

Abstract
While oxidative stress is a commonly cited toxicological mechanism, conventional methods to study it suffer from a number of shortcomings, including destruction of the sample, introduction of potential artifacts, and a lack of specificity for the reactive species involved. Thus, there is a current need in the field of toxicology for non-destructive, sensitive, and specific methods that can be used to observe and quantify intracellular redox perturbations, more commonly referred to as oxidative stress. Here, we present a method for the use of two genetically-encoded fluorogenic sensors, roGFP2 and HyPer, to be used in live-cell imaging studies to observe xenobiotic-induced oxidative responses. roGFP2 equilibrates with the glutathione redox potential (EGSH), while HyPer directly detects hydrogen peroxide (H2O2). Both sensors can be expressed into various cell types via transfection or transduction, and can be targeted to specific cellular compartments. Most importantly, live-cell microscopy using these sensors offers high spatial and temporal resolution that is not possible using conventional methods. Changes in the fluorescence intensity monitored at 510 nm serves as the readout for both genetically-encoded fluorogenic sensors when sequentially excited by 404 nm and 488 nm light. This property makes both sensors ratiometric, eliminating common microscopy artifacts and correcting for differences in sensor expression between cells. This methodology can be applied across a variety of fluorometric platforms capable of exciting and collecting emissions at the prescribed wavelengths, making it suitable for use with confocal imaging systems, conventional wide-field microscopy, and plate readers. Both genetically-encoded fluorogenic sensors have been used in a variety of cell types and toxicological studies to monitor cellular EGSH and H2O2 generation in real-time. Outlined here is a standardized method that is widely adaptable across cell types and fluorometric platforms for the application of roGFP2 and HyPer in live-cell toxicological assessments of oxidative stress.
Keywords: Cancer Research, Issue 132, Oxidative stress, Toxicology, Fluorescence, Live-cell imaging, Glutathione, Hydrogen peroxide
Introduction
The term "oxidative stress" is frequently cited as a mechanism in toxicology, yet rarely is this term described specifically. Oxidative stress can refer to several intracellular processes, including generation of reactive oxygen species, damage caused by free radicals, the oxidation of antioxidant molecules, and even the activation of specific signaling cascades. A broad range of environmental contaminants1,2 and pharmaceutical agents3,4 have been documented to induce oxidative stress by either direct action of the xenobiotic compound itself5,6 or secondarily by production of oxidant species as part of a cellular response7,8,9,10. It is therefore of great interest in toxicology to accurately observe and characterize the oxidative processes leading to adverse outcomes. Conventional methods of measuring oxidative stress involve identification of oxidized biomolecules11,12,13,14,15 or antioxidants16,17,18,19,20, or direct measurement of reactive species themselves21,22,23,24. However, these methods typically require cellular disruption, which often consumes the sample, eliminates spatial resolution, and potentially introduces artifacts25. The development of more sensitive and specific methods for the detection of oxidant species and markers of oxidative stress is broadly applicable to the investigation of the adverse effects of xenobiotic exposure.
Live-cell microscopy using a new generation of genetically-encoded fluorogenic sensors has emerged as a powerful tool to monitor the intracellular redox status of living cells. These sensors are typically expressed using a vector under the control of a viral promoter that is introduced via transfection or transduction methodologies. High expression efficiencies are not necessary, since cells expressing the fluorescent sensor can be easily identified visually. For toxicological assessments, cells expressing these sensors can be observed using fluorescence microscopy as they are exposed to xenobiotic compounds in real-time. This experimental design permits repeated measurements in the same cell, allowing each cell's established baseline to act as its own control. The high temporal resolution afforded by live-cell imaging is well-suited for the detection of oxidative events, particularly those that are modest in magnitude or transient in nature. In addition to being both sensitive and specific to their target molecules, the fluorescence of some of these sensors can be excited using two wavelengths of light. This phenomenon allows the fluorescent emission to be expressed as a ratio, which permits the discernment of signal changes associated with authentic sensor responses from those caused by artefacts such as variations in sensor expression, cell thickness, lamp intensity, photobleaching, and sensitivity of the fluorescence detector26. Another advantage of the use of fluorogenic sensors is that they can be targeted to specific cellular compartments, creating a level of spatial resolution that is unmatched by conventional methods25,26,27.
A large family of genetically-encoded sensors based on green fluorescent protein (GFP) have been developed and characterized to report on a broad variety of physiological markers, including pH, temperature, calcium concentrations, and the ATP/ADP ratio25,28,29,30,31. Included among these are sensors of the glutathione redox potential (EGSH) and hydrogen peroxide (H2O2). While these sensors were developed for applications in redox biology and physiology, they have also been adapted to study xenobiotic-induced oxidative stress. Specifically, the protocol outlined here describes the use of the EGSH sensor roGFP2 and the H2O2 sensor HyPer.
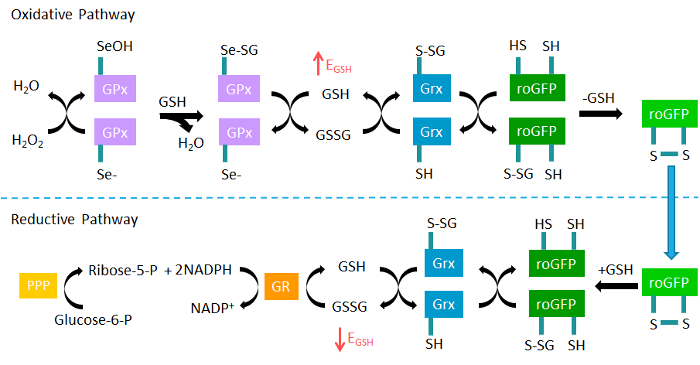
roGFP2 reports on the redox potential of intracellular reduced and oxidized glutathione (GSH/GSSG) through a redox relay involving glutathione peroxidase (GPx), glutaredoxin (Grx), and glutathione reductase (GR) (Figure 1)25,32,33. Glutathione is the predominant cellular antioxidant molecule and is present primarily in its reduced form (GSH) in millimolar concentrations in the cytosol25,34. While EGSH has not been linked to any functional outcome, it is recognized as an important indicator of intracellular oxidative status34. A relatively small increase in the concentration of GSSG results in an increase in EGSH that is detectable by roGFP2. Equally important, monitoring of EGSH using roGFP2 during xenobiotic exposures can potentially reveal much about the mechanism of action at several points in the redox relay and associated pathways, such as the pentose phosphate shunt (Figure 1)35. The second sensor discussed here, HyPer, is an intracellular H2O2 probe derived from the insertion of yellow fluorescent protein (YFP) into the regulatory domain of bacterial H2O2-sensitive transcription factor OxyR136. Although it has previously been considered a damaging reactive oxygen intermediate, H2O2 is increasingly being recognized as an important intracellular signaling molecule under physiological conditions37,38, suggesting that unrecognized roles for H2O2 exist in toxicology as well. For instance, excess H2O2 induced by a xenobiotic exposure could be a precursor to dysregulation in cellular signaling or a shift in bioenergetics.
Both genetically-encoded fluorogenic sensors have been expressed in several established cell lines, including the human epidermoid carcinoma cell line A431 and the human bronchial epithelial cell line BEAS-2B, to observe changes in EGSH and H2O2 in response to a variety of toxicological exposures. These include gaseous pollutants (ozone35), soluble components of particulate matter (1,2 naphthoquinone39,40 and zinc41), and nickel nanoparticles (unpublished data). These studies represent only a small subset of the possible applications of these two sensors. Theoretically, any cell type that is capable of receiving and expressing the DNA of these sensors through conventional molecular biology techniques can be utilized to assess the effects of xenobiotics suspected to alter the cellular oxidative state. To date, one or more of these sensors has been expressed in various prokaryotes and eukaryotes, including several mammalian, plant, bacterial, and yeast cell types25,26,36,42. The readout for both EGSH and H2O2 sensors is a change in the intensity of fluorescence emitted at 510 nm upon excitation with 488 and 404 nm light. This method is widely adaptable across fluorometric platforms, including various types of microscopy (confocal and wide-field) and plate readers. The method presented here allows for sensitive and specific observation of intracellular EGSH and H2O2 in in vitro toxicological systems.
Protocol
1. Preparation of Cells
NOTE: This procedure describes the lentiviral transduction of an immortalized cell line (BEAS-2B; ATCC, Manassas, VA) to express the desired reporter (roGFP2 or HyPer). Other cell lines/types and/or methods of gene transfer, including transfection, can be utilized as long as they result in a level of reporter expression adequate to visualize a sufficient number of sensor-expressing cells per field of view (typically 5 - 10 cells). If using transfection methods, the procedure should be performed in the dish that will ultimately be used for the method of analysis (e.g., transfect the cells in the same dish that will be presented to the microscope). The steps described below provide details for preparing cell transductions to be performed in a 6-well cell culture plate. If this format is not an appropriate size for the desired application, other vessels can be substituted.
Grow cells to approximately 40 - 60% confluence in normal growth media. NOTE: This study utilized the human bronchial epithelial cell line, BEAS-2B, grown in keratinocyte growth medium (KGM).
- Just prior to transduction, replace the growth media on the cells with 500 µL of serum-free media containing the appropriate amount of virus, as calculated by the formula:
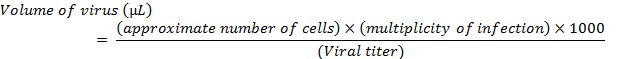
- For transduction of BEAS-2B cells, use serum-free keratinocyte basal medium (KBM) to replace the KGM growth media during viral incubations. NOTE: The above calculation is for a single transduction of one well of cells in a 6-well format. If needed, perform transductions of several wells can be performed by adjusting the formula with a multiplication by the number of wells to be transduced. For lentiviral transductions, use a multiplicity of infection (MOI) between 5 and 20. For adenoviral transductions, use an MOI between 100 and 500.
Incubate cells with the viral mixture at 37 °C for 4 h, redistributing the viral particles in the dish every 30 to 60 min with a brief rocking or swirling motion.
Add 1 mL of complete growth media to the dish and incubate at 37 °C for an additional 4 - 16 h.
Remove all media and replace with fresh complete growth media.
Continue to grow and passage cells, expand as needed. NOTE: Cells should begin appreciably expressing the sensor within 12 - 48 h. If a lentiviral vector was used for the transduction, stable expression of the desired sensor should continue across passages.
For microscopy-based assessments, seed cells into glass bottomed microscope dishes and grow to desired confluence (≥70 - 80%) prior to imaging. NOTE: Seeding density will depend on the size of the dish, growth rate of the cell-line being used, and the time of the assessment following seeding. For example, seed 300,000 BEAS-2B cells into a 35-mm dish to yield approximately 70 - 80% confluence after 1 day of growth.
2. Microscopy Set-up
NOTE: The protocol described below is performed using a confocal microscope equipped with laser lines at 404 and 488 nm. Other means of making fluorometric assessments of the sensors described within this protocol at their prescribed excitations/emission should also yield viable data. Importantly, equipment settings can vary greatly depending on the type, age, and condition of the instrument being used; thus, any instrument values mentioned may not be specific to the equipment used in other laboratories.
Perform all imaging analysis using environmental controls to maintain an appropriate temperature (e.g., 37 °C), humidity (typically >95% relative humidity), and/or gas concentration (e.g., 5% CO2) suitable for the cells throughout the duration of the experiment.
If using high values of relative humidity, keep any surfaces that may come in contact with the humidified atmosphere (e.g., the microscope objective) at or slightly above the temperature at which the humidity is generated in order to prevent condensation. This can be accomplished with the use of an objective heater and/or heating tape and an appropriate heater control.
Turn on all microscope components and set up all equipment required for sequential excitation at 488 and 404 nm with emission at 510 nm. Ensure all components of the optical configuration are set appropriately for real-time acquisition.
Set up a stage-top environmental chamber to maintain constant temperature at 37 °C, 5% CO2 atmosphere, and >95% humidity. Prior to starting image acquisition, equilibrate the environmental chamber for at least 10 min after the initial set-up of all environmental controls. NOTE: Environmental conditions may be eliminated or adjusted depending on the experiment length, cell type, and exposure being used.
Place the dish of cells (Step 1.7) on the stage-top within the environmental chamber.
With the desired objective lens, find the focal plane of cells using the eyepiece and white light, and ensure normal morphology. NOTE: A 1.4 NA 60X violet-corrected, oil-immersed objective lens is commonly used, which permits identification of intracellular compartments while maximizing the optical resolution of the confocal system.
- Check the fluorescence expression of the cells in the field of view. Do it while visualizing the cells under wide-field fluorescence illumination with an appropriate filter set, such as fluorescein isothiocyanate (FITC). At this point, using wide-field illumination while looking through the eyepiece is more convenient because it is easier to move the dish to select a field of cells to study. In general, choose a field of view that contains at least 5 to 10 cells that are expressing the sensor, as indicated by green fluorescence.
- Alternatively, perform this assessment confocally using laser excitation at a wavelength that is most compatible with the sensor being expressed (488 nm typically works best). NOTE: Due to their intrinsic fluorescent properties, it will be more difficult to visualize cells expressing HyPer than those expressing roGFP2 using either FITC or at 488 nm. However, it should still be possible to see faint cells.
Once a desired field of view is found, close the environmental chamber. NOTE: Use the focus-maintaining feature, often available on advanced microscope stands, to facilitate maintenance of a stable focal plane throughout the study.
- Set up acquisition parameters to ensure optimal assessment of the sensor of interest across the desired exposure period. Below are recommendations and approaches for confocal imaging of sensor responses:
- Adjust the laser power for excitation at 488 nm and emission at 510 nm. Choose a laser power level that allows visualization of the cells, and keep this constant between samples or replicate dishes. NOTE: For this study, 12% and 1.5% laser power were used for the 488 and 404 nm laser lines, respectively.
- Use the confocal controls of the acquisition software to ensure that the selected focal plane has been optimized for maximal fluorescence emission intensity in the center of the cells (z-axis) by scanning at 488 nm while adjusting the z-plane. This is made easier by using a high gain setting while searching for the z-plane that results in the most over-exposed cells. Once the appropriate focal plane has been found, return the gain to a setting that is most optimal for the fluorescence of the reporter being used without oversaturating the pixels being observed. NOTE: The laser and gain settings that are appropriate for finding and observing cells during experimentation is completely dependent on the confocal system being utilized. In general, once the cells have been found in the field of view, it is recommended that a minimal amount of laser power be used (typically ≤20%), as excessive scanning of cells using high-powered laser light may induce oxidative changes detectable by the sensor.
- Use the gain to fine-tune the baseline fluorescence. With roGFP2, establish the baseline near the upper limit (≈ 90% relative intensity) of the instrument without over-saturation, as these cells will lose 510 nm fluorescence induced by 488 nm excitation when EGSH increases. In contrast, the fluorescence intensity with 488 nm excitation should be low (≈ 10% relative intensity) at baseline for HyPer expressing cells, as these cells will gain 510 nm fluorescence intensity when H2O2 is detected.
- Repeat steps 2.9.2 and 2.9.3 with excitation at 404 nm and emission at 510 nm. Gain settings for the 404 nm excitation wavelength are opposite to those used with 488 nm excitation for each sensor (i.e., low baseline fluorescence (≈ 10% relative intensity) at 404 nm for roGFP2, high baseline fluorescence (≈ 90% relative intensity) for the H2O2 sensor). NOTE: In general, the fluorescence (510 emission) at 404 nm excitation will be considerably lower than that obtainable with 488 nm excitation in cells expressing both roGFP2 and HyPer, as the 404 nm peak is a relatively minor excitation maximum for both of these sensors.
3. Data Acquisition
- Set up the acquisition software to sequentially excite the two excitation wavelengths (first 488 nm and then 404 nm) and collect emissions for both at 510 nm at a predetermined time interval throughout the desired length of the experiment (e.g. capture images every 60 s for 60 min).
- Alternatively, acquire images manually before and after exposures if the approximate timing of changes in EGSH or H2O2 are known for the xenobiotic being tested. However, this may lead to loss of temporal resolution.
- For real-time assessment of experimental parameters, choose at least 5 - 10 sensor-expressing cells in the field and establish them as regions of interest ("ROIs") to monitor their fluorescence changes during the experiment. NOTE: This step is optional, and can be performed after the experiment if direct observation in real-time is undesirable or software limitations prevent continuous monitoring. Depending on the cell population, the selection of sensor expressing cells as ROIs might represent a range of expression levels across cells.
- For these studies, add the environmental toxicant 9,10-phenanthrenequinone (9,10-PQ) or hydrogen peroxide (H2O2) after a 5-min baseline period.
- Prepare all reagents for later use in this protocol. Dissolve 9,10-PQ in dimethyl sulfoxide (DMSO) to a concentration of 15 mM, and dilute in basal cell media to yield a 250 µM working solution. Additionally, prepare a working solution of hydrogen peroxide in water that will yield a final concentration of 1mM upon injection.
Once the experimental parameters have been defined, begin the time course acquisition. Establish a baseline period of at least 5 min (or 5 data points) prior to starting xenobiotic exposures.
- Expose the cells to the toxicant being investigated using conventional approaches for in vitro dosing. Prepare soluble compounds in either water or other appropriate solvent and inject directly into the media.
- If organic solvents (e.g. dimethyl sulfoxide or ethanol) are required, keep the final solvent concentration at or below 0.1%. Verify that the solvent does not produce an effect on EGSH or H2O2 production with a vehicle control in a separate dish of cells.
- For compounds with lower solubility, add a mixing step using a micropipette to the protocol after the injection is made. Pump gaseous exposures directly into the environmental chamber using the appropriate carrier gas mixture.
Monitor changes in fluorescence during the exposure period. Perform subsequent injections as needed. Take great care not to shift the dish during injections, so that the same cells are followed throughout the entire time course while imaged in the same focal plane.
- At the end of the experiment, expose the cells to appropriate controls. For both genetically-encoded fluorogenic sensors, add specific concentrations of compounds known to oxidize and reduce these sensors in a pointed demonstration of their ratiometric responsiveness. H2O2 and dithiothreitol (DTT) serve as appropriate controls to oxidize and reduce both sensors. Typically, a final concentration of 100 - 1,000 µM H2O2, followed by 1-5 mM dithiothreitol (DTT), allows the user to fully oxidize and reduce these sensors.
- For these studies, use 1 mM H2O2 followed by 5 mM DTT to determine the maximum sensor response.
- Wait at least 5 min to allow the sensor to respond, and then inject a reducing agent, such as DTT, to reduce the senor and return it to a level of fluorescence at or near its established baseline. NOTE: The use of controls in this step is crucial for normalization, and should be performed at the end of every experiment to determine the dynamic range of the sensor in each cell. For roGFP2, aldrithiol can also be added at the end of the experiment. Aldrithiol bypasses the roGFP2 redox relay to oxidize the sensor directly. This is useful to assess sensor function following xenobiotic exposure.
4. Data Analysis
If not already established, draw ROIs around the cells to be analyzed. Use the appropriate software to measure the fluorescence intensity of each ROI at each wavelength throughout the time course. Ensure that the cells did not move or the plate did not shift during the run. NOTE: The establishment of ROIs is most easily accomplished by drawing an enclosed region that follows the fluorescent expression pattern of each cell. To further assist with this process, a transmitted light (or brightfield) image of the cells within the established field of view can be overlaid onto the fluorescent image in efforts to define cell boundaries. However, the use of a transmitted light image requires the user to capture an additional channel (set of images) throughout the course of the experiment. Alternatively, certain manufacturers incorporate algorithms into their imaging software that use specific parameters such as intensity thresholds to establish ROIs in a more quantitative, less subjective, method.
Export the data to desired analysis software (e.g., electronic spreadsheet).
Calculate the ratiometric sensor response for each ROI at each time point using the formulas:
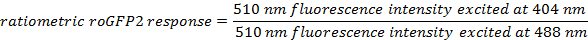
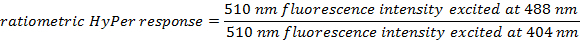
For each time point, average the calculated ratiometric values of all ROIs.
Calculate the baseline value, which will be used to normalize the data, by averaging the previously calculated ratiometric values (step 4.4) collected before addition of the xenobiotic (e.g., the first five-time points).
Normalize the ratiometric values from step 4.4 by dividing each value by the baseline value calculated in 4.5. Normalized ratios will center around a value of 1 at baseline, while increases in EGSH or H2O2 following injections will cause the ratio to increase. Assessments of xenobiotic that induce oxidative changes tend to yield values in a range of 3 - 6 times the baseline value.
To aid in comparing responses within and across experiments, express the normalized data as a percentage of maximal sensor response by defining the response to the control oxidant (e.g., 1 mM H2O2) as 100%. NOTE: If expressing data as a percentage of maximal sensor response, it is critical to ensure that the same concentration of the control oxidant is used consistently across experiments. All data derived from live-cell imaging analysis is amenable to conventional pair-wise and group-wise statistical analysis to be chosen at the discretion of the investigator.
Representative Results
The use of roGFP2 and HyPer in detecting changes in EGSH and intracellular H2O2 has been well described previously25,36,42,43 and is demonstrated here. Confocal images of cells expressing roGFP2 at baseline and following addition of H2O2 and DTT are shown in Figure 2. Data from images collected throughout the experiment were then exported and analyzed as described in section 4 of the protocol. As shown, the addition of exogenous H2O2 as a positive control produces reproducible responses when the data are normalized to their respective baseline and a defined dynamic range is established through addition of known exogenous oxidants/reductants (i.e., H2O2 and DTT) (Figures 3, 4, and 5).
Responses to toxicological stimuli are typically more variable than those induced by controls. This is to be expected, as xenobiotic compounds may have pleiotropic effects on cells, ranging from redox cycling to adduction of intracellular proteins. Examples of roGFP2 and HyPer responses induced by exposure to the toxicant 9,10-PQ, an oxidizing air pollutant formed by atmospheric reactions of phenanthrene44, are presented in Figures 6 and 7. The graphs in these figures depict the progression of each step involved in the data analysis outlined in section 4 of the protocol. As shown, normalization of each cell to its own baseline and positive control (Figure 6B), reduces intercellular variability in the magnitude of the sensor response from the raw data (Figure 6A). In cases where the variance in the cellular response is of interest, normalization of the data can be performed up to this point, with individual cells represented by their own lines on the graph. Variation in the magnitude or kinetics of the responses among cells in the same dish can reveal important mechanistic insights that reflect differences in the cell cycle, for instance. Alternatively, the averaged response of the cells in the dish can be presented along with the standard deviation (Figure 6C). Otherwise, if replicates of separate experiments are being presented, the data may be presented as the normalized relative fluorescence intensity values or a percentage of the maximum response induced by the control oxidant (Figures 3, 4, 5, and 6D), showing the average of all replicates accompanied by the standard error of the mean. The ambient air contaminant 9,10-PQ is known to be a potent redox cycler, which we hypothesize is responsible for the cyclical peaks of hydrogen peroxide production detected by HyPer (Figure 7) and stepwise EGSH increases detected by roGFP2 (Figure 6D).
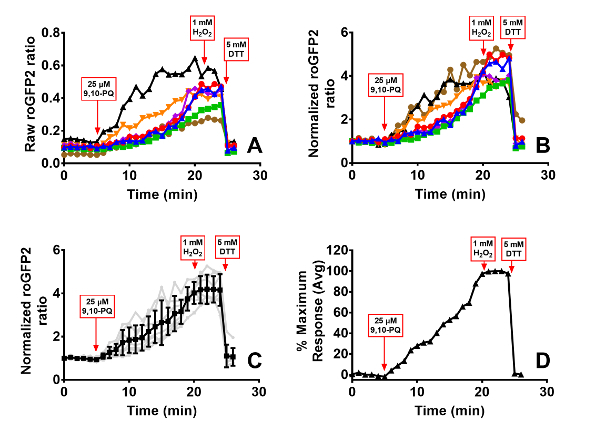
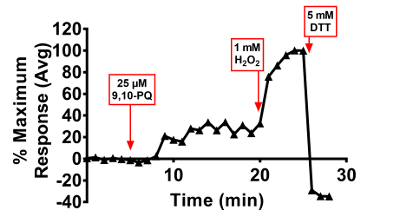
Figure 1. The roGFP2 glutathione redox relay. Glutathione peroxidases (GPx) oxidize reduced glutathione (GSH) to GSSG in response to hydrogen peroxide (H2O2), thus increasing the glutathione redox potential (EGSH). The fluorogenic sensor roGFP2 equilibrates with intracellular EGSH when oxidized by glutaredoxin (Grx). In the reductive pathway, Grx catalyzes the reduction of roGFP2 through deglutathionylation as GSSG levels decrease and normal levels of GSH are reestablished by glutathione reductase (GR) at the expense of NADPH. NADPH is supplied by glucose through the pentose phosphate pathway (PPP). Adapted from Gibbs-Flournoy et al35. Please click here to view a larger version of this figure.
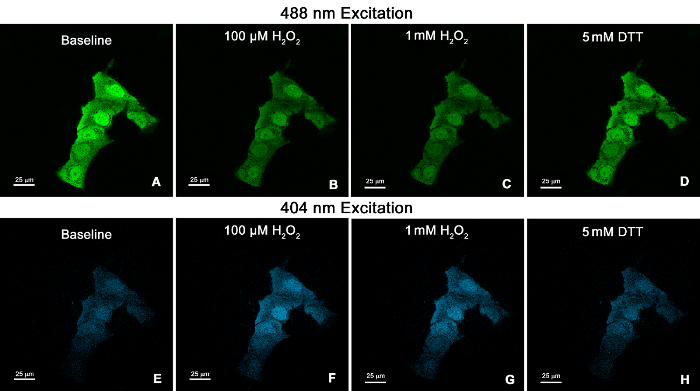
Figure 2. Confocal microscopy images of the human bronchial epithelial cell line BEAS-2B expressing cytosolic roGFP2. Cells were illuminated at 488 nm (A-D), followed by 404 nm (E-H). Images show fluorescence emitted at 510 nm for the following conditions: baseline (A, E), 100 µM H2O2(B, F), 1 mM H2O2 (C, G), and 5 mM DTT (D, H). 60X Plan Apo VC objective. Scale bars represent 25 µm. Please click here to view a larger version of this figure.
Figure 3. Dose dependent changes in roGFP2 induced by H2O2 in BEAS-2B cells. Cells were equilibrated for 30 min in phenol-red free keratinocyte basal media (KBM) prior to exposures. All additions of H2O2 occurred after a baseline period of 5 min. roGFP2 fluorescence emitted at 510 nm was collected using a 525/30 nm band pass filter following laser excitation at 404 and 488 nm. Cellular responses were normalized to their respective baseline and maximum sensor response following the addition of 1 mM H2O2. Values are presented as mean ± standard error, n=3, where n consists of an average of 5 - 10 independent cells. Please click here to view a larger version of this figure.
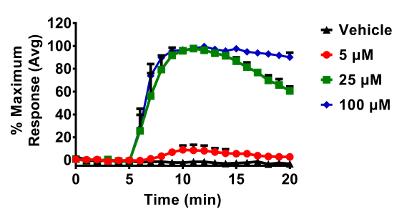
Figure 4. Response of glucose starved BEAS-2B cells expressing roGFP2 to various doses of H2O2. Cells were equilibrated for 2 hours in a minimal salt medium that does not contain glucose prior to exposures. All additions of H2O2 occurred after a baseline period of 5 min. roGFP2 fluorescence emitted at 510 nm was collected using a 525/30 nm band pass filter following laser excitation at 404 and 488 nm. Cellular responses were normalized to their respective baseline and maximum sensor response following the addition of 1 mM H2O2. Values are presented as the mean ± standard error, n = 3, where n consists of an average of 5 - 10 distinct cells. Please click here to view a larger version of this figure.
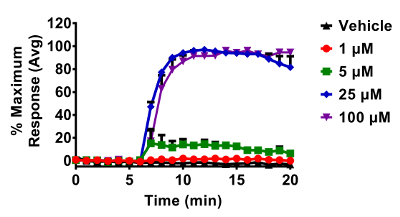
Figure 5. Response of BEAS-2B cells expressing HyPer to various doses of H2O2. Cells were equilibrated for 30 min in phenol-red free KBM prior to exposures. All additions of H2O2 occurred after a baseline period of 5 min. Fluorescence emitted at 510 nm was collected using a 525/30 nm band pass filter following laser excitation at 404 and 488 nm. Cellular responses were normalized to their respective baseline and maximum sensor response following the addition of 1 mM H2O2. Values are presented as the mean ± standard error, n = 3, where n consists of an average of 5 - 10 distinct cells. Please click here to view a larger version of this figure.
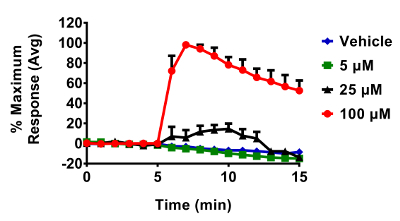
Figure 6. 9,10-PQ-induced responses of BEAS-2B cells expressing roGFP2. Cells were equilibrated for 30 min in phenol-red free KBM prior to exposure. Addition of 25 µM 9,10-PQ with mixing occurred at 5 min, followed by addition of 1 mM H2O2 at 20 min and 5 mM DTT at 24 min. Panel A depicts the raw roGFP2 ratio of individual cells in the dish, each cell represented by a separate line. Panel B shows normalization of values from each individual cell to its baseline sensor fluorescence, and the average and standard deviation of this data is superimposed on the individual normalized ratios in Panel C. The percentage of maximal sensor response averaged across all cells is depicted in Panel D. Please click here to view a larger version of this figure.
Figure 7. 9,10-PQ-induced responses of BEAS-2B cells expressing HyPer. Cells were equilibrated for 30 min in phenol-red free KBM prior to exposure. Addition of 25 µM 9,10-PQ with mixing occurred at 5 min, followed by addition of 1 mM H2O2 at 20 min and 5 mM DTT at 25 min. Fluorescence emitted at 510 nm was collected using a 525/30 nm band pass filter following laser excitation at 404 and 488 nm. Cellular responses were normalized to their respective baseline and maximum sensor response following the addition of 1 mM H2O2. Values are presented as the mean of 5 - 10 distinct cells. Please click here to view a larger version of this figure.
Discussion
The use of roGFP2 and HyPer in detecting changes in intracellular EGSH and H2O2, respectively, has been well-described in previous physiological and toxicological studies 25,35,36,39,40,41,42,43. The protocol outlined here reports an effective way to implement these genetically-encoded redox sensors in pointed toxicological studies designed to assess xenobiotic-induced perturbations of intracellular redox homeostasis. Most importantly, the proper use of these sensors links any changes observed to a specific redox pair or ROS (EGSH or H2O2), ultimately clarifying the lack of specificity with many conventional oxidative stress approaches. In assuring the quality of the data generated from these sensors, a few aspects must be taken into consideration when designing experiments and analyzing/interpreting data. They include: 1) the use of proper controls, 2) verification of appropriate sensor responses, and 3) manipulation of experimental parameters to elucidate mechanisms. Of these, the addition of exogenous H2O2 and DTT as controls at the end of the observation period serves multiple purposes. First, it affords an opportunity to assess the magnitude of the response to the toxic exposure relative to the maximal and minimal signals achievable from the sensor. This is most useful in normalizing responses for comparisons between experiments. In addition, ending the experiment with the addition of strong exogenous oxidant and reductive stimuli can evaluate the effect of the preceding exposure on sensor/relay integrity. Furthermore, aldrithiol can be added at the end of the run to bypass the relay and test the functionality of the sensor directly for potential damage that the exposure may have inflicted to the fluorophore itself.
Just as with other techniques, when applying this methodology for the specific examination of toxicological outcomes, it is important to make qualitative assessments regarding the accuracy of the observations made. When utilizing roGFP2 or HyPer with any new xenobiotic it is important to first test a range of doses, while ensuring that the fluorescence intensity (510 nm emission) from each excitation wavelength (404 nm and 488 nm) changes appropriately (i.e. increase or decrease) for the redox senor being used. The goal is to identify a dose at which a sensor response is detectable, but is not overwhelmed by overt manipulation of the sensor or other non-specific effects of the compound. Direct adduction or damage to the sensor may manifest as atypical alterations in fluorescence at either excitation wavelength (488 or 404 nm). In situations where either sensor has been observed to consistently respond inappropriately to the addition of a toxicant across exposures, it is recommended that the investigator consider examining experimental parameters in relation to dosimetry, cell viability, sensor expression, and/or optical/chromatic abnormalities of the instrumentation being used to observe the responses.
As with any toxicological readout, the data acquired are usually more meaningful when the observed responses are examined or validated using a mechanistic approach, and the experimental limitations are considered. To this end, several experimental parameters combined with the inherent properties of the sensor can be utilized to generate robust redox assessments. Investigations specific to the use of the roGFP2 sensor ultimately concern the redox relay through which roGFP2 senses EGSH (Figure 1). The relay involves intracellular enzymes, including glutaredoxin (Grx), glutathione reductase (GR), and glutathione peroxidase (GPx). Differences in endogenous levels of Grx across both cellular compartments and cell types can influence measurements of EGSH and make comparisons between experiments difficult43. To address this problem, a series of "fusion probes" have been synthesized, which link the relevant enzyme directly to the roGFP2 sensor. Most recently, human Grx-1 was linked to roGFP2 by Gutscher and colleagues32 to form Grx1-roGFP2. In addition to having a greater dynamic range than roGFP1 and being pH insensitive, Grx1-roGFP2 can monitor EGSH in cell types or compartments in which Grx activity would otherwise be limiting. Differences in NADPH levels across cell types or even among cells in the same dish can also contribute to variability in roGFP2 responses. NADPH is produced from glucose through the pentose-phosphate pathway, and can be utilized by glutathione reductase (GR) to reduce GSSG back to GSH (Figure 1). If cells in the same field begin the experiment with different reducing capacities, their EGSH changes and resulting roGFP2 responses to a stimulus may not be uniform. This problem can be surmounted through a period of glucose starvation prior to the experiment, which depletes cellular NADPH stocks and harmonizes roGFP2 responses. A decreased capacity for EGSH recovery can also be seen in the 25 µM H2O2 dose in cells that were starved of glucose as compared to cells supplemented with normal glucose concentrations (Figures 3 and 4). Thusly, manipulation of NADPH levels through glucose starvation can serve as a means to check the involvement of multiple relay components in transducing the effect of a xenobiotic exposure to the sensor. Another concern when using roGFP2 in a toxicological context is the potential for the xenobiotic to interact directly in oxidizing the sensor, rather than acting through the redox relay to alter EGSH. Direct oxidation of roGFP2 can be determined by probing each point of the redox relay and assessing its effect on the roGFP2 signal. An example of this technique using ozone as a stimulus is demonstrated in the study by Gibbs-Flournoy et al 35.
For experiments with the H2O2 sensor, the above concerns are not as relevant, as the OxyR1 domain of HyPer senses H2O2 directly rather than through a redox relay. However, unlike roGFP2, H2O2 sensor is sensitive to changes in pH, which can cause changes in fluorescence not attributable to H2O2 during an experiment. For this reason, the use of properly buffered media, an environmental chamber to maintain the desired temperature, and the use of vehicle controls are vital to any experiment utilizing the H2O2 sensor. Additionally, fluorogenic sensors have been developed to specifically monitor pH, such as pHRed, which emits fluorescence in the red channel and can be co-expressed in cells also expressing HyPer45. The ideal pH control for the H2O2 sensor is SypHer, which is a version of the H2O2 sensor that has a single point mutation rendering it unable to sense H2O2, yet preserves responsiveness to pH46. It has also been observed that cells expressing H2O2 sensor require a longer baseline period to equilibrate, particularly after being transported from an incubator to the site of analysis, during which they experience slight changes in media temperature and pH. It is recommended to acquire at least 5 data points after the baseline has stabilized before beginning any exposure.
The sensors discussed in this method, roGFP2 and HyPer, are two of many fluorogenic sensors that have a variety of emerging applications in the field of toxicology. This includes sensors designed to detect not only EGSH and H2O2, but peroxynitrite47, nitric oxide48,49, and even ozone50. These sensors can be utilized in live-cell imaging studies to specifically and sensitively monitor the oxidative species of interest. With advances in techniques such as spectral unmixing, it is also possible to co-express two sensors with similar spectral characteristics (within 5 nm of each other) in the same cell, and separate the proportion of the fluorescence emitted by each sensor51. This technique is of particular interest for roGFP2 and HyPer, as they involve excitations and emission at the same wavelengths. As briefly mentioned here, certain sensors can be targeted to specific intracellular compartments, such as the mitochondria, to observe oxidative events of interest. While these emerging sensors and techniques are beyond the scope of this method, the reader is referred to several recent publications on the topic of intracellular redox sensors, such as the review by Wages and colleagues52. The use of genetically-encoded fluorogenic sensors with live-cell imaging is a robust tool for the monitoring of oxidative responses during toxicological assessments.
Disclosures
The research described in this article has been reviewed by the National Health and Environmental Effects Research Laboratory, U.S. Environmental Protection Agency, and approved for publication. The contents of this article should not be construed to represent agency policy, nor does mention of trade names or commercial products constitute endorsement or recommendation for use.
Acknowledgments
The authors would like to thank Katelyn Lavrich for assistance with experimental design and manuscript edits.
References
- Cross CE, et al. Environmental oxidant pollutant effects on biologic systems: a focus on micronutrient antioxidant-oxidant interactions. Am J Respir Crit Care Med. 2002;166:44–50. doi: 10.1164/rccm.2206015. [DOI] [PubMed] [Google Scholar]
- Kobzik L. Environmental particulate-mediated cytokine production in lung epithelial cells (A549): role of preexisting inflammation and oxidant stress. J Toxicol Environ Health. 1998;55:31–44. doi: 10.1080/009841098158601. [DOI] [PubMed] [Google Scholar]
- Jaeschke H, McGill MR, Ramachandran A. Oxidant stress, mitochondria, and cell death mechanisms in drug-induced liver injury: lessons learned from acetaminophen hepatotoxicity. Drug Metab Reviews. 2012;44:88–106. doi: 10.3109/03602532.2011.602688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf MB, Baynes JW. The anti-cancer drug, doxorubicin, causes oxidant stress-induced endothelial dysfunction. Biochim Biophys Acta. 2006;1760:267–271. doi: 10.1016/j.bbagen.2005.10.012. [DOI] [PubMed] [Google Scholar]
- Kumagai Y, Shinkai Y, Miura T, Cho AK. The chemical biology of naphthoquinones and its environmental implications. Annu Rev Pharmacol Toxicol. 2012;52:221–247. doi: 10.1146/annurev-pharmtox-010611-134517. [DOI] [PubMed] [Google Scholar]
- Pham HT, Maccarone AT, Campbell JL, Mitchell TW, Blanksby SJ. Ozone-induced dissociation of conjugated lipids reveals significant reaction rate enhancements and characteristic odd-electron product ions. J Am Soc Mass Spectrom. 2013;24:286–296. doi: 10.1007/s13361-012-0521-9. [DOI] [PubMed] [Google Scholar]
- Abdel-Malek ZA, Kadekaro AL, Swope VB. Stepping up melanocytes to the challenge of UV exposure. Pigment Cell Melanoma Res. 2010;23:171–186. doi: 10.1111/j.1755-148X.2010.00679.x. [DOI] [PubMed] [Google Scholar]
- Berisha H, Pakbaz H, Absood A, Foda H, Said S. Nitric oxide mediates oxidant tissue injury caused by paraquat and xanthine oxidase. Ann N Y Acad Sci. 1994;723:422–425. [PubMed] [Google Scholar]
- Gao X, et al. Mitochondrial dysfunction may explain the cardiomyopathy of chronic iron overload. Free Radic Bio Med. 2010;49:401–407. doi: 10.1016/j.freeradbiomed.2010.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla A, et al. Asbestos induces mitochondrial DNA damage and dysfunction linked to the development of apoptosis. Am J Physiol Lung Cell Mol Physiol. 2003;285:1018–1025. doi: 10.1152/ajplung.00038.2003. [DOI] [PubMed] [Google Scholar]
- Charles R, Jayawardhana T, Eaton P. Gel-based methods in redox proteomics. Biochim Biophys Acta. 2014;1840:830–837. doi: 10.1016/j.bbagen.2013.04.021. [DOI] [PubMed] [Google Scholar]
- Thornalley PJ, Rabbani N. Detection of oxidized and glycated proteins in clinical samples using mass spectrometry-a user's perspective. Biochim Biophys Acta. 2014;1840:818–829. doi: 10.1016/j.bbagen.2013.03.025. [DOI] [PubMed] [Google Scholar]
- Arato S, Ito H, Miyashita K, Hayakawa K, Itabashi Y. A facile method for the detection of aldehydes in oxidized lipids using solid-phase microextraction fiber and gas chromatograph equipped with a septum-free injector. J Oleo Sci. 2009;58:17–22. doi: 10.5650/jos.58.17. [DOI] [PubMed] [Google Scholar]
- Collins AR. Measuring oxidative damage to DNA and its repair with the comet assay. Biochim Biophys Acta. 2014;1840:794–800. doi: 10.1016/j.bbagen.2013.04.022. [DOI] [PubMed] [Google Scholar]
- Churg A, Keeling B, Gilks B, Porter S, Olive P. Rat mesothelial and tracheal epithelial cells show equal DNA sensitivity to hydrogen peroxide-induced oxidant injury. Am J Physiol Lung Cell Mol Physiol. 1995;268:832–838. doi: 10.1152/ajplung.1995.268.5.L832. [DOI] [PubMed] [Google Scholar]
- Fraga CG, Oteiza PI, Galleano M. In vitro measurements and interpretation of total antioxidant capacity. Biochim Biophys Acta. 2014;1840:931–934. doi: 10.1016/j.bbagen.2013.06.030. [DOI] [PubMed] [Google Scholar]
- Niki E, Noguchi N. Evaluation of antioxidant capacity: what capacity is being measured by which method. IUBMB Life. 2000;50:323–329. doi: 10.1080/713803736. [DOI] [PubMed] [Google Scholar]
- Pharikal K, Das P, Dey C, Dasgupta S. Tissue ascorbate as a metabolic marker in cadmium toxicity. Int J Vitam Nut Res. 1987;58:306–311. [PubMed] [Google Scholar]
- Tabata M, Totani M, Murachi T. A chemiluminometric method for NADPH and NADH using a two-enzyme bioreactor and its application to the determination of magnesium in serum. Biomed Chrom. 1990;4:123–127. doi: 10.1002/bmc.1130040310. [DOI] [PubMed] [Google Scholar]
- Ballatori N, et al. Glutathione dysregulation and the etiology and progression of human diseases. Biol Chem. 2009;390:191–214. doi: 10.1515/BC.2009.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nauseef WM. Detection of superoxide anion and hydrogen peroxide production by cellular NADPH oxidases. Biochim Biophys Acta. 2014;1840:757–767. doi: 10.1016/j.bbagen.2013.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins CL, Davies MJ. Detection and characterisation of radicals in biological materials using EPR methodology. Biochim Biophys Acta. 2014;1840:708–721. doi: 10.1016/j.bbagen.2013.03.034. [DOI] [PubMed] [Google Scholar]
- Kettle AJ, et al. Measuring chlorine bleach in biology and medicine. Biochim Biophys Acta. 2014;1840:781–793. doi: 10.1016/j.bbagen.2013.07.004. [DOI] [PubMed] [Google Scholar]
- Carballal S, Bartesaghi S, Radi R. Kinetic and mechanistic considerations to assess the biological fate of peroxynitrite. Biochim Biophys Acta. 2014;1840:768–780. doi: 10.1016/j.bbagen.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer AJ, Dick TP. Fluorescent protein-based redox probes. Antioxid Redox Signal. 2010;13:621–650. doi: 10.1089/ars.2009.2948. [DOI] [PubMed] [Google Scholar]
- Dooley CT, et al. Imaging dynamic redox changes in mammalian cells with green fluorescent protein indicators. J Biol Chem. 2004;279:22284–22293. doi: 10.1074/jbc.M312847200. [DOI] [PubMed] [Google Scholar]
- Malinouski M, Zhou Y, Belousov VV, Hatfield DL, Gladyshev VN. Hydrogen peroxide probes directed to different cellular compartments. PloS One. 2011;6:14564. doi: 10.1371/journal.pone.0014564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman JN, et al. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature. 2010;468:696–700. doi: 10.1038/nature09536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breckwoldt MO, et al. Multiparametric optical analysis of mitochondrial redox signals during neuronal physiology and pathology in vivo. Nat Med. 2014;20:555–560. doi: 10.1038/nm.3520. [DOI] [PubMed] [Google Scholar]
- Wolf AM, Nishimaki K, Kamimura N, Ohta S. Real-time monitoring of oxidative stress in live mouse skin. J Invest Dermatol. 2014;134:1701–1709. doi: 10.1038/jid.2013.428. [DOI] [PubMed] [Google Scholar]
- Mishina NM, et al. Does cellular hydrogen peroxide diffuse or act locally. Antioxid Redox Signal. 2011. [DOI] [PubMed]
- Gutscher M, et al. Real-time imaging of the intracellular glutathione redox potential. Nat Methods. 2008;5:553–559. doi: 10.1038/nmeth.1212. [DOI] [PubMed] [Google Scholar]
- Lohman JR, Remington SJ. Development of a family of redox-sensitive green fluorescent protein indicators for use in relatively oxidizing subcellular environments. Biochem. 2008;47:8678–8688. doi: 10.1021/bi800498g. [DOI] [PubMed] [Google Scholar]
- Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Bio Med. 2001;30:1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- Gibbs-Flournoy EA, Simmons SO, Bromberg PA, Dick TP, Samet JM. Monitoring intracellular redox changes in ozone-exposed airway epithelial cells. Environ Health Perspect. 2013;121:312. doi: 10.1289/ehp.1206039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belousov VV, et al. Genetically encoded fluorescent indicator for intracellular hydrogen peroxide. Nat Methods. 2006;3:281–286. doi: 10.1038/nmeth866. [DOI] [PubMed] [Google Scholar]
- Wood ZA, Poole LB, Karplus PA. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science. 2003;300:650–653. doi: 10.1126/science.1080405. [DOI] [PubMed] [Google Scholar]
- Sies H. Role of Metabolic H2O2 Generation REDOX SIGNALING AND OXIDATIVE STRESS. J Biol Chem. 2014;289:8735–8741. doi: 10.1074/jbc.R113.544635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng W-Y, et al. Linking oxidative events to inflammatory and adaptive gene expression induced by exposure to an organic particulate matter component. Environ Health Perspect. 2012;120:267. doi: 10.1289/ehp.1104055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng W-Y, et al. An integrated imaging approach to the study of oxidative stress generation by mitochondrial dysfunction in living cells. Environ Health Perspect. 2010. pp. 902–908. [DOI] [PMC free article] [PubMed]
- Wages PA, et al. Role of H 2 O 2 in the oxidative effects of zinc exposure in human airway epithelial cells. Redox Biol. 2014;3:47–55. doi: 10.1016/j.redox.2014.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan B, Sobotta MC, Dick TP. Measuring E GSH and H2O2 with roGFP2-based redox probes. Free Radic Bio Med. 2011;51:1943–1951. doi: 10.1016/j.freeradbiomed.2011.08.035. [DOI] [PubMed] [Google Scholar]
- Schwarzländer M, Dick TP, Meyer AJ, Morgan B. Dissecting redox biology using fluorescent protein sensors. Antiox Redox Signal. 2016;24:680–712. doi: 10.1089/ars.2015.6266. [DOI] [PubMed] [Google Scholar]
- Wang L, Atkinson R, Arey J. Formation of 9, 10-phenanthrenequinone by atmospheric gas-phase reactions of phenanthrene. Atmos Environ. 2007;41:2025–2035. [Google Scholar]
- Tantama M, Hung YP, Yellen G. Imaging intracellular pH in live cells with a genetically encoded red fluorescent protein sensor. J Am Chem Soc. 2011;133:10034–10037. doi: 10.1021/ja202902d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matlashov ME, et al. Fluorescent ratiometric pH indicator SypHer2: applications in neuroscience and regenerative biology. Biochim Biophys Acta. 2015;1850:2318–2328. doi: 10.1016/j.bbagen.2015.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou J-T, et al. A highly selective water-soluble optical probe for endogenous peroxynitrite. Chem Commun. 2014;50:9947–9950. doi: 10.1039/c4cc04192e. [DOI] [PubMed] [Google Scholar]
- Kojima H, et al. Detection and imaging of nitric oxide with novel fluorescent indicators: diaminofluoresceins. Anal Chem. 1998;70:2446–2453. doi: 10.1021/ac9801723. [DOI] [PubMed] [Google Scholar]
- Gabe Y, Ueno T, Urano Y, Kojima H, Nagano T. Tunable design strategy for fluorescence probes based on 4-substituted BODIPY chromophore: improvement of highly sensitive fluorescence probe for nitric oxide. Anal Bioanal Chem. 2006;386:621–626. doi: 10.1007/s00216-006-0587-y. [DOI] [PubMed] [Google Scholar]
- Garner AL, et al. Specific fluorogenic probes for ozone in biological and atmospheric samples. Nat Chem. 2009;1:316–321. doi: 10.1038/nchem.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng W-Y, Larson JM, Samet JM. Monitoring intracellular oxidative events using dynamic spectral unmixing microscopy. Methods. 2014;66:345–352. doi: 10.1016/j.ymeth.2013.06.008. [DOI] [PubMed] [Google Scholar]
- Wages PA, Cheng W-Y, Gibbs-Flournoy E, Samet JM. Live-cell imaging approaches for the investigation of xenobiotic-induced oxidant stress. Biochim Biophys Acta. 2016;1860:2802–2815. doi: 10.1016/j.bbagen.2016.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]