

Abstract
One-carbon (1C) metabolism is a universal folate-dependent pathway essential for de novo purine and thymidylate synthesis, amino acid interconversion, universal methyl-donor production, and regeneration of redox cofactors. Homozygous deletion of the 1C pathway gene Mthfd1l encoding methylenetetrahydrofolate dehydrogenase (NADP+-dependent) 1-like, which catalyzes mitochondrial formate production from 10-formyltetrahydrofolate, results in 100% penetrant embryonic neural tube defects (NTDs), underscoring the central role of mitochondrially derived formate in embryonic development and providing a mechanistic link between folate and NTDs. However, the specific metabolic processes that are perturbed by Mthfd1l deletion are not known. Here, we performed untargeted metabolomics on whole Mthfd1l-null and wildtype mouse embryos in combination with isotope tracer analysis in mouse embryonic fibroblast (MEF) cell lines to identify Mthfd1l deletion–induced disruptions in 1C metabolism, glycolysis, and the TCA cycle. We found that maternal formate supplementation largely corrects these disruptions in Mthfd1l-null embryos. Serine tracer experiments revealed that Mthfd1l-null MEFs have altered methionine synthesis, indicating that Mthfd1l deletion impairs the methyl cycle. Supplementation of Mthfd1l-null MEFs with formate, hypoxanthine, or combined hypoxanthine and thymidine restored their growth to wildtype levels. Thymidine addition alone was ineffective, suggesting a purine synthesis defect in Mthfd1l-null MEFs. Tracer experiments also revealed lower proportions of labeled hypoxanthine and inosine monophosphate in Mthfd1l-null than in wildtype MEFs, suggesting that Mthfd1l deletion results in increased reliance on the purine salvage pathway. These results indicate that disruptions of mitochondrial 1C metabolism have wide-ranging consequences for many metabolic processes, including those that may not directly interact with 1C metabolism.
Keywords: mitochondrial metabolism, energy metabolism, metabolomics, folate, embryo, neural tube closure
Introduction
One-carbon metabolism is indispensable during embryonic development, and maternal deficiency of folic acid is strongly associated with neural tube defects (NTDs).2 Periconceptional intake of supplemental folic acid can reduce the incidence of NTDs by as much as 70%, and many countries now fortify their food supply with folic acid to ensure that women of child-bearing age consume adequate quantities of the vitamin. Although folic acid fortification has decreased NTD incidence in most populations, an estimated 30% of NTD cases do not respond to folic acid supplementation (1). Moreover, despite the strong clinical links between folate and NTDs, the biochemical mechanisms through which folic acid acts during neural tube development remain largely undefined (2).
Folate-dependent (1C) metabolism is highly compartmentalized in eukaryotes with mitochondria playing a central role (3). Mitochondrial 1C metabolism begins with the entry of 1C donors including serine and glycine into the mitochondria (Fig. 1). These 1C donors are processed by serine hydroxymethyltransferase (SHMT) or the glycine cleavage system to produce 5,10-methylenetetrahydrofolate (CH2-THF, reactions 4m and 5). Mitochondrial CH2-THF dehydrogenase and 5,10-methenyl-tetrahydrofolate (CH+-THF) cyclohydrolase activities are catalyzed by the bifunctional isozymes MTHFD2 and MTHFD2L (reactions 3m and 2m). The final step in the mitochondrial 1C pathway, the formation of formate from 10-formyltetrahydrofolate (10-CHO-THF), is catalyzed by the monofunctional enzyme MTHFD1L (reaction 1m). An alternative fate for 10-CHO-THF is oxidization to CO2 and THF in an NADP+-dependent reaction catalyzed by the mitochondrial 10-CHO-THF dehydrogenase activity of ALDH1L2 (reaction 9). In the cytoplasm, a single trifunctional enzyme, MTHFD1, is responsible for 10-CHO-THF synthetase, CH+-THF cyclohydrolase, and CH2-THF dehydrogenase activities (Fig. 1, reactions 1–3). The synthetase activity of MTHFD1 attaches mitochondrially derived formate to THF in the cytoplasm. This 10-CHO-THF can be used for de novo purine synthesis or reduced to CH2-THF via the cyclohydrolase and dehydrogenase activities of MTHFD1 for thymidylate synthesis (reaction 8). Finally, CH2-THF may be further reduced to 5-methyl-THF for use in the methyl cycle (reaction 6).
Figure 1.

Compartmentalization of folate-dependent one-carbon metabolism. Reactions are catalyzed by the following: 1, 2, and 3, MTHFD1; 1m, MTHFD1L; 2m and 3m, MTHFD2 or MTHFD2L; 4 and 4m, SHMT; 5, glycine cleavage system; 6, MTHFR; 7, methionine synthase; 8, thymidylate synthase; 9, ALDH1L2.
Deletion of Mthfd1l in mice has revealed the critical role of mitochondrial 1C metabolism in embryonic development. Embryos lacking MTHFD1L exhibit aberrant neural tube closure, craniofacial defects, developmental delay, and embryonic lethality by embryonic day 12.5 (E12.5) in mice (4). Homozygous deletion of Mthfd1l results in NTDs with 100% penetrance, without feeding a folate-deficient diet. The NTD phenotype is variable, including defects such as wavy neural tube, exencephaly, and craniorachischisis. Consistent with the prediction that lack of 10-CHO-THF synthetase activity would result in a loss of mitochondrial formate production, supplementation of pregnant dams with formate reduces the incidence of neural tube defects, partially rescues growth restriction, and significantly extends survival of null embryos (4).
The Mthfd1l knockout (KO) mouse provides a powerful model that can be used to identify specific metabolic processes that underlie folate-associated NTDs. Here we apply untargeted metabolomics on whole embryos in combination with isotope tracer studies in mouse embryonic fibroblast cell lines to identify disruptions in 1C and energy metabolism caused by deletion of the Mthfd1l gene.
Results
Formate production in isolated mitochondria
Isolated mitochondria are capable of producing formate from serine (5–7). Based on the known 1C pathway (Fig. 1), we anticipated that mitochondria lacking MTHFD1L would be unable to produce formate from serine. To obtain sufficient mitochondria for this assay, embryos at E14.5 or later were required. Mthfd1−/− (null) embryos do not survive past E12.5 unless the dams are supplemented with formate (4). Embryos at E14.5–E16.5 were dissected from dams that had been supplemented with 2,500 mg kg−1 d−1 of calcium formate. Isolated mitochondria from these embryos were incubated with l-[3-14C]serine and [14C]formate production was measured. Surprisingly, mitochondria from Mthfd1l−/− embryos showed significant, albeit reduced, formate production from serine (Fig. 2A). Formate production increased linearly with the l-[3-14C]serine concentration in mitochondria from both wildtype and Mthfd1l−/− embryos, indicating that serine was not saturating under these conditions. Immunoblots confirmed that MTHFD1L was undetectable in mitochondria from Mthfd1l−/− embryos (Fig. 2B).
Figure 2.
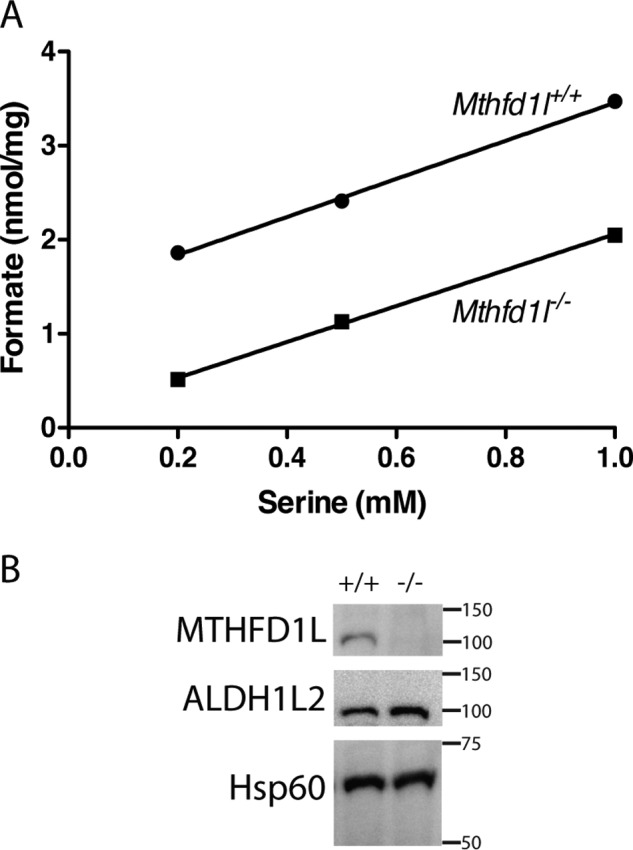
Formate production in mitochondria isolated from wildtype or Mthfd1l−/− embryos. A, [14C]formate produced from l-[3-14C]serine in mitochondria isolated from pooled E14.5 to E16.5 embryos. Due to limited amounts of mitochondria available from the pooled embryos of each genotype, each data point represents a single reaction. B, representative immunoblot of MTHFD1L and ALDH1L2 expression in mitochondria isolated from E16.5 embryos. Each lane was loaded with 20 μg of protein. Immunoblots were probed with antibodies against MTHFD1L (100 kDa), ALDH1L2 (100 kDa), or the mitochondrial matrix protein Hsp60 (60 KDa) as a loading control. Migration and size (kDa) of molecular mass markers are shown on the right of the blots.
Residual formate production from mitochondria isolated from Mthfd1l−/− embryos suggests the presence of another pathway for converting the 3-carbon of serine to formate. ALDH1L2 (mitochondrial 10-CHO-THF dehydrogenase, Fig. 1, reaction 9) has been shown to exhibit a 10-CHO-THF hydrolase activity in vitro, producing formate from 10-CHO-THF (8, 9). Immunoblots confirmed that ALDH1L2 is present in mitochondria from both wildtype and Mthfd1l−/− embryos (Fig. 2B).
Metabolomic analysis of Mthfd1l−/− embryos from unsupplemented dams
An untargeted metabolomics analysis of Mthfd1l−/− and Mthfd1l+/+ embryos (n = 5) was conducted using both nuclear magnetic resonance (NMR) and mass spectrometry (MS) to identify metabolic alterations associated with loss of MTHFD1L. A total of 98 metabolites were positively identified from NMR and MS analyses combined. Principal component analysis (PCA) indicates clear separation of the groups, primarily along PC1 (Fig. 3A). Pathway analysis was accomplished using MetaboAnalyst (10, 11), with 12 pathways identified as significantly altered in Mthfd1l−/− embryos compared with wildtype (impact > 0.2, FDR < 0.05, Table 1). Pathway perturbations included 1C metabolism, energy metabolism, and amino acid metabolism (Table 1).
Figure 3.

Metabolomic analysis of Mthfd1l−/− embryos from unsupplemented dams. A, principal component analysis of embryo metabolites. A–D, abundance of metabolites related to (B) one-carbon metabolism, (C) energy metabolism, and (D) amino acid metabolism in Mthfd1l−/− embryos relative to Mthfd1l+/+ embryos. Data (n = 5) are shown with mean ± S.D. *, indicates false discovery rate, q < 0.05. E, diagram of metabolites detected from glycolysis and TCA cycle. Thicker arrows represent multiple enzymatic steps. Not all intermediates are shown. Small up arrows (green) and down arrows (red) represent metabolites that are increased or decreased in Mthfd1l−/− embryos compared with wildtype embryos, although not all of these reached statistical significance after false discovery rate correction. Boxes represent amino acids that feed into the TCA cycle through the anaplerotic reactions.
Table 1.
Pathway analysis by MetaboAnalyst
Metabolic pathways that were found to be significantly altered (based on FDR) in Mthfd1l−/− embryos from unsupplemented dams. The rightmost column shows how each pathway has responded to formate supplementation (supplemented Mthfd1l−/− versus supplemented Mthfd1l+/+).
| Pathway | Total compounds | Hits | Unsupplemented FDR | Supplemented FDR |
|---|---|---|---|---|
| Methane metabolisma | 9 | 3 | 2.76E-06 | 5.32E-06 |
| Citrate cycle (TCA cycle)b | 20 | 6 | 0.0028 | 0.52 |
| Alanine, aspartate, and glutamate metabolismc | 24 | 8 | 0.0054 | 0.027 |
| Arginine and proline metabolismc | 44 | 10 | 0.0057 | 0.13 |
| Glyoxylate and dicarboxylate metabolism | 18 | 4 | 0.0057 | 0.39 |
| Valine, leucine and isoleucine biosynthesisc | 11 | 4 | 0.0057 | 0.63 |
| Pyruvate metabolismb | 23 | 4 | 0.0063 | 0.057 |
| Glycine, serine, and threonine metabolisma,c | 31 | 7 | 0.012 | 5.32E-06 |
| Histidine metabolismc | 15 | 2 | 0.018 | 0.020 |
| Phenylalanine metabolismc | 11 | 2 | 0.026 | 0.023 |
| Phenylalanine, tyrosine, and tryptophan biosynthesisc | 4 | 2 | 0.026 | 0.023 |
| β-Alanine metabolism | 17 | 4 | 0.028 | 0.13 |
a Pathways involved in 1C metabolism.
b Pathways for energy metabolism.
c Pathways involved in amino acids.
Metabolite differences were analyzed by t test, and p values were corrected for the FDR to yield q values (12). Of the 98 detected metabolites, 24 were significantly altered by loss of MTHFD1L (select metabolites in Fig. 3, full list in supporting Table S1). Four metabolites in the 1C metabolism pathway were detected. Of these, serine was significantly elevated, and glycine and formate were decreased significantly in null embryos (Fig. 3B). These are consistent with decreased utilization of serine for formate synthesis.
Alterations of several metabolites suggested disruptions to energy metabolism (Fig. 3C). Lactate was reduced in Mthfd1l−/− embryos, whereas glucose increased (q = 0.083), suggesting decreased flux through glycolysis. To further explore this possibility, the lactate:pyruvate ratio was used to estimate the cytoplasmic NAD+:NADH ratio using the method described by Krebs (13). This method assumes that the conversion of pyruvate + NADH to lactate + NAD+ is in chemical equilibrium (Fig. 3E). Assuming a pH of 7.0 and Keq = 1.11 × 10−4, the calculated NAD+:NADH ratio in Mthfd1l−/− embryos (116.2 ± 12.3) was significantly higher than in Mthfd1l+/+ embryos (88.4 ± 7.1, p = 0.0024), consistent with decreased flux through glycolysis in the mutant embryos (14, 15). The tricarboxylic acid (TCA) cycle also appeared to be affected. The entry metabolite, citrate, was elevated, whereas downstream metabolites, such as fumarate, were decreased in Mthfd1l−/− embryos (Fig. 3C). The adenylate energy charge was also disrupted. In Mthfd1l−/− embryos, there was a depletion of AMP and an accumulation of ADP and ATP. Leucine, isoleucine, valine, phenylalanine, tyrosine, glycine, and arginine also exhibited a trend toward depletion in Mthfd1l−/− embryos, although not all reached statistical significance after FDR correction (Fig. 3D, Table S1). Taurine was also significantly increased in Mthfd1l−/− embryos compared with Mthfd1l+/+ embryos (Table S1).
Mthfd1l−/− MEF growth curves
To determine whether deletion of Mthfd1l causes dysfunction in the synthesis of purines and thymidylate, a series of growth curves was determined using MEFs derived from Mthfd1l−/− and Mthfd1l+/+ embryos. The basal media (basal MEM) contained serine as a source of one carbon units, but lacked any nucleotides. Cell growth and viability was monitored using Cell Titer Blue. Although null MEFs were viable in basal minimal Eagle's medium (MEM), WT cells exhibited significantly more robust growth (p < 0.0001) (Fig. 4A). Addition of thymidine alone to basal MEM did not improve the growth of null MEFs as compared with WT (Fig. 4B; p = 0.0023). The addition of hypoxanthine alone (Fig. 4C, p = 0.13), hypoxanthine and thymidine together (Fig. 4D, p = 0.64), or formate alone (Fig. 4E, p = 0.30) to basal MEM rendered the growth of null MEFs indistinguishable from that of WT. Neither cell line was able to grow when methionine was replaced with homocysteine and vitamin B12 (data not shown). The absence of MTHFD1L in Mthfd1l−/− MEFs was confirmed by genotyping and immunoblotting (Fig. 4F). Immunodetection of MTHFD1L in whole cell extracts of MEFs requires long exposures, yielding higher backgrounds than immunoblots of isolated mitochondria (cf. Figs. 2B and 4F). Nonetheless, the MTHFD1L band at 100 kDa is clearly missing from the null MEF extract (Fig. 4F).
Figure 4.

Mthfd1l−/− MEF growth curves. Cell growth was monitored using the cell viability assay CellTiter Blue. A–E, cells were grown in basal MEM (described under “Experimental Procedures”) with additional 30 μm thymidine, 30 μm hypoxanthine, or 1 mm formate as indicated. p values were determined using a mixed model two-way analysis of variance for repeated measures in GraphPad Prism. Data points represent the mean of quadruplicate determinations ± S.D. F, loss of MTHFD1L expression in Mthfd1l−/− MEFs confirmed by genotyping and immunoblotting. The wildtype allele gives a 444-bp genomic fragment and the Mthfd1l−/− allele gives a 324-bp fragment. For the immunoblots (IB), each lane was loaded with 40 μg of protein from whole cell extracts. Immunoblots were probed with antibodies against MTHFD1L (100 kDa) or the mitochondrial matrix protein Hsp60 (60 kDa) as a loading control. Migration and size (kDa or bp) of molecular mass markers are shown on the right of the gels.
Deuterated serine labeling in MEFs
Despite the fact that mitochondrially derived 1C units are a major source of carbon for methionine synthesis from homocysteine (7, 16), loss of MTHFD1L did not significantly alter the concentration of methionine in Mthfd1l−/− embryos (Fig. 3B). To further explore this result, we employed a tracer experiment utilizing l-[2,3,3-2H3]serine (7, 17). This experiment relies on the principle that 1C units derived from carbon-3 of serine processed in the mitochondria before incorporation into methionine will only contain one deuterium atom (D1) due to the dehydrogenase activity of MTHFD2/2L. One-carbon units incorporated into methionine via SHMT1 in the cytosol will retain both deuterium atoms (D2). Because B12 is a required cofactor for the function of methionine synthase and basal MEM does not typically contain B12, the labeling experiment was conducted with the addition of 1 μm cyanocobalamin. We found the methionine pool in Mthfd1l+/+ cells contained approximately five times as much mitochondrially derived singly deuterated (D1) methionine as the Mthfd1l−/− cells (Table 2). Conversely, doubly deuterated (D2) methionine was nearly 5-fold higher in Mthfd1l−/− cells than Mthfd1l+/+ cells, suggesting Mthfd1l−/− cells compensate for loss of MTHFD1L by increasing flux of serine through cytosolic SHMT1. Consistent with previously reported values (7, 16), about 90% of labeled methionine in Mthfd1l+/+ cells was mitochondrially derived, whereas singly deuterated methionine accounted for only 27% of labeled methionine in Mthfd1l−/− cells (Table 2).
Table 2.
Methionine labeling with deuterated serine
MEFs were plated in triplicate in 10-cm dishes at a density of 1 × 106 cells per dish and were grown in labeling media for 4 days as described under “Experimental procedures.” Incorporation of the deuterium label into methionine was measured by LC-MS. Means of triplicate determinations ± S.D. are shown. M = unlabeled methionine, D1 = methionine + 1 deuteron, D2 = methionine + 2 deuterons. p values were calculated using two tailed t tests.
| Fraction of total (M + D1 + D2) |
Fraction of labeled (D1 + D2) |
||||
|---|---|---|---|---|---|
| D1 | D2 | D1 + D2 | D1 | D2 | |
| MTHFD1L+/+ | 0.18 ± 0.008 | 0.020 ± 0.01 | 0.20 ± 0.009 | 0.90 ± 0.05 | 0.10 ± 0.05 |
| MTHFD1L−/− | 0.033 ± 0.001 | 0.092 ± 0.009 | 0.12 ± 0.008 | 0.27 ± 0.03 | 0.73 ± 0.03 |
| p value | 6.70E-06 | 0.0011 | 5.80E-04 | 6.00-05 | 6.00E-05 |
Deuterium incorporation into hypoxanthine and IMP was used as an indicator of de novo purine synthesis. Deuterium from l-[2,3,3-2H3]serine can be incorporated into purines in one of two ways: either as 10-CHO-THF (from C3) or as glycine (from C2) produced by SHMT. Mthfd1l+/+ cells had a much higher proportion of labeled hypoxanthine and IMP than Mthfd1l−/− cells (Table 3), suggesting that Mthfd1l+/+ cells utilize de novo purine synthesis, whereas Mthfd1l−/− cells rely heavily on the salvage pathway. Although it is not possible to distinguish whether the deuterium label in the purine is derived from 10-CHO-THF or incorporated as glycine, in either case a singly deuterated species indicates purine derived from the de novo synthesis pathway rather than the salvage pathway.
Table 3.
Purine labeling with deuterated serine
MEFs were plated in triplicate in 10-cm dishes at a density of 1 × 106 cells per dish and were grown in labeling media for 4 days as described under “Experimental procedures.” Incorporation of the deuterium label into IMP and hypoxanthine (Hyp) was measured by LC-MS. Means of triplicate determinations ± S.D. are shown. IMP + D = IMP + 1 deuteron, Hyp + D = Hyp + 1 deuteron. p values were calculated using two tailed t tests.
| Fraction of total (IMP + IMP + D) |
Fraction of total (Hyp + Hyp + D) |
|||
|---|---|---|---|---|
| IMP | IMP + D | Hyp | Hyp + D | |
| MTHFD1L+/+ | 0.18 ± 0.02 | 0.82 ± 0.02 | 0.22 ± 0.01 | 0.78 ± 0.01 |
| MTHFD1L−/− | 0.90 ± 0.009 | 0.1 ± 0.009 | 0.81 ± 0.03 | 0.19 ± 0.03 |
| p value | 5.87E-06 | 5.87E-06 | 0.00024 | 0.00024 |
Finally, we used deuterium labeling to measure the effect of disruption of the mitochondrial 1C pathway on serine synthesis. Serine can be synthesized from glycine and CH2-THF via SHMT (Fig. 1, reaction 4 or 4m), generating serine with 0, 1, 2, or 3 deuterium atoms depending on the degree of labeling of glycine and CH2-THF. Serine can also be generated from protein turnover, or synthesized de novo from the glycolytic intermediate 3-phosphoglycerate. Serine derived from either of these latter pathways would likely be unlabeled. We observed that 60–70% of the serine pool was doubly or triply deuterated in both Mthfd1l+/+ and Mthfd1l−/− MEFs (Table 4). The fraction of unlabeled serine in both the intracellular and extracellular compartments was significantly higher in Mthfd1l−/− MEFs, as compared with Mthfd1l+/+ MEFs (p = 0.012 and 0.0087, respectively), suggesting that de novo synthesis of serine was increased in null cells.
Table 4.
De novo serine synthesis in Mthfd1l−/− MEFs
MEFs were plated in triplicate in 10 cm dishes at a density of 1 × 106 cells per dish and were grown in labeling media for four days as described under “Experimental procedures.” Deuterium labeling of serine from cellular extracts and serine effluxed into the media was measured by LC-MS. Means of triplicate determinations ± S.D. are shown. S = unlabeled serine, D1 = serine + 1 deuteron, D2 = serine + 2 deuterons, D3 = serine + 3 deuterons. p values were calculated using two tailed t tests.
| Fraction of total serine (S + D1 + D2 + D3) |
||||
|---|---|---|---|---|
| S | D1 | D2 | D3 | |
| Intracellular | ||||
| Mthfd1l+/+ | 0.25 ± 0.02 | 0.065 ± 0.02 | 0.29 ± 0.03 | 0.41 ± 0.04 |
| Mthfd1l−/− | 0.34 ± 0.03 | 0.055 ± 0.02 | 0.31 ± 0.02 | 0.31 ± 0.01 |
| p value | 0.012 | 0.41 | 0.23 | 0.012 |
| Media | ||||
| Mthfd1l+/+ | 0.12 ± 0.04 | NDa | ND | 0.88 ± 0.04 |
| Mthfd1l−/− | 0.34 ± 0.07 | ND | ND | 0.66 ± 0.07 |
| p value | 0.0087 | 0.0087 | ||
a ND, not detected.
Energy metabolism in Mthfd1l−/− MEFs
Metabolomic analysis of Mthfd1l−/− embryos suggests these embryos have impaired energy metabolism (Fig. 3). To further explore mitochondrial respiration and glycolytic flux, a Seahorse XFp Flux Analyzer was used to measure the oxygen consumption rate (OCR) and glycolytic flux via extracellular acidification rate (ECAR) in MEFs. Mthfd1l+/+ and Mthfd1l−/− MEFs showed similar OCRs, indicating no difference in mitochondrial respiratory function (Fig. 5, p = 0.62). Cell lines of both genotypes exhibited well-coupled mitochondria after addition of rotenone and their maximal respiration rates after addition of FCCP did not differ significantly.
Figure 5.
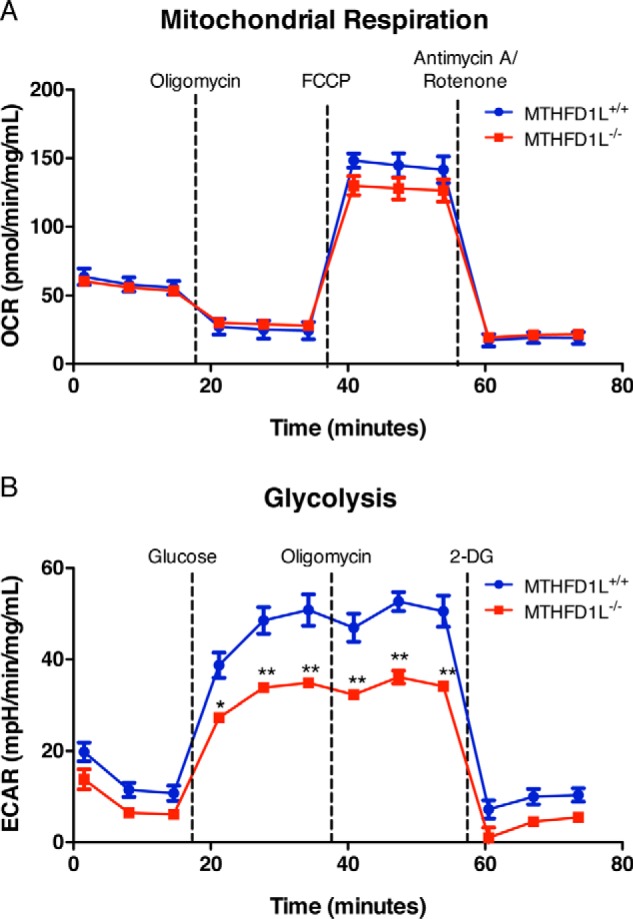
Energy metabolism in Mthfd1l−/− MEFs. Mitochondrial respiration (top) and glycolysis (bottom) were analyzed in MEFs using the Seahorse XFp Flux Analyzer as described under “Experimental procedures.” Data points represent the mean of triplicate determinations ± S.D. Dashed lines represent injections of the indicated compounds. *, p < 0.01; **, p < 0.0001 as determined by Bonferroni post test. 2-DG, 2-deoxyglucose.
Mthfd1l−/− MEFs showed lower rates of extracellular acidification, indicating a difference in glycolytic flux (Fig. 5, p = 0.010). ECAR did not increase in either cell line after the addition of oligomycin, indicating little or no glycolytic reserve in these cells. ECAR only differed significantly after glucose injection and prior to 2-deoxyglucose injection, indicating that the difference in ECAR was due to decreased glycolytic flux, rather than acidification of the media by some other cellular process. These results are consistent with the perturbations to energy metabolites observed in Mthfd1l−/− embryos (Fig. 3).
Metabolomic analysis of Mthfd1l−/− embryos from calcium formate–supplemented dams
To determine whether formate supplementation normalized the metabolome of Mthfd1l−/− embryos, extracts of Mthfd1l−/− and Mthfd1l+/+ embryos from dams supplemented with 2,500 mg kg−1 d−1 calcium formate were examined using NMR and MS. Of the 117 detected metabolites, only 9 were significantly different after correcting for the false discovery rate. Four of these 9 matched metabolites that were altered in unsupplemented embryos: serine, glycine, taurine, and AMP (select metabolites in Fig. 6, full list in supporting Table S2). Serine remained elevated, and glycine remained depleted. As expected, formate was normalized in embryos upon maternal supplementation (Fig. 6B). Taurine abundance was reversed; it was elevated in unsupplemented Mthfd1l−/− embryos, but was diminished in supplemented Mthfd1l−/− embryos. AMP remained depleted in Mthfd1l−/− embryos despite supplementation (Fig. 6C). Formate supplementation also eliminated the difference in the calculated NAD+:NADH ratio (127.8 ± 21.6 and 127.8 ± 4.8 in Mthfd1l+/+ and Mthfd1l−/− embryos, respectively). Notably, lactate levels in Mthfd1l−/− embryos were normalized by formate supplementation (Fig. 6C and Table S2), suggesting that glycolytic flux is restored upon formate supplementation. PCA indicated that despite elimination of several metabolic markers of null embryos, the groups still demonstrate distinct group separation (Fig. 6A). This suggests that whereas formate supplementation ameliorates the phenotypic and metabolic consequences of Mthfd1l knockout, it is not sufficient to completely reverse the metabolic differences between null and wildtype embryos. Pathway analysis with MetaboAnalyst was again performed, this time focusing on the pathways that had been identified as altered in unsupplemented embryos. This analysis revealed that of the 12 pathways that were altered in unsupplemented embryos, 6 were normalized by maternal formate supplementation (Table 1).
Figure 6.

Metabolomic analysis of Mthfd1l−/− embryos from calcium formate–supplemented dams. A, principal component analysis of embryo metabolites. A–D, abundance of metabolites related to (B) one-carbon metabolism, (C) energy metabolism, and (D) amino acid metabolism in Mthfd1l−/− embryos relative to Mthfd1l+/+ embryos. Data (n = 5) are shown with mean ± S.D. *, indicates false discovery rate, q < 0.05.
Discussion
The Mthfd1l knockout mouse model has revealed a central role of mitochondrially derived formate in embryonic development, providing a mechanistic link between folic acid and NTDs. Disruption of MTHFD1L function does not cause cellular folate deficiency, rather it blocks a specific metabolic step, the production and release of formate from mitochondria into the cytoplasm. This very specific metabolic defect causes aberrant neural tube closure and growth delay in 100% of Mthfd1l−/− embryos. Maternal formate supplementation partially rescues neural tube and growth defects (4). In the current study, we show that Mthfd1l−/− mitochondria retain a residual capacity to produce formate in organello (Fig. 2). Nevertheless, mitochondrial formate production in null embryos is clearly insufficient to support proper embryonic development in vivo. It is likely that the flux of formate from mitochondria in Mthfd1l−/− embryos is too low to provide for all the 1C requirements in these embryos. This hypothesis is supported by the decrease in labeled methionine observed in Mthfd1l−/− embryos (Table 2).
Because MTHFD1L is not expressed in Mthfd1l−/− embryos (Fig. 2A), what is the source of formate produced by Mthfd1l−/− mitochondria? A likely source is ALDH1L2, a mitochondrial 10-CHO-THF dehydrogenase (Fig. 1, reaction 9) that exhibits 10-CHO-THF hydrolase activity in vitro (8, 9). Instead of oxidizing the formyl group of 10-CHO-THF to CO2, this hydrolase activity releases the formyl group as formate. If ALDH1L2 exhibits this activity in vivo, it could account for the formate produced by Mthfd1l−/− mitochondria from l-[3-14C]serine. ALDH1L2 is abundantly expressed in mitochondria from Mthfd1l−/− embryos (Fig. 2B), which is consistent with involvement of this enzyme in regeneration of THF and possible production of formate.
Given the central importance of 1C metabolism, it is reasonable to expect a large global change in metabolites upon deletion of Mthfd1l. To understand the metabolic impact of this gene deletion, untargeted metabolomics and metabolic pathway analyses was performed comparing wildtype and Mthfd1l−/− embryos. These analyses revealed disruptions in several 1C pathway metabolites as well as pathways involved in energy production and amino acid metabolism (Fig. 3, B–D). Surprisingly, steady state levels of purines, thymidylate, and methionine did not differ significantly between Mthfd1l−/− and wildtype embryos. Maternal formate supplementation normalized the concentrations of a large proportion of embryo metabolites perturbed by loss of MTHFD1L, including all observed TCA cycle intermediates (Fig. 6). Calculation of the NAD+:NADH ratio in embryos from formate-supplemented dams reveals that this ratio was no longer significantly different, suggesting that glycolysis operates normally in Mthfd1l−/− embryos from dams supplemented with formate.
There are numerous ways that these sweeping changes in metabolism may interfere with neural tube closure. Diminished amino acid availability could have negative effects on protein synthesis required for rapid cell growth and division. Disruption of amino acid metabolism beyond the 1C donors serine and glycine may reflect disturbances in TCA cycle metabolites as amino acids are used to compensate for depletion in TCA intermediates through anaplerosis (Fig. 3E) (18). The impact of Mthfd1l deletion also extends to energy metabolism (Fig. 3C). Although no differences were detected in mitochondrial respiration in Mthfd1l−/− MEFs (Fig. 5A), significantly decreased flux through glycolysis was observed in Mthfd1l−/− MEFs in comparison to wildtype (Fig. 5B). This decrease in glycolytic flux may be in part explained by the observed increase in de novo serine synthesis (Table 4). Diversion of the glycolytic intermediate 3-phosphoglycerate for de novo serine synthesis would decrease the yield of the payoff phase of glycolysis. Glycolysis seems to be a critical pathway during the transition from gastrulation to the neurulation stage (E6–9 in mice). For example, mouse embryos exhibit increased glycolysis from E8.5 to E10.5 (19), which spans the time of neural tube closure. Moreover, inhibition of glycolysis is known to cause NTDs, at least in cultured embryos (20). Lactate, the end product of glycolysis, also increases from E8.5 to E10.5 in mouse embryos (19). Using mass spectroscopy imaging, these authors observed higher levels of lactate in the dorsal or posterior neural tube, somites, and head mesenchyme compared with surrounding tissues (19). We observed lower lactate levels in Mthfd1l−/− embryos compared with wildtype embryos (Fig. 3C), and formate supplementation normalized this effect (Fig. 6C). These findings suggest that the altered glycolysis associated with Mthfd1l deletion may be an important factor in the aberrant neural tube closure observed in the null embryos.
To better understand the effect of Mthfd1l deletion on flux through the 1C pathway, metabolite supplementation and isotope labeling experiments were performed using mouse embryonic fibroblasts (MEFs) derived from Mthfd1l−/− and wildtype embryos. Growth curves indicate supplementation with formate, hypoxanthine, or hypoxanthine and thymidine in combination rescue growth of Mthfd1l−/− MEFs to wildtype levels (Fig. 4). Thymidine alone did not rescue, suggesting a purine synthesis defect in Mthfd1l−/− MEFs. Purine synthesis can occur either through a de novo pathway, which requires two 1C units in the form of 10-CHO-THF, or via salvage, whereby purines derived from degradation are reutilized. Deuterium labeling experiments reveal that Mthfd1l+/+ MEFs have a much higher proportion of labeled hypoxanthine and IMP than Mthfd1l−/− cells (Table 3), which suggests that Mthfd1l+/+ cells utilize de novo purine synthesis, whereas Mthfd1l−/− cells exhibit a strong reliance on the salvage pathway. The defective mitochondrial formate production resulting from Mthfd1l deletion (Fig. 2) will cause a deficiency of cytoplasmic 10-CHO-THF, thereby limiting the de novo pathway (Fig. 1). Indeed, experiments with cultured embryos have shown that formate supplementation decreases uptake of exogenous adenine and thymidine (21), further supporting the critical role of formate for nucleotide synthesis during embryogenesis.
A stable isotope tracer experiment revealed altered methionine synthesis in Mthfd1l−/− MEFs (Table 2), indicating that deletion of this gene affects the integrity of the methyl cycle. Deuterium label incorporation suggests that Mthfd1l−/− MEFs compensate for loss of MTHFD1L by increasing flux of serine through cytosolic SHMT1 (Fig. 1, reaction 4). Analogous results have been observed with blockage of the mitochondrial 1C pathway via deletion of Shmt2, Mthfd2, or Mthfd1l in HEK293 cells, which led to increased incorporation of 1C units from serine into dTTP via the cytosolic pathway (22).
The etiology of NTDs in Mthfd1l−/− embryos is likely influenced by numerous factors including metabolism and nutrient availability. Proper function of both the methyl cycle and nucleotide synthesis are essential for development. Inhibition of SAM synthesis from methionine and inhibition of DNA methylation both lead to NTDs in mice (23, 24). Inhibition of de novo purine synthesis with lometrexol causes partially penetrant (30%) NTDs in mice (25), and defects in purine synthesis have been found in a mouse model of NTDs (26). Deletion of the glycine cleavage enzymes glycine decarboxylase and aminomethyltransferase cause NTDs that can be partially prevented either by maternal supplementation of methionine (27, 28) or by inhibition of the methionine cycle in the glycine decarboxylase KO mouse via deletion of MTHFR (28). Disruption of thymidylate synthesis by knocking out SHMT1 is also associated with NTDs (29). Finally, disruptions to both purine and thymidylate synthesis are associated with valproate-induced NTDs in mice (30). Here we have identified specific metabolite disturbances caused by loss of MTHFD1L that impact multiple metabolic pathways, including the 1C metabolic pathway, glycolysis, the TCA cycle, and amino acid metabolism. The amelioration of this deranged metabolism by formate supplementation highlights that a simple, targeted nutritional intervention can make a powerful impact on a disease phenotype.
Experimental procedures
Mice and formate supplementation
All protocols in this study were approved by the Institutional Animal Care and Use Committee of The University of Texas at Austin and conform to the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Mice were maintained on a C57BL/6 background.
The Mthfd1l knockout mouse model has been described previously (4). Homozygous null embryos (Mthfd1lz/z) carry two copies of the Mthfd1l locus disrupted by a LacZ cassette inserted between exons 4 and 6. Homozygous nulls will herein be referred to as Mthfd1l−/− or simply nulls. Where indicated, matings of heterozygous Mthfd1lz/+ mice were housed in cages equipped with a water bottle containing calcium formate. The concentration of calcium formate was adjusted to deliver a calculated dose of 2,500 mg kg−1 d−1 based on the assumption that a 25-g C57BL/6 mouse consumes water at a rate of 5 ml/day (31). Female mice had access to water supplemented with formate for a minimum of 1 day prior to the observation of a vaginal plug.
Cell lines and media
Wildtype and nullizygous Mthfd1l MEFs were derived as described below. The MEF cell lines were not immortalized, so all experiments were conducted at fewer than four cell passages. Cells were grown in either Dulbecco's modified Eagle's medium (DMEM, Sigma) supplemented with 10% fetal bovine serum (FBS) or MEM (MP Bio 1641454) supplemented with 15% dialyzed FBS. Cells were grown in a humidified cabinet at 37 °C under 5% CO2 on 100-mm dishes. Cells were typically passaged at 95% confluence at no more than a 1:4 dilution.
Mitochondrial formate synthesis assay
Embryos at E14.5–E16.5 were dissected from dams that had been supplemented with 2,500 mg kg−1 d−1 calcium formate and genotyped using yolk sack tissue as previously described (4). Embryos were pooled by genotype and mitochondria were isolated using differential centrifugation as previously described (7). Mitochondrial protein content was quantified by BCA assay (ThermoFisher Scientific). Expression of MTHFD1L and ALDH1L2 proteins was determined by immunoblotting. Mitochondrial proteins were extracted and separated by SDS-PAGE and immunoblotted as described previously (7). Blots were probed with antibodies against MTHFD1L (1:1000) (32), ALDH1L2 (1:2000) (8), or Hsp60 (1:2000; Enzo Life Sciences) and reacting bands were detected with ECL Prime (GE Healthcare Life Sciences).
Measurement of mitochondrial synthesis of formate from serine was carried out as described previously (6). Briefly, 1 mg of mitochondria was incubated with l-[3-14C]serine (1000 dpm/nmol, Moravek Biochemicals, Brea, CA) at the indicated concentration in a shaking incubator for 20 min at 37 °C. Reactions were stopped by removal of mitochondria by centrifugation at 10,000 × g at 4 °C for 10 min. The resulting supernatant was incubated at 95 °C to remove 14C-labeled CO2 that may have been formed and [14C]formate was determined by enzymatic assay as described (6).
Mouse embryonic fibroblast derivation
MEFs were derived according to established protocols (33). Briefly, dams supplemented with a calculated dose of 2,500 mg kg−1 d−1 calcium formate were euthanized at E14.5 with CO2 asphyxiation followed by cervical dislocation. Mice were thoroughly sprayed with 70% ethanol then moved to a laminar flow cabinet where uteri were dissected and washed with sterile PBS. Individual embryos were placed into ice-cold PBS, with yolk sac tissue reserved for genotyping. Embryo heads, heart, and liver were removed and the remaining tissue placed into trypsin/EDTA (0.25%) and incubated overnight at 4 °C. The following morning, excess trypsin/EDTA solution was removed, leaving approximately double the volume of tissue. Samples were incubated at 37 °C for 30 min to activate trypsin. DMEM (10% FBS, penicillin/streptomycin) was added and tissue was homogenized by pipetting, plated, and incubated at 37 °C and 5% CO2. These cells were considered passage number 0. After isolation, MEFs were genotyped as described previously (4) and tested for mycoplasma infection using a PCR test kit (SouthernBiotech). Expression of MTHFD1L protein was determined by immunoblotting. Proteins were extracted using Nonidet P-40 lysis buffer (50 mm Tris, 150 mm NaCl, 1% Nonidet P-40, pH 7.5), separated by SDS-PAGE, and immunoblotted as described above.
MEF growth curves
Mthfd1l+/+ and Mthfd1l−/− MEFs were plated onto six 96-well plates (one for each time point) at a density of 5 × 103 cells per well with four replicate wells per genotype for each condition tested. Basal media (referred to as basal MEM) was made from a formulation of modified Eagle's medium lacking methionine, cysteine, and cystine (MP Bio 1641454) and contained an additional 15% dialyzed FBS, 500 μm serine, 20 μm methionine, and the following amino acids: 25 mg/liter of alanine, 50 mg/liter of asparagine, 30 mg/liter of aspartate, 75 mg/liter of glutamate, 292 mg/liter of glutamine, 40 mg/liter of proline, 100 mg/liter of cysteine, 31 mg/liter of cystine. Supplemental 30 μm hypoxanthine, 30 μm thymidine, or 1 mm sodium formate were added to basal MEM for rescue experiments. Cell growth was monitored using the cell viability assay Cell Titer Blue (Promega).
Deuterated serine tracer experiment
Labeling of MEFs with l-[2,3,3-2H3]serine was performed as previously described (7). Briefly, cells were plated onto 10-cm dishes at a density of 1 × 106 cells/dish in triplicate. Labeled media was identical to the basal MEM used for the growth curve experiments except the methionine concentration was 10 μm, and 1 μm cyanocobalamin was added. Cells were grown in labeled media for 4 days and media was replenished after 2 days. Cells were washed with PBS and collected by trypsinization. Aliquots of spent media were collected and stored at −80 °C to be used for metabolite analysis. Cell pellets were snap frozen in liquid nitrogen and stored at −80 °C prior to extraction.
Extraction of polar metabolites
Polar metabolites were extracted from embryos (45–53 somite stage) harvested from both unsupplemented and formate-supplemented pregnant dams. Each embryo was processed individually. Prior to extraction, solutions were prechilled in a −80 °C freezer for 30 min. Metabolic extraction was performed for metabolomic analysis using both nuclear magnetic resonance (NMR) spectroscopy and mass spectrometry (MS) platforms as previously described (34–36). Briefly, embryos were suspended in 1 ml of 1:1 water/methanol and homogenized by pipetting. Lysates were added to 500 μl of chloroform, vortexed for 10 min, and centrifuged at 5250 × g for 20 min at 4 °C. The upper aqueous phase containing the polar metabolites was removed, aliquoted for NMR and MS analyses, dried in a Centrivap vacuum concentrator at 4 °C (Labconco, Kansas City, MO), and then immediately stored at −80 °C before the metabolomics analysis.
NMR-based metabolomic analysis
Polar samples for NMR analysis were resuspended in 45 μl of phosphate buffer (100 μm phosphate, 90% D2O, 1 mm TSP, 0.05% NaN3), vortexed, centrifuged at 4 °C for 10 min, and finally 35 μl of the supernatants were transferred into 1.7-mm NMR tubes (36). One-dimensional 1H NMR spectra were acquired using a Bruker Avance III 500 MHz with a 1.7-mm TCI MicroCryoProbe system (Bruker BioSpin Corp., Billerica, MA) equipped with an autosampler at 300 K as previously reported (36). The excitation sculpting pulse sequence was used to suppress the water resonance signal (37). NMRLab (38) and MetaboLab (39) were used to process the raw data. Metabolite identification and quantification were done using the Chenomx 8 NMR Suite (Chenomx Inc., Edmonton, Alberta, Canada) referencing the Birmingham Metabolite Library (40), and the Human Metabolome Database (41).
MS-based metabolomic analysis
Chromatographic separation was performed on a Thermo Scientific (Thermo Fisher Scientific, San Jose CA) Accela HPLC system equipped with a quaternary pump, vacuum degasser, and an open autosampler with a temperature controller. A 100 × 2.1 mm inner diameter, 2.6-μm particle size Kinetex biphenyl C18 column (Phenomenex Inc., Torrance, CA) was used for all experiments. Separation conditions were: solvent A, water/formic acid (99.8:0.2); solvent B, MeOH; separation gradient, initially 1% B, then linear to 100% B in 4 min, washing with 100% B for 1 min and column equilibration with 1% B for 10 min; flow rate, 0.3 ml/min; injection volume, 3 μl; autosampler temperature, 6 °C; column temperature, 22 °C. Mass spectrometry analysis was carried out on a Thermo Scientific Q Exactive benchtop Orbitrap detector (Thermo Fisher Scientific, Bremen, Germany) equipped with an electrospray ionization source operating in positive/negative switching mode. The detector was run in full scan MS analysis under the following conditions: spray voltage, 4.0 kV; capillary temperature, 300 °C; sheath gas, 55 (arbitrary units); auxiliary gas, 30 (arbitrary units); microscans, 1; AGC target, 1e6; maximum injection time, 100 ms; mass resolution, 70,000; m/z range, 50–750. The MS device was calibrated with commercial calibration solution provided by the manufacturer to maintain mass tolerance below 5 ppm. The LC-MS platform of analysis was controlled by a PC operating the Xcalibur software package (version 2.2 SP1.48, Thermo Fisher Scientific, San Jose, CA).
Following LC-MS acquisition, Thermo *.raw files were processed for untargeted analysis using two commercially available software packages from Thermo Scientific. Thermo SIEVE (version 2.2) was used for the tissue culture experiments and Compound Discoverer (version 2.0) was used for the embryos, both in conjunction with an in-house library (36), which includes 618 compounds from the IROA 300, Mass Spectrometry Metabolite Library of Standards (MSMLS; IROA Technologies, Bolton, MA). Deuterium incorporation into IMP and hypoxanthine was scored by hand in Xcalibur. Customized settings were: maximum retention time shift, 0.25 min; minimum base peak intensity 50,000; mass tolerance, 5 ppm, peak integration, ICIS; smoothing, 3 points; adduct formation, ±H. Spectral alignment above 0.9 was considered acceptable for all data sets. Retention times were used to reduce multiple assignments of spectral features and to confirm metabolite assignment. MS data were then combined to the NMR data for the subsequent post-processing, and statistical analysis.
Mitochondrial respiration and glycolysis assays
OCR and ECAR were measured using the Seahorse XFp Flux Analyzer (Seahorse Bioscience-Agilent Technologies, Billerica, MA). Cells were seeded at a density of 2 × 105 cells/ml in 80 μl of DMEM + 10% FBS + penicillin/streptomycin/glutamine) on XFp 8-well microplates and incubated for 24 h at 37 °C, 5% CO2.
For mitochondrial function analysis (based on OCR) cells were washed two times with XF media (Seahorse Bioscience-Agilent Technologies) freshly supplemented with 25 mm glucose, 2 mm glutamine, 1 mm pyruvate, adjusted to pH 7.4, and incubated in this media for 1 h at 37 °C in a CO2-free incubator. After calibration, the device measures a baseline and levels of OCR and ECAR after sequential addition of different compounds to each well: oligomycin (final concentration 2 μm), FCCP (final concentration 1 μm), and antimycin A/rotenone (final concentration 1 μm each).
For the glycolysis assay (based on ECAR), cells were washed and incubated as above except that the media lacked glucose. Treatments were as follows: glucose (final concentration 10 mm), oligomycin (final concentration 2 μm), and 2-deoxyglucose (final concentration 50 mm).
After each assay, media was removed from each well and plates were frozen at −80 °C. Cells were lysed by addition of 10 μl of lysis buffer (10 mm Tris, pH 8, 0.1% Triton X-100) to each well. A Bradford assay was carried out, and OCR and ECAR data were normalized to total protein concentration. Assays were analyzed using the Seahorse Data Analysis Software (Seahorse Bioscience-Agilent Technologies).
Statistical analysis
Growth curves and Seahorse experiments were analyzed in GraphPad Prism using a mixed model two-way analysis of variance for repeated measures, and Bonferroni post-tests were used for pairwise comparisons at individual time points. Unpaired t tests were calculated in Microsoft Excel. False discovery rates (q) for metabolite analyses were calculated using the method described by Benjamini and Hochberg (12). PCA was performed with Jmp Pro12 (SAS Institute). Pathway analysis was performed using MetaboAnalyst (10, 11). Data were log transformed and mean centered prior to PCA and pathway analysis. α was set to 0.05 for all experiments.
Author contributions
J. D. B. and S. R. S. data curation; J. D. B., S. R. S., E. S., B. X., J. M., S. T., and D. R. A. formal analysis; J. D. B., S. R. S., E. S., M. S., H. I., J. M., and S. T. methodology; J. D. B., J. M., and D. R. A. writing-original draft; J. D. B., B. X., J. M., S. T., and D. R. A. writing-review and editing; E. S., M. S., H. I., J. M., S. T., and D. R. A. investigation; B. X. and D. R. A. supervision; J. M. and D. R. A. project administration; D. R. A. conceptualization; D. R. A. funding acquisition.
Supplementary Material
Acknowledgments
We thank Dr. Sergey Krupenko for the generous gift of the antibodies against ALDH1L2. Support for the NMR facility was provided by the University of Texas Health Science Center at San Antonio (UTHSCSA) and National Institutes of Health, NCI Grant P30 CA54174 (CTRC at UTHSCSA).
This work was supported in part by National Institutes of Health Grants F32HD074428 (to J. M.), GM086856 (to D. R. A.), and HD083809 (to D. R. A. and S. T.), Welch Foundation Grant F1859 (to B. X.), and a STAR Award from the University of Texas System (to S. T.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Tables S1 and S2.
- NTD
- neural tube defect
- 1C
- one-carbon
- CH+-THF
- 5,10-methenyltetrahydrofolate
- CH2-THF
- 5,10-methylenetetrahydrofolate
- 10-CHO-THF
- 10-formyltetrahydrofolate
- ALDH1L2
- aldehyde dehydrogenase 1-like 2
- DMEM
- Dulbecco's modified Eagle's medium
- ECAR
- extracellular acidification rate
- FBS
- fetal bovine serum
- FCCP
- carbonyl cyanide-4-(trifluormethoxy)phenylhydrazone
- FDR
- false discovery rate
- KO
- knockout
- MEF
- mouse embryonic fibroblast
- MTHFD
- methylene-tetrahydrofolate dehydrogenase
- NMR
- nuclear magnetic resonance
- OCR
- oxygen consumption rate
- PCA
- principal component analysis
- SHMT
- serine hydroxymethyltransferase
- TCA
- tricarboxylic acid
- MEM
- modified Eagle's medium.
References
- 1. Crider K. S., Bailey L. B., and Berry R. J. (2011) Folic acid food fortification-its history, effect, concerns, and future directions. Nutrients 3, 370–384 10.3390/nu3030370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Au K. S., Findley T. O., and Northrup H. (2017) Finding the genetic mechanisms of folate deficiency and neural tube defects: leaving no stone unturned. Am. J. Med. Genet. Pt. A 173, 3042–3057 10.1002/ajmg.a.38478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tibbetts A. S., and Appling D. R. (2010) Compartmentalization of mammalian folate-mediated one-carbon metabolism. Annu. Rev. Nutr. 30, 57–81 10.1146/annurev.nutr.012809.104810 [DOI] [PubMed] [Google Scholar]
- 4. Momb J., Lewandowski J. P., Bryant J. D., Fitch R., Surman D. R., Vokes S. A., and Appling D. R. (2013) Deletion of Mthfd1l causes embryonic lethality and neural tube and craniofacial defects in mice. Proc. Natl. Acad. Sci. U.S.A. 110, 549–554 10.1073/pnas.1211199110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Barlowe C. K., and Appling D. R. (1988) In vitro evidence for the involvement of mitochondrial folate metabolism in the supply of cytoplasmic one-carbon units. Biofactors 1, 171–176 [PubMed] [Google Scholar]
- 6. Garcia-Martinez L. F., and Appling D. R. (1993) Characterization of the folate-dependent mitochondrial oxidation of carbon 3 of serine. Biochemistry 32, 4671–4676 10.1021/bi00068a027 [DOI] [PubMed] [Google Scholar]
- 7. Pike S. T., Rajendra R., Artzt K., and Appling D. R. (2010) Mitochondrial C1-THF synthase (MTHFD1L) supports flow of mitochondrial one-carbon units into the methyl cycle in embryos. J. Biol. Chem. 285, 4612–4620 10.1074/jbc.M109.079855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Krupenko N. I., Dubard M. E., Strickland K. C., Moxley K. M., Oleinik N. V., and Krupenko S. A. (2010) ALDH1L2 is the mitochondrial homolog of 10-formyltetrahydrofolate dehydrogenase. J. Biol. Chem. 285, 23056–23063 10.1074/jbc.M110.128843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Strickland K. C., Krupenko N. I., Dubard M. E., Hu C. J., Tsybovsky Y., and Krupenko S. A. (2011) Enzymatic properties of ALDH1L2, a mitochondrial 10-formyltetrahydrofolate dehydrogenase. Chem. Biol. Interact. 191, 129–136 10.1016/j.cbi.2011.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Xia J., and Wishart D. S. (2011) Metabolomic data processing, analysis, and interpretation using MetaboAnalyst. Curr. Protoc. Bioinformatics Chapter 14, Unit 14.10 [DOI] [PubMed] [Google Scholar]
- 11. Xia J., Psychogios N., Young N., and Wishart D. S. (2009) MetaboAnalyst: a web server for metabolomic data analysis and interpretation. Nucleic Acids Res. 37, W652–W660 10.1093/nar/gkp356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Benjamini Y., and Hochberg Y. (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. Roy. Stat. Soc. Ser. B Methodological 57, 289–300 [Google Scholar]
- 13. Williamson D. H., Lund P., and Krebs H. A. (1967) The redox state of free nicotinamide-adenine dinucleotide in the cytoplasm and mitochondrial of rat liver. Biochem. J. 103, 514–527 10.1042/bj1030514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. DeBerardinis R. J., Lum J. J., Hatzivassiliou G., and Thompson C. B. (2008) The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 7, 11–20 10.1016/j.cmet.2007.10.002 [DOI] [PubMed] [Google Scholar]
- 15. Lim J.-H., Lee Y.-M., Chun Y.-S., Chen J., Kim J.-E., and Park J.-W. (2010) Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1α. Mol. Cell 38, 864–878 10.1016/j.molcel.2010.05.023 [DOI] [PubMed] [Google Scholar]
- 16. Davis S. R., Stacpoole P. W., Williamson J., Kick L. S., Quinlivan E. P., Coats B. S., Shane B., Bailey L. B., and Gregory J. F. 3rd. (2004) Tracer-derived total and folate-dependent homocysteine remethylation and synthesis rates in humans indicate that serine is the main one-carbon donor. Am. J. Physiol. Endocrinol. Metab. 286, E272–E279 10.1152/ajpendo.00351.2003 [DOI] [PubMed] [Google Scholar]
- 17. Herbig K., Chiang E. P., Lee L. R., Hills J., Shane B., and Stover P. J. (2002) Cytoplasmic serine hydroxymethyltransferase mediates competition between folate-dependent deoxyribonucleotide and S-adenosylmethionine biosyntheses. J. Biol. Chem. 277, 38381–38389 10.1074/jbc.M205000200 [DOI] [PubMed] [Google Scholar]
- 18. Owen O. E., Kalhan S. C., and Hanson R. W. (2002) The key role of anaplerosis and cataplerosis for citric acid cycle function. J. Biol. Chem. 277, 30409–30412 10.1074/jbc.R200006200 [DOI] [PubMed] [Google Scholar]
- 19. Miyazawa H., Yamaguchi Y., Sugiura Y., Honda K., Kondo K., Matsuda F., Yamamoto T., Suematsu M., and Miura M. (2017) Rewiring of embryonic glucose metabolism via suppression of PFK-1 and aldolase during mouse chorioallantoic branching. Development 144, 63–73 10.1242/dev.138545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hunter E. S. 3rd, and Tugman J. A. (1995) Inhibitors of glycolytic metabolism affect neurulation-staged mouse conceptuses in vitro. Teratology 52, 317–323 10.1002/tera.1420520602 [DOI] [PubMed] [Google Scholar]
- 21. Sudiwala S., De Castro S. C. P., Leung K.-Y., Brosnan J. T., Brosnan M. E., Mills K., Copp A. J., and Greene N. D. E. (2016) Formate supplementation enhances folate-dependent nucleotide biosynthesis and prevents spina bifida in a mouse model of folic acid-resistant neural tube defects. Biochimie 126, 63–70 10.1016/j.biochi.2016.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ducker G. S., Chen L., Morscher R. J., Ghergurovich J. M., Esposito M., Teng X., Kang Y., and Rabinowitz J. D. (2016) Reversal of cytosolic one-carbon flux compensates for loss of the mitochondrial folate pathway. Cell Metab. 23, 1140–1153 10.1016/j.cmet.2016.04.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dunlevy L. P., Burren K. A., Mills K., Chitty L. S., Copp A. J., and Greene N. D. (2006) Integrity of the methylation cycle is essential for mammalian neural tube closure. Birth Defects Res. A Clin. Mol. Teratol. 76, 544–552 10.1002/bdra.20286 [DOI] [PubMed] [Google Scholar]
- 24. Matsuda M. (1990) Comparison of the incidence of 5-azacytidine-induced exencephaly between MT/HokIdr and Slc:ICR mice. Teratology 41, 147–154 10.1002/tera.1420410204 [DOI] [PubMed] [Google Scholar]
- 25. Xu L., Wang L., Wang J., Zhu Z., Chang G., Guo Y., Tian X., and Niu B. (2016) The effect of inhibiting glycinamide ribonucleotide formyl transferase on the development of neural tube in mice. Nutr. Metab. (Lond.) 13, 56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hansler A., Chen Q., Gray J. D., Ross M. E., Finnell R. H., and Gross S. S. (2014) Untargeted metabolite profiling of murine embryos to reveal metabolic perturbations associated with neural tube closure defects. Birth Defects Res. A Clin. Mol. Teratol. 100, 623–632 10.1002/bdra.23272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Narisawa A., Komatsuzaki S., Kikuchi A., Niihori T., Aoki Y., Fujiwara K., Tanemura M., Hata A., Suzuki Y., Relton C. L., Grinham J., Leung K.-Y., Partridge D., Robinson A., Stone V., et al. (2012) Mutations in genes encoding the glycine cleavage system predispose to neural tube defects in mice and humans. Hum. Mol. Gen. 21, 1496–1503 10.1093/hmg/ddr585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Leung K.-Y., Pai Y. J., Chen Q., Santos C., Calvani E., Sudiwala S., Savery D., Ralser M., Gross S. S., Copp A. J., and Greene N. D. (2017) Partitioning of one-carbon units in folate and methionine metabolism is essential for neural tube closure. Cell Rep. 21, 1795–1808 10.1016/j.celrep.2017.10.072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Beaudin A. E., Abarinov E. V., Noden D. M., Perry C. A., Chu S., Stabler S. P., Allen R. H., and Stover P. J. (2011) Shmt1 and de novo thymidylate biosynthesis underlie folate-responsive neural tube defects in mice. Am. J. Clin. Nutr. 93, 789–798 10.3945/ajcn.110.002766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Akimova D., Wlodarczyk B. J., Lin Y., Ross M. E., Finnell R. H., Chen Q., and Gross S. S. (2017) Metabolite profiling of whole murine embryos reveals metabolic perturbations associated with maternal valproate-induced neural tube closure defects. Birth Defects Res. 109, 106–119 10.1002/bdra.23583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bachmanov A. A., Reed D. R., Beauchamp G. K., and Tordoff M. G. (2002) Food intake, water intake, and drinking spout side preference of 28 mouse strains. Behav. Genet. 32, 435–443 10.1023/A:1020884312053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Prasannan P., and Appling D. R. (2009) Human mitochondrial C1-tetrahydrofolate synthase: submitochondrial localization of the full-length enzyme and characterization of a short isoform. Arch. Biochem. Biophys. 481, 86–93 10.1016/j.abb.2008.10.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Michalska A. E. (2007) Isolation and propagation of mouse embryonic fibroblasts and preparation of mouse embryonic feeder layer cells. Curr. Protoc. Stem Cell Biol. Chapter 1, Unit 1C.3 [DOI] [PubMed] [Google Scholar]
- 34. Tiziani S., Kang Y., Harjanto R., Axelrod J., Piermarocchi C., Roberts W., and Paternostro G. (2013) Metabolomics of the tumor microenvironment in pediatric acute lymphoblastic leukemia. PLoS ONE 8, e82859 10.1371/journal.pone.0082859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lodi A., Saha A., Lu X., Wang B., Sentandreu E., Collins M., Kolonin M. G., DiGiovanni J., and Tiziani S. (2017) Combinatorial treatment with natural compounds in prostate cancer inhibits prostate tumor growth and leads to key modulations of cancer cell metabolism. NPJ Precis. Oncol. 1, 18 10.1038/s41698-017-0024-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lu X., Solmonson A., Lodi A., Nowinski S. M., Sentandreu E., Riley C. L., Mills E. M., and Tiziani S. (2017) The early metabolomic response of adipose tissue during acute cold exposure in mice. Sci. Rep. 7, 3455 10.1038/s41598-017-03108-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hwang T.-L., and Shaka A. (1995) Water suppression that works. Excitation sculpting using arbitrary wave-forms and pulsed-field gradients. J. Magn. Res. Ser. A 112, 275–279 10.1006/jmra.1995.1047 [DOI] [Google Scholar]
- 38. Gunther U. L., Ludwig C., and Ruterjans H. (2000) NMRLAB: advanced NMR data processing in MATLAB. J. Magn. Reson. 145, 201–208 10.1006/jmre.2000.2071 [DOI] [PubMed] [Google Scholar]
- 39. Ludwig C., and Gunther U. L. (2011) MetaboLab: advanced NMR data processing and analysis for metabolomics. BMC Bioinformatics 12, 366 10.1186/1471-2105-12-366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ludwig C., Easton J. M., Lodi A., Tiziani S., Manzoor S. E., Southam A. D., Byrne J. J., Bishop L. M., He S., Arvanitis T. N., Gunther U. L., and Viant M. R. (2012) Birmingham Metabolite Library: a publicly accessible database of 1-D H-1 and 2-D H-1 J-resolved NMR spectra of authentic metabolite standards (BML-NMR). Metabolomics 8, 8–18 10.1007/s11306-011-0347-7 [DOI] [Google Scholar]
- 41. Wishart D. S., Tzur D., Knox C., Eisner R., Guo A. C., Young N., Cheng D., Jewell K., Arndt D., Sawhney S., Fung C., Nikolai L., Lewis M., Coutouly M. A., Forsythe I., et al. (2007) HMDB: the human metabolome database. Nucleic Acids Res. 35, D521–D526 10.1093/nar/gkl923 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.