

Abstract
CDH1a, a non-canonical transcript of the CDH1 gene, has been found to be expressed in some gastric cancer (GC) cell lines, whereas it is absent in normal gastric mucosa. Recently, we detected CDH1a transcript variant in fresh-frozen tumor tissues obtained from patients with GC. The expression of this variant in tissue samples was investigated by the chip-based digital PCR (dPCR) approach presented here. dPCR offers the potential for an accurate, robust, and highly sensitive measurement of nucleic acids and is increasingly utilized for many applications in different fields. dPCR is capable of detecting rare targets; in addition, dPCR offers the possibility for absolute and precise quantification of nucleic acids without the need for calibrators and standard curves. In fact, the reaction partitioning enriches the target from the background, which improves amplification efficiency and tolerance to inhibitors. Such characteristics make dPCR an optimal tool for the detection of the CDH1a rare transcript.
Keywords: Cancer Research, Issue 132, CDH1a, dPCR, rare transcript variant, gastric cancer, fresh-frozen tissues, tumor tissue, normal tissue
Introduction
The CDH1 gene encodes for E-cadherin, a key factor involved in the maintenance of the normal gastric epithelium through the regulation of cell adhesion, survival, proliferation, and migration1. Loss of E-cadherin protein as a result of deleterious germline or somatic alterations of CDH1 has been associated with the development of GC2,3. Non-canonical transcripts arising from intron 2 of the gene have also been hypothesized to play a role in gastric carcinogenesis4,5. In particular, one such transcript, CDH1a, has been shown to be expressed in GC cell lines but is absent from the normal stomach4. We recently detected CDH1a in GC tissue samples from GC patients using chip-based dPCR5.dPCR was used to evaluate, for the first time, the presence of the CDH1a gene transcript in intestinal GC and in normal tissue.
The gold standard method to determine gene expression is Real-Time quantitative PCR (qPCR). However, the resulting data can sometimes be variable and of poor quality, especially when the level of target in the sample is low. This variability can be caused by contaminants, which inhibit polymerase activity and primer annealing, leading to non-specific amplification and competitive side reactions6.
Although the basic biochemical principles of dPCR are similar to those of qPCR, dPCR shows some advantages, allowing for very precise measurements of genomic DNA (gDNA)/complementary DNA (cDNA) molecules. Indeed, dPCR is an end-point reaction that relies on the calibrated partitioning of a sample into thousands of wells, so that each well contains zero or a single target molecule. Amplification then occurs only in the wells containing a copy of the target and is indicated by a fluorescent signal. The absolute number of target molecules in the original sample can then be calculated by determining the ratio of positive to total partitions using binomial Poisson statistics7.
In addition, the dPCR technique eliminates the need for running a standard curve and, hence, the associated bias and variability, allowing for a direct quantification of targets8,9; it produces more precise and reproducible results independently of contaminants and efficiency due to its high tolerance to inhibitors10; it is more sensitive and specific than qPCR, and is thus a reliable method for the detection of a rare target. Finally, the partitioning of the sample into multiple reactions reduces the competition with background molecules and improves the limit of target detection, making amplification possible and facilitating the detection of single molecules of gDNA/cDNA6. The detection and quantification of nucleic acids by chip-based dPCR has been increasingly applied to copy number variation, quantification of DNA fragments, and mutation analyses11,12,13, given the precision and low material input requirement of the method. In addition, dPCR has recently been integrated into the analysis of both microRNAs14 and gene transcripts5,15.
Protocol
The protocol follows the guidelines of the IRST Human Research Ethics Committee.
NOTE: This procedure is specifically designed for the detection of a low number of cDNA molecules in human fresh-frozen tissues. The tissue sections have been cut on dry ice, while still frozen, from previously validated patient-derived gastric tumor or normal tissue samples.
1. RNA Isolation and Purification
- RNA extraction Note: RNA isolation is performed under the hood using a specific product (see Table of Materials). However, a variety of isolation kits are commercially available.
- Homogenize 50 - 100 mg of finely cut fresh-frozen tissue sample in a 1.5 mL tube with 1 mL of the RNA isolation reagent and vortex vigorously for 15 s. Incubate the tubes at -80 °C overnight.
- Incubate the tube containing the sample at room temperature for 5 min and mix by vortexing for 15 s.
- Keep the tube on ice, add 200 µL of chloroform, and vortex vigorously for 15 s.
- Incubate the tubes at room temperature for 60 s and centrifuge at 12,000 x g for 15 min at 4 °C.
- Transfer the aqueous phase in a 1.5 mL tube and add 20 µg of glycogen. NOTE: It is important to carefully avoid transferring any of the interphase or organic layer in order to reduce contamination.
- Add 500 µL of isopropanol, inverting the tube to mix, and incubate on ice for 10 min.
- Centrifuge the tube at 12,000 x g for 15 min at 4 °C to precipitate RNA. NOTE: RNA will be present in a gel-like pellet on the side and bottom of the tube, often invisible after centrifugation.
- Remove the supernatant without disturbing the precipitate and wash it with 1 mL of 75% ethanol by pipetting.
- Centrifuge the tube at 7,500 x g for 5 min at 4 °C and remove the supernatant without disturbing the pellet.
- Let the pellet dry until the precipitate becomes transparent and dissolve it in 50 µL of RNase-free water.
- Freeze the samples at -80 °C at least overnight before quantification.
- Genomic DNA elimination and RNA purification Note: A DNase digestion step, followed by a column-based RNA purification, is recommended for analyses of low-abundance targets in order to digest contaminating DNA.
- Dissolve the lyophilized DNase I (1,500 Kunitz units) in 550 µL of RNase-free water using a syringe, mix gently by inverting the vial, and divide the reconstituted stock solution into aliquots.
- Transfer in a 1.5 mL tube the calculated volume of sample with 15 µg of RNA, 10 µL of DNase Digestion buffer from the kit (listed in Table of Materials), 2.5 µL of DNase I stock solution, and RNase-free water to 100 µL. Incubate at room temperature for 10 min.
- Add 350 µL of tissue lysis buffer (from the kit, see Table of Materials) and mix by pipetting.
- Add 250 µL of 100% ethanol and mix by pipetting.
- Transfer the entire volume (700 µL) into a new spin column and centrifuge at 12,000 x g for 15 s.
- Transfer the filter into a new collection tube, add 500 µL of 80% ethanol to the column and centrifuge at 12,000 x g for 2 min.
- Open the lid of the spin column and centrifuge at 12,000 x g for 5 min with a new collection tube to dry the filter; then transfer the filter to a 1.5 mL tube.
- Add 14 µL of RNase-free water directly to the filter column and centrifuge at 12,000 x g for 1 min.
- Keep the samples on ice and proceed to quantification using 2 µL of the sample on a bench-top spectrophotometer or store the RNA at -80 °C until use.
2. cDNA Synthesis
Place 1,000 ng of RNA, 4 µL of master mix, 1 µL of reverse transcriptase, and RNase-free water to a final volume of 20 µL in a sterile PCR tube. Mix gently and spin down.
Incubate the reaction mix in a thermal cycler, applying the following conditions: 5 min at 25 °C, 30 min at 42 °C, 5 min at 85 °C.
Centrifuge the tubes briefly and proceed to dPCR analysis or store at -20 °C until needed.
3. Digital PCR Reaction Set Up
- dPCR reaction mix and sample preparation
- Thaw the master mix and the assay at room temperature for at least 20 min.
- Dilute the cDNA samples to a concentration of 300 ng in 6 µL of water.
- Gently vortex the master mix and prepare the mix in a sterile tube with 8.7 µL of master mix, 0.87 µL of the CDH1a custom designed assay primer, and 1.83 µL of nuclease-free water for a final volume of 11.4 µL. NOTE: For CDH1a custom designed assay primer details see the Table of Materials.
- Transfer 11.4 µL of the prepared mix to the diluted cDNA sample, mix gently, and briefly centrifuge. NOTE: Volumes include 20% excess to compensate for volume loss from pipetting. Prepare the mix for all samples and a no template control (NTC).
- Chip preparation Note: For optimal results load the chips as soon as possible.
- Plug in the chip loader and wait until the indicator light turns green.
- Remove the cap of the immersion fluid syringe by gently pulling back the plunger 1 - 2 mm and releasing it to facilitate this step, and replace it with a tip.
- Take a new chip and take note of the code written on the lid to associate it with the sample.
- Hold the lid carefully by its side, peel away the protective film, and place the lid with the sticky face up in the correct orientation.
- Carefully pick up a chip, taking care not to touch the internal part, and load it onto the chip nest in the correct position by pressing down the lever to open the clamp.
- Load a new loading blade onto the loader and push it gently to ensure that it is firmly in place.
- Transfer 14.5 µL of the dPCR reaction mix onto the loading blade without making air bubbles or deflecting the blade, after which press the black loading button to distribute the volume on the chip.
- Use the immersion fluid syringe to transfer about 20 drops onto the chip surface taking care not to touch the surface with the tip. NOTE: It is important to cover the entire surface without spilling fluid over the edges.
- Rotate the loader arm to make the lid come into contact with the chip and press down for 15 s.
- Press the lid button to release the chip and return the arm to its position.
- Hold the assembled chip at a 45° angle and carefully dispense the immersion fluid with the syringe through the fill port, rotate the chip slightly to make sure there are no air bubbles, and remove any excess fluid with a sterile wipe.
- Seal the chip case by gently peeling away the label on top of the chip lid and press over the fill port for at least 5 s.
- Store the chip in the dark until ready to load in the thermal cycler. NOTE: Prepared chips should be used within 2 h.
- dPCR reaction
- Open the lid and install the adapters to both blocks, even when a single block is used.
- Place the chips on the sample block in the correct position. NOTE: The fill port must be oriented towards the front of the thermal cycler in an elevated position to allow any air bubbles to float to the top without disturbing the window of the chip. Use empty chips to balance the two blocks.
- Lay the thermal pad on the sample block to completely cover the chips.
- Close the lid and start the PCR run, applying the following conditions: hold at 96 °C for 10 min; 45 cycles of 60 °C for 2 min and 98 °C for 30 s; hold at 60 °C for 2 min; hold at 4 °C. Turn off the thermal cycler and thaw the chips at room temperature for at least 10 min. NOTE: Chip analysis must be performed within one hour.
- Chip analysis
- Open the lid of the instrument and remove the thermal pads, then remove the chips from the adapters. NOTE: Store the chips in a dark, clean location until analysis.
- Clean the chip surface with isopropanol and a sterile wipe. NOTE: Inspect each chip for leaks or potential problems.
- Insert the USB into the detector system to save the data.
- Open the chip tray of the detector system, load the chip face-up in the correct position and then close the tray.
- Wait 30 s for processing, then remove the chip and insert the next one.
- Wait for the analysis to be completed for all the processed chips then insert the USB into a computer to transfer the files. NOTE: The total time for analysis is around 2 to 3 min/chip.
4. Data Analysis and Interpretation
Connect to the cloud-based software platform required to carry out all the downstream analyses.
Create a project and import all data files of the chips of interest.
Enter the sample name; select the dye and assay used in the "Define Chips" tab.
Determine if the chip is acceptable by visualizing it in the "Review Data" tab, checking how thoroughly the sample was loaded onto the chip and how many data points are evaluable. NOTE: Reject chips with less than 13,000 evaluable data points.
Move on to the scatter plot of the selected chip on the right side of the screen; apply a threshold of 6,000 for the fluorescein amidite (FAM) reporter dye signals (Y-axis) to all the chips. NOTE: The threshold set up may vary on the basis of the assays used.
Remove any dubious positive signals to prevent false positive results by selecting the relative spot on the scatter plot using the lasso tool and pressing on "undetermined". All remaining positive spots indicate the presence of cDNA copies of the rare target analyzed.
Representative Results
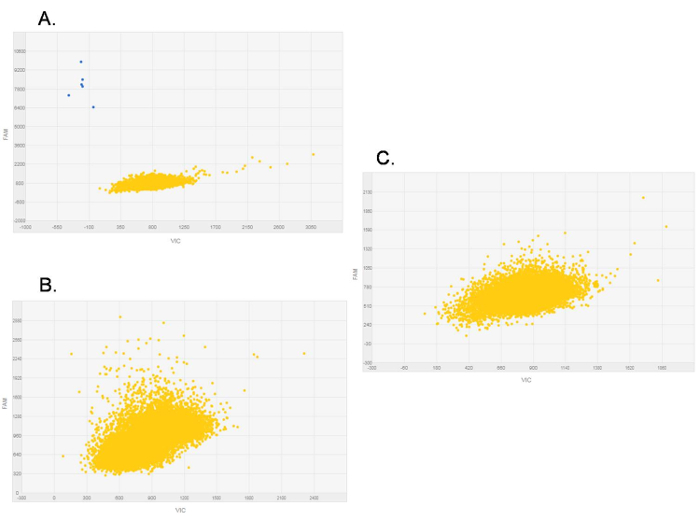
Using the procedure presented here, we checked for the expression of the rare transcript variant CDH1a in gastric fresh-frozen tissues. The analysis by dPCR was performed on 21-paired normal and cancer tissue samples and in 11 additional tumor samples. CDH1a was detectable in 15 out of 32 (47%) tumors, whereas no normal tissue samples showed the presence of this rare transcript5. In our analysis, chips with less than 13,000 data points were rejected, as were chips with non-homogeneous loading. Figure 1 shows a chip view for both evaluable (Figure 1A) and non-evaluable (Figure 1B, C) samples. In the case of the latter, the chip must be discarded due to the presence of multiple bubbles (Figure 1B), a single big bubble (Figure 1C), or as a result of an insufficient number of data points above threshold (Figure 1B). These samples must be run again to obtain interpretable results. With regard to the scatter plot provided by the software (Figure 2), it normally depicts the signals from the FAM reporter dye (Y-axis) against signals from the VIC (manufacturer's proprietary) reporter dye (X-axis). The data points presented on the scatter plot are color-coded, and here, we only visualized the FAM reporter dye signals in blue (Figure 2A), indicating the wells in which the target was amplified. These signals are located closer to the Y-axis and further from the origin of the plot. The second color seen on the plot is yellow and is indicative of the wells in which no amplification occurred. A reliable threshold for the positive signals was set and applied to all the chips to prevent call bias. Here, the selected threshold was 6,000 based on the range of signals observed for the various chips in these experiments. CDH1a cDNA copies (blue signals) were found in a number of tumor samples, showing a clear difference in amplitude from negative signals (Figure 2A). Conversely, no signals above the selected threshold were found in samples negative for CDH1a transcript, i.e., all normal tissue samples and some tumor samples (Figure 2B). Similarly, no positive signals were detected in the NTC in which water was added instead of cDNA, hence serving as a negative control for the dPCR reaction (Figure 2C).

A fine chip analysis must be applied to filter the low-quality data points and eliminate the risk of ambiguous results. This was done by viewing the chip to determine the exact location of the corresponding positive signal: if the blue signal is located at the very borders of the chip (Figure 3A) or around bubbles (Figure 1B), the signal is dismissed from the analysis. To further prevent the risk of false positives, signals above 6,000 for the FAM channel and on the far right of the plot were considered as probably aspecific and were thus filtered out, even though they were localized well within the chip (Figure 3B). In this way, a sample is considered positive for the expression of the rare transcript, CDH1a, even if it shows a single amplification signal, as long as that signal complies with the aforementioned conditions.
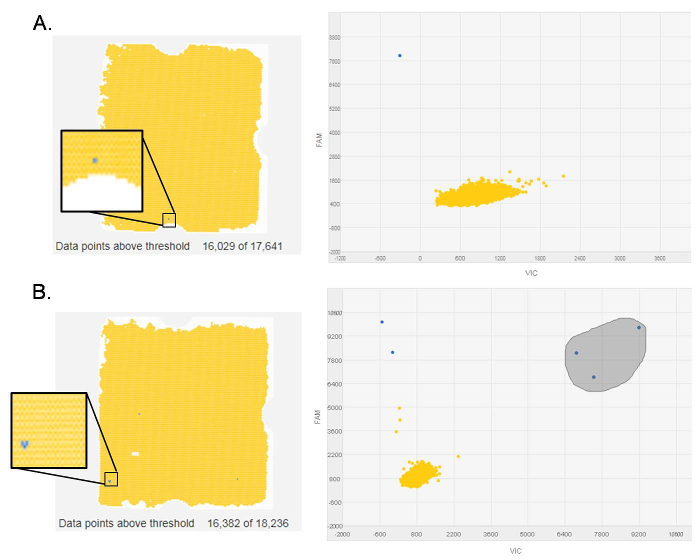
Figure 1. Representative evaluable and non-evaluable chips. The blue dots are wells in which the CDH1a transcript was amplified; the yellow dots are wells in which no amplification occurred; the white dots are empty wells. Data points above threshold are indicated underneath each chip. (A) An evaluable chip with blue signals localized well within the chip's borders. (B) A non-evaluable chip with less than 13,000 data points above threshold. (C) A non-evaluable chip with a large bubble. Please click here to view a larger version of this figure.
Figure 2.Representative digital PCR scatter plots of CDH1a expression. Plots depict signals from the FAM reporter dye (Y-axis) against signals from the VIC reporter dye (X-axis). The blue dots are CDH1a positive expression signals, while the yellow dots are the "No amplification" signals. (A) CDH1a-positive sample. (B) CDH1a-negative sample. (C) No template control (NTC) showing zero CDH1a expression. Please click here to view a larger version of this figure.
Figure 3.Representative fine-chip analysis of digital PCR scatter plots. On the left is the chip view with the signals of interest zoomed-in in a black square, while on the right is the corresponding scatter plot. (A) A blue signal localized on the correct fraction of the plot but at the border of the chip. (B) Three blue signals highlighted in grey localized on the far right of the plot but within the chip. Please click here to view a larger version of this figure.
Discussion
dPCR was originally developed for DNA molecular measurements10,11,12,13 and in time this technology was adapted for the quantification of microRNAs and RNA transcripts5,14,15. In this protocol we have extended the list of applications to include detection of rare transcripts derived from fresh-frozen tissue samples. To that purpose we utilized a rather high amount of cDNA (300 ng). Although this amount is not the maximum that can be used in dPCR, it was sufficient for setting up standardized conditions to compare all our samples. By setting the florescence signal threshold at 6,000 and minding the locations of positive amplification signals on both the chip and scatter plot, we successfully detected this rare target in gastric tumor tissue samples.
By dPCR, we could not absolutely quantify CDH1a, due to a very low amount of the target, but we could reliably assess the presence/absence of this rare transcript. Indeed, the quantitative nature of this technique is limited by the initial number of targets present in the analyzed samples7. Of relevance, the technique itself remains to be fairly expensive and time intensive. Thus, when the analysis of numerous samples and/or of several targets is required, alternative techniques should be considered7,8.
That being said, dPCR undoubtedly provides the ultimate platform for sensitive measurement and quantification of nucleic acids, as it improves precision and reproducibility with respect to qPCR7,8. Moreover, it could be considered as the only method available for analyzing gene expression of low-abundant nucleic acids that near the limit of qPCR sensitivity6.
The protocol here described can be adapted to RNAs from any tissues, enabling the detection of rare targets, as in our study. In addition, the assay could be extended onto absolute quantification of transcripts. In such cases, the number of copies/µL reported by the instrument for a given sample must be simply multiplied by the loaded reaction volume and divided by the initial amount of cDNA. To overcome variability produced by reverse-transcription, gene-specific calibrators should be included, when possible6. Looking to the future, dPCR holds considerable potential in becoming the gold standard platform, not only for rare sequence detection, but also rare mutation detection and precise copy number quantification7. This is especially relevant in genetic mosaicism, as well as in cancer research where tumor heterogeneity can impact a patient's clinical outcome and response to treatment.
From the practical point of view, when setting up the dPCR protocol there are multiple critical steps that must be highlighted. First of all, the reaction mix containing the appropriate amount of cDNA based on the target expression level expected, should be transferred with great care in the loading blade so as not to create air bubbles, which could interfere with sample homogeneity. In addition, the blade itself should first be positioned appropriately; otherwise it could result in an uneven sample distribution. Furthermore, sufficient coating with immersion fluid and accurate sealing of the lid should always be performed. Regarding chip processing and analysis, it is important to be aware of the possibility of accumulating condensation on the chip; in such a scenario, the chip should be re-wiped with isopropanol and re-analyzed. Moreover, as dPCR is based on Poisson statistics, a minimum number of evaluable data points (>10,000) must be present on the chip, otherwise the sample should be run again. Finally, for low target copy detection experiments, a critical confounding factor could be the signal-to-noise ratio, but a reliable threshold positioning can help in identifying real positive signals.
Disclosures
The authors report no conflicts of interest for this work.
Acknowledgments
The authors wish to thank Gráinne Tierney for editorial assistance.
References
- van Roy F, Berx G. The cell-cell adhesion molecule E-cadherin. Cell Mol Life Sci. 2008;65(23):3756–3788. doi: 10.1007/s00018-008-8281-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carneiro P, et al. E-cadherin dysfunction in gastric cancer: cellular consequences, clinical applications and open questions. FEBS Lett. 2012;586(18):2981–2989. doi: 10.1016/j.febslet.2012.07.045. [DOI] [PubMed] [Google Scholar]
- Corso G, et al. Somatic mutations and deletions of the E-cadherin gene predict poor survival of patients with gastric cancer. J Clin Oncol. 2013;31(7):868–875. doi: 10.1200/JCO.2012.44.4612. [DOI] [PubMed] [Google Scholar]
- Pinheiro H, et al. Transcription initiation arising from E-cadherin/CDH1 intron2: a novel protein isoform that increases gastric cancer cell invasion and angiogenesis. Hum Mol Genet. 2012;21(19):4253–4269. doi: 10.1093/hmg/dds248. [DOI] [PubMed] [Google Scholar]
- Abou Khouzam R, et al. Digital PCR identifies changes in CDH1 (E-cadherin) transcription pattern in intestinal-type gastric cancer. Oncotarget. 2017;8(12):18811–18820. doi: 10.18632/oncotarget.13401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SC, Laperriere G, Germain H. Droplet Digital PCR versus qPCR for gene expression analysis with low abundant targets: from variable nonsense to publication quality data. Sci Rep. 2017;7(1) doi: 10.1038/s41598-017-02217-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu AS. Digital Assays Part I: Partitioning statistics and digital PCR. SLAS Technol. 2017;22(4):369–386. doi: 10.1177/2472630317705680. [DOI] [PubMed] [Google Scholar]
- Huggett JF, Whale A. Digital PCR as a novel technology and its potential implications for molecular diagnostics. Clin Chem. 2013;59(12):1691–1693. doi: 10.1373/clinchem.2013.214742. [DOI] [PubMed] [Google Scholar]
- Alikian M, et al. RT-qPCR and RT-Digital PCR: A comparison of different platforms for the evaluation of residual disease in chronic myeloid leukemia. Clin Chem. 2017;63(2):525–531. doi: 10.1373/clinchem.2016.262824. [DOI] [PubMed] [Google Scholar]
- Hoshino T, Inagaki F. Molecular quantification of environmental DNA using microfluidics and digital PCR. Syst Appl Microbiol. 2012;35(6):390–395. doi: 10.1016/j.syapm.2012.06.006. [DOI] [PubMed] [Google Scholar]
- Salvi S, et al. Circulating AR copy number and outcome to enzalutamide in docetaxel-treated metastatic castration-resistant prostate cancer. Oncotarget. 2016;7(25):37839–37845. doi: 10.18632/oncotarget.9341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessitore MV, et al. Detection of newly produced T and B lymphocytes by digital PCR in blood stored dry on nylon flocked swabs. J.Transl.Med. 2017;15(1) doi: 10.1186/s12967-017-1169-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sho S, et al. Digital PCR Improves mutation analysis in pancreas fine needle aspiration biopsy specimens. PLoS One. 2017;12(1):e0170897. doi: 10.1371/journal.pone.0170897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conte D, et al. Novel method to detect microRNAs using chip-based QuantStudio 3D digital PCR. BMC Genomics. 2015;16:849. doi: 10.1186/s12864-015-2097-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders R, Mason DJ, Foy CA, Huggett JF. Evaluation of digital PCR for absolute RNA quantification. PLoS One. 2013;8(9):e75296. doi: 10.1371/journal.pone.0075296. [DOI] [PMC free article] [PubMed] [Google Scholar]