-resolution single-cell analysis of macromolecular dynamics in response to laser-induced damage confined to a submicrometer region in the cell nucleus. However, various laser conditions have been used without appreciation of differences in the types of damage induced. As a result, the nature of the damage is often not well characterized or controlled, causing apparent inconsistencies in the recruitment or modification profiles. We demonstrated that different irradiation conditions (i.e., different wavelengths as well as different input powers (irradiances) of a femtosecond (fs) near-infrared (NIR) laser) induced distinct DDR and repair protein assemblies. This reflects the type of DNA damage produced. This protocol describes how titration of laser input power allows induction of different amounts and complexities of DNA damage, which can easily be monitored by detection of base and crosslinking damages, differential poly (ADP-ribose) (PAR) signaling, and pathway-specific repair factor assemblies at damage sites. Once the damage conditions are determined, it is possible to investigate the effects of different damage complexity and differential damage signaling as well as depletion of upstream factor(s) on any factor of interest.
Keywords: Genetics, Issue 131, Laser microirradiation, lasers in medicine, DNA damage, DNA damage response, DNA repair, poly (ADP-ribose) polymerase (PARP), complex damage, strand breaks, base damage
Introduction
In Vivo DNA Damage Signaling is Not Well UnderstoodIn vivo, DNA is complexed with histones and other factors to form chromatin fibers. Regulation of chromatin structure is of paramount importance for DNA metabolism. For example, the histone variant H2AX is phosphorylated by ataxia-telangiectasia mutated (ATM) and other kinases following double-strand breaks (DSB) induction, and is important for DSB damage signal amplification as well as providing a docking site for other factors. Spreading of damage signaling and repair pathway choice appear to be critically influenced by local chromatin structure at damage sites1. A number of chromatin remodeling factors, histone chaperones, and histone modifying enzymes are indeed recruited to damage sites and are important for efficient DNA repair, highlighting the significance of chromatin regulation in the DDR and repair2,3,4. Furthermore, damage site clustering or repositioning was observed in yeast and drosophila5,6,7,8, reminiscent of relocalization of gene loci in the subnuclear compartment associated with gene regulation9,10. Recent studies in mouse and human cells also revealed mobilization of DSB sites, which influences repair fidelity and pathway choice11,12. This raises the possibility that DDR/repair may also be intimately linked to nuclear architecture, higher-order chromatin organization, and chromosome dynamics in the cell nucleus. Thus, it is critical to develop high-resolution methods to study DDR and repair processes in the context of the endogenous nuclear environment in a live cell in order to understand short- and long-term consequences of DNA damage.
Critical Role of PAR Polymerase (PARP) in Gauging the Extent and Type of Damage and Regulating Protein Assembly at the Damage Site PARP1 is a DNA nick sensor rapidly activated by DNA damage that plays a critical role in DNA repair13. PARP1 was originally thought to function together with X-Ray Repair Cross Complementing 1 (XRCC1) to facilitate base excision repair (BER), but recent studies reveal its role in other DNA repair pathways, including DSB repair14. Activated PARP1 uses nicotinamide adenine dinucleotide (NAD+) as a substrate to ADP-ribosylate multiple target proteins, including itself. This enzyme and other family members have attracted much attention in recent years as PARP inhibitors have emerged as promising therapeutic drugs for cancers. Although PARP inhibitors were initially found to be effective in breast cancer gene (BRCA)-mutated breast cancer cells, there is now a plethora of evidence for their effects in mono- and combination therapies together with DNA damaging agents/irradiation against a broad spectrum of cancers with mutations not limited to BRCA15,16,17,18,19,20.
At the molecular level, PARP activation was shown to play critical roles in organizing the local chromatin structure at damage sites. PAR-dependent recruitment of chromatin modification enzymes facilitates DSB repair and dictates repair pathway choices, suggesting the important scaffolding role of PAR modification at damage sites. 13,21,22,23,24,25,26,27,28,29,30,31 We recently demonstrated the exclusion of p53-binding protein 1 (53BP1) from damage sites by PAR 32, providing an alternative explanation for 53BP1-dependent hyperactivation of non-homologous endjoining (NHEJ) by PARP inhibitor and highlighting the significance of PARP in DSB repair pathway choice33,34. PARP1 also directly PARylates and affects the activity of multiple DNA repair factors14.
Using Laser Microirradiation as a Tool to Study DDR/Repair In Vivo Laser microirradiation to produce sub-micron alterations on individual chromosomes was first described in 196935 and reviewed in detail in 198136. Several decades later, laser microirradiation was shown to induce DNA damage at a defined submicrometer region in the cell nucleus, and was proven to be a valuable technique to study recruitment or modifications of various factors to DNA lesions in vivo13,37,38,39,40,41. This method allows detection of those factors that do not form distinct irradiation-induced foci (IRIF) at damage sites39,42. It is also possible to examine the spatiotemporal dynamics of chromatin structural changes both at damage sites and in the rest of the nucleus. We carefully compared the DDRs induced by different laser systems and input powers to evaluate the relationship between the type of DNA damage and the microirradiation conditions32,43,44,45. The aberrant recruitment patterns of 53BP1 and telomeric repeat binding factor 2 (TRF2) were observed in the previous laser damage studies, which provided the basis for the recurring concern for the "non-physiological" nature of laser-induced damage46,47,48,49. We found that these apparent discrepancies could now be explained by differential PARP signaling which gauges the amount and complexity of induced damage32. We confirmed that: 1) laser-microirradiated cells (even after high input-power irradiation) are arrested in interphase in a damage checkpoint control-dependent manner and remain viable (at least up to 48 h)32,50; and 2) repair factor recruitment/modifications faithfully recapitulate those observed with treatment with conventional DNA damaging agents and DSB induction by endonucleases32,39,42,44,50,51,52. These results strongly support the physiological relevance of studying laser damage-induced cellular responses.
Protocol
1. Basic Cell Preparation
Note: This step is for standard immunofluorescence detection of the endogenous protein recruitment or modification, and for the use of a cell line stably expressing a fluorescently tagged recombinant protein. For example, Potorustridactylus (PtK2) marsupial kidney epithelial cells stably expressing EGFP-53BP11220-1711 or TRF2-YFP were used in our previous study (Figure 1)32. In the former case, the focus forming region of 53BP1 (amino acid 1220-1711) that contains the oligomerization domain, the Tudor domain, and the ubiquitylation-dependent recruitment (UDR) motif, was fused to enhanced GFP (EGFP-53BP11220-1711). This fusion protein recapitulates the damage site recruitment of the full-length 53BP153,54,55. In HeLa cells, fluorescently tagged subunits of cohesin (e.g., hSMC1-GFP51, GFP-Scc1 (Rad21)52, and GFP-SA2 51) must be stably expressed to ensure efficient incorporation into the complex and to recapitulate the S/G2 cell cycle phase-specific damage site recruitment of the endogenous cohesin51.
- Seed any adherent cell type onto a 35 mm tissue culture dish with a gridded coverslip to allow for individual cell identification. Adjust the initial cell number to achieve 60% confluency in 36-48 h based on the proliferation rate of the particular cell line. For example:
- For PtK2 marsupial kidney epithelial cells (either wild type or those stably expressing EGFP-53BP11220-1711 or TRF2-YFP): plate 2.0 x 104 cells in 2 mL Advanced Minimum Essential Medium (MEM) supplemented with 2 mM L-Glutamine, 4% fetal bovine serum (FBS), and antibiotics (100 U/mL penicillin and 100 µg/mL streptomycin), and incubate at 37 °C with 7% CO2.
- For HeLa cells with stably expressing fluorescently tagged recombinant proteins GFP-SA2: seed 1.0 x 105 cells in 2 mL Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% FBS, 2 mM L-Glutamine, 100 U/mL penicillin, and 100 µg/mL streptomycin, and incubate at 37 °C with 5% CO2. Other HeLa cells may also be used including unmodified cells or cells expressing hSMC1-GFP.
Allow cells to adhere and proliferate for 36-48 h before DNA damage induction at 30-60% confluence (to allow visualization of the grid number). Proceed to Section 4.
2. Transient Transfection
Note: Sometimes good antibodies are not available to detect the endogenous proteins. If cell lines stably expressing marker proteins are not available, carry out transient transfections to monitor fluorescently labeled proteins for live cell analyses.
For HeLa cells, on day 1, seed 6-8 x 104 cells in 0.5 mL culture media in one well of a 24-well plate and incubate in a 100% humidified incubator at 37 °C and with 5% CO2. See step 1.1 for culture media conditions.
Day 2, use a mammalian expression plasmid encoding a fluorescently tagged DNA repair factor (e.g., GFP-NTH1 or GFP-OGG1 for base damage32,44,56, GFP-Ku for high dose DSBs57, GFP-53BP132 for relatively simple DSBs, or other proteins of interest fused to fluorescent tag) to perform liposome-mediated DNA transfection. Dilute 0.3 µg plasmid DNA into 25 µL of serum-free culture media in a 1.5 mL tube.
In another 1.5 mL tube, dilute 1 µL liposome-based DNA transfection reagent into 25 µL of serum-free culture media and mix gently.
Mix the diluted DNA and transfection agent gently, and incubate for 10 min at room temperature (RT) to form DNA-lipid complexes.
Rinse cells with 0.5 mL culture media once, remove the media, and add 0.5 mL fresh culture media to the cells (as in step 1.1).
Add the DNA-lipid complexes to the cells. Mix gently by rocking the plate a few times. Put the cells back in the incubator as in step 2.1.
After 4-6 h, remove the culture media from the cells, and add 300 µL 0.05% Trypsin-EDTA. Remove it, add 200 µL Trypsin-EDTA, and incubate in the 37 °C incubator for 3-4 min.
After confirming that the cells are detached, add 800 µL culture media and resuspend the cells by gently pipetting to break up the cell clumps. Transfer the cell suspension to a 35 mm culture dish with a gridded coverslip, and add culture media to a final volume of 2 mL.
Incubate cells in the incubator as in step 2.1 for 48 h, then proceed to Section 4. Note: If the expression of the recombinant protein is toxic (transfected cells show round or irregular morphology after incubation in step 2.8), shorten the expression time, and perform laser microirradiation (Section 4) at 16-20 h after transfection as long as the protein expression can be confirmed (for fluorescence detection, see step 4.3). In this case, perform transfection directly in the 35 mm dish. Seed 3.0 x 105 HeLa cells in a 35 mm dish with a gridded coverslip to achieve approximately 50-70% confluence after 30 h. Transfect DNA plasmids, and change media 2-6 h after transfection. Proceed with laser microirradiation 12-20 h later if protein expression is reasonable and cells appear healthy.
3. Cell Cycle Synchronization
Note: For homologous recombination repair (HR) proteins, such as Rad51 and cohesin, the recruitment is more prominent in S/G2 phase of the cell cycle, in which HR repair takes place51. Thus, to monitor the recruitment of these proteins, cell synchronization to S/G2 phase is necessary.
- S/G2 phase synchronization
- Rinse the cells with 1 mL thymidine-free culture media (as in step 1.1) twice, and incubate the cells in 2 mL thymidine-free culture media for 9 h as in step 1.1.
- Add 200 µL of 27.5 mM thymidine stock solution (dissolved in culture media) into the culture media with the cells to a final concentration of 2.5 mM thymidine. Incubate the cells as in step 3.1.1 for another 15 h. Rinse and incubate the cells in thymidine-free culture media for 4-6 h and induce DNA damage (Section 4).
- Confirm synchronization efficiency by using cell cycle markers (Cyclin A2 and B1 for S/G2 and G2, respectively), S/G2 phase-specific DNA damage repair factors (e.g., Rad51 or cohesin), or by IdU/EdU incorporation (for S phase cells)51.
- G1 phase synchronization
- Seed 6-8 x 104 HeLa cells in a 35 mm culture dish with gridded coverslip (step 2.1).
- After 2 days, identify metaphase cells, which are slightly lifted and round-shaped, under an inverted microscope using 10X objective and 10X ocular lenses. Acquire images to record the position of metaphase cells on the gridded coverslip.
- After 3-4 h, confirm cell division into two daughter cells (in G1 phase), and induce DNA damage (Section 4).
4. Titration of Laser Input Power
Note: In this protocol, we induce DNA damage using a 780 nm pulsed fs NIR laser system attached to a confocal microscope, or an 800 nm pulsed fs NIR laser coupled to an inverted epi-fluorescence microscope. Although input power (actual irradiance at the laser focal point in the specimen) needed to induce comparable DDR is different in these systems, the basic principle of input power titration is applicable to both, or any, NIR systems32,57. The in situ energy per laser pulse and peak irradiance at the focal point for the two laser systems were in the range of 5.33 × 10-2-4 × 10-1 nJ and 7 × 1010-5.24 × 1011 W/cm2, respectively32.
Turn on the NIR laser and allow the laser system to warm-up for 1 h before use.
Place a dish of cells on a heated (37 °C) stage in a chamber that allows CO2 (5%) and humidity (100%) control. Note: If a CO2 chamber is not available, use CO2-independent culture media for up to 5-6 h. If a heating stage is not available, keep the cells under the microscope for < 20 min and immediately replace into the tissue culture incubator with proper temperature, CO2, and humidity control. These culture conditions may vary depending on the specific cell type used as well as the organism of derivation (e.g., mammals versus amphibians).
For live cell analysis of fluorescently tagged proteins, select a GFP filter (450-490 nm excitation, 515-586 nm emission) or Cy3 filter (540-552 nm excitation, > 590 nm emission) for GFP or mCherry-fused protein, respectively, using a 100X or 63X oil immersion objective.
Search for cells that have comparable fluorescence signals, reflecting comparable levels of protein expression. Avoid cells that have too weak or too strong expression in order to reduce cell-to-cell variability in fluorescence measurements.
- Titrate the laser input power.
- 780 nm NIR laser attached to a confocal microscope
- Use the software bleach function to target linear tracks inside the cell nucleus for exposure to single laser scans (resolution 512 x 512 pixels, zoom X1, bleached region 50 x 4 pixels, 12.8 µs pixel dwell time, iteration x1) through the oil objective (100X/1.3 NA).
- Adjust the laser transmission percentage (using the software/hardware built into the laser microscope system by the manufacturer) to 5% without changing any other settings.
- Place a cell in the center of the field. Click the region of interest (ROI) tool, and draw a line or box (approximately 50 x 4 pixels) in the nucleus using the mouse, and click the "Bleach" button to induce DNA damage. Note: In subsequent irradiation steps, "bleaching" should be increased in 5% increments to 40% or up to 100% if necessary.
- 800 nm NIR laser attached to epi-fluorescence microscope
- Adjust the polarizer angle (°) and measure the input power (mW) entering the microscope, using a laser power meter. Determine the polarizer angles that provide input powers that range from 20 mW-155 mW at 5-10 mW increments.
- Set the polarizer angle that corresponds to 20 mW input power. Note: For our laser system, if the polarizer angle is 40 °, the input power is 65 mW. Increase input power with 5-10 mW increments.
For live cell analysis, go to steps 6.1 and 6.3. For immunofluorescence detection of the endogenous proteins or modifications proceed to Section 5. After the analysis, go to step 4.7 for further titration of the laser power. To analyze the upstream factor requirement, go to Section 7. Note: Recruitment of BER and NHEJ factors is rapid (within a few minutes) and transient (< ~ 30 min-1 h depending on the factor and < ~4 h post damage induction, respectively). In contrast, the recruitment of HR factors Rad51 and cohesin is detectable at ~30 min-1 h and they tend to persist more than 8 h at unrepaired DNA lesions44,50,51. Thus, it is necessary to examine different time points post irradiation for different repair factors.
Increase input power by desired increments (i.e., 5% for 780 NIR laser and 5-10 mW for 800 NIR) as in step 4.4, and repeat steps 4.5-4.7 on a new set of cells to determine the threshold (50% of damaged cells show recruitment) and peak (100% of cells show recruitment) for each protein.
5. Immunofluorescence Staining
Note: For detection of covalent protein modification, such as PAR (complex damage marker) and H2AX phosphorylation (γH2AX) (DSB marker), as well as cyclobutane pyrimidine dimer (CPD)/(6-4) photoproducts (6-4PP) and 8-oxoguanine (8-oxoG) (crosslinking and base damage markers, respectively), cells must be fixed and stained with specific antibodies. In addition, it is best to confirm the results obtained with the fluorescently tagged recombinant proteins by antibody detection of the endogenous proteins, to distinguish artifacts induced by overexpression of recombinant fusion proteins.
Fix cells at different time points after damage induction (step 4.5) depending on the factors by replacing the culture media (see step 1.1) with 1.5 mL of 4% paraformaldehyde/1X PBS for 10 min at RT. Caution: Paraformaldehyde fumes are toxic and all work should be done in a ventilated fume hood.
Remove 4% paraformaldehyde/PBS and add 1 mL of 0.5% detergent/PBS for 10 min at RT.
Remove 0.5% detergent/PBS and rinse cells with 2 mL of PBS for 5 min twice, and incubate cells in 2 mL of blocking solution (10% calf serum, 1% BSA/PBS) for 1 h at RT.
Remove blocking solution and add 2 mL of PBS to cells, and replace PBS with 250 µL primary antibody solution of 3% BSA/PBS overnight at 4 °C or 1 h at RT. Use a 1:200-1:500 dilution (typical concentration approximately 1 µg/mL, empirically determined) for antibodies specific for different DNA damage markers (e.g., γH2AX/53BP1 (DSB); Ku (DSB, NHEJ); Rad51/cohesin (DSB, HR); CPD/6-4PP (crosslinking damage); 8-oxoG/DNA glycosylases (e.g., NTH1) (base damage); PAR (high dose complex damage)).
Remove the antibody solution and rinse with 2 mL of PBS for 5 min twice, remove PBS, and incubate with 250 µL 0.1% secondary antibody in 3% BSA/PBS for 1 h in the dark at RT.
Remove the secondary antibody solution and add 2 mL of PBS for 5 min twice and keep cells in 1.5-2 mL PBS at 4 °C for imaging in steps 6.1 and 6.2.
6. Quantitative Fluorescence Analysis of Immunostained or Live Cells
Capture images of stained or live cells (n >10) using microscope used for irradiation. For the epi-fluorescence microscope system, use the resolution of 1,344 x 1,024 pixels and save images as TIFF 16-bit files. For a confocal microscope, use 1X magnification; Argon laser for GFP/FITC, HeNe 594 laser for Cy3/mCherry; 512 x 512 or 1,024 x 1,024 pixels for image resolution; scan speed 5, number: 1 for quick scan or 2+ for averaging over multiple scans to improve signal to noise ratio.
- Use an image analysis programs designed to measure pixel intensity.
- Use the Volume Rectangle Tool to draw a ROI around the damage site in the individual cell image. Use the same size ROI to measure the background fluorescence signal in the nucleoplasm adjacent to, but not overlapping with the damage sites. This is important for normalization of photobleaching associated with the non-confocal microscope system.
- To measure fluorescence signals using the image analysis program, click "Volume Analysis Report" found in the tool box. A new window will appear. Under the "Data to Display" section, check only the "Name" and "Density" boxes. Then click "Done" to visualize the measurements.
- Export the values to a spreadsheet program by clicking on the table icon found at the bottom of the Volume Report window and select "Tabs (excel format)" for Field Separators and "File" for Export Destination.
- To obtain relative signals, use the spreadsheet program to divide the fluorescence intensity at the damage site (column 1) by that in the control background region (column 2) in the nucleoplasm and subtract by 1 for normalization.
- Real-time quantitative fluorescence time-course analysis using the confocal microscope
- Identify fluorescently tagged protein-positive cells using the microscope as in step 4.3.
- Perform time-lapse fluorescence imaging before damage and at different time points after damage induction in Section 4. First draw the bleach ROI (for damage induction), then select the "Acquire>Time Series" submenu, set "Number" as 20, and the " cycle delay" as 30 s, choose "bleach once after first scan", and then clicking "start B". In this case, the first image is acquired immediately before damage induction, followed by 19 image acquisitions at a 30 s interval. Intervals ("cycle delay") can be changed depending on the purpose of the study.
- Once image acquisition is complete, click "Mean", and draw the ROI at the damage region. Click "Show Table" to get a table listing the intensity within the selected ROIs. Normalize the data by dividing the fluorescence intensity of the damage site at different time points by the intensity of the same region before damage, and subtract by 1.
7. Small Interfering RNA (siRNA) Transfection
Note: Depletion of the target protein is an effective way to delineate the upstream factor requirement for the observed protein recruitment to damage sites (Section 4). Furthermore, the strategy is the best way to address the specificity of an antibody used for immunofluorescence detection (see Section 5).
Day 1: Prepare 2 wells of HeLa cells in a 24-well plate by seeding 6-8 x 104 HeLa cells in 0.5 mL culture media in each well, one for control siRNA transfection and one for PARP1 siRNA (5'-CCG AGA AAT CTC TTA CCT CAA-3').
Day 2: Confirm that cells are approximately 40-50% confluent. Perform the first round of siRNA transfection: Dilute 5 pmol siRNA into 25 µL of serum-free culture media, add 3 µL lipids based transfection reagent into the tube, mix gently, and incubate for 5-10 min at RT.
Rinse the cells with 0.5 mL fresh culture media once, and keep the cells in 0.5 mL fresh culture media (see step 1.1).
Add the RNA-lipid complexes to the cells, and mix gently by tilting the dish back and forth. Put the cells back in the incubator as in step 2.1.
Aspirate the transfection mix, and add 0.5 mL fresh culture media (step 1.1) after 6 h.
Day 3: Perform the second round of siRNA transfections (see step 7.2). Then, seed 60-100% of the cells into a 35 mm gridded coverslip dish as described in step 2.3.
Day 5: Proceed with laser microirradiation using the optimal laser power condition for maximum recruitment of the factor of interest, as determined by Section 4. Typically, efficient depletion can be observed in approximately 60-90% of HeLa cells. Note: As an alternative and complementary approach, inhibitors, if available, can be used to inhibit the function of the factor of interest32,44. For analyzing the effect of inhibiting DDR signaling, add 20 µM PARP inhibitor, 1 µM PAR glycohydrolase (PARG) inhibitor, or 10 µM DNA-dependent protein kinase with catalytic subunit (DNA-PKcs) inhibitor and 10 µM ATM inhibitor in dimethyl sulfoxide (DMSO) to the dishes 1 h prior to damage induction. Add DMSO only to control cells.
Representative Results
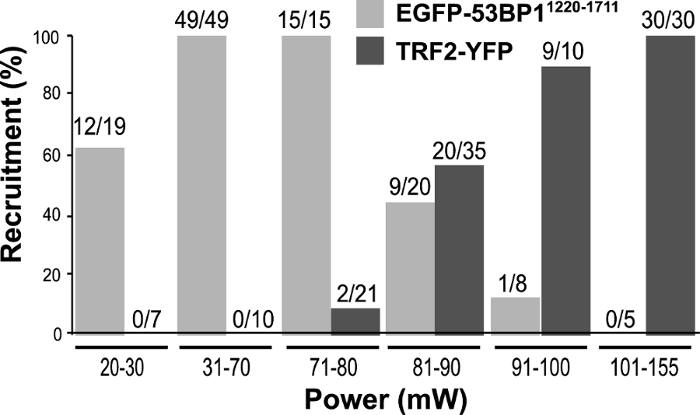
Using PtK2 cells stably expressing EGFP-53BP11220-1711 or TRF2-YFP, laser input power titration was carried out to determine the optimal condition for their recruitment (Figure 1)32.
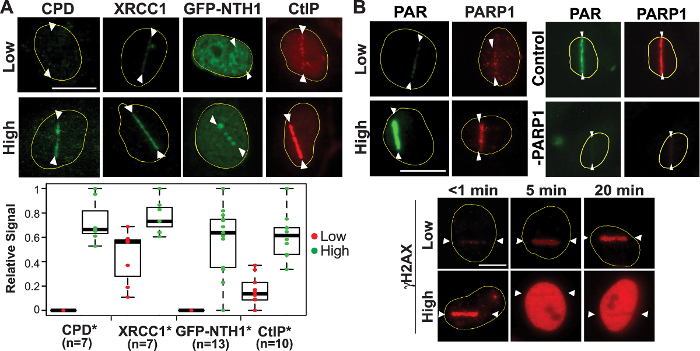
At high input-power laser damage sites, the significant clustering of CPD (crosslinking damage typically generated by ultraviolet (UV) damage) as well as GFP-NTH1 DNA glycosylase that specifically recognizes base damage were observed. In addition, higher XRCC1 and CtIP signals were observed, reflecting the increased number of strand breaks, and demonstrating that complex DNA damage is induced under this condition (Figure 2A). Other DNA glycosylases fused to fluorescent proteins (e.g., GFP-nei endonuclease VIII-like 2 (NEIL2) and GFP-8-Oxoguanine glycosylase (OGG1)) as well as condensin I can also be used as base damage markers44,59. Consistent with this, we found that the PAR response is induced significantly by high input-power laser (i.e., energy and irradiance in the focal spot in the specimen), but only weakly by low laser power in the focal spot (Figure 2B, top left). PAR signals, but not PARP1 protein localization, at damage sites were sensitive to the PARP inhibitor (data not shown). Additionally, siRNA specific for PARP1 diminished both PAR and PARP1 at damage sites (Figure 2B, top right)32. Under the high input power condition, γH2AX initially appears at the damage sites, and rapidly spreads to the entire nucleus in an ATM/DNA-PK-dependent manner (Figure 2B, bottom). Similar ATM/DNA-PK-dependent spreading of γH2AX was reported for γ-irradiation60. Consistent results were obtained with both 800 and 780 nm NIR laser systems32. Taken together, the results reveal that it is possible to titrate laser input power (and therefore, energy and irradiance in the focal spot in the cell) to find the conditions that induce different degrees of PARP response correlating with the number of DSBs and the complexity of the DNA damage.
The above results initially suggested that the presence of complex DNA damage might have caused differential recruitment of 53BP1 and TRF2. Interestingly, however, the 53BP11220-1711 recruitment to a low-power damage site was effectively inhibited by the presence of the second damage site induced by high-input-power in the same cell nucleus (Figure 3A in contrast to Figure 3B). The results indicate that high-input-power laser damage suppresses 53BP1 recruitment in trans. Inhibition of PAR by PARP inhibitor as well as inhibition of nuclear-wide γH2AX spreading by ATM/DNA-PK inhibitors restore this recruitment even to the high input power damage site (Figure 3A). This is distinct from the recruitment of Mediator of DNA Damage Checkpoint 1 (MDC1) to damage sites, which was primarily inhibited by γH2AX dispersion, and therefore, was restored by ATM/DNA-PK inhibitors, not PARP inhibitor (Figure 3A, bottom). Importantly, hyperactivation of PAR by PARG inhibition (without affecting γH2AX status) effectively suppressed initial 53BP1 recruitment even at the low input-power damage sites that normally recruit 53BP1 efficiently (Figure 3B)32. Taken together, these results indicate that PAR signaling, and not the type of damage per se, interferes with the recruitment of 53BP132. This is an example of the versatility of laser microirradiation, which allows us to examine how multiple damage sites influence and interact with each other.
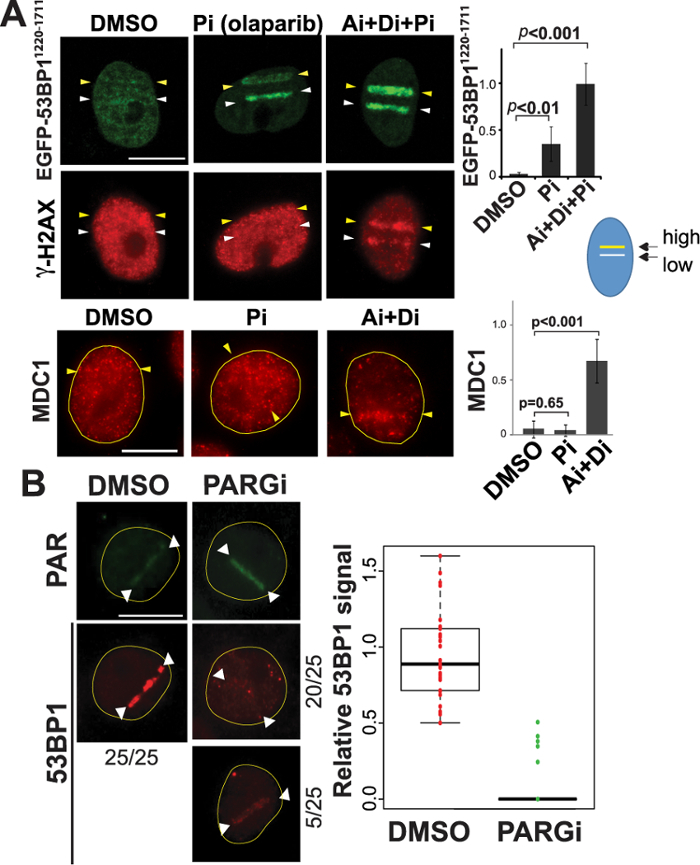
Figure 1: Titration of laser input power. To determine the laser power to use for future experiments, PtK2 cells stably expressing EGFP-53BP1 and TRF2-YFP were subjected to laser microirradiation with input powers ranging between 20 mW and 155 mW using the 800 nm NIR laser/inverted epi-fluorescence microscope system. EGFP-53BP1 recruitment was monitored for 15 min post irradiation (p.i.), while TRF2-YFP cells were followed for 6 min p.i. The numbers directly above the bars represent the number of cells that showed positive recruitment of EGFP-53BP1 or TRF2-YFP to the damage sites over the total number of cells tested. This figure was modified from Saquilabon Cruz32. Please click here to view a larger version of this figure.
Figure 2: Induction of complex damage and robust DDR signaling by high input-power laser. (A) High-input-power laser microirradiation induces complex damage including strand breaks (both SSBs and DSBs), crosslinking, and base damage. CPD, XRCC1 (SSB and DSB repair), NTH1 DNA glycosylase (BER), and CtIP (alternative NHEJ and HR) are shown (only GFP-NTH1 is a live cell image and the rest are observed following immunofluorescence staining). Quantitative fluorescence intensity measurements of the damage site recruitment were done as stated in protocol Section 7 and are shown in the boxplots. The total number of cells (n) tested can be seen under each quantified protein. *p-value <0.05. (B) Top (left): Robust PAR and PARP1 signals at high input-power damage sites. Top (right): PARP1 siRNA depletion is sufficient to abolish the PAR signal32,44. Bottom: Time course analysis of γH2AX in cells with low (top) and high (bottom) input power damage as indicated. Scale bars = 10 µm. This figure was modified from Saquilabon Cruz32. Please click here to view a larger version of this figure.
Figure 3: Differential activation of DDR signaling affects 53BP1 recruitment to damage sites. (A) Top: PtK2 cells stably expressing EGFP-53BP11220-1711 were microirradiated with both low (15%) and high (25%) input power (white and yellow arrowheads, respectively) in the same nucleus using the 780 nm NIR/confocal microscope system. This was performed in the presence of DMSO, PARP inhibitor (Pi), or the combination of ATM, DNA-PK, and PARP inhibitors (Ai+Di+Pi) as indicated. Live-cell imaging of EGFP-53BP11220-1711 was captured at 30 min after DSB induction. Cells were then fixed and stained with an antibody specific for γH2AX as a DSB marker. Changes of fluorescence intensity of EGFP-53BP11220-1711 at damage sites are shown on the right with p-values (mean values are: DMSO, 0.03 ± 0.02; Pi, 0.34 ± 0.19; Ai+Di+Pi, 0.98 ± 0.23). Bottom: The effect of Pi and Ai+Di treatment on MDC1 localization with high input-power damage as indicated. Scale bar is 10 µm. Right: Changes of fluorescence signals of MDC1 at high input-power damage sites in the presence of DMSO, Pi, or Ai+Di with p-values (mean values are: DMSO, 0.07 ± 0.10; Pi, 0.05 ± 0.07; Ai+Di, 0.86 ± 0.26). Scale bar = 10 µm. N = 10 for each intensity measurement. (B) The effect of enhancement of PAR signals by PARGi treatment on the endogenous 53BP1 recruitment to low input-power damage sites. Right: Boxplots representing the quantitative analysis of endogenous 53BP1 recruitment (detected by immunofluorescence staining) with low input-power laser (60 mW in the 800 nm NIR laser/inverted epi-fluorescence microscope system) at 15 min post irradiation in the presence of DMSO or PARGi as indicated. Left: PAR and 53BP1 immunofluorescence staining pictures of cells damaged under the same conditions with DMSO or PARGi treatment. For DMSO treatment, 100% of cells examined (N = 25) exhibited robust 53BP1 recruitment (right). In contrast, 20/25 showed no 53BP1 recruitment in the presence of PARGi (bottom right), and 5 cells showed weak recruitment. Scale bar = 10 µm. This figure was modified from Saquilabon Cruz32. Please click here to view a larger version of this figure.
Discussion
The advantages of using laser microirradiation for DDR research are:
1. It is feasible to induce different types and amounts of DNA damage from simple strand breaks to complex DNA damage, and to detect different repair factors at the DNA damage sites by adjusting the laser irradiation parameters. It is also possible to inflict damage multiple times in the same cell nucleus in order to evaluate trans effects (as in Figure 3).
2. Importantly, events happening at damage sites and the secondary events that occur elsewhere in the nucleus can easily be distinguished.
3. It is possible to carry out high-resolution real-time measurements of fluorescently tagged protein dynamics in response to damage at a single cell level, including, but not limited to: Förster Resonance Energy Transfer (FRET), Fluorescence Lifetime Imaging (FLIM), Pair Correlation Function (pCF), etc., to capture DDR in live cells.
4 .Combining with RNA interference or inhibitor treatment, it is relatively straightforward to test upstream factor requirement in a small number of cells.
5. It should also be noted that NIR (or green) laser radiation does not require pre-sensitization of DNA with nucleotide analogs or DNA-intercalating dyes, thus avoiding unwanted side effects on chromatin packing. This is in contrast to the UV laser system that requires sensitization at low dose and causes aberrant DDR at high dose39,49,61.
Critical Steps Within the Protocol/Modifications and Troubleshooting It is important that the cells are in healthy conditions when DDR analyses are performed in order to minimize artifact and experimental variations. DNA- or siRNA-transfected cells usually are more sensitive or easily detached when growing on the gridded coverslip. It helps to keep cells for shorter times in the transfection reagent as long as the transfection works, and seed more transfected cells onto the gridded coverslip dish compared to the untransfected cells. This is necessary in order to achieve comparable cell confluences for the experiments.
It has been reported that the thymidine block leads to increased levels of γH2AX in the synchronized cells62, which may affect DDR and skew the results. Furthermore, the base line γH2AX signal was shown to be higher in S and G2/M phase even without any exogenous damage63,64. We, however, do not observe any significant γH2AX signal in the nucleoplasm that would affect or interfere with the specific γH2AX response at laser-induced damage sites in the cells synchronized in S/G2 phase by a double thymidine block51.
In order to obtain consistent results, it is necessary to periodically check the laser system output parameters (every 2 to 4 weeks) and optical alignment (every 2 months). Over time, laser system components may need to be re-aligned, objective lens may be damaged and need to be replaced, and the stability of the total input power may change. The key is to evaluate the presence of damage (crosslinking (CPD and/or 6-4PP) and base damage (8-oxoG)), marker proteins (e.g., DNA glycosylases, Rad51, cohesin, and Ku), and PAR modification described in this protocol to determine the dosage and complexity of DNA damage induced by the laser microscope system used.
Limitations of the Technique Laser microirradiation has limitations. DNA damage induced by the laser beam is highly clustered in one area in the nucleus compared to naturally occurring damage that may be spread throughout the nucleus. This may change how damage signaling is propagated in the neighboring chromatin or in the nucleus. Since a relatively low number of cells can be irradiated at one time, population-based biochemical assays (e.g., Western blot, protein complex purification, chromatin immunoprecipitation (ChIP)) cannot be easily performed. Reliance on fluorescently tagged proteins (with over expression of the exogenous recombinant proteins or even with those expressed endogenously using CRISPR-mediated tag insertion) for dynamic imaging makes the studies vulnerable to protein fusion-induced artifacts. The unique nature of laser-induced damage and potential artifactual effects of tagged proteins, therefore, need to be evaluated by comparison with the results using conventional DNA damaging methods (IR, chemical damaging agents) and with the endogenous untagged proteins (though sometimes it is not possible to perform complete parallel experiments).
Significance with Respect to Existing Methods The use of sequence-specific endonucleases such as I-SceI, FokI, AsiSI, and I-PpoI provides an alternative strategy to induce site-specific DSBs (strictly simple DSBs). Factor recruitment or modifications can be analyzed by site-specific or genome-wide ChIP analysis65,66,67,68,69. Relatively synchronous induction of DSBs can be achieved by tagging the endonuclease with a steroid hormone receptor and a degradation signal (degron) for rapid nuclear translocation and degradation, respectively65,66,67,68,69. One must take into consideration, however, that the efficiency of endonuclease expression and target site accessibility, and ongoing repair status can be variable in different cells and at different target sites. These heterogeneities will be averaged in the population-based ChIP analysis, which may skew the results. Laser microirradiation, on the other hand, offers the highest possible temporal resolution (milliseconds) as well as spatial resolution (submicrometer) of damage response dynamics, which can be analyzed at the single-cell level. High damage density, however, may influence the outcome. Both strategies can be sensitive to artifacts caused by antibody accessibility and/or peptide tagging. It is, therefore, important to understand the pros and cons of both strategies, which may potentially be used to complement each other.
Future Applications With rapid advancement of fluorescence imaging and dynamics techniques, versatility of laser systems to induce different amounts and types of DNA damage at specific locations and cell cycle stage provides a valuable opportunity to interrogate the cellular and molecular responses to DNA damage with high spatiotemporal resolution at a single cell level. It will be possible, for example, to tease out damage site-specific and nucleus- and cell-wide DDR signal transduction with millisecond-resolution (e.g., detect molecular interactions or modifications that occur exclusively at damage sites, examine dynamic changes of chromatin structure in real-time, or observe real-time spreading of damage signaling from damage sites to the whole nucleus). It is also possible to follow the same cell over a long period of time to examine the effects of DNA damage on cell fate. We envision that the laser microirradiation methods, if used properly, will continue to contribute significantly to advancement of the DNA repair field.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We thank Dr. Akira Yasui at Tohoku University, Japan for the GFP-NTH1 expression plasmid, and Dr. Eros Lazzerini Denchi at the Scripps Research Institute, La Jolla, California for the TRF2-YFP and EGFP-53BP11220-1711 expression plasmids. This work was supported by the Air Force Office of Scientific Research (FA9550-04-1-0101) and the Beckman Laser Institute Inc. Foundation (to M.W.B), the Ford Foundation Fellowship from the National Academy of Sciences (to B.A.S), and NSF MCB-1615701 and CRCC CRR-17-426665 (to K. Y.).
References
- Altmeyer M, Lukas J. To spread or not to spread--chromatin modifications in response to DNA damage. Curr. Opin. Genet. Dev. 2013;23(2):156–165. doi: 10.1016/j.gde.2012.11.001. [DOI] [PubMed] [Google Scholar]
- Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol. Cell. 2010;40(2):179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misteli T, Soutoglou E. The emerging role of nuclear architecture in DNA repair and genome maintenance. Nat. Rev. Mol. Cell. Biol. 2009;10(4):243–254. doi: 10.1038/nrm2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Attikum H, Gasser SM. Crosstalk between histone modifications during the DNA damage response. Trends Cell Biol. 2009;19(5):207–217. doi: 10.1016/j.tcb.2009.03.001. [DOI] [PubMed] [Google Scholar]
- Chiolo I, et al. Double-strand breaks in heterochromatin move outside of a dynamic HP1a domain to complete recombinational repair. Cell. 2011;144(5):732–744. doi: 10.1016/j.cell.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisby M, Mortensen UH, Rothstein R. Colocalization of multiple DNA double-strand breaks at a single Rad52 repair centre. Nat. Cell Biol. 2003;5(6):572–577. doi: 10.1038/ncb997. [DOI] [PubMed] [Google Scholar]
- Ryu T, et al. Heterochromatic breaks move to the nuclear periphery to continue recombinational repair. Nat. Cell Biol. 2015;17(11):1401–1411. doi: 10.1038/ncb3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Rosell J, et al. The Smc5-Smc6 complex and SUMO modification of Rad52 regulates recombinational repair at the ribosomal gene locus. Nat. Cell Biol. 2007;9(8):923–931. doi: 10.1038/ncb1619. [DOI] [PubMed] [Google Scholar]
- Chakalova L, Debrand E, Mitchell JA, Osborne CS, Fraser P. Replication and transcription: Shaping the landscape of the genome. Nat. Rev. Genet. 2005;6(9):669–678. doi: 10.1038/nrg1673. [DOI] [PubMed] [Google Scholar]
- Schoenfelder S, et al. Preferential associations between co-regulated genes reveal a transcriptional interactome in erythroid cells. Nat. Genet. 2010;42(1):53–61. doi: 10.1038/ng.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelot C, et al. The Cohesin Complex Prevents the End Joining of Distant DNA Double-Strand Ends. Mol. Cell. 2015;61(1):15–26. doi: 10.1016/j.molcel.2015.11.002. [DOI] [PubMed] [Google Scholar]
- Lottersberger F, Karssemeijer RA, Dimitrova N, de Lange T. 53BP1 and the LINC Complex Promote Microtubule-Dependent DSB Mobility and DNA Repair. Cell. 2015;163(4):880–893. doi: 10.1016/j.cell.2015.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball AR, Jr, Yokomori K. Damage site chromatin: open or closed? Curr. Opin. Cell Biol. 2011;23(3):277–283. doi: 10.1016/j.ceb.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck C, Robert I, Reina-San-Martin B, Schreiber V, Dantzer F. Poly(ADP-ribose) polymerases in double-strand break repair: Focus on PARP1, PARP2 and PARP3. Exp. Cell Res. 2014;329(1):18–25. doi: 10.1016/j.yexcr.2014.07.003. [DOI] [PubMed] [Google Scholar]
- Basu B, Sandhu SK, de Bono JS. PARP inhibitors: mechanism of action and their potential role in the prevention and treatment of cancer. Drugs. 2012;72(12):1579–1590. doi: 10.2165/11635510-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Deeks ED. Olaparib: first global approval. Drugs. 2015;75(2):231–240. doi: 10.1007/s40265-015-0345-6. [DOI] [PubMed] [Google Scholar]
- Sonnenblick A, de Azambuja E, Azim HA, Jr, Piccart M. An update on PARP inhibitors--moving to the adjuvant setting. Nat. Rev. Clin. Oncol. 2015;12(1):27–41. doi: 10.1038/nrclinonc.2014.163. [DOI] [PubMed] [Google Scholar]
- Garber K. PARP inhibitors bounce back. Nat. Rev. Drug Discov. 2013;12(10):725–727. doi: 10.1038/nrd4147. [DOI] [PubMed] [Google Scholar]
- Lord CJ, Tutt AN, Ashworth A. Synthetic lethality and cancer therapy: lessons learned from the development of PARP inhibitors. Annu. Rev. Med. 2015;66:455–470. doi: 10.1146/annurev-med-050913-022545. [DOI] [PubMed] [Google Scholar]
- Feng FY, de Bono JS, Rubin MA, Knudsen KE. Chromatin to Clinic: The Molecular Rationale for PARP1 Inhibitor Function. Mol. Cell. 2015;58(6):925–934. doi: 10.1016/j.molcel.2015.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AG, Sarkaria JN, Kaufmann SH. Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc. Natl. Acad. Sci. 2011;108(8):3406–3411. doi: 10.1073/pnas.1013715108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahel D, et al. Poly(ADP-ribose)-dependent regulation of DNA repair by the chromatin remodeling enzyme ALC1. Science. 2009;325(5945):1240–1243. doi: 10.1126/science.1177321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottschalk AJ, et al. Poly(ADP-ribosyl)ation directs recruitment and activation of an ATP-dependent chromatin remodeler. Proc. Natl. Acad. Sci. 2009;106(33):13770–13774. doi: 10.1073/pnas.0906920106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, et al. Histone H3 methylation links DNA damage detection to activation of the tumour suppressor Tip60. Nat. Cell Biol. 2009;11(11):1376–1382. doi: 10.1038/ncb1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayrapetov MK, Gursoy-Yuzugullu O, Xu C, Xu Y, Price BD. DNA double-strand breaks promote methylation of histone H3 on lysine 9 and transient formation of repressive chromatin. Proc. Natl. Acad. Sci. 2014;111(25):9169–9174. doi: 10.1073/pnas.1403565111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoury-Haddad H, et al. PARP1-dependent recruitment of KDM4D histone demethylase to DNA damage sites promotes double-strand break repair. Proc. Natl. Acad. Sci. 2014;111(7):E728–E737. doi: 10.1073/pnas.1317585111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou DM, et al. A chromatin localization screen reveals poly (ADP ribose)-regulated recruitment of the repressive polycomb and NuRD complexes to sites of DNA damage. Proc. Natl. Acad. Sci. 2010;107(43):18475–18480. doi: 10.1073/pnas.1012946107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen DH, et al. The chromatin-remodeling factor CHD4 coordinates signaling and repair after DNA damage. J. Cell Biol. 2010;190(5):731–740. doi: 10.1083/jcb.200912135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polo SE, Kaidi A, Baskcomb L, Galanty Y, Jackson SP. Regulation of DNA-damage responses and cell-cycle progression by the chromatin remodelling factor CHD4. EMBO J. 2010;29(18):3130–3139. doi: 10.1038/emboj.2010.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeenk G, et al. The NuRD chromatin-remodeling complex regulates signaling and repair of DNA damage. J. Cell Biol. 2010;190(5):741–749. doi: 10.1083/jcb.201001048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izhar L, et al. A Systematic Analysis of Factors Localized to Damaged Chromatin Reveals PARP-Dependent Recruitment of Transcription Factors. Cell Rep. 2015;11(9):1486–1500. doi: 10.1016/j.celrep.2015.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saquilabon Cruz GM, et al. Femtosecond near-infrared laser microirradiation reveals a crucial role for PARP signaling on factor assemblies at DNA damage sites. Nuc. Acids Res. 2015;44(3):e27. doi: 10.1093/nar/gkv976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouwman P, et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Struc. Mol. Biol. 2010;17(6):688–695. doi: 10.1038/nsmb.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaspers JE, et al. Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated mouse mammary tumors. Cancer Discov. 2013;3(1):68–81. doi: 10.1158/2159-8290.CD-12-0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berns MW, Olson RS, Rounds DE. In vitro production of chromosomal lesions with an argon laser microbeam. Nature. 1969;221(5175):74–75. doi: 10.1038/221074a0. [DOI] [PubMed] [Google Scholar]
- Berns MW, et al. Laser microsurgery in cell and developmental biology. Science. 1981;213(4507):505–513. doi: 10.1126/science.7017933. [DOI] [PubMed] [Google Scholar]
- Botchway SW, Reynolds P, Parker AW, O'Neill P. Laser-induced radiation microbeam technology and simultaneous real-time fluorescence imaging in live cells. Methods Enzymol. 2012;504:3–28. doi: 10.1016/B978-0-12-391857-4.00001-X. [DOI] [PubMed] [Google Scholar]
- Ferrando-May E, et al. Highlighting the DNA damage response with ultrashort laser pulses in the near infrared and kinetic modeling. Front Genet. 2013;4:135. doi: 10.3389/fgene.2013.00135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong X, et al. Comparative analysis of different laser systems to study cellular responses to DNA damage in mammalian cells. Nucleic Acids Res. 2009;37(9):e68. doi: 10.1093/nar/gkp221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruhlak MJ, Celeste A, Nussenzweig A. Monitoring DNA breaks in optically highlighted chromatin in living cells by laser scanning confocal microscopy. Methods Mol. Biol. 2009;523:125–140. doi: 10.1007/978-1-59745-190-1_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter J, Cremer T, Miyagawa K, Tashiro S. A new system for laser-UVA-microirradiation of living cells. J. Microsc. 2003;209(2):71–75. doi: 10.1046/j.1365-2818.2003.01117.x. [DOI] [PubMed] [Google Scholar]
- Kim JS, et al. In situ analysis of DNA damage response and repair using laser microirradiation. Methods Cell Biol. 2007;82:377–407. doi: 10.1016/S0091-679X(06)82013-3. [DOI] [PubMed] [Google Scholar]
- Kim JS, Krasieva TB, LaMorte VJ, Taylor AMR, Yokomori K. Specific recruitment of human cohesin to laser-induced DNA damage. J. Biol. Chem. 2002;277(47):45149–45153. doi: 10.1074/jbc.M209123200. [DOI] [PubMed] [Google Scholar]
- Kong X, et al. Condensin I Recruitment to Base Damage-Enriched DNA Lesions Is Modulated by PARP1. PLoS One. 2011;6(8):e23548. doi: 10.1371/journal.pone.0023548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heale JT, et al. Condensin I interacts with the PARP-1-XRCC1 complex and functions in DNA single-stranded break repair. Mol. Cell. 2006;21(6):837–848. doi: 10.1016/j.molcel.2006.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradshaw PS, Stavropoulos DJ, Meyn MS. Human telomeric protein TRF2 associates with genomic double-strand breaks as an early response to DNA damage. Nat. Genet. 2005;37(2):193–197. doi: 10.1038/ng1506. [DOI] [PubMed] [Google Scholar]
- Huda N, et al. Recruitment of TRF2 to laser-induced DNA damage sites. Free Radic. Biol. Med. 2012;53(5):1192–1197. doi: 10.1016/j.freeradbiomed.2012.07.024. [DOI] [PubMed] [Google Scholar]
- Williams ES, et al. DNA double-strand breaks are not sufficient to initiate recruitment of TRF2. Nat. Genet. 2007;39:696–698. doi: 10.1038/ng0607-696. [DOI] [PubMed] [Google Scholar]
- Bekker-Jensen S, et al. Spatial organization of the mammalian genome surveillance machinery in response to DNA strand breaks. J. Cell Biol. 2006;173(2):195–206. doi: 10.1083/jcb.200510130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JS, et al. Independent and sequential recruitment of NHEJ and HR factors to DNA damage sites in mammalian cells. J. Cell Biol. 2005;170(3):341–347. doi: 10.1083/jcb.200411083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong X, et al. Distinct functions of human cohesin-SA1 and cohesin-SA2 in double-strand break repair. Mol. Cell Biol. 2014;34(4):685–698. doi: 10.1128/MCB.01503-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu N, et al. Scc1 sumoylation by Mms21 promotes sister chromatid recombination through counteracting Wapl. Genes Dev. 2012;26(13):1473–1485. doi: 10.1101/gad.193615.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fradet-Turcotte A, et al. 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature. 2013;499:50–54. doi: 10.1038/nature12318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryde F, et al. 53BP1 exchanges slowly at the sites of DNA damage and appears to require RNA for its association with chromatin. J. Cell Sci. 2005;118:2043–2055. doi: 10.1242/jcs.02336. [DOI] [PubMed] [Google Scholar]
- Zgheib O, Pataky K, Brugger J, Halazonetis TD. An oligomerized 53BP1 tudor domain suffices for recognition of DNA double-strand breaks. Mol. Cell. Biol. 2009;29:1050–1058. doi: 10.1128/MCB.01011-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan L, et al. In situ analysis of repair processes for oxidative DNA damage in mammalian cells. Proc. Natl. Acad. Sci. 2004;101(38):13738–13743. doi: 10.1073/pnas.0406048101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinde E, Kong X, Yokomori K, Gratton E. Chromatin dynamics during DNA repair revealed by pair correlation analysis of molecular flow in the nucleus. Biophys. J. 2014;107(1):55–65. doi: 10.1016/j.bpj.2014.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva BA, Stambaugh JR, Yokomori K, Shah JV, Berns MW. DNA damage to a single chromosome end delays anaphase onset. J. Biol. Chem. 2014;289(33):22771–22784. doi: 10.1074/jbc.M113.535955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinde E, Yokomori K, Gaus K, Hahn KM, Gratton E. Fluctuation-based imaging of nuclear Rac1 activation by protein oligomerisation. Sci. Rep. 2014;4:4219. doi: 10.1038/srep04219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer B, et al. Clustered DNA damage induces pan-nuclear H2AX phosphorylation mediated by ATM and DNA-PK. Nuc. Acids Res. 2013;41(12):6109–6118. doi: 10.1093/nar/gkt304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 1999;146(5):905–915. doi: 10.1083/jcb.146.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurose A, Tanaka T, Huang X, Traganos F, Darzynkiewicz Z. Synchronization in the cell cycle by inhibitors of DNA replication induces histone H2AX phosphorylation: an indication of DNA damage. Cell Prolif. 2006;39(3):231–240. doi: 10.1111/j.1365-2184.2006.00380.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Tanaka T, Kurose A, Traganos F, Darzynkiewicz Z. Constitutive histone H2AX phosphorylation on Ser-139 in cells untreated by genotoxic agents is cell-cycle phase specific and attenuated by scavenging reactive oxygen species. Int. J. Oncol. 2006;29(2):495–501. [PubMed] [Google Scholar]
- MacPhail SH, Banáth JP, Yu Y, Chu E, Olive PL. Cell cycle-dependent expression of phosphorylated histone H2AX: reduced expression in unirradiated but not X-irradiated G1-phase cells. Radiat. Res. 2003;159(6):759–767. doi: 10.1667/rr3003. [DOI] [PubMed] [Google Scholar]
- Aymard F, et al. Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nat. Struc. Mol. Biol. 2014;21(4):366–374. doi: 10.1038/nsmb.2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkovich E, Monnat RJJ, Kastan MB. Roles of ATM and NBS1 in chromatin structure modulation and DNA double-strand break repair. Nat. Cell Biol. 2007;9(6):683–690. doi: 10.1038/ncb1599. [DOI] [PubMed] [Google Scholar]
- Caron P, et al. Cohesin protects genes against γH2AX Induced by DNA double-strand breaks. PLoS Genet. 2012;8(1):e1002460. doi: 10.1371/journal.pgen.1002460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein M, Derheimer FA, Tait-Mulder J, Kastan MB. Nucleolin mediates nucleosome disruption critical for DNA double-strand break repair. Proc. Natl. Acad. Sci. 2013;110(42):16874–16879. doi: 10.1073/pnas.1306160110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacovoni JS, et al. High-resolution profiling of gammaH2AX around DNA double strand breaks in the mammalian genome. EMBO J. 2010;29(8):1446–1457. doi: 10.1038/emboj.2010.38. [DOI] [PMC free article] [PubMed] [Google Scholar]