

Abstract
The creation of mutant lines by genome editing is accelerating genetic analysis in many organisms. CRISPR/Cas9 methods have been adapted for use in the African clawed frog, Xenopus, a longstanding model organism for biomedical research. Traditional breeding schemes for creating homozygous mutant lines with CRISPR/Cas9-targeted mutagenesis have several time-consuming and laborious steps. To facilitate the creation of mutant embryos, particularly to overcome the obstacles associated with knocking out genes that are essential for embryogenesis, a new method called leapfrogging was developed. This technique leverages the robustness of Xenopus embryos to "cut and paste" embryological methods. Leapfrogging utilizes the transfer of primordial germ cells (PGCs) from efficiently-mutagenized donor embryos into PGC-ablated wildtype siblings. This method allows for the efficient mutation of essential genes by creating chimeric animals with wildtype somatic cells that carry a mutant germline. When two F0 animals carrying "leapfrog transplants" (i.e., mutant germ cells) are intercrossed, they produce homozygous, or compound heterozygous, null F1 embryos, thus saving a full generation time to obtain phenotypic data. Leapfrogging also provides a new approach for analyzing maternal effect genes, which are refractory to F0 phenotypic analysis following CRISPR/Cas9 mutagenesis. This manuscript details the method of leapfrogging, with special emphasis on how to successfully perform PGC transplantation.
Keywords: Genetics, Issue 132, Xenopus, genetics, mutants, CRISPR/Cas9, TALEN, genome editing, mutagenesis, loss of function
Introduction
How genotype encodes phenotype has been a major question in biology since the rediscovery of Mendel's laws. An understanding of the roles of genes, their regulation and interactions within gene networks, and the functions of encoded products promises to provide tools for uncovering new biology and ameliorating disease states. For over half a century1, the African clawed frog, Xenopus, has been a leading model for studies on a wide variety of topics in basic biology and biomedicine, including the genetic control of development. Historically, most research on Xenopus has used the allotetraploid frog, X. laevis, but more recently, due to its diploidy, X. tropicalis has been developed as an amphibian genetic model. Complete genome sequences have been assembled from both Xenopus species2,3. The "frog community" is now at a turning point where basic genome modification technology permits the study of gene function, virtually at will. Programmable CRISPR/Cas9 endonucleases have made the mutagenesis of genes highly efficient, with biallelic mutation possible in most cells of the animal4,5,6,7,8,9,10,11. These studies, underscored by Bhattacharya et al.12 and Shigeta et al.13, have shown that the function of many genes can be studied by mutagenesis in F0 mosaic animals. This approach has many advantages; however, Cas9-sgRNA-microinjected embryos often display variable phenotypes due to incomplete loss of function (LOF). More significantly, the generation of mutant lines is highly advantageous for some applications—in particular when studying genes that have a maternal mRNA contribution. Maternal mRNAs and proteins, and their influences on epigenetics, persist for an extended period into embryogenesis14,15,16, rendering the early developmental contributions of many genes refractory to F0 analyses. Therefore, other LOF approaches are required.
When seeking to create mutant lines, the path to obtaining homozygous LOF embryos has several obstacles. First, efficient mutagenesis to produce biallelic mutations can be disadvantageous because loss of essential gene functions results in failure to survive to sexual maturity, interfering with the production of a viable line. A common solution is the careful titration of the amount of Cas9-sgRNA delivered. Here, the goal is to achieve a balance between reducing lethality while also maximizing germline mutagenesis efficiency. A second problem arises from the standard breeding scheme, where phenotypic analyses are deferred until the F2 generation. Using the standard approach, sexually mature F0 animals that transmit mutant alleles through the germline are outcrossed to produce F1 heterozygous "carriers", which are then grown to sexual maturity. Two F1 heterozygotes are then intercrossed to produce F2 mutant embryos at an expected Mendelian frequency of 25%. Thus, two generations of breeding are necessary for analysis of mutant phenotypes. Mutant animals could be either homozygotes or compound heterozygotes (i.e., progeny containing two different mutant alleles, which depends on the genotypes of the parental animals used in the F1 intercross).
These obstacles can be overcome by confining programmable nuclease-mediated mutagenesis to germ cells, which underlies a new method called leapfrogging17. Leapfrogging has two main components: (1) the microinjection of Cas mRNA together with sgRNA, or nuclease-sgRNA complexes (or, in principle, TALENs or zinc finger nucleases) at the single-cell stage to efficiently mutagenize embryonic genomes, followed by (2) the transplantation of PGCs into wildtype sibling embryos, where the endogenous PGCs were removed. When both steps are efficient, complete germline replacement with mutant germ cells can be obtained. Blackler demonstrated in the early 1960s that Xenopus PGCs could be transplanted between embryos at the late neurula and early tailbud stages18,19. For leapfrogging, Blackler's approach was modified by performing the transplantations at the blastula stage17, when PGCs are localized in the vegetal pole of the embryo20,21. Transplantation before gastrulation has two main advantages. First, the engraftment of transplants and the subsequent normal development was found to be more efficient when the transplantation is performed at the blastula stage (unpublished observations). Second, by performing blastula-stage transplantations shortly after zygotic transcription has begun, one can avoid the lethality arising from developmental gene mutations that disrupt gastrulation or that otherwise lead to malformed late neurulae. PGC transplant-bearing ("leapfrogged") embryos are grown to sexual maturity, and intercrosses between these F0 animals have demonstrated that, in many cases, 100% of the F1 progeny display the LOF phenotype (most being compound heterozygotes), indicating complete germline replacement with the targeted mutations.
It is expected that leapfrogging will accelerate genetic approaches in Xenopus. Leapfrogging also provides an alternative to the "host transfer" method22 for the LOF analysis of maternal-effect genes (unpublished observations). In the current publication, a detailed description of the method, especially focusing on PGC transplantation, is presented in X. tropicalis (with minor modifications for X. laevis). The transplantation of PGCs is demonstrated here to facilitate a more rapid transfer of this technology to other laboratories working with Xenopus. The principles of this method should be successful in other amphibians (e.g., urodeles), and organism-specific modifications in the methodology should allow for application to many other animals in which efficient mutagenesis can be accomplished and PGCs are readily transplantable.
Protocol
All methods described here have been approved by the Institutional Animal Care and Use Committee of the University of California, Irvine.
1. Preparations for PGC Transplantation
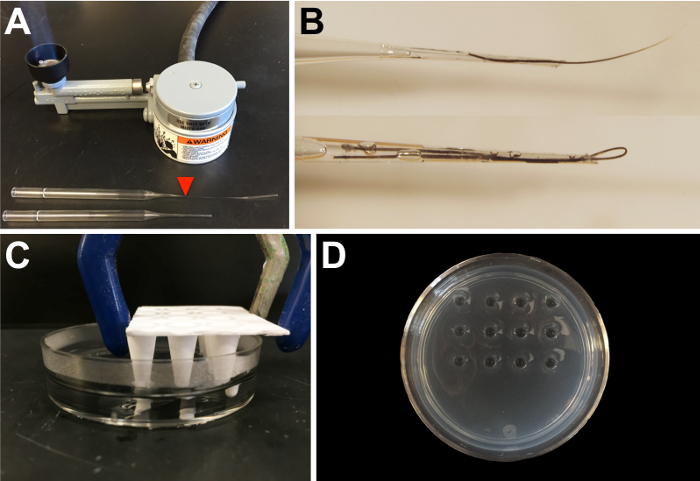
Prepare dissection tools in advance, as previously described23. NOTE: Eyebrow hair knives are used to make incisions with the assistance of a hair loop to stabilize the embryo while performing the surgeries. Eyebrow hairs and hair loops are glued into borosilicate glass Pasteur pipettes that have first been drawn in a flame and broken on the thinned portion (see Figure 1A, arrowhead) to create a shorter, narrower tip (Figure 1B) for greater dexterity when performing surgeries.
Generate sgRNAs using procedures previously described24 . NOTE: Briefly, short DNA templates coding for sgRNAs are created using overlapping deoxyoligonucleotides containing 5' bacteriophage T7 promoters, which are annealed and "filled in" by an error-free, thermostable DNA polymerase. In vitro sgRNA synthesis reactions are incubated for several hours to overnight (depending on the kit used). Reactions are DNAse-treated to remove the template. The sgRNAs are purified by standard methods of phenol/chloroform extraction and ammonium acetate/isopropanol precipitation, according to the kit manufacturer's instructions24.
Shortly before performing microinjections, prepare Cas9-sgRNA complexes by first denaturing ~250 ng of sgRNA in a 3 µL total volume of RNAse-free (diethylpyrocarbonate-treated) H2O at 60-65 °C for 5 min, followed by quick-cooling on ice for 5 min. Centrifuge the sgRNA for a few seconds, add 1 µL of 1 µg/µL Cas9 protein, and incubate for 10 min at 37 °C to promote complex formation. NOTE: Following this incubation, the tube can be kept on ice while preparing the embryos for microinjection.
Obtain X. tropicalis embryos by in vitro fertilization using standard methods, as previously described25. De-jelly25,26 the embryos at 10 min post-fertilization. NOTE: Fertilization time is considered the time after the sperm suspension is added to the eggs and the dish is flooded with 1/9thX Marc's Modified Ringers (MMR)26. For X. tropicalis25, unlike X. laevis, perform the de-jellying in 1/9x MMR containing 3% cysteine free base (not the HCl salt), pH adjusted to 7.8-8.0, followed by multiple washes in 1/9x MMR. Transfer the embryos to an agarose (1%)-coated dish containing 1/9x MMR at room temperature.
Immediately transfer the embryos to be injected to a 1% agarose-coated dish containing 1x MMR using a Pasteur pipette.
- Inject at the 1-cell stage. See references26,27 for detailed descriptions of Xenopus microinjection methods.
- Inject each embryo at a single site in the animal pole, with 4 nL of Cas9-sgRNA complex (final amount = 1 ng of Cas9 and ~250 pg of sgRNA; see the Discussion)24. After 10-20 min of injection site healing, transfer the embryos to an agarose-coated dish containing 1/9x MMR and incubate at 25 °C. Also create a dish of uninjected sibling embryos that will serve as graft recipients.
- Prepare Petri plates (60 mm) that contain a ~5 mm-thick layer of 1% agarose made in 0.3x MMR, if doing transplantations in X. tropicalis, or 1x MMR for X. laevis.
- Create depressions ~3-4 mm deep by inserting a 3- x 4-well mold (created by cutting a 96-well PCR plate) into the molten agarose when pouring the plates (see Figure 1Cand D). Hold the mold several millimeters above the bottom of the Petri dish using a clamp attached to a ring stand until the agarose has hardened.
- Additionally, make a 24-well plate to house individual embryos by coating the wells with a thin layer of 1% agarose made in 1/9x MMR. NOTE: The culture medium added to these wells is 1/9x MMR supplemented with 50 µg of gentamycin sulfate/mL.
2. Transplantation of PGCs
When the embryos just reach Nieuwkoop and Faber28 blastula stage 9 (Figure 2A), ~4.5 h post-fertilization (hpf) for X. tropicalis, remove them from the 25 °C incubator and allow them to equilibrate to room temperature for an additional 0.5 h.
To begin the transplantation at 5 hpf (Figure 2B), use a Pasteur pipette to transfer one PGC donor embryo (injected with Cas9-sgRNA) to the 60 mm agarose dish containing depressions (created in step 1.7) in 0.3x MMR (or 1x MMR if using X. laevis). Also transfer one uninjected sibling embryo, the graft recipient, to this dish. NOTE: It is critical that the identities of each of these embryos are not confused.
Manually remove the vitelline envelopes from each embryo using forceps and rotate both embryos so that their vegetal poles are in view and accessible for surgery. NOTE: A description of manual de-vitellination of embryos has previously been described26.
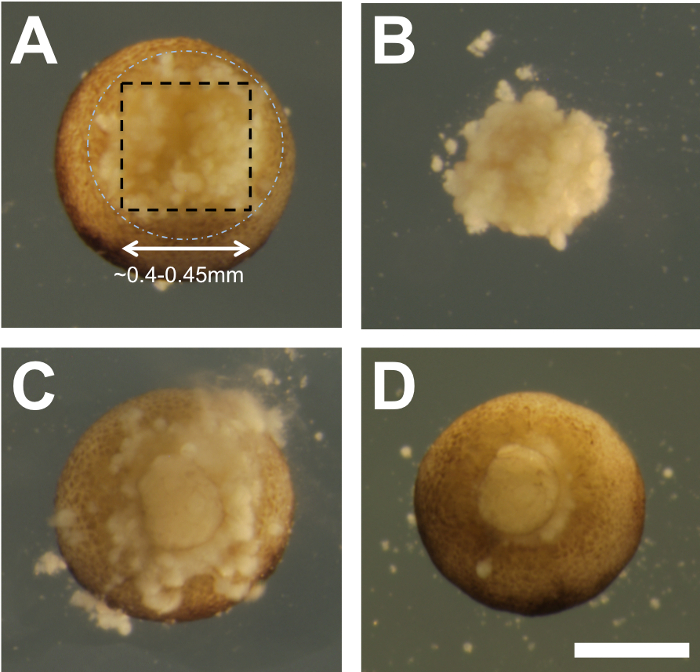
Using the sharp tip of an eyebrow hair knife, make four shallow incisions in the shape of a square on the vegetal pole of the recipient embryo, inside the zone where future bottle cells that mark the blastopore will form. Make the incisions by inserting the tip of the eyebrow hair knife into the embryo, just below the surface, and make upward slicing movements while stabilizing the embryo with the hair loop. NOTE: Figure 2 shows vegetal views of an embryo at half-hourly intervals from 4.5-7.0 hpf to show the cell size and to provide a guide for estimating where the bottles cells will form (dashed white circle). The aim is to make incisions where the dashed black box is indicated.
Once the four sides of the square are delineated by incisions, deepen each incision with the eyebrow hair knife using similar cutting motions to reach a depth of approximately 1/3 to 1/2 of the distance to the blastocoel floor.
Free the vegetal tissue explant from the recipient embryo (Figure 3Aand B) by making a horizontal (parallel to the vegetal surface) cut(s) in the deep vegetal region. NOTE: The size of the PGC-containing vegetal explant is approximately 0.4-0.45 mm per side and 0.25-0.3 mm in depth. This vegetal explant from the recipient embryo is no longer needed and should therefore be set aside to be discarded.
Working quickly, repeat this procedure (steps 2.4-2.6) on the PGC donor embryo to create a similarly sized vegetal tissue fragment for transplantation. NOTE: It is important to carry out this second dissection with little delay to minimize the time for the recipient embryo to heal its open wound.
Once the tissue containing PGCs is removed from the donor embryo, use the eyebrow hair knife and hair loop to move this explant into position in the opening created in the recipient embryo. NOTE: The interior surface of the donor graft must be facing the interior of the recipient embryo. It is not necessary to match the orientations of the dorsal-ventral and left-right axes of the graft with the recipient embryo.
Use the long edge of the shaft of the eyebrow hair knife, held parallel to the surface of the graft, to gently press the graft into the opening in the vegetal surface of the recipient embryo.
Once the graft has been placed, use a hairloop or the shaft of an eyebrow hair knife to gently slide the "carcass" of the donor embryo across the agarose surface and into an agarose depression. Make sure that the open wound of the carcass is facing the bulk liquid. If not, use the eyebrow hair knife to rotate the embryo to achieve this orientation.
Slide the recipient embryo, with the graft healing in place, into an adjacent depression and likewise make sure that the grafted vegetal pole tissue is facing the bulk liquid.
Repeat this procedure (steps 2.1-2.11) using another pair of donor and recipient embryos to create another transplant/carcass pair; transfer these to empty depressions. NOTE: It is critical that one makes notations to keep track of matched pairs of carcasses and transplant-bearing embryos. The donor carcasses will be used as a proxy for the efficiency of Cas9-sgRNA-induced mutagenesis in the transplanted PGCs, which cannot be easily assayed until the transplant-bearing animals reach sexual maturity (see step 3.3, below, and the Discussion).
Continue making transplants until the embryos reach approximately early gastrula stage 10, which is approximately 6.5 hpf in X. tropicalis (see Figure 2E).

3. Post-healing Care of Embryos
NOTE: Grafts heal into place within ~30 min, and it is normal to observe some yolky debris from cell lysis exuding from the embryo (Figure 3C).
- Once healed, use a Pasteur pipette to very gently transfer embryos from the depressions to individual wells of an agarose-coated 24-well plate. The exudate will be removed (Figure 3D) by fluid mixing during the transfer.
- Rotate the embryos so that they are placed vegetal-pole up, facing the bulk solution. Again, place the donor carcasses and graft recipients in adjacent wells to assist in keeping track of embryo pairs; record this information.
Transfer the 24-well plate containing embryos to a 25 °C incubator for overnight culture. NOTE: The next day, the embryos will have reached tailbud stages.
Move the graft recipients to clean agarose-coated 6-well plates containing 1/9x MMR supplemented with 50 µg of gentamycin sulfate/mL, with 1 embryo per well. Maintain a clear record of donor carcasses that match these graft recipients.
To assess mutagenesis efficiency by sequencing PCR amplicons5,17,24 or using other methods that rely on PCR amplicons (see the Discussion), move individual carcasses to 0.2 mL PCR strip tubes. Remove most of the medium and homogenize in 100 µL lysis buffer24 containing proteinase K (PK) by repeated up-and-down pipetting using a P200 pipette.
Perform embryo lysis24 at 56 °C for 6 h to overnight to permit PK digestion. Inactivate PK by heating it to 90-95 °C for 10 min, followed by quick cooling to 4 °C. Use lysates without further cleanup steps to seed the PCR reactions to obtain short amplicons for direct Sanger DNA sequencing24. Store the lysates at -20 °C. NOTE: Within several days, the tadpoles will reach feeding stages (approximately stage 45-46) and can be fed a suspension of planktonic powder (sera Micron).
After ~1 week, with daily changes of culture medium to maintain cleanliness and minimize microbial overgrowth, move tadpoles to small tanks in a circulating aquatic system with drip flow. Use the sequencing data to segregate tadpoles with highly efficiently mutagenized PGCs from tadpoles with lower mutagenesis efficiency. Once metamorphosis is complete, move the froglets to the adult aquatic system in the lab. NOTE: Regimens developed by the National Xenopus Resource (Marine Biology Laboratory, Woods Hole, MA) provide a guide for tadpole feeding29 and adult frog maintenance30.
Representative Results
Following transplantation, the qualitative determination of the efficacy of CRISPR/Cas9 mutagenesis should be performed before expending the effort in animal husbandry to grow and maintain the animals to sexual maturity. Because animals carrying leapfrog transplants are somatically wildtype and the germline is difficult to access for direct measurements, saving the carcasses of donor embryos becomes important. DNA analysis from the carcasses serves as a proxy for the extent of mutagenesis that occurs in the transplanted germline17.
One approach to assess the success of mutagenesis is to directly sequence PCR amplicons spanning the genomic region being targeted. This becomes especially costly and inefficient if one wishes to sequence numerous individually-cloned DNA fragments from many animals. Instead, sequencing amplicons heterogeneous at the target site, as they are derived from genetically mosaic embryos, is used to assess mutagenesis efficiency in each embryo19. Visualization of chromatograms shows the wild type sequence (peaks) upstream of the target sequence and peaks become "scrambled" (i.e., the co-occurrence of peaks corresponding to multiple bases appearing at each nucleotide position) downstream of the site of the Cas9-catalyzed double-strand break. Examples of such profiles can be found in Figure S1 of Blitz et al.17. Accurate quantitation of the extent of mutagenesis represented by these scrambled peaks can be obtained using a sequence decomposition algorithm, TIDE (https://tide-calculator.nki.nl/)31. Screening individual embryos using this technique gives confidence that each embryo was properly injected and targeted for mutagenesis. A tracer, such as a fluorescently tagged dextran, can be used to confirm successful injection24 and can also aid in establishing that a tadpole is indeed carrying the transplanted tissue (unpublished data). It is important to note that the formal possibility that the coinjection of a fluorescent dextran together with Cas9-sgRNA might alter Cas9 mutagenesis efficiency has not been explored. Any "leapfrogged" embryos containing transplants from poorly or unmutagenized donors can be discarded.
Leapfrogging circumvents the need for F2 generation analysis in standard genetic breeding schemes, by permitting efficient phenotypic analyses in the F1 embryos. When using the standard approach to mutate essential genes, careful titration of Cas9-sgRNA dose is required to ensure the viability of founder embryos, resulting in a reduced mutagenesis efficiency. Surviving F0 animals from this standard scheme are then outcrossed with wildtypes to obtain viable F1 embryos, which are screened to identify carriers. Finally, these F1 heterozygotes are grown to sexual maturity and intercrossed to obtain homozygous F2 individuals displaying mutant phenotypes. In contrast, because leapfrogging produces animals that have the complete replacement of their germlines with mutant alleles, intercrossing these F0 animals produces mutant phenotypes in the F1 generation. Blitz et al.17 reported that a majority of F0 animals targeted at the tyrosinase (tyr) locus, which is required for pigmentation of melanocytes and retina, showed 100% germline transmission (measured by outcrossing with tyr-/- albino animals) of donor-derived mutant genomes (see Figure 4A-B and Table 1). Thus, intercrossing two animals produced by leapfrogging would yield homozygous or compound heterozygous mutant F1 embryos.
Similar results were obtained17 by intercrossing F0 leapfrogged animals carrying mutations in the homeobox (encoding the DNA-binding homeodomain) of the goosecoid (gsc) gene (see Figure 4A, C-H). Knockdowns of Gsc expression using morpholino antisense oligonucleotides in X. laevis suggested that the gsc gene is necessary for the complete development of the anterior head32, and Cas9-sgRNA injected tadpoles also show embryonic lethal phenotypes17. However, the leapfrogged F0 animals, being somatically wild type, are viable and robust, and the F1 intercross produced embryos with identical LOF phenotypes to those published in X. laevis knockdowns17. These data provided genetic corroboration of the morpholino phenotype, as well as a proof of principle that leapfrogging can be used to overcome embryonic lethal phenotypes.
Figure 1: Materials needed for transplantation. (A) Borosilicate glass Pasteur pipettes are used to make eyebrow hair knives and hair loops for microsurgery on early embryos. Drawing out the glass using a Bunsen burner allows for a narrower end for the insertion of the hair. The red arrowhead marks the approximate position at which to break the glass. (B) An example of an eyebrow hair knife (top) and hair loop (bottom), fixed into pipettes with fast-drying glue. (C) A portion of a 96-well PCR plate is used to create depressions in an agarose plate by allowing the 1% agarose to solidify around the mold (agarose is made in 0.3x or 1x MMR, depending on whether the transplantations are being performed in X. tropicalis or X. laevis, respectively). The thickness of the agarose is ~5 mm, with 3-4 mm-deep wells. (D) An example of an agarose plate for transplantation, with 12 depressions, as shown in panel C. Please click here to view a larger version of this figure.
Figure 2: Morphology of the vegetal pole during the late blastula to early gastrula stages and the strategy for dissection. (A-F) Xenopus tropicalis embryos are shown in vegetal view at half-hourly time intervals, from the beginning of blastula stage 9 (4.5 hpf) to early gastrula stage ~10.25 (7 hpf). Prior to 5 hpf, blastomeres are large and more easily damaged. The white, dashed circle marks the expected position of bottle cell formation during gastrulation, which can be seen forming beginning on the dorsal side (top) in panels (E and F). For leapfrogging, four incisions are made within a square, depicted by black dashed lines, with a final cut made parallel to the surface of the tissue (not illustrated) to free the vegetal explant. Scale bar = 400 µm. Please click here to view a larger version of this figure.
Figure 3: Transplantation. (A) An example of a ~5.5 hpf embryo after the removal of the vegetal PGC-containing tissue. The black dashed box and white dashed circle are the same relative dimensions as those shown in Figure 2. (B) The vegetal explant removed from the embryo in panel A, inner surface facing the viewer, is approximately 0.4-0.45 mm per side and 0.25-0.3 mm in depth. (C) An embryo with the transplanted vegetal tissue. The image was acquired ~10 min after transplantation. (D) 30 min after transplantation, the vegetal tissue is seen to have healed into place (gentle swirling with an eyebrow hair knife above the embryo was used to create a vortex that swept away the debris seen in panel C). After transfer to 1/9x MMR and incubation at 25 °C, most embryos complete gastrulation normally. Scale bar = 400 µm. Please click here to view a larger version of this figure.
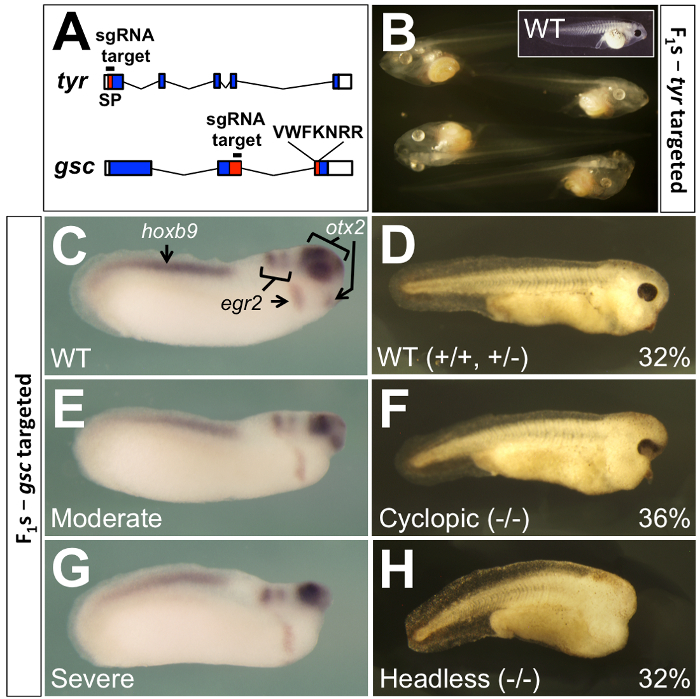
Figure 4: Germline transmission from F0 animals produced by leapfrogging. (A) For leapfrogging, two genes were targeted by CRISPR/Cas9, tyrosinase (top) and goosecoid (bottom). The tyr gene was targeted4,5 within the coding sequencing (blue shading) for an N-terminal signal peptide (SP, red shading), while the gsc gene was targeted17 within coding sequence at the beginning of the homeobox (red shading), which is split between two exons. Untranslated 5’ and 3’ exonic regions are unshaded. (B) All 160 F1 tadpoles from (see also Table 1) a mating of a leapfrogged F0 male (LF1) and an albino (tyr-/-) female were albino, indicating that the entire germline from the leapfrogged male had been replaced with tyr mutant germ cells. The inset shows a wild-type tadpole with normal retinal and melanocyte pigmentation. (C-H) F1 embryos derived from an intercross between two F0 leapfrogged Xenopus targeting the homeobox gene gsc. Approximately 2/3rds of the embryos were phenotypically mutant for gsc, with the phenotype being varying degrees of anterior truncation of the head (E-H). Half of the mutants were either cyclopic or had closely-spaced eyes (F), while the other half were eyeless (H). Genotyping showed these to be homozygotes or compound heterozygotes. The 1/3rd of embryos showing wild-type phenotype (C, D) were either homozygous wild type or heterozygotes. In situ hybridization for otx2 (a marker of forebrain, midbrain, and eyes), egr2 (a marker of hindbrain rhombomeres 3 and 5, and a stream of neural crest), and hoxb9 (a marker of the spinal cord) shows that loss of gsc function affects only the anterior portion of the otx2 domain. These data are reproduced from Blitz et al.17 with permission from the Company of Biologists. Please click here to view a larger version of this figure.
| Cross | Male | Female | Albino | Wild type | Total | % Albino |
| 1 | LF1 | tyr-/- | 160 | 0 | 160 | 100 |
| 2 | LF2 | tyr-/- | 148 | 0 | 148 | 100 |
| 3 | tyr-/- | LF3 | 409 | 0 | 409 | 100 |
| 4 | tyr-/- | LF4 | 0 | 519 | 519 | 0 |
| 5 | tyr-/- | LF5 | 223 | 0 | 223 | 100 |
| 6 | LF6 | tyr-/- | 493 | 703 | 1196 | 41.2 |
| 7 | tyr-/- | LF7 | 1 | 94 | 95 | 1.1 |
| 8 | tyr-/- | LF8 | 125 | 0 | 125 | 100 |
| 9 | tyr-/- | LF9 | 0 | 417 | 417 | 0 |
| 10 | tyr-/- | LF10 | 252 | 0 | 252 | 100 |
Table 1: Efficient germline replacement by leapfrog transplants carrying mutations in tyrosinase. Targeting tyrosinase, transplant-bearing animals of both sexes were obtained. These animals were crossed with albinos, which are homozygous tyr-/-, to test for the efficiency of the germline transmission of mutant alleles, and the percent of the F1 embryos that showed albinism was assayed at the tadpole stage. Entries in the columns labeled "Male" and "Female" indicate which parent is either tyr-/- or the test animal derived from leapfrogging (LF). The percent of embryos within a cross showing the albino phenotype are indicated under "% Albino." Note that 6 of 10 animals with leapfrog transplants showed complete (100%) replacement with mutant alleles. This table is reproduced from Blitz et al.17, with permission.
Discussion
This report provides a detailed protocol for the transplantation of vegetal tissue containing PGCs. Transplantation of PGCs is used in conjunction with genome-editing technologies (e.g., CRISPR/Cas9) to modify the germline of an animal while maintaining nearly all of its somatic tissues as genetically wild type. For leapfrogging to be successful, there are a number of critical factors to consider prior to performing performing transplantations.
To ensure the complete replacement of the germline with mutant alleles, it is recommended that one first determines the in vivo efficiency of sgRNAs. Experience suggests that most sgRNAs designed by CRISPRDirect (https://crispr.dbcls.jp/) or CRISPRScan (http://www.crisprscan.org/) are efficient when used at 250 pg/embryo. However, in some cases, it may be necessary to use higher concentrations (e.g., the gsc sgRNA published by Blitz et al17, required ~1 ng/embryo). Assessing the efficiency of mutagenesis of the target site is essential and the use of TIDE permits a quantitative assessment31. While the method emphasized here uses the direct Sanger sequencing of PCR amplicons (representing the population of mutant alleles in F0 embryos), numerous other methods have been used in the genome-editing literature for this purpose. Other low-to-moderate-cost techniques that are commonly used include fragment length polymorphism analysis using either the loss of restriction enzymes33 or Cas9/TALEN cleavage sites34, the T7 endonuclease 135 and Surveyor36 assays, the heteroduplex mobility assay37, the high-resolution melt assay38, fragment analysis by capillary electrophoresis39,40, and qPCR-based methods41,42.
A second consideration is the choice of target site location within the target gene. To permit the efficient recovery of mutant phenotypes in the F1 generation, it is important to maximize the chance for complete LOF alleles. When making mutant lines using classical approaches it is a common strategy to target Cas9 cleavage near the 5' end of coding regions, to elicit premature translation termination near the beginning of the protein coding region. F1 heterozygotes would be identified with frameshift mutations, and these animals would be selected for further work because they are expected to carry null alleles. Because the leapfrogging strategy uses F0 intercrosses, the approach of targeting the 5' end of the coding sequence would be highly inefficient. F0 embryos have mosaic germlines, and intercrossing results in populations of F1 mutants that are mostly compound heterozygotes17. Since large-scale analysis verifies that, on average, the expected ~1/3rd of mutations produced by double-strand breaks are in-frame43, most F1 embryos produced in this fashion would have at least one allele with an in-frame mutation that might retain wild type activity. Therefore, a different strategy is needed to ensure that even these in-frame mutant alleles are likely to be nulls. Targeting double strand breaks within protein-folding domains that are essential for normal protein function is expected to result in a higher chance of complete LOF (null alleles)17,44. Careful consideration to the atomic-level structure(s) of protein domains, when available, can assist in identifying regions of secondary structure that, even when containing a single amino acid in-frame deletion, might be expected to disrupt domain structure (e.g., the Δ3bp mutation in gsc published by Blitz et al17). If CRISPR/Cas9 target sites can be identified within these regions, one has a higher chance of successfully creating null alleles. Small deletions within solvent-exposed loops between protein secondary structural components are less desirable because mutations in more flexible regions might still result in functional proteins.
A third consideration for leapfrogging is the efficiency of PGC removal from the recipient embryo. This step needs to be efficient in order to ensure complete germline replacement. It is currently unclear whether all graft recipient embryos produced by the method described here have complete removal of their PGCs. Whole-mount in situ hybridizations show variability in the localization of PGC markers in blastula-stage embryos. In many cases, all or nearly all the signal is found near the vegetal-most end of the embryo and would therefore be removed by the surgery outlined here (Blitz et al.17 and citations therein). However, some embryos show PGC marker expression in a few cells closer to the blastocoel floor. Germ plasm first coalesces at the vegetal pole after fertilization and then is swept along cleavage furrows during early cleavages. Some germ plasm may become distributed in cells in the middle of the vegetal hemisphere and not be effectively removed without using transplants that extend to the blastocoel. However, it is also important to note that these deep cells containing germ cell markers might not contribute to the germline, as microRNAs are known to clear PGC mRNAs from somatic cells45,46. These deep cells staining positive for PGC markers might be somatic cells undergoing such clearance. Methods to ensure complete PGC removal from recipient embryos are currently being developed.
Finally, it is also important to consider how many F0 animals to generate by leapfrogging. As a variable number of animals will not survive to sexual maturity, it is recommended that more than 20 embryos containing transplants are created. It will be necessary to have enough surviving animals to ensure (1) that sufficient numbers of both sexes will be obtained and (2) that these transmit mutant alleles at a sufficiently high frequency to be suitable for the analyses planned by the researcher.
Using the protocol described here, with some practice, amphibian embryologists should be able to master the PGC transplantations technique with some practice. Leapfrogging should be possible, not only in other amphibians, but also other organisms containing PGCs that are readily transplantable between individuals at early stages of embryogenesis.
Disclosures
The author has nothing to disclose.
Acknowledgments
This work was performed with the support of a grant, 5R21HD080684-02, from the National Institute of Child Health and Human Development. The author wishes to thank Ken Cho for his continuing enthusiasm and support. The author would also like to acknowledge Bruce Blumberg for use of his camera, Rebekah Charney, for the critical reading of the manuscript, and Sean McNamara and Marcin Wlizla at the National Xenopus Resource (RRID:SCR_013731), for the valuable conversations regarding X. tropicalis feeding and animal care regimens.
References
- Gurdon JB, Hopwood N. The introduction of Xenopus laevis into developmental biology: of empire, pregnancy testing and ribosomal genes. Int J Dev Biol. 2000;44(1):43–50. [PubMed] [Google Scholar]
- Hellsten U, et al. The genome of the Western clawed frog Xenopus tropicalis. Science. 2010;328(5978):633–636. doi: 10.1126/science.1183670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Session AM, et al. Genome evolution in the allotetraploid frog Xenopus laevis. Nature. 2016;538(7625):336–343. doi: 10.1038/nature19840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blitz IL, Biesinger J, Xie X, Cho KW. Biallelic genome modification in F(0) Xenopus tropicalis embryos using the CRISPR/Cas system. Genesis. 2013;51(12):827–834. doi: 10.1002/dvg.22719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama T, Fish MB, Fisher M, Oomen-Hajagos J, Thomsen GH, Grainger RM. Simple and efficient CRISPR/Cas9-mediated targeted mutagenesis in Xenopus tropicalis. Genesis. 2013;51(12):835–843. doi: 10.1002/dvg.22720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, et al. Efficient RNA/Cas9-mediated genome editing in Xenopus tropicalis. Development. 2014;141(3):707–714. doi: 10.1242/dev.099853. [DOI] [PubMed] [Google Scholar]
- Wang F, Shi Z, Cui Y, Guo X, Shi YB, Chen Y. Targeted gene disruption in Xenopus laevis using CRISPR/Cas9. Cell Biosci. 2015;5:15. doi: 10.1186/s13578-015-0006-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe KM, et al. c21orf59/kurly Controls Both Cilia Motility and Polarization. Cell Rep. 2016;14(8):1841–1849. doi: 10.1016/j.celrep.2016.01.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, et al. Efficient genome editing of genes involved in neural crest development using the CRISPR/Cas9 system in Xenopus embryos. Cell Biosci. 2016;6:22. doi: 10.1186/s13578-016-0088-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naert T, et al. CRISPR/Cas9 mediated knockout of rb1 and rbl1 leads to rapid and penetrant retinoblastoma development in Xenopus tropicalis. Sci Rep. 2016;6(35264) doi: 10.1038/srep35264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratzan W, Falco R, Salanga C, Salanga M, Horb ME. Generation of a Xenopus laevis F1 albino J strain by genome editing and oocyte host-transfer. Dev Biol. 2016;426(2) doi: 10.1016/j.ydbio.2016.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya D, Marfo CA, Li D, Lane M, Khokha MK. CRISPR/Cas9: An inexpensive, efficient loss of function tool to screen human disease genes in Xenopus. Dev Biol. 2015;408(2):196–204. doi: 10.1016/j.ydbio.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigeta M, et al. Rapid and efficient analysis of gene function using CRISPR-Cas9 in Xenopus tropicalis founders. Genes Cells. 2016;21(7):755–771. doi: 10.1111/gtc.12379. [DOI] [PubMed] [Google Scholar]
- Hontelez S, et al. Embryonic transcription is controlled by maternally defined chromatin state. Nat Commun. 2015;6:10148. doi: 10.1038/ncomms10148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peshkin L, et al. On the Relationship of Protein and mRNA Dynamics in Vertebrate Embryonic Development. Dev Cell. 2015;35(3):383–394. doi: 10.1016/j.devcel.2015.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens ND, et al. Measuring Absolute RNA Copy Numbers at High Temporal Resolution Reveals Transcriptome Kinetics in Development. Cell Rep. 2016;14(3):632–647. doi: 10.1016/j.celrep.2015.12.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blitz IL, Fish MB, Cho KW. Leapfrogging: primordial germ cell transplantation permits recovery of CRISPR/Cas9-induced mutations in essential genes. Development. 2016;143(15):2868–2875. doi: 10.1242/dev.138057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackler AW. Transfer of germ-cells in Xenopus laevis. Nature. 1960;185:859–860. doi: 10.1038/185859a0. [DOI] [PubMed] [Google Scholar]
- Blackler AW, Fischberg M. Transfer of primordial germ-cells in Xenopus laevis. J Embryol. Exp Morph. 1961;9:634–641. [PubMed] [Google Scholar]
- Bounoure L. L'origine des cellules reproductrices et le problem de la lignee germinale. Paris: Gauthier-Villars; 1939. [Google Scholar]
- Nieuwkoop PD, Sutasurya LA. Primordial Germ Cells in the Chordates: Embryogenesis and phylogenesis. Cambridge: Cambridge University Press; 1979. [Google Scholar]
- Heasman J, Holwill S, Wylie CC. Fertilization of cultured Xenopus oocytes and use in studies of maternally inherited molecules. Methods Cell Biol. 1991;36:213–230. doi: 10.1016/s0091-679x(08)60279-4. [DOI] [PubMed] [Google Scholar]
- Sive HL, Grainger RM, Harland RM. Embryo dissection and micromanipulation tools. CSH Protoc. 2007. [DOI] [PubMed]
- Nakayama T, et al. Cas9-based genome editing in Xenopus tropicalis. Methods Enzymol. 2014;546:355–375. doi: 10.1016/B978-0-12-801185-0.00017-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogino H, McConnell WB, Grainger RM. High-throughput transgenesis in Xenopus using I-SceI meganuclease. Nat Protoc. 2006;1(4):1703–1710. doi: 10.1038/nprot.2006.208. [DOI] [PubMed] [Google Scholar]
- Sive HL, Grainger RM, Harland RM. Early development of Xenopus laevis.: a laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2000. [Google Scholar]
- Kay BK. Injection of oocytes and embryos. In: Kay BK, Peng HB, editors. Xenopus laevis: Practical Uses in Cell and Molecular Biology (Methods in Cell Biology Vol. 36) San Diego: Academic Press; 1991. [Google Scholar]
- Nieuwkoop P, Faber J, editors. Normal Table of Xenopus laevis. (Daudin): A Systematical and Chronological Survey of the Development from the Fertilized Egg till the End of Metomorphosis. New York: Garland Publishing Inc; 1994. [Google Scholar]
- Xenopus Tadpole Feeding. 2017. Available from: http://www.mbl.edu/xenopus/files/2016/06/Xenopus-Tadpole.pdf.
- Xenopus tropicalis Animal Care. 2017. Available from: http://www.mbl.edu/xenopus/files/2016/06/Xenopus-Tropicalis.pdf.
- Brinkman EK, Chen T, Amendola M, van Steensel B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014;42(22):e168. doi: 10.1093/nar/gku936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander V, Reversade B, De Robertis EM. The opposing homeobox genes Goosecoid and Vent1/2 self-regulate Xenopus patterning. EMBO J. 2007;26(12):2955–2965. doi: 10.1038/sj.emboj.7601705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedell VM, et al. In vivo genome editing using a high-efficiency TALEN system. Nature. 2012;491(7422):114–118. doi: 10.1038/nature11537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JM, Kim D, Kim S, Kim JS. Genotyping with CRISPR-Cas-derived RNA-guided endonucleases. Nat Commun. 2014;5:3157. doi: 10.1038/ncomms4157. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Lee HJ, Kim H, Cho SW, Kim JS. Targeted genome editing in human cells with zinc finger nucleases constructed via modular assembly. Genome Res. 2009;19(7):1279–1288. doi: 10.1101/gr.089417.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu P, Shandilya H, D'Alessio JM, O'Connor K, Durocher J, Gerard GF. Mutation detection using Surveyor nuclease. Biotechniques. 2004;36(4):702–707. doi: 10.2144/04364PF01. [DOI] [PubMed] [Google Scholar]
- Ota S, et al. Efficient identification of TALEN-mediated genome modifications using heteroduplex mobility assays. Genes Cells. 2013;18(6):450–458. doi: 10.1111/gtc.12050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlem TJ, et al. Simple Methods for Generating and Detecting Locus- Specific Mutations Induced with TALENs in the Zebrafish Genome. PLoS Genet. 2012;8(8):e1002861. doi: 10.1371/journal.pgen.1002861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramlee MK, Yan T, Cheung AM, Chuah CT, Li S. High-throughput genotyping of CRISPR/Cas9-mediated mutants using fluorescent PCR-capillary gel electrophoresis. Sci Rep. 2015;5:15587. doi: 10.1038/srep15587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya D, Marfo CA, Li D, Lane M, Khokha MK. CRISPR/Cas9: An inexpensive, efficient loss of function tool to screen human disease genes in Xenopus. Dev Biol. 2015;408(2):196–204. doi: 10.1016/j.ydbio.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C, Zhang Y, Yao S, Wei Y. A PCR Based Protocol for Detecting Indel Mutations Induced by TALENs and CRISPR/Cas9 in Zebrafish. PLoS ONE. 2014;9(6):e98282. doi: 10.1371/journal.pone.0098282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mock U, Hauber I, Fehse B. Digital PCR to assess gene-editing frequencies (GEF-dPCR) mediated by designer nucleases. Nat Protoc. 2016;11(3):598–615. doi: 10.1038/nprot.2016.027. [DOI] [PubMed] [Google Scholar]
- Varshney GK, et al. High-throughput gene targeting and phenotyping in zebrafish using CRISPR/Cas9. Genome Res. 2015;25(7):1030–1042. doi: 10.1101/gr.186379.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Wang E, Milazzo JP, Wang Z, Kinney JB, Vakoc CR. Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nat Biotechnol. 2015;33(6):661–667. doi: 10.1038/nbt.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koebernick K, Loeber J, Arthur PK, Tarbashevich K, Pieler T. Elr-type proteins protect Xenopus Dead end mRNA from miR-18-mediated clearance in the soma. Proc Natl Acad Sci USA. 2010;107:16148–16153. doi: 10.1073/pnas.1004401107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Aguero T, King ML. The Xenopus maternal-to-zygotic transition from the perspective of the germline. Curr Top Dev Biol. 2015;113:271–303. doi: 10.1016/bs.ctdb.2015.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]