

Abstract
The arrest of rolling leukocytes on various target vascular beds is mediated by specialized leukocyte integrins and their endothelial immunoglobulin superfamily (IgSF) ligands. These integrins are kept in largely inactive states and undergo in situ activation upon leukocyte-endothelial contact by both biochemical and mechanical signals from flow-derived shear forces. In vivo and in vitro studies suggest that leukocyte integrin activation involves conformational alterations through inside-out signaling followed by ligand-induced rearrangements accelerated by external forces. This activation process takes place within fractions of seconds by in situ signals transduced to the rolling leukocyte as it encounters specialized endothelial-displayed chemoattractants, collectively termed arrest chemokines. In neutrophils, selectin rolling engagements trigger intermediate affinity integrins to support reversible adhesions prior to chemokine triggered arrest. Different leukocyte subsets appear to use different modalities of integrin activation during rolling and arrest at distinct endothelial sites.
Keywords: integrins, chemokines, selectins, leukocytes, signal transduction, inflammation
Introduction
Leukocyte emigration at specific target sites is regulated by adhesive cascades mediated by three sequential and partially overlapping steps initiated by selectin mediated capturing and rolling, followed by chemokine triggered activation and integrin- dependent arrest on endothelial immunoglobulin superfamily (IgSF) ligands [1]. Different selectin and integrin family members, together with diverse endothelial displayed chemokines, provide large combinatorial specificity to this process. Although adapted to operate under shear flow conditions [2,3] selectin and integrin bonds are differently regulated at leukocyte-endothelial contacts. Whereas the adhesiveness of the selectins and their cognate glycoprotein ligands is generally not modulated by in situ endothelial signals, the avidity of leukocyte integrins to their endothelial ligands is rapidly and reversibly regulated by chemokine-triggered signals [4] transduced via specialized G-protein coupled receptors, GPCRs (for a detailed review on chemokines please refer to [5]). In many cases, endothelial chemokines trigger conformational integrin switches within a fraction of a second and at the actual site of leukocyte arrest by triggering inside-out signals and facilitating additional ligand-induced integrin activation events [6]. Recent studies suggest that shear forces exerted on the arrested leukocyte at the endothelial contacts facilitate chemokine-stimulated integrin activation [7]. In addition to this apparently universal modality of leukocyte integrin activation, neutrophils and perhaps other myeloid leukocytes use their rolling interactions on specific endothelial selectins to ligate glycoproteins that stimulate specific tyrosine kinases to partially activate integrins during the rolling period [8]. Whereas in situ integrin activation by arrest chemokines is abrupt and highly localized within the direct endothelial site of arrest [9], these weaker selectin-triggered kinase signals appear to stimulate integrins on the entire surface area of the rolling leukocyte in a gradual manner. Selectin-mediated rolling also increases the probability of chemokine and integrin ligand encounter by leukocytes and thereby facilitate integrin activation even without triggering signaling effectors. In this review, we focus on recent works which shed light on the molecular and mechanical basis of leukocyte integrin activation in blood vessels and discuss their physiological outcome during lymphocyte and neutrophil rolling and arrest on various endothelial targets.
Initial selectin-mediated leukocyte capturing to blood vessels- a force regulated process
Selectins are the main receptors that mediate the initial capture of circulating leukocytes to vascular endothelial surfaces [10]. Leukocyte capture is followed by rolling adhesions, which proceed from seconds (for most leukocytes) to a few minutes (neutrophils) [11]. The selectins comprise a three member family with highly conserved N-terminal C-type lectin and epidermal growth factor (EGF) -like tandem domains which bind sialyl-Lewisx-like carbohydrate ligands [10]. L-selectin is expressed on most circulating leukocytes and is the key receptor that initiates leukocyte capture events in high endothelial venules in secondary lymphoid tissues and at peripheral sites of injury and inflammation. L-selectin can also bind leukocyte ligands, particularly PSGL-1, and this interaction can enhance the capture of leukocytes by intravascular adherent leukocytes. P- and E- selectins are inducibly expressed in both acutely and chronically inflamed endothelial beds, and in many of these settings their contribution is redundant.
Selectin-mediated adhesions are characterized by fast on- and off-rates and exceptional resistance to disruptive shear forces exerted on the leukocyte at the vessel wall [12]. Selectins undergo conformational changes upon ligand binding that decrease their off-rate under tensile forces, making them “catch bonds” [13]. Recent evidence suggests that the lectin-EGF interdomain hinge in L-selectin and P-selectin may critically regulate this stabilization via force-stabilized extension [2,3]. While E-selectin may also need to utilize shear forces to stabilize extension, this selectin often generates instantaneous multivalent bonds with ligands presented on closely spaced multivalent glycans [10]. Increasing evidence suggests that in order to load forces, L-selectin, P-selectin and their glycoprotein ligands need to be properly anchored to the cytoskeleton either directly or indirectly through engagements with submembranal assemblies [14,15]. Selectins and their ligands are localized to microvillus-like projections, favorable sites of leukocyte-endothelial collisions [16] and possibly major sites of integrin activation (Fig. 1). The topographic distribution of selectins and ligands on both leukocyte and endothelial cell microvilli is thought to be important not only for increasing their effective association rates [16], but also as a means to dissipate a portion of the dissociation energy exerted on individual adhesive bonds along the microvillus axis.
Figure 1.
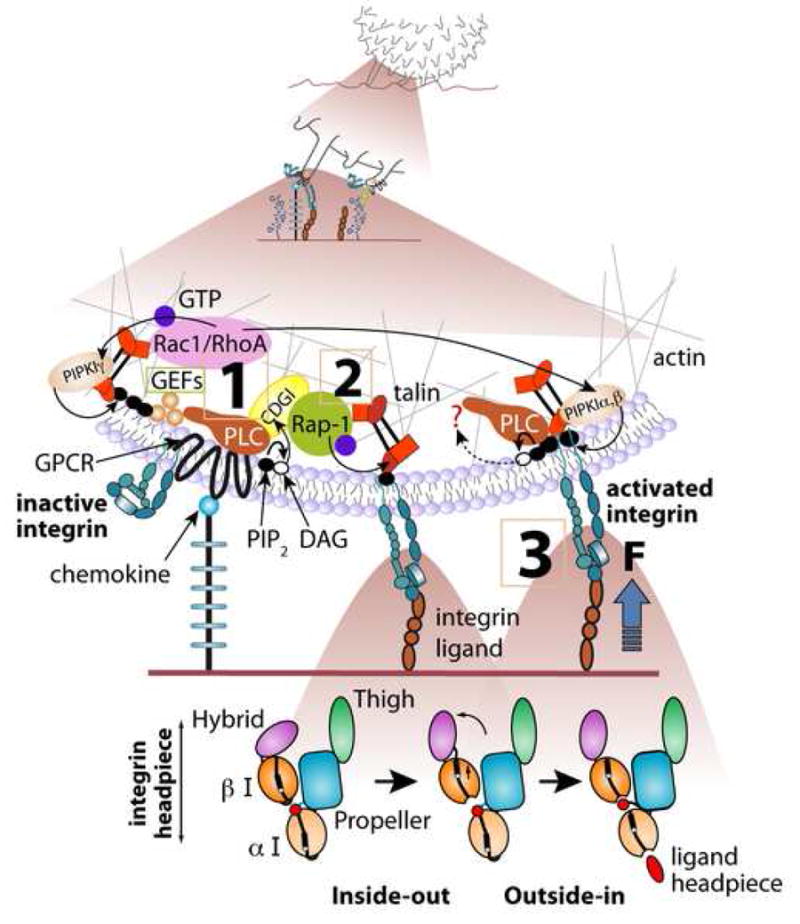
Rapid GPCR mediated integrin activation by endothelial-immobilized chemokine; involvement of Rap-1 and Rho GTPases. A proposed intracellular signaling cascade from chemokine (blue ball) bound to GPCR to the LFA-1 integrin (blue heterodimer) within a single microvillus of a lymphocyte rolling on L-selectin ligands. The front microvillus is occupied by L-selectin ligand whereas integrin activation takes place during occupancy and stretching of the rear microvillus, which may be co-occupied via L-selectin (not drawn). Canonical bidirectional activation of LFA-1 from inactive bent state to a semi-active extended state (unbent ectodomain with the two headpiece I-domains in closed conformation with low affinity to ligand) and to high affinity extended state (unbent integrin with both of the I-domains stabilized in an open state, see the ribbon diagram of the various LFA-1 headpiece states in the lower panels) is shown. Upon initial encounter of the proper endothelial-bound chemokine, the leukocyte GPCR is activated within milliseconds and transmits signals converting a nearby inactive (folded) integrin to its extended conformation, likely through Gβγ-triggered phospholipase C (PLC) mediated hydrolysis of PIP2, activation of CalDAG-GEFI (CDGI), its target Rap-1 and the downstream effector talin (steps 1,2). GPCR-triggered GEFs of RhoA and possibly of Rac1 co-activate downstream PIP2-generating kinases (PIPKIα,β,γ). Local increase of PIP2 directly activates talin binding to and unclasping of the inactive integrin, rendering it extended (step 2) and facilitating its full outside in activation by ICAM-1 (step 3). Force exerted on the ligand-occupied integrin facilitates this outside-in activation by opening the I-domains on both the α and β subunits (lower panels) and both CDG-I activated Rap-1 and PIP2 activated talin may further stabilize the unclasped integrin. Integrin homodimers may interact with preexistent ligand dimers (not shown) and increase binding avidity. Similar events can take place in lymphocytes and other leukocytes rolling on P and E selectins (not shown). The degree of integrin extension, headpiece activation by ligand, force loading and headpiece rearrangements, and ligand induced dimerization is likely to differ among distinct integrins, GPCRs, cell types and species. The lower panels were modified from [17].
Leukocyte integrin activation at endothelial contacts
Chemokine signals to leukocyte integrins- conformational changes in the ectodomain triggered by and coupled to cytoskeletal associations
Under normal flow, selectin bonds cannot arrest, on their own, rolling leukocytes. Leukocyte arrest is nearly exclusively mediated by members of the integrin superfamily [17]. Both in vivo and in vitro studies in numerous leukocyte-endothelial model systems have established that leukocyte arrest results from an abrupt and local activation of integrin adhesiveness to cognate endothelial ligands [18] [19,20]. This rapid triggering of integrin adhesiveness is preferentially transduced by leukocyte GPCRs through local triggering of heterotrimeric G-proteins, primarily of the Gi/o subtype, and in neutrophils specifically Gαi2 [21], as they encounter immobilized, endothelial-presented chemoattractants, primarily chemokines and some lipid attractants (For reviews please refer to [20,22]). As in other cell types, integrin conformation regulation by GPCRs involves bidirectional activation initiated by inside-out conformational activation and reinforced by ligand binding (outside-in activation, Fig. 1) [17].
In vivo analysis suggests that integrin-activating endothelial-displayed chemokines and chemoattractants transduce their signals to integrins on rolling lymphocytes and monocytes in an abrupt rather than in a stepwise manner [19,23]. Integrin heterodimers on the majority of circulating leukocytes are kept in an inactive conformation via a cytoplasmic clasp which can be disrupted by the binding of the talin head domain to the integrin β subunit tail [24]. The most detailed information about integrin conformational activation comes out from structural and kinetic analysis of αvβ3 and LFA-1-mediated interactions with cognate ligands [17] (Fig. 1). The LFA-1 α and β subunits were shown to move apart in response to overexpression of the talin head or chemokine exposure [25], a change which results in release of multiple constraints on the integrin headpiece [17]. LFA-1 activation by chemokine signals as well as by ICAM-1 under shear flow was shown to also depend on intact talin1 [19]. Introduction of mAb probes for integrin ectodomain extension and headpiece activation also allowed to probe chemokine-induced conformational LFA-1 switches in real time [19]. These and other studies suggest that stimulatory chemokines capable of triggering integrin-mediated leukocyte arrest ideally function when encountered by the stimulated leukocyte in juxtaposition to the integrin ligand, since upstream exposure of rolling leukocytes to immobilized chemokine signals is insufficient to trigger integrin adhesiveness at a downstream field [26]. These results collectively suggest that GPCR engagement by an arrest chemokine can trigger a signaling cascade that activates neighbor integrin(s) in situ in an instantaneous manner via a talin dependent mechanism, and that prior encounters of chemokine signals during the rolling phase are not necessary for integrin activation [9,19]. In T lymphocytes interacting with chemokines and ICAM-1 under shear flow, full stimulation of high LFA-1 affinity by prototypic arrest chemokines occurs within < 0.4 sec of cell contact with surface bound ICAM-1 [19] and appears to involve only small subsets of the entire surface expressed integrin. The chemokine signal is thought to first trigger a reversible extension of the LFA-1 integrin which dramatically increases the association rate of its headpiece for a nearby surface immobilized ICAM-1 [17]. This, and subtle conformational changes in the integrin β subunit hybrid domain [17] locally prime the integrin headpiece for a subsequent and instantaneous ligand-induced rearrangement resulting in full activation (opening) of the headpiece I-domains [19](Fig.1).
Recent studies on T lymphocytes indicate that certain chemokines can trigger VLA-4 and LFA-1 adhesiveness even in the absence of integrin extension and induction of headpiece conformational epitopes associated with high affinity recognition [27,28], suggesting that chemokine signals may facilitate integrin activation also via rapid post-ligand-binding stabilization rather than by an a priori conformational activation of the integrin ectodomain. With the increasing evidence that integrins undergo direct activation by forces [29,30], it becomes apparent that the ability to rapidly respond to and undergo activation by shear forces relies also on proper anchorage of the integrin to the cell cytoskeleton. Anchorage of the α4β1 heterodimer to the actin cytoskeleton facilitates mechanical stabilization of VLA-4-VCAM-1 bonds under external strain without increasing VLA-4 affinity to soluble ligand [31]. A recent study indicates that immobilized chemokines locally stiffen membrane compartments nearby VCAM-occupied VLA-4 and thereby strengthen VLA-4-VCAM-1 bonds (Schmitz, submitted). Extended LFA-1 is thought to be pre-anchored to the lymphocyte cytoskeleton and can undergo further anchorage upon occupancy by ICAM-1 [32], highlighting the possibility that chemokine signals can stimulate integrin adhesiveness under shear flow by anchoring integrins right after they bound their cognate extracellular ligands. Following arrest, chemokine signals may increase within seconds integrin clustering with additional endothelial ligand molecules probably by acting on mobile integrin subsets [26]. The consequence of these versatile activation modalities is that subsets of VLA-4-VCAM-1 and LFA-1-ICAM-1 as well as Mac-1-ICAM-1 bonds can support both rolling and firm adhesions depending on their basal affinity states [26,27,33] as well as on the degree by which the integrin ligand can rearrange and activate the headpiece of its cognate integrin [9]. This capacity is likely to depend on the exposure time and local availability of the activating chemokine [26].
Selectin signaling to integrins- a global activation modality prior to leukocyte arrest
Recently, engagements of neutrophil PSGL-1 during active rolling on E-selectin and to a lesser extent on P-selectin was demonstrated to activate the tyrosine kinase SYK which rapidly transmits weak reversible activation signals to subsets of LFA-1 on rolling neutrophils [8]. The PSGL-1 triggered signals stabilize these LFA-1 subsets at intermediate affinity states (i.e., unbent extended integrins with their headpiece in a closed conformation with low affinity to ICAM-1) which mediate rolling adhesions rather than firm arrests. Unlike the abrupt integrin activation events transduced by local GPCR signals, E-selectin triggered signals require seconds of rolling during which LFA-1 conformations are globally triggered over the entire surface of the rolling leukocyte (Fig. 2). These selectin engagements lower the threshold for subsequent arrest induction by low concentrations of immobilized chemokine [8]. Under these conditions, neutrophils can roll slowly for minutes without becoming adherent. Although LFA-1 clearly engages its ligand ICAM-1, PSGL-1-mediated LFA-1 activation does not result in full ligand-induced LFA-1 activation. Neutrophils rolling for minutes on inflamed endothelial cells also accumulate cytosolic Ca2+, followed by a large rise in Ca2+, upon arrest and integrin engagement [34]. It is unclear, however, whether Syk and Ca2+ dependent signals triggered by rolling engagements of neutrophils cooperate in this integrin activation modality. Another unresolved question is why selectins do not contribute to integrin activation in lymphocytes, including memory lymphocyte subsets that express E-selectin binding PSGL-1 glycoforms but fail to activate their LFA-1 without chemokine signals (Alon, unpublished). One possibility is that lymphocyte ZAP-70, Syk and Src kinases cannot substitute for neutrophil Syk and Src kinases (Zarbock and Ley, unpublished). Another open question is to what extent lymphocytes as well as neutrophils and monocytes utilize L-selectin to signal and activate leukocyte integrins prior to leukocyte arrest on target endothelium. L-selectin signaling to integrins, including integrin translocation to the plasma membrane, has been argued to control various adhesion strengthening events in neutrophils [35]. There is no doubt, however, that in both myeloid and lymphoid cells, selectin-mediated rolling facilitates integrin mediated leukocyte arrest by topographical means i.e., by microvilli flattening which enhances chemokine encounter by the rolling leukocyte.
Figure 2. Accumulating vs, abrupt switches in integrin avidity states in rolling leukocytes.
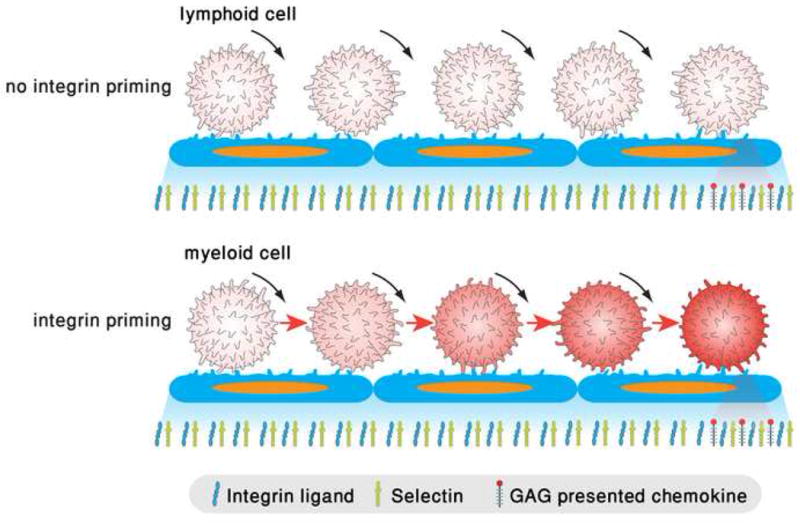
Upper panel: A lymphocytes rolls via PSGL-1 or other endothelial selectin ligands (not shown) but fails to undergo integrin activation by successive rolling engagements due to insufficient triggering of kinases, PLCs and secondary messengers. When lymphocytes encounter high density of arrest chemokines juxtaposed to integrin ligands, their integrins are locally and instantaneously activated by Gαiβγ signals as delineated in figure 1. Lower panel: a rolling neutrophil and possibly other myeloid leukocytes can integrate weak integrin activation signals through engagements of E-selectin ligands including PSGL-1, which trigger phosphorylation and activation of spleen tyrosine kinase Syk [8,58]. Within seconds, LFA-1 and possibly other integrins are stabilized in an extended intermediate affinity state on the entire plasma membrane. These integrins can form reversible adhesive bonds with endothelial ICAM-1 sufficient to slow down selectin mediated rolling. Like the lymphocyte, when the rolling neutrophil encounters high density of arrest chemokines, its integrins can undergo robust bidirectional activation via Gi signals. Post arrest, ligand-driven integrin microclustering can immediately follow to further stabilize the integrin-mediated contact. Co-ligation of multiple selectin ligands and integrins trigger both Src and Syk kinases to further activate integrin avidity and adhesion strengthening [59].
GTPases implicated in spontaneous and in chemokine-stimulated integrin adhesiveness under shear flow
Both inside-out and outside-in leukocyte integrin activation stimulated by arrest chemokines are triggered by specialized chemokine-occupied G-protein coupled receptors (GPCRs) [20,36,37]. Increasing evidence suggests that full integrin activation requires the GPCR-transduced signals to activate integrin affinity and clustering as well as to stabilize integrin-ligand complexes in properly anchored states to allow these complexes to undergo optimal activation by shear forces and develop high mechanical resistance [9]. The relative contribution of each of these modalities to the activation of specific integrins by specific GPCRs is likely to vary between different types of integrins (LFA-1 versus VLA-4), GPCRs, leukocytes and species [20,36,37]. Two conserved players in GPCR signaling to integrins at leukocyte endothelial contacts are Rap and Rho small GTP-binding proteins, which undergo direct activation by chemokine signals [36] and talin-1, a key integrin cytoskeleton linker that controls both integrin affinity and anchorage states [19,24,27,38] (Fig. 1). The most upstream element of chemokine signaling to integrins is PLC [37]. Although knockouts of the major Gi activated PLCs in neutrophils, PLCβ2 and PLCβ3 do not exhibit major trafficking defects possibly due to redundancy, pharmacological blockage of total PLC activity in platelets, monocytes and neutrophils impair integrin activation by GPCR agonists [39-41]. A key target for PLC activity underlying GPCR mediated integrin activation in these cells as well as in lymphocytes is CalDAG-GEFI [39,41-43], a Ca2+ and DAG dependent GEF for the ras-like small GTPase Rap1. This GTPase has emerged over the past few years as a key regulator of rapid integrin activation by a variety of inside out signals [44]. The central role of its activator, CalDAG-GEFI, in integrin activation in platelets, neutrophils and lymphocytes is highlighted by patients suffering from a rare adhesion deficiency syndrome termed LAD-III [41]. LAD-III patient cells exhibit reduced levels of this GEF and suffer from global defects in integrin activation by multiple GPCR agonists. These defects include abrogated GPCR-transduced affinity modulation of the platelet integrin αIIbβ3 and the lymphocyte integrin LFA-1, the loss of β2 integrin mediated neutrophil arrest on endothelial ligands under shear flow [41] and deficiencies in both LFA-1 and VLA-4 mediated lymphocyte arrest. Loss of CalDAG-GEFI in mouse platelets and neutrophils mimic the LAD-III syndrome [42], but murine lymphocytes lack this GEF [39,41], highlighting species dependent differences in GPCR/Rap-1 mediated inside out integrin activation.
GPCR-stimulated Rap-1 appears to trigger high integrin affinity by activating talin [45] (Figure 1). Talin1, the major leukocyte talin isoform, is recruited by activated Rap-1 and its effector RIAM to the vicinity of β3 and β2 integrins and switch these integrins into high affinity and high avidity states [45]. Although this Rap-1 regulation of talin was so far established only in prolonged inside out integrin activation processes, it is plausible that CalDAG-GEFI activated Rap-1 can also locally modulate talin-integrin associations during the earliest integrin activation events underlying GPCR triggered platelet activation [46] and GPCR stimulated leukocyte arrest on vascular endothelium. In addition to being recruited by RIAM, talin can be directly activated to bind and stimulate integrins by local changes in the plasma membrane lipid PtdIns(4,5)P2 (PIP2). This phosphoinositide not only activates talin [47] but is a key substrate for PLC-mediated Cal-DAG-GEFI activation and Rap-1 stimulation [39]. In addition, Rap1 can upregulate LFA-1 avidity through recruitment of a direct effector, RAPL, to the cytoplasmic interface of the LFA-1 α chain [48]. Consistent with this possibility, initial LFA-1 mediated arrest of RAPL null T cells on HEVs is normal whereas subsequent adhesion strengthening is reduced (Kinashi, personal communication). Interestingly, PIP2 production can be driven by combined activities of a talin-associated PtdIns(4)P 5 kinase, PIPKIγ [49] and two other PIP2 generating enzymes, PIPKIα and PIPKIβ, which may be activated by GPCR-stimulated Rho proteins. Indeed, rapid lymphocyte and neutrophil integrin activation by endothelial chemokine signals require intact RhoA [50]. In situ chemokine-activated RhoA may locally elevate PIP2 near integrins and thereby activate talin both directly and indirectly i.e. via a PLC-Cal-DAG-GEFI-Rap-1-RIAM axis [45]. Rho GTPases can also remodel actin filaments and elevate their bundling within integrin-GPCR signalosomes thereby stiffening the cortical cytoskeleton near ligand-occupied integrins, which in turn may facilitate mechanical activation of these integrins (Fig. 1). Lastly, Rap-1 and RhoA may activate target integrins by sequestering integrin partners that impose constraints on integrin unclasping and activation (Fig. 1). It is therefore possible that full integrin activation only occurs when both Rap-1 and RhoA are coactivated by chemokine signals. Recent in vitro results further suggest that the relative contribution of Rap-1 and RhoA signaling to integrin activation can substantially vary with the type of integrin and with the stimulatory GPCR [28,43] within distinct integrin-GPCR signalosomes.
Rapid integrin activation machineries: preformed GPCR assemblies on leukocytes?
As integrin activation by surface-bound chemokines under physiological conditions is a highly localized event which takes place over a fraction of a second, chemokines must be able to organize a localized network of interacting proteins, possibly within supramolecular structures localized on leukocyte-surface microvilli, preferential sites of leukocyte-endothelial contacts under shear flow [16]. Indeed some integrin-activating GPCRs are clustered on lymphocyte microvilli [51]. Each supramolecular assembly of a given GPCR may consist of distinct combinations of regulatory GTPases, their GEFs and GAPs, as well as of GPCR scaffolds like filamins, myosins and β-arrestins together with integrin effectors like talin and talin-binding cytoskeletal adaptors like vinculin and α-actinin. Notably, VLA-4 and LFA-1 are topographically segregated on the leukocyte surface [52]. It is therefore possible that a given GPCR scaffold and an integrin target communicate in distinct membranal compartments. In addition, GPCRs may exist as distinct homo- or hetero-dimers. Different GPCR assemblies are known to associate with different heterotrimeric G-proteins and their associated effectors and may therefore signal differently to their integrin targets. For example, a single chemokine, RANTES, transduces rapid integrin avidity stimulatory signals to monocytes and effector T cells through its GPCR CCR1 but fails to activate integins through its second receptor, CCR5 on the same cells [53].
Conclusions and perspectives
Chemokine signals are the most efficient endothelial-displayed triggers of integrin activation on leukocytes. Myeloid cells and subsets of activated lymphocytes can also integrate signals from endothelial selectins [35], immunoreceptors [54] neighbor integrin ligands [55], integrin-associated adhesion receptors [56] and regulatory integrin partners [57] to further strengthen integrin adhesiveness at endothelial sites of arrest. In spite of increasing information on numerous potential effectors involved in rapid integrin activation processes on different leukocyte subsets, we still don’t know how these effectors cooperate in facilitating post arrest adhesion strengthening events. Reversible changes both in integrin conformation, cytoskeletal associations and outside in signaling to different actomyosin effectors must be co-regulated during the first few seconds of these critical post arrest processes. Future studies must dissect these complex activation programs and their variations among different types of leukocytes and endothelial beds in order to generate a road map of molecular networks that control initial integrin-mediated arrest, subsequent adhesion strengthening, as well as motility from the arrest site to the final site of transendothelial migration.
Acknowledgments
We wish to thank Valentin Grabovsky, Ronit Pasvolsky, Adi Sagiv and Alexander Zarbock for sharing unpublished data. We also thank Dr. Sara Feigelson for fruitful discussions. R. Alon is the The Linda Jacobs Chair in Immune and Stem Cell Research and his research is supported by the Israel Science Foundation and the Minerva foundation, and MAIN, the EU6 Program for Migration in Inflammation. K. Ley is Professor and Head of the Division of Inflammation Biology, La Jolla Institute for Allergy and Immunology, and is supported by the Public Health Service: National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and recommended reading
- 1.Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: The multistep paradigm. Cell. 1994;76:301–314. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 2.Lou J, Yago T, Klopocki AG, Mehta P, Chen W, Zarnitsyna VI, Bovin NV, Zhu C, McEver RP. Flow-enhanced adhesion regulated by a selectin interdomain hinge. J Cell Biol. 2006;174:1107–1117. doi: 10.1083/jcb.200606056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Phan UT, Waldron TT, Springer TA. Remodeling of the lectin-EGF-like domain interface in P- and L-selectin increases adhesiveness and shear resistance under hydrodynamic force. Nat Immunol. 2006;7:883–889. doi: 10.1038/ni1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Constantin G, Majeed M, Giagulli C, Piccio L, Kim JY, Butcher EC, Laudanna C. Chemokines trigger immediate β2 integrin affinity and mobility changes: differential regulation and roles in lymphocyte arrest under flow. Immunity. 2000;13:759–769. doi: 10.1016/s1074-7613(00)00074-1. [DOI] [PubMed] [Google Scholar]
- 5.Rot A, von Andrian UH. Chemokines in innate and adaptive host defense: basic chemokinese grammar for immune cells. Annu Rev Immunol. 2004;22:891–928. doi: 10.1146/annurev.immunol.22.012703.104543. [DOI] [PubMed] [Google Scholar]
- 6.Laudanna C. Integrin activation under flow: a local affair. Nat Immunol. 2005;6:429–430. doi: 10.1038/ni0505-429. [DOI] [PubMed] [Google Scholar]
- 7*.Woolf E, Grigorova I, Sagiv A, Grabovsky V, Feigelson SW, Shulman Z, Hartmann T, Sixt M, Cyster JG, Alon R. Lymph node chemokines promote sustained T lymphocyte motility without triggering stable integrin adhesiveness in the absence of shear forces. Nat Immunol. 2007;8:1076–1085. doi: 10.1038/ni1499. First indication that chemokine-primed LFA-1 and VLA-4 require shear forces to undergo full activation and support firm adhesion. [DOI] [PubMed] [Google Scholar]
- 8*.Zarbock A, Lowell CA, Ley K. Spleen tyrosine kinase Syk is necessary for E-selectin-induced alpha(L)beta(2) integrin-mediated rolling on intercellular adhesion molecule-1. Immunity. 2007;26:773–783. doi: 10.1016/j.immuni.2007.04.011. The paper identifies a novel E-selectin triggered pathway for LFA-1 priming on neutrophils. E-selectin engaged PSGL-1 triggers Syk signals that stabilize LFA-1 in an intermediate affinity state that supports reversible rolling adhesions and slow down neutrophils rolling on E-selectin. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alon R, Dustin ML. Force as a facilitator of integrin conformational changes during leukocyte arrest on blood vessels and antigen-presenting cells. Immunity. 2007;26:17–27. doi: 10.1016/j.immuni.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 10.McEver RP. Selectins: lectins that initiate cell adhesion under flow. Curr Opin Cell Biol. 2002;14:581–586. doi: 10.1016/s0955-0674(02)00367-8. [DOI] [PubMed] [Google Scholar]
- 11.Ley K. Integration of inflammatory signals by rolling neutrophils. Immunol Rev. 2002;186:8–18. doi: 10.1034/j.1600-065x.2002.18602.x. [DOI] [PubMed] [Google Scholar]
- 12.Alon R, Hammer DA, Springer TA. Lifetime of the P-selectin-carbohydrate bond and its response to tensile force in hydrodynamic flow. Nature. 1995;374:539–542. doi: 10.1038/374539a0. [DOI] [PubMed] [Google Scholar]
- 13.Marshall BT, Long M, Piper JW, Yago T, McEver RP, Zhu C. Direct observation of catch bonds involving cell-adhesion molecules. Nature. 2003;423:190–193. doi: 10.1038/nature01605. [DOI] [PubMed] [Google Scholar]
- 14.Dwir O, Kansas GS, Alon R. The cytoplasmic tail of L-selectin regulates leukocyte capture and rolling by controlling the mechanical stability of selectin:ligand tethers. J Cell Biol. 2001;155:145–156. doi: 10.1083/jcb.200103042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Setiadi H, McEver RP. Clustering endothelial E-selectin in clathrin-coated pits and lipid rafts enhances leukocyte adhesion under flow. Blood. 2008;111:1989–1998. doi: 10.1182/blood-2007-09-113423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.von Andrian UH, Hasslen SR, Nelson RD, Erlandsen SL, Butcher EC. A central role for microvillous receptor presentation in leukocyte adhesion under flow. Cell. 1995;82:989–999. doi: 10.1016/0092-8674(95)90278-3. [DOI] [PubMed] [Google Scholar]
- 17.Luo BH, Carman CV, Springer TA. Structural basis of integrin regulation and signaling. Annu Rev Immunol. 2007;25:619–647. doi: 10.1146/annurev.immunol.25.022106.141618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Campbell JJ, Hedrick J, Zlotnik A, Siani MA, Thompson DA. Chemokines and the arrest of lymphocytes rolling under flow conditions. Science. 1998;279:381–384. doi: 10.1126/science.279.5349.381. [DOI] [PubMed] [Google Scholar]
- 19.Shamri R, Grabovsky V, Gauguet JM, Feigelson S, Manevich E, Kolanus W, Robinson MK, Staunton DE, von Andrian UH, Alon R. Lymphocyte arrest requires instantaneous induction of an extended LFA-1 conformation mediated by endothelium-bound chemokines. Nat Immunol. 2005;6:497–506. doi: 10.1038/ni1194. [DOI] [PubMed] [Google Scholar]
- 20.Laudanna C, Alon R. Right on the spot. Chemokine triggering of integrin-mediated arrest of rolling leukocytes. Thromb Haemost. 2006;95:5–11. [PubMed] [Google Scholar]
- 21.Zarbock A, Deem TL, Burcin TL, Ley K. Galphai2 is required for chemokine-induced neutrophil arrest. Blood. 2007;110:3773–3779. doi: 10.1182/blood-2007-06-094565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ley K. Arrest chemokines. Microcirculation. 2003;10:289–295. doi: 10.1038/sj.mn.7800194. [DOI] [PubMed] [Google Scholar]
- 23.Huo Y, Schober A, Forlow SB, Smith DF, Hyman MC, Jung S, Littman DR, Weber C, Ley K. Circulating activated platelets exacerbate atherosclerosis in mice deficient in apolipoprotein E. Nat Med. 2003;9:61–67. doi: 10.1038/nm810. [DOI] [PubMed] [Google Scholar]
- 24.Tadokoro S, Shattil SJ, Eto K, Tai V, Liddington RC, de Pereda JM, Ginsberg MH, Calderwood DA. Talin binding to integrin beta tails: a final common step in integrin activation. Science. 2003;302:103–106. doi: 10.1126/science.1086652. [DOI] [PubMed] [Google Scholar]
- 25.Kim M, Carman CV, Springer TA. Bidirectional transmembrane signaling by cytoplasmic domain separation in integrins. Science. 2003;301:1720–1725. doi: 10.1126/science.1084174. [DOI] [PubMed] [Google Scholar]
- 26.Grabovsky V, Feigelson S, Chen C, Bleijs R, Peled A, Cinamon G, Baleux F, Arenzana-Seisdedos F, Lapidot T, van Kooyk Y, et al. Subsecond induction of α4 integrin clustering by immobilized chemokines enhances leukocyte capture and rolling under flow prior to firm adhesion to endothelium. J Exp Med. 2000;192:495–505. doi: 10.1084/jem.192.4.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Manevich E, Grabovsky V, Feigelson SW, Alon R. Talin 1 and paxillin facilitate distinct steps in rapid VLA-4-mediated adhesion strengthening to vascular cell adhesion molecule 1. J Biol Chem. 2007;282:25338–25348. doi: 10.1074/jbc.M700089200. [DOI] [PubMed] [Google Scholar]
- 28.Pasvolsky R, Grabovsky V, Giagulli C, Shulman Z, Shamri R, Feigelson SW, Laudanna C, Alon R. RhoA Is Involved in LFA-1 Extension Triggered by CXCL12 but Not in a Novel Outside-In LFA-1 Activation Facilitated by CXCL9. J Immunol. 2008;180:2815–2823. doi: 10.4049/jimmunol.180.5.2815. [DOI] [PubMed] [Google Scholar]
- 29*.Astrof NS, Salas A, Shimaoka M, Chen J, Springer TA. Importance of Force Linkage in Mechanochemistry of Adhesion Receptors. Biochemistry. 2006;45:15020–15028. doi: 10.1021/bi061566o. The paper suggests that shear force application on ligand-bound I –domain stabilizes this domain in an open state favorable for adhesion. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Puklin-Faucher E, Gao M, Schulten K, Vogel V. How the headpiece hinge angle is opened: new insights into the dynamics of integrin activation. J Cell Biol. 2006;175:349–360. doi: 10.1083/jcb.200602071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alon R, Feigelson SW, Rose DM, Schmitz J, Manevich E, Overby DR, Winter E, Grabovsky V, Shinder V, Matthews BD, et al. Integrin α4β1(VLA-4)-dependent T cell tethering and adhesion strengthening under shear stress requires paxillin binding to the α4 cytoplasmic domain. J Cell Biol. 2005;171:1073–1084. doi: 10.1083/jcb.200503155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32*.Cairo CW, Mirchev R, Golan DE. Cytoskeletal regulation couples LFA-1 conformational changes to receptor lateral mobility and clustering. Immunity. 2006;25:297–308. doi: 10.1016/j.immuni.2006.06.012. A first demonstration for the existence of multiple conformational states of LFA-1 with distinct mobility states in the plasma membrane. The work suggests that subsets of high affinity LFA-1 are anchored prior to ligand binding and that integrin anchorage is further enhanced upon ligand binding. [DOI] [PubMed] [Google Scholar]
- 33.Chan JR, Hyduk SJ, Cybulsky MI. Chemoattractants induce a rapid and transient upregulation of monocyte α4 integrin affinity for vascular cell adhesion molecule 1 which mediates arrest: an early step in the process of emigration. J Exp Med. 2001;193:1149–1158. doi: 10.1084/jem.193.10.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kunkel EJ, Dunne JL, Ley K. Leukocyte arrest during cytokine-dependent inflammation in vivo. J Immunol. 2000;164:3301–3308. doi: 10.4049/jimmunol.164.6.3301. [DOI] [PubMed] [Google Scholar]
- 35.Green CE, Pearson DN, Camphausen RT, Staunton DE, Simon SI. Shear-dependent capping of L-selectin and P-selectin glycoprotein ligand 1 by E-selectin signals activation of high-avidity beta2-integrin on neutrophils. J Immunol. 2004;172:7780–7790. doi: 10.4049/jimmunol.172.12.7780. [DOI] [PubMed] [Google Scholar]
- 36.Kinashi T. Intracellular signalling controlling integrin activation in lymphocytes. Nat Rev Immunol. 2005;5:546–559. doi: 10.1038/nri1646. [DOI] [PubMed] [Google Scholar]
- 37.Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7:678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- 38*.Wegener KL, Partridge AW, Han J, Pickford AR, Liddington RC, Ginsberg MH, Campbell ID. Structural basis of integrin activation by talin. Cell. 2007;128:171–182. doi: 10.1016/j.cell.2006.10.048. X-ray crystal structure and NMR analysis of the talin PTB domain interacting with the NPXY motif on the cytoplasmic tail of the β3 subunit. The authors suggest a two-step model for talin mediated activation of integrins. [DOI] [PubMed] [Google Scholar]
- 39.Crittenden JR, Bergmeier W, Zhang Y, Piffath CL, Liang Y, Wagner DD, Housman DE, Graybiel AM. CalDAG-GEFI integrates signaling for platelet aggregation and thrombus formation. Nat Med. 2004;10:982–986. doi: 10.1038/nm1098. [DOI] [PubMed] [Google Scholar]
- 40.Hyduk SJ, Chan JR, Duffy ST, Chen M, Peterson MD, Waddell TK, Digby GC, Szaszi K, Kapus A, Cybulsky MI. Phospholipase C, calcium, and calmodulin are critical for alpha4beta1 integrin affinity up-regulation and monocyte arrest triggered by chemoattractants. Blood. 2007;109:176–184. doi: 10.1182/blood-2006-01-029199. [DOI] [PubMed] [Google Scholar]
- 41*.Pasvolsky R, Feigelson SW, Kilic SS, Simon AJ, Tal-Lapidot G, Grabovsky V, Crittenden JR, Amariglio N, Safran M, Graybiel AM et al. A LAD-III syndrome is associated with defective expression of the Rap-1 activator CalDAG-GEFI in lymphocytes, neutrophils, and platelets. J Exp Med. 2007;204:1571–1582. doi: 10.1084/jem.20070058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42*.Bergmeier W, Goerge T, Wang H, Crittenden JR, Baldwin ACW, Cifuni SM, Housman DE, Graybiel AM, Wagner DD. Mice lacking the signaling molecule CalDAG-GEFI represent a model for leukocyte adhesion deficiency type III. J Clin Invest. 2007;117:1699–1707. doi: 10.1172/JCI30575. Ref. 41 describes the identification of a rare leukocyte adhesion deficiency syndrome, LAD-III, with multiple defects in integrin activation by GPCR signals on platelets, neutrophils and lymphocytes resulting from impaired expression of the Rap-1 GEF, CalDAG-GEFI. Ref. 42 describes dramatic effects of CalDAG-GEF-I deficiency on integrin activation in murine neutrophils and demonstrates inability of GEF null neutrophils to arrest on inflamed vessels in vivo. The two papers underscore the key role of Rap-1 activation by chemokine signals in integrin activation in all major blood borne cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ghandour H, Cullere X, Alvarez A, Luscinskas FW, Mayadas TN. Essential role for Rap1 GTPase and its guanine exchange factor CalDAG-GEFI in LFA-1 but not VLA-4 integrin-mediated human T cell adhesion. Blood. 2007 doi: 10.1182/blood-2007-03-077628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shimonaka M, Katagiri K, Nakayama T, Fujita N, Tsuruo T, Yoshie O, Kinashi T. Rap1 translates chemokine signals to integrin activation, cell polarization, and motility across vascular endothelium under flow. J Cell Biol. 2003;161:417–427. doi: 10.1083/jcb.200301133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Han J, Lim CJ, Watanabe N, Soriani A, Ratnikov B, Calderwood DA, Puzon-McLaughlin W, Lafuente EM, Boussiotis VA, Shattil SJ, et al. Reconstructing and deconstructing agonist-induced activation of integrin alphaIIbbeta3. Curr Biol. 2006;16:1796–1806. doi: 10.1016/j.cub.2006.08.035. [DOI] [PubMed] [Google Scholar]
- 46.Petrich BG, Fogelstrand P, Partridge AW, Yousefi N, Ablooglu AJ, Shattil SJ, Ginsberg MH. The antithrombotic potential of selective blockade of talin-dependent integrin alpha IIb beta 3 (platelet GPIIb-IIIa) activation. J Clin Invest. 2007;117:2250–2259. doi: 10.1172/JCI31024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nayal A, Webb DJ, Horwitz AF. Talin: an emerging focal point of adhesion dynamics. Curr Opin Cell Biol. 2004;16:94–98. doi: 10.1016/j.ceb.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 48.Katagiri K, Maeda A, Shimonaka M, Kinashi T. RAPL, a Rap1-binding molecule that mediates Rap1-induced adhesion through spatial regulation of LFA-1. Nat Immunol. 2003;4:741–748. doi: 10.1038/ni950. [DOI] [PubMed] [Google Scholar]
- 49.Di Paolo G, Pellegrini L, Letinic K, Cestra G, Zoncu R, Voronov S, Chang S, Guo J, Wenk MR, De Camilli P. Recruitment and regulation of phosphatidylinositol phosphate kinase type 1 gamma by the FERM domain of talin. Nature. 2002;420:85–89. doi: 10.1038/nature01147. [DOI] [PubMed] [Google Scholar]
- 50.Giagulli C, Scarpini E, Ottoboni L, Narumiya S, Butcher EC, Constantin G, Laudanna C. RhoA and zeta PKC control distinct modalities of LFA-1 activation by chemokines. Critical role of LFA-1 affinity triggering in lymphocyte in vivo homing. Immunity. 2004;20:25–35. doi: 10.1016/s1074-7613(03)00350-9. [DOI] [PubMed] [Google Scholar]
- 51.Singer II, Scott S, Kawka DW, Chin J, Daugherty BL, DeMartino JA, DiSalvo J, Gould SL, Lineberger JE, Malkowitz L, et al. CCR5, CXCR4, and CD4 are clustered and closely apposed on microvilli of human macrophages and T cells. J Virol. 2001;75:3779–3790. doi: 10.1128/JVI.75.8.3779-3790.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Abitorabi MA, Pachynski RK, Ferrando RE, Tidswell M, Erle DJ. Presentation of integrins on leukocyte microvilli: a role for the extracellular domain in determining membrane localization. J Cell Biol. 1997;139:563–571. doi: 10.1083/jcb.139.2.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weber C, Weber KS, Klier C, Gu S, Wank R, Horuk R, Nelson PJ. Specialized roles of the chemokine receptors CCR1 and CCR5 in the recruitment of monocytes and Th1-like/CD45RO+ T cells. Blood. 2001;97:1144–1146. doi: 10.1182/blood.v97.4.1144. [DOI] [PubMed] [Google Scholar]
- 54.Abram CL, Lowell CA. Convergence of immunoreceptor and integrin signaling. Immunol Rev. 2007;218:29–44. doi: 10.1111/j.1600-065X.2007.00531.x. [DOI] [PubMed] [Google Scholar]
- 55.Chan JR, Hyduk SJ, Cybulsky MI. Alpha 4 beta 1 integrin/VCAM-1 interaction activates alpha L beta 2 integrin-mediated adhesion to ICAM-1 in human T cells. J Immunol. 2000;164:746–753. doi: 10.4049/jimmunol.164.2.746. [DOI] [PubMed] [Google Scholar]
- 56.Nandi A, Estess P, Siegelman M. Bimolecular complex between rolling and firm adhesion receptors required for cell arrest; CD44 association with VLA-4 in T cell extravasation. Immunity. 2004;20:455–465. doi: 10.1016/s1074-7613(04)00077-9. [DOI] [PubMed] [Google Scholar]
- 57.Ticchioni M, Raimondi V, Lamy L, Wijdenes J, Lindberg FP, Brown EJ, Bernard A. Integrin-associated protein (CD47/IAP) contributes to T cell arrest on inflammatory vascular endothelium under flow. Faseb J. 2001;15:341–350. doi: 10.1096/fj.99-0833com. [DOI] [PubMed] [Google Scholar]
- 58.Simon SI, Hu Y, Vestweber D, Smith CW. Neutrophil tethering on E-selectin activates β2 integrin binding to ICAM-1 through a mitogen-activated protein kinase signal transduction pathway. J Immunol. 2000;164:4348–4358. doi: 10.4049/jimmunol.164.8.4348. [DOI] [PubMed] [Google Scholar]
- 59.Giagulli C, Ottoboni L, Caveggion E, Rossi B, Lowell C, Constantin G, Laudanna C, Berton G. The Src family kinases Hck and Fgr are dispensable for inside-out, chemoattractant-induced signaling regulating beta 2 integrin affinity and valency in neutrophils, but are required for beta 2 integrin-mediated outside-in signaling involved in sustained adhesion. J Immunol. 2006;177:604–611. doi: 10.4049/jimmunol.177.1.604. [DOI] [PubMed] [Google Scholar]