

Abstract
Vasculogenesis is a complex process by which endothelial stem and progenitor cells undergo de novo vessel formation. Quantitative assessment of vasculogenesis has become a central readout of endothelial progenitor cell functionality, and therefore, several attempts have been made to improve both in vitro and in vivo vasculogenesis models. However, standard methods are limited in scope, with static measurements failing to capture many aspects of this highly dynamic process. Therefore, the goal of developing this novel protocol was to assess the kinetics of in vitro vasculogenesis in order to quantitate rates of network formation and stabilization, as well as provide insight into potential mechanisms underlying vascular dysfunction. Application of this protocol is demonstrated using fetal endothelial colony forming cells (ECFCs) exposed to maternal diabetes mellitus. Fetal ECFCs were derived from umbilical cord blood following birth, cultured, and plated in slides containing basement membrane matrix, where they underwent vasculogenesis. Images of the entire slide wells were acquired using time-lapse phase contrast microscopy over 15 hours. Images were analyzed for derivation of quantitative data using an analysis software called Kinetic Analysis of Vasculogenesis (KAV). KAV uses image segmentation followed by skeletonization to analyze network components from stacks of multi-time point phase contrast images to derive ten parameters (9 measured, 1 calculated) of network structure including: closed networks, network areas, nodes, branches, total branch length, average branch length, triple-branched nodes, quad-branched nodes, network structures, and the branch to node ratio. Application of this protocol identified altered rates of vasculogenesis in ECFCs obtained from pregnancies complicated by diabetes mellitus. However, this technique has broad implications beyond the scope reported here. Implementation of this approach will enhance mechanistic assessment and improve functional readouts of vasculogenesis and other biologically important branching processes in numerous cell types or disease states.
Keywords: Developmental Biology, Issue 131, Vasculogenesis, endothelial colony forming cells, microscopy, gestational diabetes, time-lapse imaging, kinetic, network formation
Introduction
The ability of endothelial progenitor cells to undergo vasculogenesis, or de novo vessel formation, is critical in establishing embryonic vasculature during development1. Additionally, further vessel formation and maturation of pre-existing vessels, which is known as angiogenesis, is also a key process in development and in postnatal life to maintain blood flow and homeostasis throughout the body2. Every organ in the body is dependent on the vascular system for delivery of oxygen and nutrients, and for the removal of waste3. If vascular homeostasis is not maintained, such that blood vessel formation and repair are either insufficient or in excess, vascular diseases can result4. Therefore, vascular formation and adaptation are commonly studied, as they are essential in the maintenance of organ health and are implicated in the development of numerous pathologic states.
Due to an increased understanding of the involvement of the vascular system in development, as well as in disease manifestation and progression, assays have been developed to model vasculogenesis and angiogenesis in vitro and in vivo5,6,7. Modeling vessel formation involves plating vascular cells, such as endothelial cells, in basement membrane matrix that promotes cell organization and formation of vessel networks8,9,10. Typically, following overnight incubation, images of cell networks are captured at a single time point, resulting in a small number of images for analysis11,12,13. Therefore, analytic approaches have largely been developed and optimized for single time point assessments10,14. However, static analyses are simply insufficient at capturing the dynamic process of vessel formation and provide limited insight into potential mechanisms involved. Although increasing imaging frequency would likely provide the necessary data to identify formation kinetics, application of previously developed analytics to a multi-time point imaging approach would be inefficient and labor intensive14. Additionally, despite development of commercially available analyses, a pay-per-image fee renders this option cost-prohibitive for kinetic studies in which thousands of images are generated15. Therefore, there is a need in the field for a streamlined and efficient approach to capture and quantify vasculogenesis in vitro, including the ability to analyze large image sets generated by time-lapse live cell microscopy.
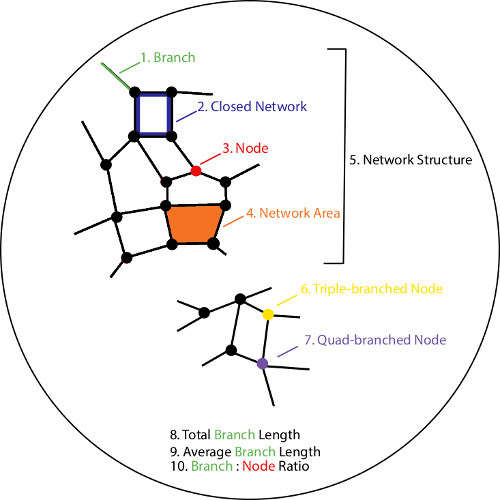
To overcome these limitations, a new approach was developed with the purpose of expanding single time point imaging to enable dynamic assessment of vasculogenesis with images acquired every 15 min16. By capturing multiple time points over several hours, this approach provides a more detailed depiction of the process of vasculogenesis, as well as insight into potential mechanisms contributing to the formation and maintenance of vessel networks. In addition to improving the frequency and quality of image acquisition, this approach incorporates novel software in the form of an open-source plug-in17. The software, referred to as Kinetic Analysis of Vasculogenesis (KAV), is a streamlined application that incorporates image processing and analysis specifically for large image sets generated from multi-time point acquisitions. KAV analyzes phase contrast images through image segmentation followed by skeletonization18. Ten parameters of network structure are quantified by KAV including: branches, closed networks, nodes, network areas, network structures, triple-branched nodes, quad-branched nodes, total branch length, average branch length, and the branch to node ratio (see schematic in Figure 1)16. Application of the KAV approach includes a novel, calculated phenotype of in vitro vasculogenesis, referred to as the branch to node ratio. Our recent work demonstrated that this ratio is indicative of network connectivity and may be associated with other cellular processes involved in network formation, such as motility16.
Although fetal endothelial colony forming cell (ECFC) vasculogenesis was assessed in these studies, this image acquisition and analysis approach can be readily applied to evaluate any cell types that undergo vasculogenesis or angiogenesis. Additionally, this approach can be used to identify altered vascular function resulting from a variety of pathologic states, such as gestational diabetes mellitus, as shown in these studies. Furthermore, this method could be adapted to assess network formation and branching, which are important for other biologically relevant processes. Thus, the potential impact of applying this novel approach to unique biological systems is yet to be determined.
Protocol
1. Preparations
- Culture endothelial colony forming cells (ECFCs) NOTE: Fetal ECFC samples used in these experiments were isolated from human umbilical cord blood and cultured by the Angio BioCore (ABC) at the Indiana University School of Medicine as previously described6. Routine quality control phenotyping was conducted within the ABC to confirm expression of endothelial antigens as previously described19,20,21. Samples used in experiments were passaged minimally (2-3 times). All tissue culture work was performed in a tissue culture hood. All reagents, including tissue culture plates and solutions, were sterile.
- Two days prior to the experiment, coat 100 mm round tissue culture plates with 6 mL of 0.05 mg/mL type 1 collagen, and incubate at 37 °C overnight. NOTE: Type 1 collagen is diluted in double-distilled water containing 20 nM acetic acid to a working concentration of 0.05 mg/mL.
- The day prior to the experiment, trypsinize and replate ECFCs on the type 1 collagen coated plates in 1.1.1. (See Note under 1.1).
- To trypsinize ECFCs, aspirate media from plated cells and wash with 7 mL of PBS. Aspirate PBS, then add 2-3 mL of 0.5% trypsin and incubate cells for 2-5 min at 37 °C.
- Immediately following incubation with trypsin, add 3 mL of EGM2 and pipet up and down to dislodge all adherent cells. Transfer cell suspension to a 15-mL conical tube.
- Centrifuge all samples at 500 x g for 5 min at room temperature.
- Aspirate supernatant and re-suspend cell pellets in 1-2 mL of EGM2. Mix the cell suspension thoroughly and remove a small aliquot (20-40 μL) for counting.
- Add equal parts trypan blue to the cell aliquot and pipette 10 µL onto a hemocytometer. Count the number of cells in the four primary quadrants of the hemocytometer. NOTE: Use both sides of the hemocytometer for all sample counts to increase accuracy.
- Wash the collagen-coated tissue culture plates, which were prepared in step 1.1.1, twice with 7 mL of PBS. After aspirating the second PBS wash, add 10 mL of endothelial growth medium 2 (EGM2) culture media supplemented with 10% fetal bovine serum to the plates.
- Using the cell counts obtained in step 1.1.7, calculate the volume of cell suspension required to plate 4 x 105 cells. Plate 4 x 105 ECFCs on the tissue culture plates prepared in step 1.1.8, and incubate at 37 °C and 5% carbon dioxide (CO2) overnight.
- Prior to the experiment, store supplies at 4 °C overnight
- Store a 3" x 2" metal plate, slide (keep in package until open in hood), a box of sterile 20 microliter (μL) tips, and basement membrane matrix (matrix). NOTE: The matrix is kept at -80 °C for long-term storage; always thaw it at 4 °C overnight or for several hours prior to use.
Confirm availability of adequate space for the images on the computer's hard-drives. NOTE: Using the configuration outlined in this protocol, approximately 60 gigabytes (GB) of space were required per experiment (4 GB/well). However, space estimates are based on hardware configuration and will need to be determined for each system.
2. Protocol 1: Experimental Setup: Preparing the Slide
- Gather and prepare supplies
- The day of the experiment, gather supplies for plating cells on the slide, including a bucket of ice, a small container of dry ice, a pair of scissors, and a 20 μL pipette. Spray all items with 70% ethanol, wipe dry with paper towels, and place items into a sterile tissue culture hood.
- Gather all supplies stored at 4 °C (see step 1.2) including the metal plate, box of tips, slide, and matrix. Spray the box of tips with 70% ethanol and place the box in the container of dry ice. NOTE:Remove matrix from the 4 °C fridge only when ready to use and always keep the matrix on ice.
- Spray the metal plate with 70% ethanol and embed it into the ice in the ice bucket. Open the package containing the slide, and place the slide on the metal plate to keep it cold.
- Load matrix onto the slide
- Set the pipette to 10 μL and remove the tip from cold tip box with the pipette. Cut approximately 1 cm off the end of the tip with the scissors. NOTE:Cut perpendicular to the tip to reduce bubbles in the matrix. The pipette can be set to a volume slightly greater than 10 μL to account for matrix sticking to the inside of the tip.
- Slowly draw the matrix into the pipette tip. Immediately place the pipette tip on the center of the well. Gently touch the pipette tip to the bottom of the well and slowly push the matrix out. NOTE:Do not over pipette, as this will cause bubbles to form. If a bubble forms, pop it immediately using a small tip.
- The slide contains 15 individual wells. Repeat the previous step for each well. Use a new pipette tip for each well to maintain consistency and reduce matrix solidifying inside the tip.
- Once the slide is loaded with all wells containing matrix, push the lid all the way onto the slide, and incubate at 37 °C for 30-60 min until the matrix solidifies. NOTE: Do not let the matrix dry out. Add a small volume (2-3 mL) of PBS in the cap of a 50-mL conical tube near the slide in the incubator to reduce the risk of matrix drying.
3. Protocol 2: Plating ECFCs on the Matrix
- Remove adherent ECFCs from tissue culture plates and collect in suspension
- Obtain tissue culture plates containing the ECFCs plated the previous day (see section 1.1.9). Aspirate EGM2 media and wash adherent ECFCs once with 7 mL of PBS.
- Aspirate PBS, add 2-3 mL of trypsin, and incubate adherent cells for 2-5 min at 37 °C.
- Immediately following incubation, add 3 mL of EGM2 and pipet up and down to dislodge all adherent cells. Transfer cell suspension to a 15-mL conical tube.
- Repeat steps 3.1.1-3.1.3 for the remaining three ECFC samples until all four cell samples are collected in suspension.
- Prepare master mixes
- Centrifuge all samples at 500 x g for 5 min at room temperature.
- Aspirate supernatant and re-suspend cell pellets in 1-2 mL of EGM2.
- Mix the cell suspension thoroughly and remove a small aliquot (20-40 μL) for counting.
- Add equal parts trypan blue to the cell aliquot and pipette 10 μL onto a hemocytometer. Count the number of cells in the four primary quadrants of the hemocytometer. NOTE: Use both sides of the hemocytometer for all sample counts to increase accuracy.
- Using the cell counts from above, calculate the volume of each cell sample that contains 1.4x104 cells. Mix all cell suspensions thoroughly and aliquot 1.4 x 104 cells into a fresh tube to prepare a master mix for each experimental condition. Add EGM2 media so that the final volume of each master mix is 175 μL.
- Mix each master mix by gently pipetting and aliquot 50 μL of master mix into individual wells on the slide. NOTE:All samples are plated in triplicate, with master mixes calculated for 3.5 wells to ensure sufficient volume for 3 wells.
- Incubate the slide
- Incubate the slide at 37 °C until it can be moved into the microscope chamber for imaging. NOTE: The time between plating and imaging should be as brief as possible to limit data loss during the early stages of vasculogenesis.
4. Protocol 3: Microscope Setup and Image Acquisition
NOTE: For our studies, Protocols 2 and 3 were conducted simultaneously by separate individuals. If Protocols 2 and 3 must be completed by the same individual, Protocol 3 Steps 4.1 and 4.2 below should be completed prior to initiating Protocol 2 above.
- Turn on the microscope and computer
- Approximately one to two hours prior to starting the experiment, turn on and preheat the stage top incubator to 37 °C, set the CO2 to 5%, and set the humidity to 85%. Fill the incubator's humidity reservoir, if empty.
- Place an empty chambered slide in one of the two open positions, and fill with distilled water. Secure the feedback thermometer for chamber temperature control, if available. NOTE: If the feedback thermometer is available, use this "incubator temperature" to verify that the chamber has equilibrated.
- Turn on a secondary blower set to 39-42 °C to heat the slides from below and minimize condensation.
- Add a second slide as a blank until the experimental slide can be added. This slide serves as a location placeholder for configuration of the image acquisition settings for individual wells during setup.
- Add water to the reservoir surrounding the wells on the second slide to mimic the conditions during the imaging experiment. The water surrounding the wells enables assessment of humidity during microscope setup and preheating. NOTE: Pre-equilibration of the stage-top incubator and secondary blower is critical for maintaining a stable culturing environment. Most live cell chamber controllers give real-time feedback of temperature, CO2, and humidity to assess the readiness of the system. Prior to continuing, check for the accumulation of humidity on the slide and excessive drying of the reservoir as filled in 4.1.5. Humidity may need to be adjusted if there is excessive drying or condensation in the chamber. To increase the humidity, add wipes wetted with distilled water. To decrease condensation, either decrease the humidity setting as adjusted in 4.1.1, or as condensation could be a sign of low temperature, check the chamber temperature settings and the positioning of the secondary blower.
- Configure basic image acquisition settings during equilibration. NOTE:Image acquisition is configured through software that controls the stage, shutter, and camera. A multidimensional setup tool is the easiest way to configure the software. Furthermore, the software will need to have autofocus routines to ensure focus control over the duration of the experiment.
- Use the software to set multiple stage positions for each well. For this protocol, select the center of the wells.
- Configure the image acquisition such that at each stage position, the entire well is imaged. For example, use a tilescan with 10-20% image overlap and approximately 5 x 5 fields, depending upon camera and final magnification.
- Configure the time lapse for imaging every 15 min. NOTE: Using the configuration outlined in this protocol, all 15 wells of the slide are imaged within a 15 min interval.
- Load the experimental slide
- Following equilibration of the incubator chamber, replace the blank slide with the experimental slide. NOTE:An equilibrated chamber will have stable temperature, CO2, and humidity for at least 20 min. Once the experimental slide is ready, it is imperative that steps are completed quickly to minimize the "dead-time" between plating and the initial time point captured by the microscope. Furthermore, to facilitate comparison between imaging experiments, this time should be consistent. In these experiments, all steps outlined in Protocol 3 Section 4.3 required approximately 30 min to complete.
- Remove slide lid and add water to the reservoir surrounding the wells on the slide. NOTE: Lid is removed for the duration of the imaging experiment.
- Configure final image acquisition settings
- Place the CFI Plan Fluor DL 10X objective in position and select the Ph1 phase ring on the condenser. With the first well in focus, adjust the diascopic light path for Köhler illumination and configure phase optics by checking alignment of phase ring in the condenser with the objective phase mask.
- Configure the exposure time and adjust the lamp intensity for phase contrast, which is typically less than 500 ms.
- Cycle through each stage position set in 4.2.1 and confirm that the center of the well is selected, and adjust it, if necessary.
- At each stage position, focus on the cells plated in the well and set the stage position's z position used for imaging in the software.
- Test the autofocus mechanism for the microscope. If the autofocus is hardware based, ensure it is on and positioned correctly for all stage positions. NOTE:As the specimen and stage may drift vertically during the experiment, it is important that the software check the autofocus at each stage position and every time-point to ensure reliable imaging during the time-course. If possible, an autofocus position determined in the previous time point should be used as the starting point for the subsequent autofocus routine. This way, on-going drift can be adjusted for easily.
- Reduce or extinguish the lights in the microscope room and start the imaging experiment.
5. Protocol 4: Image Processing and Kinetic Analysis of Vasculogenesis (KAV)
Save images from individual time points as an image stack for each well NOTE:Most software for image capture can export multipage tiffs (stacks) of time series. KAV is designed to work on datasets that may include multiple time points. Thus, analysis occurs on the entire image stack for a given stage position. If required, the image processing software can import single images from each time point to reconstruct an image stack.
Open the image processing software on the computer
- Add KAV to the image processing software
- Drag the KAV file from a folder into the grey bar at the bottom of the software window. Click 'Save' to add the software to the list.
- Open the image stack to be analyzed
- Click 'File' then 'Open' through the dropdown menu in the image processing software, or drag the image file into the grey bar as in step 5.3.1.
- Convert the images to 8 bit by clicking 'Image', 'Type', '8 bit'.
- Create a region of interest (ROI)
- Open the ROI Manager by clicking on 'Analyze' in the tool bar. Then select 'Tools' followed by 'ROI Manager'.
- Create a new ROI by clicking on the circle or rectangle selection tools in the Toolbar and drawing the desired selection area on the image. Click 'Add' in the ROI manager window to save that shape. NOTE: A previously created ROI can be opened by dragging saved ROIs (zipped folder) into the 'Drag and Drop' bar to open in Manager.
- Click on the ROI label listed in the ROI Manager to view it on image. NOTE: If the ROI won't appear on the image, create a second ROI (any size/shape) and add it to the manager. Once both ROIs appear in the list, click back and forth between the two and the desired ROI will appear on the image.
- Align the ROI if needed. Once the ROI is in place on the image in the desired location, go to the Menu and click 'Edit' then 'Clear Outside'. This will delete the image data outside of the ROI (useful for non-rectangular shapes) for every image in the stack. NOTE: You can also click on 'Image' and 'Crop' if you are using a rectangularly shaped ROI.
- Run the KAV software
- Click 'Plug-ins' in the menu bar.
- Select the 'Analyze Network' Plug-in. This will open up a window with options to alter the settings in the Plug-in.
- Change the thresholding method using the drop down arrow.
- Once all of the appropriate settings are selected, click 'OK' to initiate software processing. NOTE: Appropriate settings are determined through visualization of skeleton and mask renditions generated by KAV, which are indicative of the accurary of the software to identify and discern cells and networks from background in the phase contrast image.
- Save the KAV-generated data
- Once the plug-in has finished the analysis, two windows will pop-up on the screen including a data table with numerical values and a stack of fused images depicting skeleton and mask renditions.
- Save the numerical values by navigating to the upper left corner of the data table and clicking 'Edit' then 'Copy' or 'Cut' to paste values into an excel spreadsheet.
- Click on the fused skeleton and mask image. Save the fused skeleton/mask image stack as a Tagged Image File Format (TIFF) file by clicking 'File,' 'Save As,' 'TIFF.'
- Save the cropped phase contrast image stack (created using ROI) by clicking 'File,' 'Save As,' 'TIFF.'
- Click on the ROI Manager window and select the ROI used to crop the images. Click 'More' followed by 'Save' to save the ROI for future use. NOTE: Using the same ROI will help maintain consistency across analyses.
Representative Results
KAV produces visual representations of network structure The contrast between the ECFCs and the matrix background in the phase contrast images enables KAV to identify cell-specific structures. ECFC networks identified by KAV are represented pictorially as skeleton and mask renditions to illustrate structures used by the software for quantification (Figure 2A). Importantly, qualitative assessment of the skeleton and mask renditions enables rapid identification of KAV sensitivity and accuracy, which can be useful in determining optimal threshold settings and interpreting analysis outcomes. When the quality of the phase contrast images is high and sufficient contrast is achieved, KAV accurately identifies ECFC networks as indicated by similarity between the phase contrast image and the KAV-generated skeleton and mask renditions (Figure 2A and Video 1).
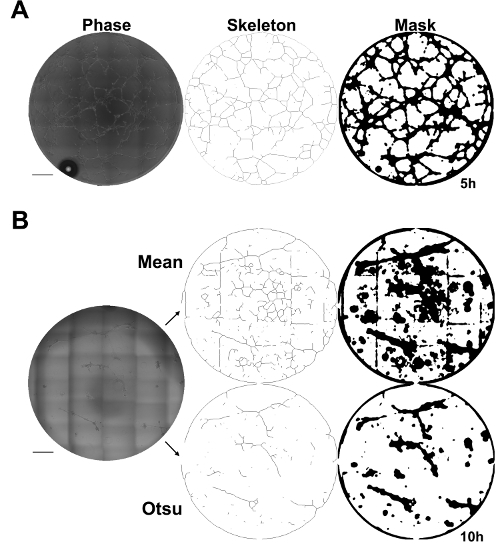
Alternatively, if phase contrast images do not have high contrast and/or artefacts of imaging such as gridding occur, network detection accuracy is reduced and outcomes become ambiguous (Figure 2B). Additionally, formation of air bubbles within the cell media can also obscure detection accuracy (Figure 2). However, problems with image quality can often be overcome through selection of different thresholding methods included in the KAV software. For example, the phase contrast image in Figure 2B was analyzed using identical image processing settings except for the thresholding method. From the Skeleton and Mask renditions, it is evident that the Otsu thresholding resulted in a more accurate detection of the ECFC networks shown in the phase contrast image (Figure 2B). Therefore, image quality is critical to achieving accurate and meaningful results in this assay. However, different thresholding methods included in the KAV user interface allow adjustment of image analysis based on the quality of the input images.
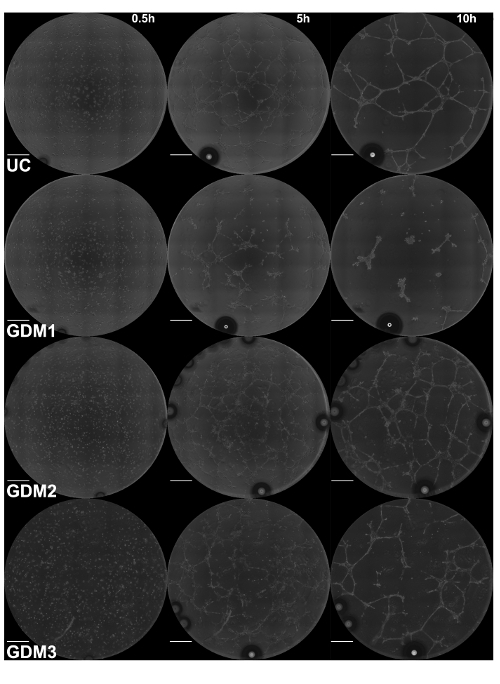
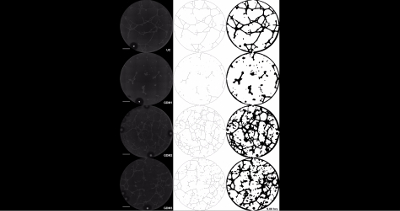
Time-lapse microscopy identifies qualitative and quantitative differences in ECFC vasculogenesis following intrauterine gestational diabetes mellitus exposure Recently, the KAV analytic approach was applied to assess fetal ECFC function following exposure to maternal type 2 diabetes mellitus (T2DM) in utero16. Using KAV, altered kinetics of vasculogenesis were identified in fetal ECFCs exposed to T2DM. However, in addition to T2DM exposure, which occurs throughout the entire gestation, gestational diabetes mellitus (GDM), or the development of glucose intolerance commonly in the third trimester of pregnancy, also impairs ECFC function13. Therefore, KAV was applied to determine if GDM-exposed ECFCs also display altered kinetics of ECFC network formation. Phase 1 (0-5 h) and Phase 2 (5+ h) were assessed using time lapse microscopy coupled with KAV analysis (Figure 3). Representative phase contrast images acquired at the start of image acquisition (0.50 h) and throughout the time course (5.00 and 10.00 h) depict ECFC network formation in the four samples tested from a single experimental day (Figure 3 and Video 1). Despite equivalent cell loading, as observed in the phase contrast images at 0.50 hours, qualitative differences in network structure are evident at the 5.00 and 10.00-hour time points. ECFCs from the uncomplicated pregnancy (UC) form a complex and intricate network 5 hours post-plating, similar to our previously published data16. Conversely, ECFCs from GDM sample 1 (GDM1) form very few network structures that are not interconnected. However, this pattern is not reflected in all ECFC samples obtained from GDM pregnancies, indicative of heterogeneity between samples. Samples GDM2 and GDM3 display greater network formation compared to GDM1, although the patterns of connectivity appear altered compared to the UC sample. Importantly, KAV measures several structural components of ECFC networks to identify both obvious and subtle phenotypes.
In addition to generating skeleton and mask renditions of network structure, KAV quantitates ten metrics of network structure, including the number of individual network structures, nodes, triple-branched nodes, quadruple-branched nodes, branches, total branch length, average branch length, branch to node ratio, total closed networks, and network area16. KAV compiles the data for each sample into a single table of values for all images of the time course. Table 1 includes representative raw data from one imaging study for one ECFC sample. Parameters of network structure measured by KAV are organized into columns, and the data for sequential images acquired over time are organized into rows. For example, in our studies, image 1 was obtained 30 min post-plating with subsequent images being collected every 15 min. Raw values generated by KAV can then be graphed or further analyzed using more complex statistical approaches16.
Five graphs depicting mean values of network structure from three separate experiments are shown for a single uncomplicated sample (UC) and the three GDM samples (Figure 4). The UC sample in these experiments performed similarly to previously analyzed UC samples16. Previously, it was identified that ECFC vasculogenesis in vitro is bi-phasic, consisting of Phase 1 (0 - 5 h) and Phase 2 (5 - 10 h)16. This pattern is consistent in the current studies, as evidenced by the graph depicting closed network data (Figure 4). The UC sample formed a greater number of closed networks compared to all three of the GDM samples. Interestingly, three of the four samples appear to have a similar time to maximal number of closed networks, which occurs between 2.5 and 3 hours. However, the GDM2 sample was slower to reach maximal closed networks, which occurred 5 hours post-plating. And, despite the GDM2 sample forming fewer maximal networks compared to the UC sample, the networks it forms were maintained similarly to the UC sample over time. Conversely, GDM1 and GDM3, which formed fewer networks compared to the UC sample, also exhibited an overall reduction in network number over time. Overall, from the graph depicting closed network number, it is evident that all ECFC samples displayed a bi-phasic pattern of network formation, however the rate of formation and the maximal number of networks achieved vary across samples.
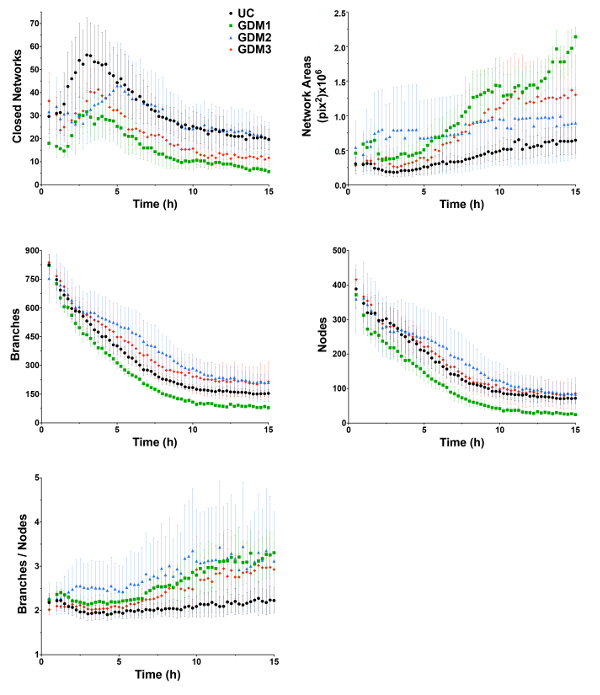
Network area represents the average area within the closed networks formed by the ECFCs. Therefore, the greater the closed network number, the smaller the network areas. This pattern is reflected in the network area graphs where the UC sample, which formed a larger number of closed networks, had smaller network areas over time compared to the three GDM samples (Figure 4). GDM2, which reached maximal closed networks more slowly, exhibited high network area initially, however the area stabilized over time, and this was most similar to the UC sample at 15 hours. Over time, the average network area in all samples increased due to network de-stabilization. However, some samples, like GDM1 and GDM3, exhibited a more rapid increase in network area, which is likely indicative of decreased stability compared to other samples exhibiting a more gradual increase.
GDM-exposed ECFCs exhibit reduced network stability The ratio of branches to nodes is a novel phenotype calculated by KAV and identified in our previous studies, and this is indicative of network connectivity16. In the current study, the UC sample had a low ratio, which represented a high level of network connectivity that was maintained over time (Figure 4). Conversely, the three GDM samples had a higher ratio of branches to nodes, especially in Phase 2 of network formation. This observation demonstrates reduced connectivity and de-stabilization of network structures.
Overall, GDM1 formed fewer nodes and branches compared to the other samples, with the reduction maintained over the course of the experiment. GDM2 and GDM3 formed and maintained a greater number of branches compared to the UC sample, especially between 5-15 h. The numbers of nodes detected in the GDM2 and GDM3 networks were more similar to the number of nodes in the UC sample, especially between 10-15 h. A greater number of branches, but a similar number of nodes, could account for the increased branch to node ratio evident at the later time points in the GDM samples. Importantly, simultaneous changes in branch and node number can be difficult to interpret in separate graphs. However, the novel branch to node ratio offers a way to assess how changes, including slight changes difficult to detect in the individual graphs, in both branch and node number, result in altered network connectivity.
Figure 1: Schematic outlining 10 parameters quantified by Kinetic Analysis of Vasculogenesis (KAV). KAV quantitates ten distinct parameters of network structure. All parameters are color-coded and outlined in the schematic numerically (1-10). Parameters include the number of branches (green, 1), closed networks (blue, 2), and nodes (red, 3), average network area (orange, 4), the number of network structures (black, 5), triple-branched nodes (yellow, 6), and quad-branched nodes (purple, 7), as well as the total (9) and average (10) branch length, and the ratio of the number of branches to the number of nodes (10). Please click here to view a larger version of this figure.
Figure 2: Kinetic Analysis of Vasculogenesis (KAV) quantitates network structure. (A) Skeleton and mask renditions of network structures identified by KAV using phase contrast images provide visual representation of network structures identified and quantified by KAV. Scale bar = 500 µm. (B) A representative phase contrast image of ECFC networks 10 h post-plating that has low contrast and grid marks from stitching individual images together. Different thresholding methods, such as Mean or Otsu, can be selected in the KAV plug-in to improve quantitation accuracy, if phase contrast images have low contrast or if gridding occurs. Skeleton and mask renditions of the phase contrast image are shown for both Mean and Otsu thresholding methods. Phase contrast images were captured using a 10X objective. Scale bar = 500 µm. Please click here to view a larger version of this figure.
Figure 3: Intrauterine GDM exposure alters ECFC network formation. Images of ECFC network formation were captured at 15 min intervals for 15 h by phase contrast microscopy. Representative phase contrast images at 5 h increments, starting at the time of plating (0.5 h), are shown for the UC and three GDM samples. Phase contrast images were captured using a 10X objective. Scale bar = 500 µm. Please click here to view a larger version of this figure.
Figure 4: ECFCs exposed to intrauterine GDM exhibit altered network formation kinetics. ECFCs were obtained from an uncomplicated pregnancy (UC, black) and three pregnancies complicated by gestational diabetes mellitus (GDM 1-3, red). Phase contrast images were captured at 15 min intervals for 15 h. Kinetic Analysis of Vasculogenesis (KAV) software quantitated closed networks, network areas, branches, nodes, and the ratio of branches to nodes. The data illustrated represent the mean ± standard error of the mean (SEM) of three separate experiments for each sample. Please click here to view a larger version of this figure.
 Video 1: Intrauterine GDM exposure alters kinetics of ECFC network formation. Images of ECFC network formation were captured at 15 min intervals for 15 h by phase contrast microscopy. Phase contrast images are shown for the UC and three GDM samples over 15 h starting at 0.5 h. The scale bar represents 500 µm. Please click here to view this video. (Right-click to download.)
Video 1: Intrauterine GDM exposure alters kinetics of ECFC network formation. Images of ECFC network formation were captured at 15 min intervals for 15 h by phase contrast microscopy. Phase contrast images are shown for the UC and three GDM samples over 15 h starting at 0.5 h. The scale bar represents 500 µm. Please click here to view this video. (Right-click to download.)
| Image | Total Branch Networks | Nodes | Triples | Quadruples | Branches | Total Branch Length* | Avg Branch Length* | Branch to Node Ratio | Closed Networks | Network area** |
| 1 | 382 | 290 | 278 | 11 | 943 | 47210 | 124 | 3.25 | 9 | 480214 |
| 2 | 284 | 345 | 337 | 8 | 940 | 50699 | 179 | 2.72 | 16 | 264469 |
| 3 | 205 | 376 | 366 | 9 | 910 | 55728 | 272 | 2.42 | 27 | 150621 |
| 4 | 162 | 422 | 407 | 15 | 947 | 59692 | 368 | 2.24 | 40 | 98713 |
| 5 | 132 | 454 | 441 | 12 | 967 | 61923 | 469 | 2.13 | 53 | 72951 |
| 6 | 122 | 435 | 419 | 14 | 907 | 62587 | 513 | 2.09 | 68 | 56948 |
| 7 | 88 | 429 | 411 | 17 | 852 | 63983 | 727 | 1.99 | 74 | 51429 |
| 8 | 68 | 451 | 437 | 13 | 874 | 63960 | 941 | 1.94 | 74 | 51780 |
| 9 | 93 | 437 | 417 | 18 | 905 | 60901 | 655 | 2.07 | 49 | 82919 |
| 10 | 126 | 511 | 487 | 24 | 1075 | 61981 | 492 | 2.1 | 37 | 116857 |
| 11 | 49 | 397 | 384 | 12 | 737 | 61823 | 1262 | 1.86 | 79 | 48805 |
| 12 | 81 | 376 | 364 | 10 | 751 | 56817 | 701 | 2 | 63 | 64431 |
| 13 | 98 | 509 | 482 | 26 | 1028 | 60799 | 620 | 2.02 | 44 | 98056 |
| 14 | 93 | 449 | 416 | 31 | 909 | 58282 | 627 | 2.02 | 45 | 94081 |
| *rounded to nearest micron, **rounded to nearest micron^2 |
Table 1: Representative raw data from one imaging study for one ECFC sample.
Discussion
KAV enables evaluation of large data sets with multiple time points Traditionally, quantitation of vasculogenesis in vitro has consisted of a single, or a few time point measurements. This static approach is simply inadequate for capturing and quantifying a dynamic and complex process. Therefore, this novel method was developed to enable efficient analyses of network formation kinetics to gain insight into potential molecular mechanisms involved in the dynamic process of vasculogenesis. Efficiency and automation are important components in the development of novel techniques to generate and analyze large quantities of imaging data. KAV was developed specifically to analyze large image stacks consisting of hundreds of images to decrease the time and labor required to derive biologically meaningful conclusions from time-lapse data sets. Importantly, KAV conducts image processing and data generation in a matter of seconds for small (less than 100 images) image stacks and minutes for larger (greater than 100 images) image stacks, resulting in unparalleled efficiency. Additionally, data organization into spreadsheets by time and parameters measured enables rapid generation of graphical presentation and statistical analysis.
Challenges in successful application of this approach Although this assay includes improvements to both image acquisition and analysis, some challenges may impede successful implementation. The four main obstacles include humidity stability, timing precision from plating to image acquisition, software and hardware reliability, and management of large data sets generated for each experiment. Stable live cell imaging over several hours can be challenging. Specifically, appropriate and consistent humidification is required for the imaging chambers. Angiogenesis slides are used in this assay, which are designed for parallelizing matrix-based angiogenesis assays. Their low volume, approximately 50 µL, makes evaporation and condensation significant considerations for extended culture conditions. To combat this experimental issue, it is necessary to use an incubator that regulates humidity. However, it may also be required to augment this regulation if excessive drying of the imaging chamber occurs over time. We found that the simplest approach to increase humidity is to increase water surface area in the chamber. To accomplish this goal, our protocol suggests the following three water sources: a second chambered slide filled with water, which is also where the incubator temperature is measured, water in the area surrounding the wells on the slides, and wetted filter paper or wipes. Conversely, condensation occurs when the air temperature in the room cools the bottom of the slide and the humidity inside the chamber condenses on the slides and the wells. This complication can lead to a dilution of the cell media and a lensing effect that interferes with the phase contrast. In these studies, condensation was minimized through application of heated air (39-42 °C) onto the bottom of the slide.
A key consideration for any time-lapse study is consistency between experiment initiation and imaging. The timing of when imaging starts is critical for interpretation of downstream events. To ensure the timing precision of this assay, the time between initial plating and the first imaged time point should be tightly controlled. In practice, this "dead time" can be minimized, but more importantly, it needs to be consistent to allow experiments on different days to be compared. For instance, in this protocol we expect a dead time of approximately 30 min. This can be facilitated by the proximity of experimental preparation and imaging facilities.
What might seem like mundane details at first glance are important for software, hardware stability, and data management. The software and associated hardware drivers, network environment and automated software updates all affect the stability of the software. It is worth testing this protocol in a "dry run" to identify bottlenecks and potential sources of problems in the image acquisition; for instance, check for unreliable live cell setup, stage, shutter, and camera reliability, and the institutional information technology policies on automated operating system and software updates.
Data management is an ongoing concern of any imaging experiment that generates hundreds to thousands of images. Fortunately, commercial software often checks for available space prior to starting an imaging experiment. However, this assay also includes post-capture image processing and analysis that can add to the hard drive space burden and interfere with imaging experiments, if available space is limited. Further, the security and stability of the computer cannot be guaranteed, even with hardware solutions such as a redundant array of identical disks. Thus, a data management strategy to ensure space on the computer's hard drives for ongoing experiments and a robust remote archiving, for instance with an institutional archiving resource, is necessary.
Strategies to Maximize Image Quality Although the ability of KAV to accurately discern ECFCs from the matrix background is robust, the sensitivity of the assay is dependent on image quality. For example, if the image has low contrast, the software will detect cell networks with less accuracy (Figure 2B). The quality of the phase contrast is impacted by several factors, including the settings of the microscope used for image acquisition, loading of the matrix and the cell suspension media during assay preparation, and maintenance of media levels within the wells throughout imaging. To optimize the phase contrast for these assays, minor adjustments to phase ring alignment in the microscope were made to improve contrast. Additionally, sample preparation was rigorously tested to ensure equal and consistent media loading. If the amount of matrix and/or media loaded into the wells is either below or above the optimum range, a meniscus can form leading to altered phase contrast. Finally, due to the small volume of liquid in the wells, it is critical to maintain media levels by minimizing evaporation and condensation, as noted above. Overall, assay optimization is critical to generate high quality contrast images for analysis.
In addition to phase contrast quality, other factors can also impact assay outcomes including media contamination, imperfections in the matrix, and particles or debris in the matrix or media. Contamination is a hazard of this assay since it involves live cell imaging over several hours in a microscopy chamber. To reduce the risk of contamination, the matrix and cell suspensions are loaded onto the slide in a sterile hood. In addition, each sample is typically plated in duplicate or triplicate for an experiment to minimize the risk of data loss due to unforeseen issues such as debris or particles confounding the analysis.
Sample heterogeneity drives need for increased assay sensitivity Heterogeneity has been observed and reported in previous studies of ECFCs exposed to GDM13. A high level of heterogeneity in cell function observed in these samples is speculated to be attributable to several factors including severity of maternal disease, duration of disease, and management style used to regulate blood glucose levels. Importantly, this analytic approach captures the dynamic range of ECFC vasculogenesis, making it feasible to identify phenotypic differences due to functional heterogeneity in samples from GDM pregnancies. Overall, the use of primary patient samples, such as the ones used in these studies, can introduce larger variability compared to a cell line. As greater emphasis is placed on translational studies involving multiple primary animal or human samples with functional variability, assay sensitivity is essential for detection and derivation of biologically meaningful measurements and conclusions. Therefore, development of approaches like KAV will improve the quantity and quality of data generated by in vitro vasculogenesis and angiogenesis assays to enable more robust observations and conclusions. Furthermore, these data will facilitate future investigation of underlying molecular mechanisms that contribute to altered ECFC vasculogenesis following intrauterine GDM exposure.
Future applications of this approach Although this method was applied to assess fetal ECFC vasculogenesis in vitro in these studies, the potential applications of this approach are numerous. This technique can be readily implemented to study any cell population that participates in the processes of vasculogenesis or angiogenesis. Specifically, it can be used to study individual cell populations, as was demonstrated in this study, but it could also be applied to co-culture systems. In the future, it would be beneficial to expand this approach beyond the two-dimensional in vitro assay to assess three-dimensional (3D) models. Although the current version of KAV would be insufficient for quantitating 3D imaging data, a similarly designed time-lapse, multi-parametric analytic approach specifically for 3D or in vivo models would inform if observations made in two-dimensions in vitro are representative of cell function in a more biologically relevant setting.
Disclosures
The authors have nothing to disclose.
Acknowledgments
The authors acknowledge Lucy Miller, Leanne Hernandez, Dr. David Haas, Brittany Yeley (Indiana University School of Medicine), Dr. Karen Pollok, Julie Mund, Matthew Repass, and Emily Sims (Angio BioCore at the Indiana University Simon Cancer Center) for excellent technical assistance in deriving ECFC samples. The authors also acknowledge Drs. Maureen A. Harrington, Edward F. Srour, Richard N. Day, Mervin C. Yoder, and Matthias A. Clauss (Indiana University School of Medicine) for scholarly discussion as well as Janice Walls (Indiana University School of Medicine) for administrative support. All imaging was performed at the Indiana Center for Biological Microscopy, Indiana University School of Medicine. This work was supported by the National Institutes of Health (R01 HL094725, P30 CA82709, and U10 HD063094) and the Riley Children's Foundation. Additionally, this publication was made possible with partial support from the National Heart, Lung, and Blood Institute of The National Institutes of Health under Award # T32 HL007910.
References
- Risau W. Mechanisms of angiogenesis. Nature. 1997;386(6626):671–674. doi: 10.1038/386671a0. [DOI] [PubMed] [Google Scholar]
- Tung JJ, Tattersall IW, Kitajewski J. Tips, stalks, tubes: notch-mediated cell fate determination and mechanisms of tubulogenesis during angiogenesis. CSH Med. 2012;2(2):006601. doi: 10.1101/cshperspect.a006601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473(7347):298. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P. Angiogenesis in health and disease. Nat Med. 2003;9(6):653–660. doi: 10.1038/nm0603-653. [DOI] [PubMed] [Google Scholar]
- Arnaoutova I, Kleinman HK. In vitro angiogenesis: endothelial cell tube formation on gelled basement membrane extract. Nat Protoc. 2010;5(4):628–635. doi: 10.1038/nprot.2010.6. [DOI] [PubMed] [Google Scholar]
- Prasain N, Meador JL, Yoder MC. Phenotypic and Functional Characterization of Endothelial Colony Forming Cells Derived from Human Umbilical Cord Blood. J Vis Exp. 2012. [DOI] [PMC free article] [PubMed]
- Simons M, et al. State-of-the-Art Methods for Evaluation of Angiogenesis and Tissue Vascularization: A Scientific Statement From the American Heart Association. Circ Res. 2015;116(11):99–132. doi: 10.1161/RES.0000000000000054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabtree B, Subramanian V. Behavior of endothelial cells on Matrigel and development of a method for a rapid and reproducible in vitro angiogenesis assay. In Vitro Cell Dev-An. 2007;43(2):87–94. doi: 10.1007/s11626-007-9012-x. [DOI] [PubMed] [Google Scholar]
- Khoo CP, Micklem K, Watt SM. A comparison of methods for quantifying angiogenesis in the Matrigel assay in vitro. Tissue Eng Part C-Me. 2011;17(9):895–906. doi: 10.1089/ten.tec.2011.0150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allier CP, et al. Video lensfree microscopy of 2D and 3D culture of cells. SPIE BiOS. 2014;8947:89471. [Google Scholar]
- Wang Y, Chen Q, Zhang Z, Jiang F, Meng X, Yan H. Interleukin-10 overexpression improves the function of endothelial progenitor cells stimulated with TNF-α through the activation of the STAT3 signaling pathway. Int J Mol Med. 2015;35(2):471–477. doi: 10.3892/ijmm.2014.2034. [DOI] [PubMed] [Google Scholar]
- Ingram DA, et al. In vitro hyperglycemia or a diabetic intrauterine environment reduces neonatal endothelial colony-forming cell numbers and function. Diabetes. 2008;57(3):724–731. doi: 10.2337/db07-1507. [DOI] [PubMed] [Google Scholar]
- Blue EK, et al. Gestational diabetes induces alterations in the function of neonatal endothelial colony-forming cells. Pediatr Res. 2014;75(2):266–272. doi: 10.1038/pr.2013.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpentier G. ImageJ contribution: Angiogenesis Analyzer. 2012.
- WimTube. 2012. Available from: https://www.wimasis.com/en/products/13/WinTube.
- Varberg KM, et al. Kinetic analyses of vasculogenesis inform mechanistic studies. Am J Physiol-Cell Ph. 2017. [DOI] [PMC free article] [PubMed]
- Schindelin J, et al. Fiji: an open-source platform for biological-image analysis. Nat Met. 2012;9(7):676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arganda-Carreras I, Fernández-González R, Muñoz-Barrutia A, Ortiz-De-Solorzano C. 3D reconstruction of histological sections: Application to mammary gland tissue. Microsc Res Techniq. 2010;73(11):1019–1029. doi: 10.1002/jemt.20829. [DOI] [PubMed] [Google Scholar]
- Ingram DA, et al. Identification of a novel hierarchy of endothelial progenitor cells using human peripheral and umbilical cord blood. Blood. 2004;104(9):2752–2760. doi: 10.1182/blood-2004-04-1396. [DOI] [PubMed] [Google Scholar]
- Mead LE, Prater D, Yoder MC, Ingram DA. Isolation and characterization of endothelial progenitor cells from human blood. Current Protocols in Stem Cell Biology. 2008. Chapter 2, Unit 2C.1. [DOI] [PubMed]
- Yoder MC, et al. Redefining endothelial progenitor cells via clonal analysis and hematopoietic stem/progenitor cell principals. Blood. 2007;109(5):1801–1809. doi: 10.1182/blood-2006-08-043471. [DOI] [PMC free article] [PubMed] [Google Scholar]