

Abstract
Correct disulfide bond formation is essential for proper folding of many proteins, including bacterial virulence factors. The suppressor of copper sensitivity (Scs) proteins have roles in dithiol/disulfide interchange and the bacterial response to copper stress. Encoded in a four-gene cassette (ScsABCD) present in many Gram-negative bacteria, the Scs proteins are enigmatic and poorly characterized. Here, we show that the periplasmic α-domain of the membrane protein ScsB in the Gram-negative bacterium Proteus mirabilis forms a redox relay with the soluble periplasmic protein PmScsC. We also found that the periplasmic α-domain is sufficient to activate the disulfide isomerase activity of PmScsC. The crystal structure of PmScsBα at a resolution of 1.54 Å revealed that it comprises two structurally similar immunoglobulin-like folds, one of which includes a putative redox-active site with the sequence CXXXC. We confirmed the importance of these cysteine residues for PmScsBα function, and in addition, we engineered cysteine variants that produced a stable complex between PmScsC and PmScsBα. Using small-angle X-ray and neutron scattering analyses with contrast variation, we determined a low-resolution structure of the PmScsC–PmScsBα complex. The structural model of this complex suggested that PmScsBα uses both of its immunoglobulin-like folds to interact with PmScsC and revealed that the highly dynamic PmScsC becomes ordered upon PmScsBα binding. These findings add to our understanding of the poorly characterized Scs proteins.
Keywords: protein structure, oxidation-reduction (redox), immunoglobulin-like domain, copper, protein disulfide isomerase, bacterial copper sensitivity, CXXXC active site, thioredoxin fold, disulfide bond, Scs protein
Introduction
The correct formation of disulfide bonds is an essential component in the folding of many proteins, including bacterial virulence factors. In bacteria, disulfide bond–forming (Dsb)5 proteins are responsible for introducing disulfide bonds into substrate proteins (DsbA and -B) (1, 2) as well as reducing and isomerizing (proofreading and shuffling) disulfide bonds that have been incorrectly introduced (DsbC and -D) (3, 4). Dsb proteins function through the redox action of two catalytic cysteines that are often embedded in a thioredoxin fold. These systems are well characterized in Escherichia coli; however, homologues of Dsb proteins exist in a range of Gram-negative bacteria. A group of related and poorly studied proteins found most commonly in Proteobacteria are the suppressor of copper sensitivity (Scs) proteins (5). The Scs proteins contribute to the bacterial virulence trait of resistance to copper stress (6, 7).
Originally identified in Salmonella enterica serovar Typhimurium (6), the scs locus encodes four proteins, ScsA–D, all of which have predicted catalytic motifs consisting of two cysteines (CXXC or CXXXC). Three of these (ScsB–D) incorporate a predicted thioredoxin fold, and one of the four (ScsA) has a predicted copper-binding motif. ScsD has a predicted N-terminal membrane anchor linked to a predicted periplasmic thioredoxin-fold domain (6) reminiscent of proteins involved in cytochrome c biogenesis, such as DsbE/CcmG (8, 9). The best studied of the four Scs proteins is the soluble periplasmic protein ScsC (5, 7, 10). An extraordinary feature is the variation in structure and function of ScsC proteins across different bacteria. Caulobacter crescentus ScsC (CcScsC) is reported to be a dimeric disulfide isomerase (5), S. enterica serovar Typhimurium ScsC (StScsC) is a monomeric redox protein with no disulfide isomerase activity (7), and Proteus mirabilis ScsC (PmScsC) is a highly dynamic “shape-shifting” trimeric disulfide isomerase (10).
The present work focuses on the structure and function of P. mirabilis ScsB, a putative redox partner of P. mirabilis ScsC. ScsB resembles E. coli DsbD in many respects (5). EcDsbD is the membrane protein partner of the archetypal disulfide isomerase E. coli DsbC. Like EcDsbD, the ScsB protein is predicted to comprise three domains: an N-terminal periplasmic domain (DsbDα/ScsBα), a transmembrane domain (DsbDβ/ScsBβ) of eight helices, and a periplasmic C-terminal thioredoxin-fold domain (DsbDγ/ScsBγ) (Fig. 1, A and B). Each of the three domains in both EcDsbD and ScsB have two catalytic cysteine residues. In DsbD, these cysteine pairs in each domain form a reduction cascade that originates from thioredoxin reductase and NADPH in the cytoplasm, is passed through thioredoxin to DsbDβ, DsbDγ, and then DsbDα, which reduces the cysteines in specific substrate proteins, such as DsbC (11, 12) (Fig. 1A). The similarity between the predicted architectures of EcDsbD and ScsB suggests the ScsB protein forms a similar redox relay system. Indeed, C. crescentus ScsB has been shown to interact with CcScsC and to maintain CcScsC in its reduced state, which is necessary for disulfide isomerase activity (5). A major difference between EcDsbD and ScsB is the size of their N-terminal α-domains. DsbDα comprises ∼165 residues, whereas ScsBα is much larger at ∼255 residues, and there is no detectable sequence identity between the α-domains of DsbD and ScsB. Because the α-domain of EcDsbD interacts directly with substrates, such as EcDsbC, the different size and sequence of the ScsB α-domain suggests that the proteins interact with different substrates (5).
Figure 1.
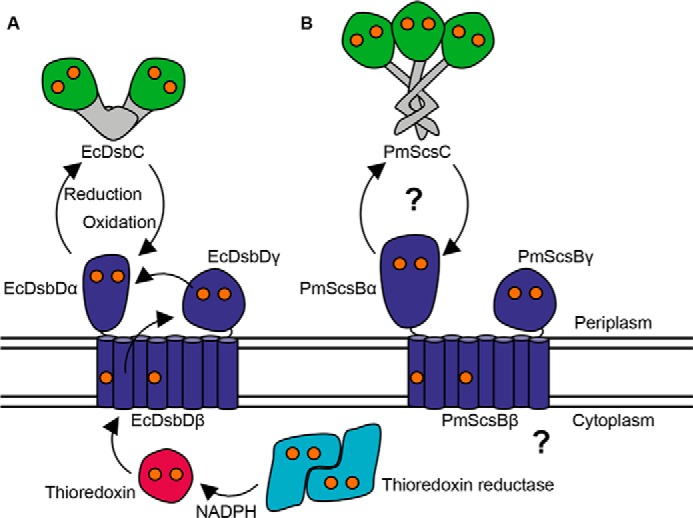
Schematic representation of bacterial disulfide isomerases and their redox relay partners. A, EcDsbC is reduced by EcDsbDα, which obtains its reducing power from a reduction cascade that starts with thioredoxin reductase in the bacterial cytoplasm. B, PmScsC and PmScsBα are putative interaction partners, and the source of PmScsB's reducing power is yet to be determined (represented by the question marks). The catalytic cysteines in each protein are shown as orange spheres.
In the present study, we investigated the structure and function of P. mirabilis ScsBα, showing that it forms a functional redox relay with the highly dynamic, trimeric disulfide isomerase PmScsC. We also report the first crystal structure of any ScsBα, revealing unexpectedly the presence of tandem immunoglobulin folds in PmScsBα and showing that this same arrangement is likely to be shared in ScsBs encoded by other organisms. We created a stable PmScsC–PmScsBα (trimer–monomer) complex and analyzed small-angle X-ray scattering (SAXS) data and small-angle neutron scattering (SANS) data with contrast variation to produce a low-resolution model of the complex. This model revealed that the highly dynamic PmScsC protein becomes more symmetric and ordered upon interaction with PmScsBα and that both immunoglobulin folds of PmScsBα interact with one protomer of PmScsC.
Results
PmScsC and PmScsBα form a specific redox relay system
From sequence analogy with CcScsB and CcScsC, PmScsBα is predicted to be the redox partner of PmScsC. If this functional relationship holds true, PmScsBα would specifically reduce oxidized PmScsC to convert it to the active reduced form. To confirm that a redox relay occurs between PmScsC and PmScsBα, we performed a redox gel-shift assay. First, we mixed PmScsC with PmScsBα and then analyzed the redox states of the two proteins after an incubation period. The redox state was defined by analyzing the proteins by SDS-PAGE after reaction with 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid (AMS) (Fig. 2A). When AMS binds to a reduced thiol, the mass is increased by 0.5 kDa, thus allowing discrimination between reduced and oxidized cysteine forms of a protein on mass-based gel separation. The results showed that when oxidized PmScsC was incubated with reduced PmScsBα, both proteins changed their redox states, confirming that disulfide exchange occurs between the two proteins (Fig. 2B). The reverse redox reaction did not occur: reduced PmScsC remained reduced when mixed with oxidized PmScsBα. Moreover, we were able to confirm the residues involved in the change in redox state because addition of the variant PmScsBα C114A to oxidized PmScsC did not catalyze disulfide exchange. However, there was some evidence for formation of a PmScsC–PmScsBα C114A complex (Fig. 2B, band marked X).
Figure 2.

PmScsBα reduces and activates PmScsC. A, schematic diagrams of the gel-shift and scRNase A experiments. B, a representative gel from the gel-shift assay showing the interaction between PmScsC and PmScsBα. Dotted lines are a visual aide to delineate lanes on the gel. The band marked X is likely to be 1:1 PmScsC–PmScsBα C114A complex as the usually trimeric PmScsC runs as a monomer on SDS-PAGE. C, results from the modified scrambled RNase A assay. Activity is shown relative to native RNase A (100%) and scrambled RNase A only (0%) controls. For each sample, the activity of three or five replicates and the mean with S.D. (error bars) is shown. D, schematic showing the redox interaction that occurs between PmScsC and PmScsBα. red, reduced; ox, oxidized.
Control experiments using PmScsC or PmScsBα alone showed that, under the conditions of the experiment, these proteins did not spontaneously change their redox state. These results support the notion that PmScsC and PmScsBα form a specific redox relay system (Fig. 2B) and that the residues Cys114 and Cys118 of PmScsBα are responsible for the redox interaction.
PmScsBα primes the disulfide isomerase activity of PmScsC
We have previously shown that PmScsC is a disulfide isomerase (10). We tested the hypothesis that the redox interaction between PmScsBα and PmScsC might contribute to this function of PmScsC. We measured the disulfide isomerase activity of PmScsC using scrambled RNase A as substrate (Fig. 2A). Reduced PmScsC, but not oxidized PmScsC, was able to refold scrambled RNase A and thereby activate the enzyme (10) (Fig. 2C). PmScsBα on its own (oxidized or reduced) had no effect on the activity of scrambled RNase A. However, in the presence of reduced PmScsBα, the inactive oxidized PmScsC was able to restore the activity of RNase A. This result supports the hypothesis that PmScsBα reduces the disulfides of oxidized PmScsC, thereby enabling its disulfide isomerase activity (Fig. 2D). This experiment also confirmed that disulfide exchange in this system does not flow in the reverse direction: reduced PmScsC did not lose its ability to shuffle scrambled RNase A disulfides when it is incubated with oxidized PmScsBα. This may suggest that reduced PmScsC is unable to interact with PmScsBα. Furthermore, the cysteine variant in which Cys114 is mutated to Ala (PmScsBα C114A) does not activate oxidized PmScsC, showing that this cysteine is required for the redox relay to operate.
Crystal structure of PmScsBα
After removal of the periplasmic signal sequence, the predicted PmScsB α-domain comprises 255 residues (residues 21–275 of the reported PmScsB protein sequence, UniProt entry B4EV20). The crystal structure of this domain was solved to a resolution of 1.54 Å by using selenomethionine (SeMet)-labeled protein together with SAD phasing (Protein Data Bank code 6C29; Table 1 and Fig. 3, A–D). The structure revealed a monomer consisting of two immunoglobulin-like folds linked by an α-helix. This arrangement was consistent across all three molecules in the asymmetric unit (r.m.s.d., 0.7–1.0 Å, 247 residues aligned). Curiously, the two subdomains, A and B, are reasonably similar in structure (Fig. 3D; r.m.s.d., 2.5 Å, 81 residues aligned), although the sequence identity for such an alignment is very low (∼7%). The subdomains each comprise two β-sheets, one with three β-strands and the other with four β-strands (Fig. 3, A and B). Subdomain A has additional features, including the cysteine motif (114CXXXC118), two β-strands forming a β-hairpin that links the two β-sheets of the canonical immunoglobulin fold, and a loop between β2 and β3 located on one side of the catalytic cysteines (Fig. 3D, arrows). The cysteines are present in the reduced form in all three molecules in the asymmetric unit.
Table 1.
Crystallography statistics for PmScsBα
a/u, asymmetric unit; NA, not applicable.
| Protein Data Bank code 6C29 (SeMet) | |
|---|---|
| Data collection | |
| Wavelength (Å) | 0.9786 |
| Resolution (Å) | 97.06–1.538 (1.544–1.538)a |
| Space group | P212121 |
| Unit cell dimensions | |
| a, b, c (Å) | 69.98, 97.06, 110.20 |
| α, β, γ (°) | 90, 90, 90 |
| No. measured reflections | 1,761,650 |
| No. of unique reflections | 111,511 |
| Rmerge | 0.146 (2.114) |
| Rp.i.m. | 0.038 (0.531) |
| Mean I/σI | 16.0 (2.1) |
| Redundancy | 15.8 (16.3) |
| Completeness (%) | 99.7 (98.3) |
| Wilson B | 15.96 |
| SAD phasing | |
| No. of sites | 14 |
| Figure of merit | 0.42 |
| Refinement | |
| No. of monomers in a/u | 3 |
| Resolution used in refinement (Å) | 55.10–1.538 |
| No. of reflections | 111,388 |
| Rfree (%) | 19.8 |
| Rwork (%) | 17.3 |
| No. of protein atoms | 5,860 |
| No. of ligand atoms | 0 |
| No. of waters | 669 |
| B factors (Å2) | |
| Average | 25.5 |
| Protein atoms | 24.8 |
| Ligands | NA |
| Waters | 31.8 |
| r.m.s.d. bond length (Å) | 0.011 |
| r.m.s.d. bond angles (°) | 1.332 |
| Ramachandran favored/outlier (%) | 97/0 |
a Values in parentheses refer to the highest resolution shell.
Figure 3.

Structural characterization of PmScsBα. A, schematic representation of the crystal structure of PmScsBα (Protein Data Bank code 6C29). B, topology diagram of PmScsBα showing the secondary structure features of the protein. C, the omit electron density around the catalytic loop in chain A of the PmScsBα crystal structure is displayed. The wire mesh represents the likelihood-weighted Fo − Fc electron density difference map, which is contoured to 3σ and was generated using phenix.refine with the region of interest omitted from the structure. D, superimposition of subdomain A and B structures showing the structural similarity between the domains. Arrows indicate the inserted β-hairpin structure and the extended loop of subdomain A. In all panels, subdomain A is colored purple, and subdomain B is magenta with the linking α-helix shown in gray. The catalytic cysteines are represented as orange spheres. The N and C termini are labeled in A and B.
The presence of an immunoglobulin-like fold in both PmScsBα subdomains resulted in many hits in a DALI search against the Protein Data Bank (13). Notably, the top hit was the functionally equivalent E. coli DsbDα (Protein Data Bank code 1JPE; r.m.s.d., 2.5 Å, 103 residues aligned), although it has only one immunoglobulin-like fold, which can be aligned with subdomain A of PmScsBα (Fig. 4A). Like PmScsBα subdomain A, E. coli DsbDα has two catalytic cysteines and a β-hairpin close to the active site, but it does not have the β2-β3 loop of PmScsBα. Two other hits in the DALI search were the mouse γ-adaptin appendage domain from clathrin-binding adaptor AP-1 (Protein Data Bank code 2A7B; r.m.s.d., 2.4 Å, 106 residues aligned) and the GAE domain of the clathrin-binding adaptor GGA protein from Saccharomyces cerevisiae (Protein Data Bank code 3MNM; r.m.s.d., 2.4 Å, 102 residues aligned) (Fig. 4, B and C). These proteins have typical immunoglobulin-like folds with no catalytic cysteines, and like EcDsbDα they have one immunoglobulin fold that aligns with subdomain A of PmScsBα. These three proteins were also hits in a DALI search against PmScsBα subdomain B although with much higher r.m.s.d. values (Protein Data Bank code 1JPE: r.m.s.d., 3.8 Å, 75 residues aligned; Protein Data Bank code 2A7B: r.m.s.d., 3.0 Å, 84 residues aligned; Protein Data Bank code 3MNM: r.m.s.d., 2.9 Å, 86 residues aligned). Curiously, there were no high-scoring DALI hits that had a tandem immunoglobulin-fold arrangement like that of PmScsBα. We did find lower-scoring DALI hits with two immunoglobulin-like folds (mostly antigen-binding fragments of antibodies; e.g. Protein Data Bank code 5JUE; Z-score, 4.6; r.m.s.d., 3.0 Å, 80 residues aligned), but they lacked the connecting α-helix of PmScsBα.
Figure 4.
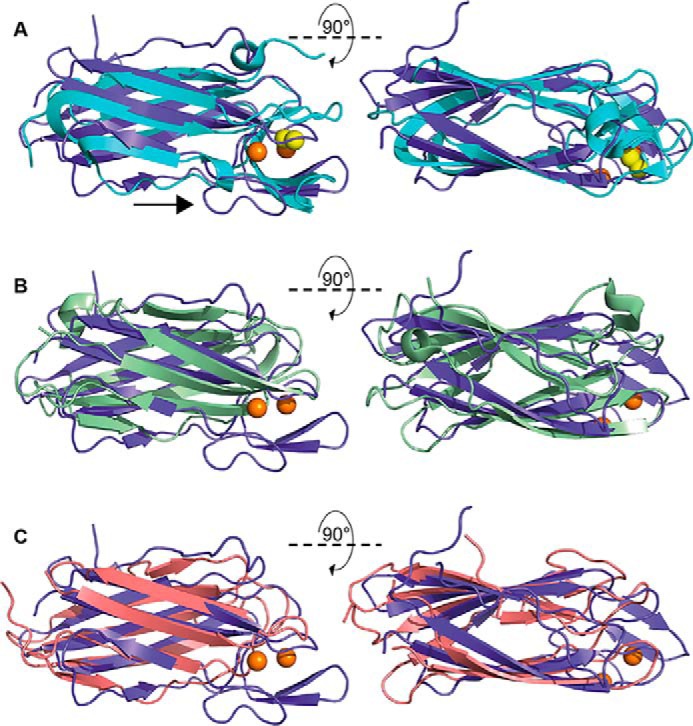
Structural overlay of PmScsBα subdomain A with selected DALI hits. A, PmScsBα subdomain A (purple) aligned with EcDsbDα (Protein Data Bank code 1JPE) (cyan). The catalytic cysteines of both PmScsBα subdomain A and EcDsbDα are shown as spheres and colored in orange and yellow, respectively. An arrow indicates the extended loop in the PmScsBα structure. B, PmScsBα subdomain A (purple) aligned with Protein Data Bank code 2A7B (light green). C, PmScsBα subdomain A (purple) aligned with Protein Data Bank code 3MNM (light pink). Neither 2A7B nor 3MNM have catalytic cysteines so in both B and C the orange spheres correlate to the cysteines of PmScsBα.
Structure-based sequence alignment
The amino acid sequence of PmScsBα was aligned with that of EcDsbDα using structure-based alignment and the sequences of CcScsBα and StScsBα (Fig. 5). ScsBα from S. enterica Typhimurium and C. crescentus were chosen as these are the only other organisms where the interaction partner, ScsC, has been characterized. EcDsbDα was chosen because of the known structural and functional relationship. PmScsBα shares 44% sequence identity with StScsBα (280 residues), 21% identity with CcScsBα (291 residues), and just 12% identity with EcDsbDα (116 residues of Protein Data Bank code 1JPE). There are several conserved residues across the three ScsBα and EcDsbDα protein sequences aside from the catalytic cysteines. Other conserved residues include Leu34 and Ile58 (PmScsBα numbering); their side chains interact with each other in the hydrophobic core of both PmScsBα and EcDsbDα so their conservation suggests a key role in folding or stability. Interestingly, the tyrosine hydroxyl of the conserved residue Tyr45 (Tyr42 in EcDsbDα) is within 3.5–3.8 Å of the active-site sulfurs of Cys114 and Cys118, suggesting a potential role in catalysis or substrate binding. Two other conserved residues, Tyr87 and Pro72, are located at either end of the β-hairpin. The hydroxyl group of Tyr87 is within hydrogen-bonding distance of the main-chain oxygen of Pro72 (∼2.6 Å), suggesting that their conservation may be important in preserving the structure of the β-hairpin. The β2-β3 loop sequence is also highly conserved across the ScsBα sequences, suggesting an important function.
Figure 5.

Sequence alignments. The sequence alignment of the PmScsBα crystal structure with StScsBα and CcScsBα sequences and the structure-based sequence alignment with EcDsbDα crystal structure (Protein Data Bank code 1JPE) are shown. Secondary structure elements of PmScsBα are shown above the corresponding sequence and are colored according to the scheme in Fig. 3. Residues that are conserved between all four proteins are highlighted in red; those that are conserved only between the ScsBα proteins are colored blue.
Formation of a 3:1 PmScsC–PmScsBα complex
The interaction between the catalytic cysteines of PmScsC and PmScsBα is transient. For this reason, the catalytic cysteine variants PmScsC C87S and PmScsBα C114A were produced. These variants allowed the capture of a stable PmScsC–PmScsBα complex for structural studies. Size exclusion chromatography (SEC) and subsequent SDS-PAGE analysis suggested that the predominant complex species had a 3:1 ratio of PmScsC to PmScsBα, corresponding to one PmScsC trimer bound to one PmScsBα monomer (Fig. 6). The predicted molecular mass calculated from SAXS and SANS analysis (Table 2) also provided evidence to support this stoichiometry. A peak corresponding to a larger species was seen on the SEC chromatogram (Fig. 6A, peak marked Z), suggesting that a smaller amount of 3:2 and/or 3:3 complex may have also formed.
Figure 6.
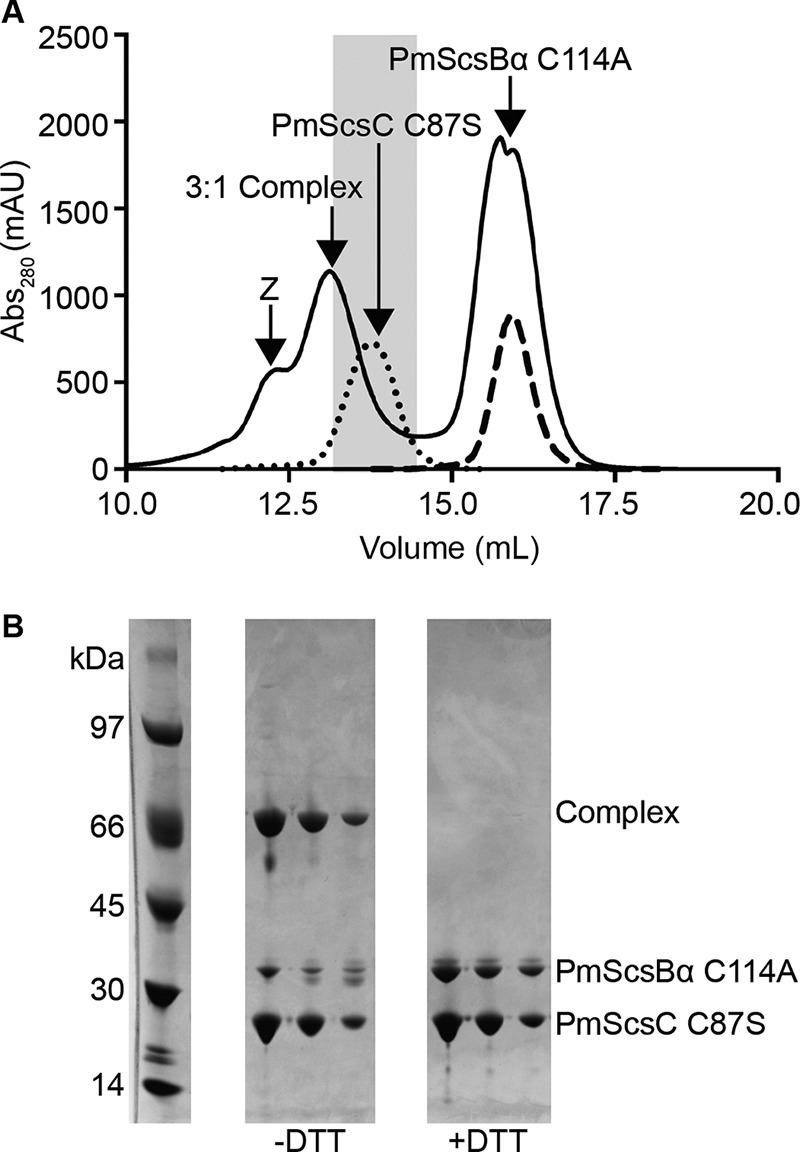
Formation of 3:1 PmScsC C87S–PmScsBα C114A complex. A, SEC chromatographs from the purification of the 3:1 PmScsC–PmScsBα complex (solid line), purified PmScsC C87S (dotted line), and PmScsBα C114A (dashed line). In the purification of the complex, excess PmScsC was removed prior to SEC using an immobilized metal ion affinity chromatography step. The leading shoulder (Z) suggests the presence of a 3:2 and/or 3:3 PmScsC C87S–PmScsBα C114A complex. The shaded region highlights the fractions of the 3:1 PmScsC–PmScsBα peak (solid line) that were pooled for structural analysis. B, the fractions from the shaded region of the 3:1 PmScsC–PmScsBα complex peak run on SDS-PAGE without and with DTT. PmScsC is a trimer that separates and runs as a monomer on SDS-PAGE. The lane labeled −DTT shows that the trimer–monomer complex separates into complex (covalently linked protomer of PmScsC with monomer of PmScsBα; 55.1 kDa) and uncomplexed PmScsC C87S (24.8 kDa) in an approximate 1:1 ratio. When run with DTT (lane labeled +DTT), the higher band dissociates into the two complex components in an approximate 3:1 ratio. Abs, absorbance; mAU, milliabsorbance units.
Table 2.
SAS data collection and analysis details for the PmScsC–PmScsBα complex
ANSTO, Australian Nuclear Science and Technology Organisation; %S, percentage of D2O in the sample; %B, percentage of D2O in the buffer.
| SAXS | SANS | |
|---|---|---|
| Data collection parameters | ||
| Instrument | SAXS-WAXS (Australian Synchrotron) | QUOKKA (ANSTO) |
| Beam geometry | Point | Point |
| Wavelength (Å) | 1.033 | 5.00 |
| Sample to detector distance (m) | 2.680 | 2.000 (short); 10.000 (long) |
| q-range (Å−1) | 0.007–0.35 | 0.03–0.40 (short); 0.01–0.09 (long) |
| Exposure time (s) | 13 (13 × 1-s exposures) | 0%S, 7,200; 20%S, 10,800; 42%S, 21,600; 80%S, 7,200; 100%S, 3,600; 0%B, 10,800; 100%B, 7,200 |
| Measurement type | Concentration series from 96-well plate; 1.0-mm quartz capillary | Neutron contrast variation; Hellma 120-QS 1.0-mm quartz cells |
| Protein concentration range (mg/ml) | 0.56–4.50 | |
| Temperature (K) | 283 | 289 |
| Absolute intensity calibration | Water | Incident beam intensity |
| Sample details | ||
| Extinction coefficient (A280, 0.1%, w/v) | 0.979 | 0.979 |
| Partial specific volume (cm3 g−1) | 0.737 | 0.737 |
| Contrast, Δρ (1010 cm−2) | 2.86 | 3.36 (0%); 2.23 (20%); 1.10 (42%); −1.17 (80%); −2.30 (100%) |
| Molecular mass (from sequence) (kDa) | 105.5 | 105.5 |
| Protein concentration (mg/ml)a | 0.56 | 5.30 (0%S); 5.20 (20%S); 5.10 (42%S); 5.00 (80%S); 4.90 (100%S) |
| Structural parameters | ||
| I(0) (cm−1) (from Guinier) | 0.04060 ± 0.00010 | |
| Rg (Å) (from Guinier) | 39.2 ± 0.2 | |
| I(0) (cm−1) (from p(r)) | 0.04026 ± 0.00010 | 0.5020 ± 0.0019 (0%); 0.2217 ± 0.0013 (20%); 0.0357 ± 0.0007 (42%); 0.0561 ± 0.0008 (80%); 0.2211 ± 0.0010 (100%) |
| Rg (Å) (from p(r)) | 38.7 ± 0.1 | 36.3 ± 0.2 (0%); 34.6 ± 0.3 (20%); 24.3 ± 1.1 (42%); 35.6 ± 0.5 (80%); 37.0 ± 0.2 (100%) |
| Dmax (Å) | 115 ± 3 | 110 ± 3 |
| Porod volume (Å3) | 131,300 ± 7,000 | 116,000 ± 6,000b |
| Molecular mass determination | ||
| Molecular mass from I(0) (kDa) | 97 ± 5 | 95 ± 5b |
| Molecular mass from Porod (kDa) | 108 ± 5 | 95 ± 5 |
a I(0) = 0.0854 ± 0.0002 cm−1, Rg = 39.0 ± 0.1 Å, M = 103 ± 5 kDa (1.13 mg/ml); I(0) = 0.1779 ± 0.0002 cm−1, Rg = 39.0 ± 0.1 Å, M = 107 ± 5 kDa (2.25 mg/ml); I(0) = 0.3476 ± 0.0003 cm−1, Rg = 38.8 ± 0.1 Å, M = 105 ± 5 kDa (4.50 mg/ml). There is a small systematic change in Rg but no systematic change in M; thus, it was deemed that the complex is largely free of concentration-dependent effects up to a concentration of ∼5.0 mg/ml.
b The Porod volume calculation is not valid for systems where the Δρ of the particle is not homogeneous (such as is the case for the neutron contrast variation experiment reported here). Instead, the composite scattering functions (Fig. 8D) were summed together to give the scattering curve of the protein complex with homogeneous contrast (i.e. Ihomogeneous = I11 + I22 + I12), and the Porod volume was determined from this curve. The molecular mass derived from the I(0) was also taken from this curve.
SAXS and SANS provide structural insights into the PmScsC–PmScsBα interaction
SANS contrast variation experiments were performed on the 3:1 PmScsC–PmScsBα complex formed using unlabeled PmScsC C87S and deuterium-labeled PmScsBα C114A (DPmScsBα C114A) (Fig. 7). Deuterium labeling changes the neutron scattering length density of PmScsBα. It is possible to tune the neutron scattering length density of the solvent by changing its deuterium content such that it matches one of the components of the protein complex. This is said to be the match point of that component, and the measured scattering data can then be interpreted as being from the unmatched component of the complex alone. Data collected close to the DPmScsBα C114A match point (100% D2O, where PmScsC C87S dominates the scattering) indicate that the highly dynamic PmScsC protein becomes comparatively rigid and adopts a conformation that, at low-resolution, appears symmetrical upon interaction with PmScsBα. Specifically, the pair-distance distribution function (p(r)) is bimodal (Fig. 8A, green curve) with two well defined peaks and similar to the p(r) generated from the extended symmetrical crystal structure (Protein Data Bank code 5ID4) (Fig. 8A, black dashed curve) (10). An asymmetric arrangement of the PmScsC trimer would yield an asymmetrically shaped second peak in the p(r), whereas significant flexibility would see this same peak broaden.
Figure 7.
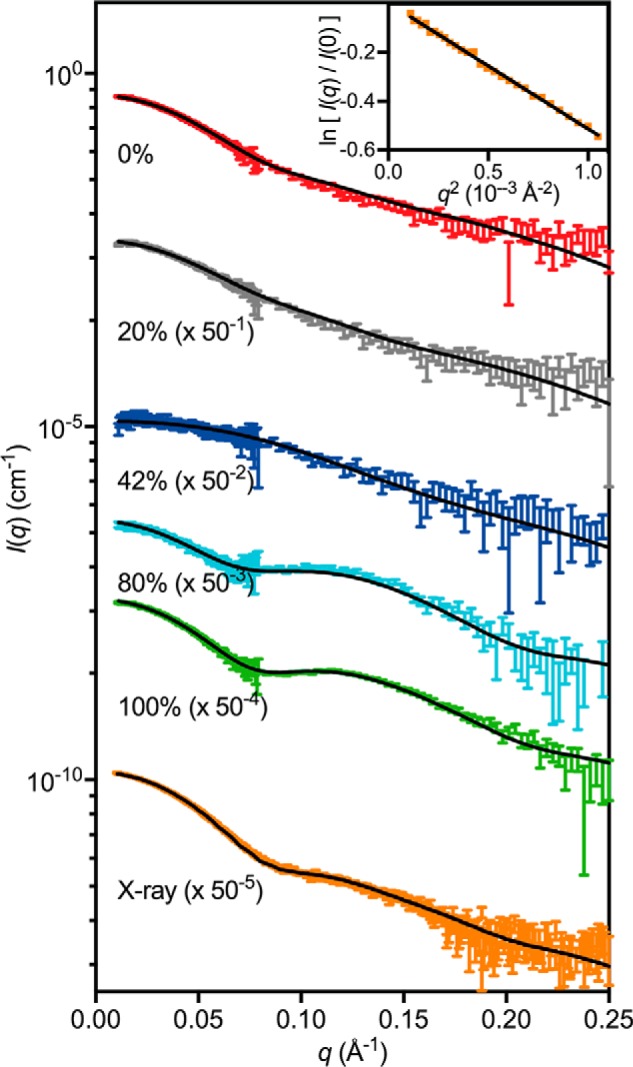
SAXS/SANS scattering curves. SAXS and SANS data (offset for clarity) collected from the 3:1 PmScsC C87S–DPmScsBα C114A complex with the model scattering curves overlaid (solid black line): 0% (χ2 = 1.16; red; on absolute scale), 20% (χ2 = 1.20; gray; offset by a factor of 50−1), 42% (χ2 = 1.00; blue; offset by a factor of 50−2), 80% (χ2 = 1.11; cyan; offset by a factor of 50−3), 100% (χ2 = 0.86; green; offset by a factor of 50−4), and X-ray (χ2 = 1.80; orange; offset by a factor of 50−5). A Guinier plot for the SAXS data is shown in the inset. The match point of PmScsC C87S is ∼42% D2O (blue) where DPmScsBα C114A dominates the scattering, whereas the match point of DPmScsBα C114A is ∼100% D2O (green) where PmScsC C87S dominates the scattering. Error bars represent the S.E.
Figure 8.

Other SAXS/SANS results. A, p(r) derived from the experimental X-ray scattering data (orange) and the 100% scattering data (green), which should approximate the p(r) for PmScsC C87S alone. The p(r) derived from the extended (ext.) crystal structure (Protein Data Bank code 5ID4) is shown as a black dotted line for reference. B, a plot of I(0) normalized by concentration as a function of D2O content of the supporting solvent. The plot is parabolic in shape and reveals that the match point of the entire complex is 59% D2O (vertical dotted line). C, Stuhrmann plot for the 3:1 PmScsC C87S–DPmScsBα C114A complex, conforming to Rg2 = Rm2 + αΔρ−1 − βΔρ−2 where Rm is the radius of gyration of that object with homogenous contrast and α and β are related to the contrast variations within the object. The values obtained from a fit to the plot (solid black line) are: Rm = 37.5 ± 0.4, α = −210 ± 50, and β = 520 ± 100. The negative value of α reveals that the region with higher contrast (i.e. DPmScsBα C114A) lies toward the center of the molecule. Error bars represent the S.E. D, the composite scattering functions determined from the neutron contrast variation data. The I11 curve (green; left y axis) corresponds to scattering from PmScsC C87S, the I22 curve (blue; left y axis) corresponds to scattering from DPmScsBα C114A, and the I12 curve (gray; right y axis) is related to the arrangement of DPmScsBα C114A relative to PmScsC C87S. Ihomogeneous = I11 + I22 + I12 (red; left y axis) is the scattering curve of an object with the same shape as the PmScsC C87S–DPmScsBα C114A complex but with homogeneous contrast and is used for estimating the Porod volume and molecular mass from the SANS data.
Analysis of the SANS data showed that a plot of the scattering intensity at zero angle (I(0)) normalized by protein concentration versus deuterium content of the solvent was well represented by a parabolic function, indicating that the samples were pure (Fig. 8B). A Stuhrmann plot revealed that the DPmScsBα C114A molecule was located toward the center of the complex (Fig. 8C). A rigid body model of the complex was optimized simultaneously against the SAXS data set and five SANS contrast variation data sets. The resulting model provided an excellent fit to all of the scattering data (Fig. 9A). The configuration adopted by PmScsBα and PmScsC in the model precludes the simultaneous binding of two (or three) PmScsBα molecules to PmScsC, helping to explain the preferential formation of a trimer–monomer complex (Fig. 6). The model also highlights two potential interfaces between PmScsC and PmScsBα (Fig. 9A). The most obvious interface is that between chain A of PmScsC and subdomain A of PmScsBα for which the intermolecular disulfide bond is modeled. The resolution of the model is too low to be definitive, but the interface includes residues that could form hydrogen bond interactions. The second interface is between chain A of PmScsC and subdomain B (the second immunoglobulin domain) of PmScsBα.
Figure 9.
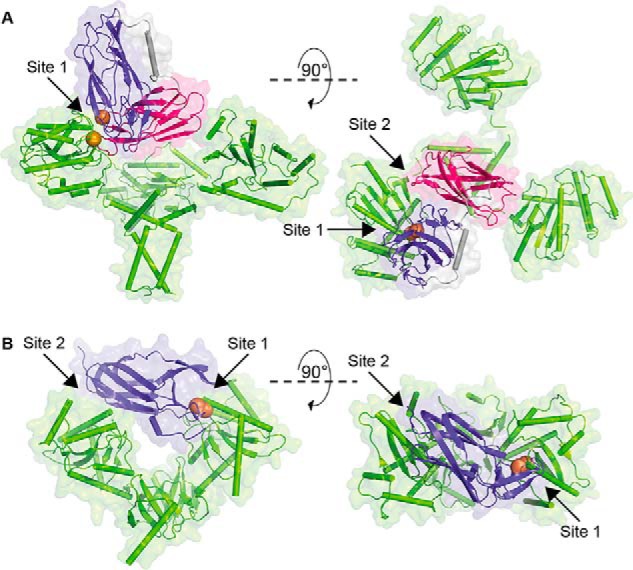
Binding sites in the PmScsC–PmScsBα SANS model and EcDsbC-EcDsbDα structure (Protein Data Bank code 1JZD). A, model of the 3:1 complex generated from the SAXS/SANS data shown in backbone and surface representation. PmScsBα binds to one PmScsC (green) protomer with both subdomains A (purple; Site 1) and B (magenta; Site 2). B, crystal structure of the EcDsbC–EcDbsDα complex. EcDsbDα (purple) binds to both protomers of EcDsbC (green; binding Sites 1 and 2). In both A and B, the catalytic cysteines involved in the intermolecular disulfide bond between the partner proteins are shown as orange spheres.
Discussion
Suppressor of copper sensitivity proteins play a role in the response to copper stress of the important human pathogens S. enterica Typhimurium and P. mirabilis (7, 10). At least two of the four Scs proteins are homologues of Dsb proteins (6), which are essential for the correct formation of disulfide bonds in virulence factors of many Gram-negative bacteria (14–17). P. mirabilis ScsC was recently shown to be a trimeric, highly dynamic disulfide isomerase (10). The present study aimed to characterize PmScsBα, the predicted interaction partner of PmScsC, and to investigate the nature of the interaction between the two.
The domain architecture of full-length ScsB resembles that of E. coli DsbD, the major difference being the size of the periplasmic N-terminal domain. This difference suggested that ScsBα would be structurally distinct from DsbDα perhaps because they interact with different substrates. We confirmed that PmScsC and PmScsBα are redox partners and are therefore functionally similar to EcDsbC and EcDsbDα. We also reported the structure of PmScsBα, showing that it consists of two immunoglobulin-like folds connected by an α-helix. The structures of the two immunoglobulin-like subdomains of PmScsBα are broadly similar with the exception that subdomain A has a catalytic CXXXC motif as well as a β-hairpin and extended loop that both cover the CXXXC active site. The positioning of the β-hairpin and the loop could potentially shield the active site from nonspecific interactions or facilitate interactions with partner proteins. Unexpectedly, we found that domain A of PmScsBα shares the same features of the immunoglobulin fold of EcDsbDα (Fig. 4; r.m.s.d., 2.5 Å, 103 residues aligned) despite a sequence identity of just 12%. The key difference between the structures of subdomain A and EcDsbDα is that EcDsbDα lacks the β2-β3 loop of PmScsBα. The sequence of this β2-β3 loop is conserved in S. enterica Typhimurium and C. crescentus ScsBα, suggesting that it plays a key functional role. A residue that is conserved across the three ScsBαs and EcDsbDα is Tyr45 (PmScsBα numbering). In EcDsbDα, the equivalent Tyr42 hydroxyl is thought to be involved in nucleophilic attack of Cys103, which resolves the intermolecular disulfide formed between EcDsbDα and its substrate (e.g. EcDsbC) (18). The nucleophilic attack in EcDsbDα is proposed to also require Asp68 (18), a residue that is not conserved in Pm-, Cc-, or StScsBα. It is unclear from the PmScsBα structure which residue, if any, might play a role equivalent to that of Asp68.
The discovery of tandem immunoglobulin-like folds in PmScsBα raises the question as to the function of the second or noncatalytic immunoglobulin-like fold. EcDsbDα has a single immunoglobulin-like fold, which, in the E. coli DsbC-DsbDα crystal structure, spans the central cleft of the V-shaped DsbC substrate to interact with both domains of the disulfide isomerase (Fig. 9B) (19). This binding mode is thought to favor binding to dimeric EcDsbC; EcDsbDα is unable to interact with monomeric EcDsbC (3, 19). By comparison, the SANS-derived model of the PmScsC–PmScsBα complex suggests that PmScsBα subdomain A interacts with just one of the three PmScsC protomers in the trimer. Subdomain B of PmScsBα forms a secondary binding site to PmScsC that may stabilize the primary interaction or add specificity (Fig. 9A). Subdomain B could also be required for interactions with other proteins that have yet to be identified.
EcDsbC–EcDsbDα is the redox relay complex formed between dimeric EcDsbC and monomeric EcDsbDα. The crystal structure revealed a 2:1 (rather than a 2:2) complex between the dimer of EcDsbC and a monomer of EcDsbDα (3, 19). On this basis, we hypothesized that PmScsC–PmScsBα would form a 3:1 complex between trimeric PmScsC and monomeric PmScsBα, and the SANS structure of the complex appears to preclude higher stoichiometry. Nevertheless, we did find in vitro evidence suggestive of PmScsC forming 3:2 and 3:3 complexes with PmScsBα (Fig. 6A), showing that different arrangements might form with PmScsBα in the absence of the PmScsB β- and γ-domains. We expect that these higher order complexes would not form in the context of full-length PmScsB.
How do our findings on PmScsBα and PmScsC inform on interactions between ScsC and ScsB in other organisms? A functional interaction between ScsC and ScsB has already been reported for C. crescentus (5). With two examples now confirmed, the ScsB-C redox relay system is likely to be present in all organisms that encode the four-gene scs cassette. However, there is likely to be a wide diversity of interactions. ScsC is reported to be monomeric for StScsC (7), dimeric for CcScsC (5), and trimeric for PmScsC (10). By contrast, ScsB shares similar features across the same organisms, suggesting that StScsBα and CcScsBα both incorporate the tandem immunoglobulin-fold arrangement present in PmScsBα. As indicated above, both subdomains of PmScsBα interact with one PmScsC protomer, suggesting that this tandem immunoglobulin-fold arrangement could facilitate the interaction with ScsCs regardless of the ScsC oligomeric state. Future studies will be needed to establish whether it is the tandem immunoglobulin-fold architecture that permits interactions with monomeric, dimeric, and trimeric PmScsCs. Curiously, a BLAST search of E. coli DsbD or S. enterica Typhimurium DsbD against the P. mirabilis genome did not identify any hits, suggesting the possibility that PmScsB may also act as the redox partner for P. mirabilis DsbC proteins.
Experimental procedures
Sequence analysis
The TMHMM Server (version 2.0) was used to determine the region of the ScsB proteins that formed the N-terminal periplasmic domain. EMBOSS Needle (20, 21) with residues 1–275, 1–279, and 1–290 of UniProt entries B4EV20 (PmScsB), AAL20046 (StScsB), and Q9ABL0 (CcScsB), respectively, was used to calculate the sequence identities reported among these proteins. The DALI sever (13) was used to calculate r.m.s.d. values and structure-based sequence identities. Coot (22) was used to create visuals of the structural alignments. The sequences of all four proteins were aligned using PROMALS3D (23) with the Protein Data Bank files of PmScsBα and EcDsbDα (code 1JPE) instead of the sequence alone. Residue numbers quoted in the text were taken from the deposited Protein Data Bank files for the PmScsC and PmScsBα protein structures, which are numbered based on the tobacco etch virus protease–cleaved protein sequence (starting S1N2A3 …).
Molecular biology
A codon-optimized gene encoding P. mirabilis ScsBα without the predicted signal peptide (UniProt entry B4EV20, residues 21–275; Integrated DNA Technologies) was inserted into pMCSG7 (24) using ligation-independent cloning. The PmScsC (UniProt entry C2LPE2/B4EV21) construct used throughout this study was created previously (10). The constructs for the variants PmScsC C87S and PmScsBα C114A were created using the QuikChange® Lightning site-directed mutagenesis kit (Agilent Technologies). The WT PmScsC and PmScsBα constructs were used as templates, and primers 5′-gcggatcgaaacgtttagagtacgggcaattatag-3′, 5′-ctataattgcccgtactctaaacgtttcgatccgc-3′, 5′-gtcagaatgcatacattgctagcggtactcaatgtaaggacacc-3′, and 5′-ggtgtccttacattgagtaccgctagcaatgtatgcattctgac-3′, respectively, were used to introduce the point mutations. PmScsBα was also subcloned into pET24a for deuterated bioreactor expression using restriction enzymes EcoRI and NdeI. All constructs contained an N-terminal His6 tag followed by a tobacco etch virus protease cleavage site and were confirmed by sequencing.
Protein production
Except for the selenomethionine-labeled PmScsBα and deuterated PmScsBα C114A, all proteins were expressed and purified as outlined previously (10). The redox state of the cysteines was confirmed spectrophotometrically with 5,5′-dithiobis(2-nitrobenzoic acid) (Ellman's reagent) as described previously (25). Yields for PmScsC and PmScsBα derivatives were around 80 and 40 mg/liter of culture (∼15 g cell weight harvested), respectively. SDS-PAGE (NuPAGE® system, 4–12% BisTris gel, Invitrogen) with Coomassie Blue stain was used to assess the protein quality.
Expression of SeMet-labeled PmScsBα
To express SeMet-labeled PmScsBα, needed for experimental phasing in crystallographic structure determination, a 10-ml starter culture of E. coli BL21 (DE3) pLysS cells containing pMCSG7-PmScsBα was grown in LB medium supplemented with the appropriate antibiotics at 37 °C for 4–5 h at 220 rpm. This culture was then used to inoculate 1 liter of M9 minimal medium supplemented with antibiotics and amino acids (100 mg of l-methionine, l-lysine, l-threonine, and l-phenylalanine and 50 mg of l-leucine, l-isoleucine, and l-valine) and then incubated overnight at 37 °C at 220 rpm. Cells were harvested from the overnight culture, washed with 1 liter of sterile water, and resuspended in water to an A600 of ∼1. M9 minimal medium containing antibiotics but no amino acids was then inoculated with the cells to a final A600 of 0.1. The cells were cultured at 30 °C at 220 rpm for 6–8 h until an A600 of 0.5 was reached. Amino acids, including 100 mg of l-selenomethionine instead of l-methionine, were then added to the culture flasks, and after 15 min at 30 °C isopropyl 1-thio-β-d-galactopyranoside was added to a final concentration of 1 mm to induce expression. The cultures were incubated at 30 °C at 220 rpm overnight. Cells were harvested, and the protein was purified as described previously (10) with the addition of 5 mm DTT to all purification buffers. The yield of SeMet-labeled PmScsBα was 5 mg/liter of culture (∼5 g of cell pellet).
Expression of deuterated PmScsBα C114A
Deuterated PmScsBα C114A was expressed for SANS analysis of the complex between PmScsC C87S and PmScsBα C114A. The deuterated protein PmScsBα C114A was produced at the National Deuteration Facility, Australian Nuclear Science and Technology Organisation in 1 liter of “ModC1” medium (26) containing 90% (v/v) D2O and unlabeled glycerol (40 g/liter). Miniprep plasmid DNA “pET24a-PmScsBα C114A” was used to transform 50 μl of Invitrogen competent BL21*(DE3) cells, which were then incubated with 250 μl of SOC medium for 2 h. The 300-μl culture was added to 10 ml of ModC1 medium containing 50% D2O and 40 μg/ml kanamycin, shaking at 200 rpm at 37 °C. After 16 h (A600, 1.2), 9 ml was added to 36 ml of fresh medium with kanamycin at 100% D2O (i.e. 45 ml at 90% D2O). After another 5 h (A600, 0.9), the culture was diluted with fresh 90% D2O medium to 102 ml. After another 5 h, 100 ml (A600, 0.87) was used to inoculate 900 ml of fresh 90% D2O medium with kanamycin in a 2-liter Real Time Engineering bioreactor aerated with air at 0.5 liter/min and with pH held to a minimum of 6.2 by addition of 28% NH4OH in H2O (Sigma). Dissolved oxygen tension was set to 75% and was above 60% throughout. At an A600 of 14.2, the temperature was lowered to 20 °C, and expression was induced by the addition of 1 mm isopropyl 1-thio-β-d-galactopyranoside. At 17 h postinduction, with an A600 of 32, the culture was harvested with a wet weight yield of 79.4 g and with excellent expression as evident by SDS-PAGE. The deuteration level was determined by partial trypsin digest MALDI-TOF comparison of unlabeled and labeled samples and was found to be 70.8%. Deuterated PmScsBα C114A was purified as described for the unlabeled protein except the His6 tag was not cleaved as it was needed for the purification of the complex. The yield of purified deuterated PmScsBα was ∼240 mg/liter of bioreactor culture (∼40 g of cell pellet).
Determination of protein concentration
The reported protein concentrations were determined using the A280 of the sample (read using a Thermo Scientific NanoDrop 2000c spectrophotometer) and calculated extinction coefficients from ProtParam (27).
Crystal structure of PmScsBα
A number of conditions from commercial screens resulted in PmScsBα crystals. These conditions were replicated in 24-well plates, and the best diffracting SeMet-labeled PmScsBα crystals were obtained in a drop containing 1 μl of 43 mg/ml protein in 10 mm HEPES, pH 7.4, 5 mm DTT and 1 μl of 0.1 m MES monohydrate, pH 6, 6% (v/v) Tacsimate, pH 6, 24% PEG 4000. A commercial additive screen (Hampton Research) was used to optimize crystals, and those grown over 2 days to ∼400 μm with the addition of 4% 2,5-hexanediol or 4% 1,3-butanediol resulted in the best diffraction. Crystals were cryoprotected in the crystallization condition plus 20% glycerol and flash frozen in liquid nitrogen.
Because of the lack of a suitable model for molecular replacement, experimental phasing was required to solve the crystal structure of PmScsBα. A fluorescence scan was used to determine the absorption edge of SeMet-labeled crystals, and a data set (Table 1) was collected near the peak absorption wavelength (0.9786 Å) at 100 K. Data processing in autoPROC (28), which utilizes XDS (29) and AIMLESS (30), determined the space group to be P212121. SAD phasing using the SHELX workflow (31) in HKL2MAP (32) found 14 selenium sites with an occupancy >0.2. We expected only 12 selenium sites; the extra two sites were positioned very close to other sites, suggesting alternate conformations, although only one SeMet was modeled with an alternate conformation in the final structure. SAD phasing resulted in an initial incomplete alanine model of the protein structure (figure of merit, 0.42). This initial model was then used for molecular replacement against the same data in Phaser (33) in the CCP4 suite (34). Two rounds of automated building and refinement using Buccaneer (35) with REFMAC (36), as implemented in CCP4 (34), completed the structure, revealing three molecules in the asymmetric unit. Then several rounds of manual adjustment in Coot (22) and refinement in PHENIX (37) were performed with reference to the validation program MolProbity (38). Multiple residues in the crystal structure have alternate conformers, and occupancies of the selenium atoms in the SeMet residues were all <1.0. The electron density for some loop regions is poor in chains B and C, particularly residues 79–82, 99–100, and 51–52 (chain B only). R/Rfree values are 17.3 and 19.8% at a resolution of 1.54 Å. All structural figures were made in PyMOL (39).
Redox interaction assay
To confirm the redox interaction between PmScsC and PmScsBα, oxidized PmScsC and reduced PmScsBα were incubated together at final concentrations of 10 μm in 100 mm sodium phosphate, pH 7, 1 mm EDTA for 2 h. As controls, oxidized and reduced forms of both proteins, a sample containing reduced PmScsC and oxidized PmScsBα, and a sample with oxidized PmScsC with PmScsBα C114A were also incubated for 2 h. All samples were then treated with 10% TCA to precipitate the protein, and after centrifugation the pellets were washed with 100% cold acetone and dried. The protein pellets were resuspended in a solution of 200 μm AMS in 1% SDS, 50 mm Tris, pH 7.0. AMS binds to the reduced cysteines of proteins, increasing the molecular weight and allowing the differentiation between the oxidized and reduced forms of a protein when run on a gel. Therefore, all samples were run on a nonreducing, nondenaturing 12% NuPAGE BisTris gel for 2.5 h at 100 V. The gel was Coomassie-stained and imaged using an Odyssey® Fc imaging system (LI-COR® Biosciences) with the 700-nm channel.
RNase A isomerase assay
To determine whether PmScsBα activates the isomerase PmScsC, an isomerase assay utilizing scrambled RNase A (scRNase A) and the RNase A model substrate cCMP was performed. Bovine RNase A (purchased from Sigma-Aldrich) was scrambled as described previously (40). The various oxidized and reduced protein samples described above for the gel-shift redox interaction assay were again prepared but with the addition of 40 μm scRNase A to each sample. After 2 h, 50 μl of the protein/scRNase A reactions was taken and added to 150 μl of 4 mm cCMP in a UV-Star 96-well plate (Greiner Bio-One, Austria). The increase in absorbance at 296 nm was monitored over 3 min with a Synergy H1 multimode plate reader (BioTek) and used to calculate the rate of cCMP hydrolysis. Native RNase A– and scRNase A–only samples were included as controls and used to determine the relative activity of the RNase A in each sample. The results shown are the average and S.D. of three technical replicates.
Complex formation
The PmScsC and PmScsBα cysteine variants were designed following the same approach used for the EcDsbC–EcDsbDα interaction (19). Mutation of cysteines in EcDsbC and EcDsbDα resulted in a stable complex between the two proteins (3). Cysteines Cys87 of PmScsC and Cys114 of PmScsBα are the least accessible of the two cysteines in each active site, suggesting they facilitate resolution of intermolecular disulfide bonds. Variants PmScsC C87S and PmScsBα C114A were therefore prepared and used in complex formation studies.
The 3:1 PmScsC C87S–PmScsBα C114A complex was formed by mixing the two proteins together in a 3:1 ratio at concentrations of ∼200 mg/ml and incubating the mixture at 37 °C for 3 days. To separate the complex and free PmScsBα from PmScsC, PmScsBα was left His-tagged, and the protein mixture was then run over 2 ml of fresh TALON resin twice. Columns were washed with 25 mm Tris, pH 7.4, 150 mm NaCl, and the complex and free PmScsBα were eluted with 25 mm Tris, pH 7.4, 150 mm NaCl, 250 mm imidazole. This was then run over a Superdex 200 10/300 Increase size exclusion column on an ÅKTA system to separate PmScsBα from complex. A peak on SEC eluted earlier than for ScsC, suggesting that a complex between the two proteins had formed (Fig. 6A). Running the fractions from the peak corresponding to the 3:1 complex on nonreducing SDS-PAGE resulted in three bands (Fig. 6B). The lower bands correlated to free PmScsC C87S (24.8 kDa) and a small amount of PmScsBα C114A (30.3 kDa), and the upper band corresponded to the molecular mass of one PmScsC C87S molecule bound to one PmScsBα C114A molecule (55.1 kDa). The upper band dissociated when reducing agent was added, leaving two bands correlating to monomeric PmScsC C87S and PmScsBα C114A in an approximate 3:1 ratio (Fig. 6B). There was also evidence for the formation of small amounts of complex containing PmScsC C87S and PmScsBα C114A in ratios of 3:3 and 3:2, but only those SEC fractions containing 3:1 complex were collected for further analysis. The complex formation process was repeated multiple times to gain enough sample for SAXS/SANS analysis as the overall yield of 3:1 complex was ∼2.5%.
Small-angle solution scattering
Small-angle neutron scattering data shown in Table 2 were collected on the QUOKKA instrument at the Australian Nuclear Science and Technology Organisation (Table 2) (41). Two aliquots of PmScsC C87S–DPmScsBα C114A complex at ∼5 mg/ml were dialyzed, one against a buffer with a D2O content of 0% and the other against a buffer with a D2O content of ∼100%. Samples with D2O contents of 20, 42, and 80% were obtained by mixing the two original solutions at the appropriate ratios. The two-dimensional data were normalized to a common incident neutron count and corrected for sample transmission, background radiation, empty cell scattering, and detector sensitivity and radially averaged to produce I(q) versus q profiles. Scattering data from the two different sample–detector distances were then merged, and buffer scattering data were then subtracted from the protein + buffer data to give the protein scattering profiles (the 20, 42, and 80% buffer scattering curves were taken as a linear combination of the 0 and 100% buffer scattering curves). To correct for the effects of incoherent scattering by 1H-rich samples, backgrounds levels were adjusted by a small constant such that the high-q scattering displayed q−4 dependence.
Small-angle X-ray scattering data were collected on the SAXS-WAXS beamline at the Australian Synchrotron (Table 2) (42). Serial dilutions of the stock solution of PmScsC C87S–DPmScsBα C114A complex were made to give four concentrations between 0.55 and 4.5 mg/ml that were centrifuged at 10,000 × g immediately before loading into a 96-well plate. To reduce radiation damage, samples (∼100 μl) were drawn into a capillary from the 96-well plate and flowed past the beam. All measured two-dimensional data were averaged and corrected for sample transmission, solvent scattering, and detector sensitivity and radially averaged to produce I(q) versus q profiles using Scatterbrain (version 2.7.1) (43).
The estimated molecular masses were calculated using values for contrast and partial specific volume predicted from the protein sequence using MULCh (version 1.1) (44). Data processing and Guinier analysis were performed using PRIMUS (version 3.2) (45). The p(r) was generated from the experimental data using GNOM (version 4.6) (4) from which I(0), Rg, and Dmax were determined. For the SANS data, analysis of the dependence of Rg upon contrast and calculation of the composite scattering functions were also calculated using MULCh (Fig. 8D). Corefinement of a rigid-body model against SANS and SAXS data was performed using SASREF (version 7.d) (46) where the crystal structure of PmScsC (Protein Data Bank code 4XVW, chain A) was broken into three rigid sections: 1) residues Ala3–Gln43 were fixed to preserve the structure of the trimerization domain observed in the crystal structure, 2) residues Gln44–Phe47 were defined as a short linker with a restraint between residues Gln43 and Gln44 to keep them in close proximity, and 3) residues Arg48–Lys224 were defined as the catalytic domain free to move with a restraint between residues Phe47 and Arg48 to keep them in close proximity. The additional PmScsC chains were generated by symmetry (assuming a C3 point group). The crystal structure of PmScsBα (residues Ala2–Ala248) was included in the optimization with a distance restraint between Cys84 of a PmScsC monomer and Cys118 of PmScsBα of 8 Å. Thus, although the complex overall is not symmetrical, the PmScsC portion of the model possesses 3-fold symmetry. The optimization was run 16 times, and the model that provided the best global fit to the scattering data is reported here; however, all models obtained are similar in structure.
Author contributions
E. J. F., F. K., and J. L. M. conceptualization; E. J. F., A. E. W., and J. L. M. data curation; E. J. F., F. K., and A. E. W. formal analysis; E. J. F., A. E. W., and J. L. M. validation; E. J. F., H. G. C., F. K., and A. E. W. investigation; E. J. F. and A. E. W. visualization; E. J. F., H. G. C., F. K., A. P. D., and A. E. W. methodology; E. J. F. writing-original draft; E. J. F., H. G. C., F. K., A. P. D., A. E. W., and J. L. M. writing-review and editing; H. G. C., F. K., A. E. W., and J. L. M. supervision; F. K., A. P. D., A. E. W., and J. L. M. resources; J. L. M. funding acquisition; J. L. M. project administration.
Acknowledgments
We acknowledge the support of the Australian Centre for Neutron Scattering, the National Deuteration Facility, and the SAXS-WAXS and MX beamlines at the Australian Synchrotron (Australian Nuclear Science and Technology Organisation), each partly supported by the National Collaborative Research Infrastructure Strategy – an initiative of the Australian Government, in providing the facilities used in this work. We thank the staff and facilities of the University of Queensland Remote Operation Crystallisation and X-ray (UQ ROCX) facility for support. We also acknowledge access to the University of Sydney Mass Spectrometry Core Facility, which was used to produce partial trypsin digest MALDI-TOF spectra. We thank Dr. Róisín M. McMahon and Signe Christensen for helpful discussions.
This work was supported by Australian Research Council Laureate Fellowship FL0992138 (to J. L. M.), Australian Research Council Discovery Project DP130100576 (to J. L. M.; H. G. C. salary), an Australian postgraduate award (to E. J. F.), and an Institute for Molecular Bioscience research advancement award (to E. J. F.). The authors declare that they have no conflicts of interest with the contents of this article.
This article was selected as one of our Editors' Picks.
The atomic coordinates and structure factors (code 6C29) have been deposited in the Protein Data Bank (http://wwpdb.org/).
SAXS/SANS data and the model of the 3:1 PmScsC–PmScsBα complex have been submitted to the Small Angle Scattering Biological Data Bank (SASBDB) under ID SASDC48.
- Dsb
- disulfide bond–forming
- Scs
- suppressor of copper sensitivity
- Pm
- P. mirabilis
- Cc
- C. crescentus
- St
- S. enterica serovar Typhimurium
- Ec
- E. coli
- r.m.s.d.
- root mean square deviation
- SAXS
- small-angle X-ray scattering
- SANS
- small-angle neutron scattering
- WAXS
- wide-angle X-ray scattering
- AMS
- 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid
- SAD
- single-wavelength anomalous diffraction
- SEC
- size exclusion chromatography
- DPmScsBα C114A
- deuterium-labeled PmScsBα C114A
- p(r)
- pair-distance distribution function
- I(0)
- scattering intensity at zero angle
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- scRNase A
- scrambled RNase A
- Rg
- radius of gyration.
References
- 1. Martin J. L., Bardwell J. C., and Kuriyan J. (1993) Crystal structure of the DsbA protein required for disulfide bond formation in vivo. Nature 365, 464–468 10.1038/365464a0 [DOI] [PubMed] [Google Scholar]
- 2. Inaba K., Murakami S., Suzuki M., Nakagawa A., Yamashita E., Okada K., and Ito K. (2006) Crystal structure of the DsbB-DsbA complex reveals a mechanism of disulfide bond generation. Cell 127, 789–801 10.1016/j.cell.2006.10.034 [DOI] [PubMed] [Google Scholar]
- 3. Goldstone D., Haebel P. W., Katzen F., Bader M. W., Bardwell J. C., Beckwith J., and Metcalf P. (2001) DsbC activation by the N-terminal domain of DsbD. Proc. Natl. Acad. Sci. U.S.A. 98, 9551–9556 10.1073/pnas.171315498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. McCarthy A. A., Haebel P. W., Törrönen A., Rybin V., Baker E. N., and Metcalf P. (2000) Crystal structure of the protein disulfide isomerase, DsbC, from Escherichia coli. Nat. Struct. Biol. 7, 196–199 10.1038/73295 [DOI] [PubMed] [Google Scholar]
- 5. Cho S. H., Parsonage D., Thurston C., Dutton R. J., Poole L. B., Collet J. F., and Beckwith J. (2012) A new family of membrane electron transporters and its substrates, including a new cell envelope peroxiredoxin, reveal a broadened reductive capacity of the oxidative bacterial cell envelope. mBio 3, e00291–11 10.1128/mBio.00291-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gupta S. D., Wu H. C., and Rick P. D. (1997) A Salmonella typhimurium genetic locus which confers copper tolerance on copper-sensitive mutants of Escherichia coli. J. Bacteriol. 179, 4977–4984 10.1128/jb.179.16.4977-4984.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shepherd M., Heras B., Achard M. E., King G. J., Argente M. P., Kurth F., Taylor S. L., Howard M. J., King N. P., Schembri M. A., and McEwan A. G. (2013) Structural and functional characterization of ScsC, a periplasmic thioredoxin-like protein from Salmonella enterica serovar Typhimurium. Antioxid. Redox Signal. 19, 1494–1506 10.1089/ars.2012.4939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Edeling M. A., Ahuja U., Heras B., Thöny-Meyer L., and Martin J. L. (2004) The acidic nature of the CcmG redox-active center is important for cytochrome c maturation in Escherichia coli. J. Bacteriol. 186, 4030–4033 10.1128/JB.186.12.4030-4033.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Edeling M. A., Guddat L. W., Fabianek R. A., Thöny-Meyer L., and Martin J. L. (2002) Structure of CcmG/DsbE at 1.14 Å resolution: high-fidelity reducing activity in an indiscriminately oxidizing environment. Structure 10, 973–979 10.1016/S0969-2126(02)00794-3 [DOI] [PubMed] [Google Scholar]
- 10. Furlong E. J., Lo A. W., Kurth F., Premkumar L., Totsika M., Achard M. E. S., Halili M. A., Heras B., Whitten A. E., Choudhury H. G., Schembri M. A., and Martin J. L. (2017) A shape-shifting redox foldase contributes to Proteus mirabilis copper resistance. Nat. Commun. 8, 16065 10.1038/ncomms16065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Krupp R., Chan C., and Missiakas D. (2001) DsbD-catalyzed transport of electrons across the membrane of Escherichia coli. J. Biol. Chem. 276, 3696–3701 10.1074/jbc.M009500200 [DOI] [PubMed] [Google Scholar]
- 12. Collet J. F., Riemer J., Bader M. W., and Bardwell J. C. (2002) Reconstitution of a disulfide isomerization system. J. Biol. Chem. 277, 26886–26892 10.1074/jbc.M203028200 [DOI] [PubMed] [Google Scholar]
- 13. Holm L., and Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 10.1093/nar/gkq366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ireland P. M., McMahon R. M., Marshall L. E., Halili M., Furlong E., Tay S., Martin J. L., and Sarkar-Tyson M. (2014) Disarming Burkholderia pseudomallei: structural and functional characterization of a disulfide oxidoreductase (DsbA) required for virulence in vivo. Antioxid. Redox Signal. 20, 606–617 10.1089/ars.2013.5375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Heras B., Shouldice S. R., Totsika M., Scanlon M. J., Schembri M. A., and Martin J. L. (2009) DSB proteins and bacterial pathogenicity. Nat. Rev. Microbiol. 7, 215–225 10.1038/nrmicro2087 [DOI] [PubMed] [Google Scholar]
- 16. Miki T., Okada N., and Danbara H. (2004) Two periplasmic disulfide oxidoreductases, DsbA and SrgA, target outer membrane protein SpiA, a component of the Salmonella pathogenicity island 2 type III secretion system. J. Biol. Chem. 279, 34631–34642 10.1074/jbc.M402760200 [DOI] [PubMed] [Google Scholar]
- 17. Totsika M., Heras B., Wurpel D. J., and Schembri M. A. (2009) Characterization of two homologous disulfide bond systems involved in virulence factor biogenesis in uropathogenic Escherichia coli CFT073. J. Bacteriol. 191, 3901–3908 10.1128/JB.00143-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Goulding C. W., Sawaya M. R., Parseghian A., Lim V., Eisenberg D., and Missiakas D. (2002) Thiol-disulfide exchange in an immunoglobulin-like fold: structure of the N-terminal domain of DsbD. Biochemistry 41, 6920–6927 10.1021/bi016038l [DOI] [PubMed] [Google Scholar]
- 19. Haebel P. W., Goldstone D., Katzen F., Beckwith J., and Metcalf P. (2002) The disulfide bond isomerase DsbC is activated by an immunoglobulin-fold thiol oxidoreductase: crystal structure of the DsbC-DsbDα complex. EMBO J. 21, 4774–4784 10.1093/emboj/cdf489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rice P., Longden I., and Bleasby A. (2000) EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 16, 276–277 10.1016/S0168-9525(00)02024-2 [DOI] [PubMed] [Google Scholar]
- 21. Li W., Cowley A., Uludag M., Gur T., McWilliam H., Squizzato S., Park Y. M., Buso N., and Lopez R. (2015) The EMBL-EBI bioinformatics web and programmatic tools framework. Nucleic Acids Res. 43, W580–W584 10.1093/nar/gkv279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 23. Pei J., Kim B. H., and Grishin N. V. (2008) PROMALS3D: a tool for multiple protein sequence and structure alignments. Nucleic Acids Res. 36, 2295–2300 10.1093/nar/gkn072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stols L., Gu M., Dieckman L., Raffen R., Collart F. R., and Donnelly M. I. (2002) A new vector for high-throughput, ligation-independent cloning encoding a tobacco etch virus protease cleavage site. Protein Expr. Purif. 25, 8–15 10.1006/prep.2001.1603 [DOI] [PubMed] [Google Scholar]
- 25. Walden P. M., Halili M. A., Archbold J. K., Lindahl F., Fairlie D. P., Inaba K., and Martin J. L. (2013) The α-proteobacteria Wolbachia pipientis protein disulfide machinery has a regulatory mechanism absent in γ-proteobacteria. PLoS One 8, e81440 10.1371/journal.pone.0081440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Duff A. P., Wilde K. L., Rekas A., Lake V., and Holden P. J. (2015) Robust high-yield methodologies for 2H and 2H/15N/13C labeling of proteins for structural investigations using neutron scattering and NMR. Methods Enzymol. 565, 3–25 10.1016/bs.mie.2015.06.014 [DOI] [PubMed] [Google Scholar]
- 27. Gasteiger E., Hoogland C., Gattiker A., Duvaud S., Wilkins M. R., Appel R. D., and Bairoch A. (2005) Protein identification and analysis tools on the ExPASy server, in The Proteomics Protocols Handbook (Walker J. M., ed) pp. 571–607, Humana Press, New York [Google Scholar]
- 28. Vonrhein C., Flensburg C., Keller P., Sharff A., Smart O., Paciorek W., Womack T., and Bricogne G. (2011) Data processing and analysis with the autoPROC toolbox. Acta Crystallogr. D Biol. Crystallogr. 67, 293–302 10.1107/S0907444911007773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 12 10.1107/S09074449090473375–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Evans P. R., and Murshudov G. N. (2013) How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 69, 1204–1214 10.1107/S0907444913000061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sheldrick G. M. (2010) Experimental phasing with SHELXC/D/E: combining chain tracing with density modification. Acta Crystallogr. D Biol. Crystallogr. 66, 479–485 10.1107/S0907444909038360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pape T., and Schneider T. R. (2004) HKL2MAP: a graphical user interface for macromolecular phasing with SHELX programs. J. Appl. Crystallogr. 37, 843–844 10.1107/S0021889804018047 [DOI] [Google Scholar]
- 33. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 10.1107/S0907444910045749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cowtan K. (2006) The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr. D Biol. Crystallogr. 62, 1002–1011 10.1107/S0907444906022116 [DOI] [PubMed] [Google Scholar]
- 36. Vagin A. A., Steiner R. A., Lebedev A. A., Potterton L., McNicholas S., Long F., and Murshudov G. N. (2004) REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr. D Biol. Crystallogr. 60, 2184–2195 10.1107/S0907444904023510 [DOI] [PubMed] [Google Scholar]
- 37. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 10.1107/S0907444909042073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. DeLano W. L. (2015) The PyMOL Molecular Graphics System, version 1.6, Schrödinger, LLC, New York [Google Scholar]
- 40. Hillson D. A., Lambert N., and Freedman R. B. (1984) Formation and isomerisation of disulfide bonds in proteins: protein disulfide-isomerase. Methods Enzymol. 107, 281–294 10.1016/0076-6879(84)07018-X [DOI] [PubMed] [Google Scholar]
- 41. Gilbert E. P., Schulz J. C., and Noakes T. J. (2006) ‘Quokka’—the small-angle neutron scattering instrument at OPAL. Physica B Condens. Matter 385–386, 1180–1182 [Google Scholar]
- 42. Kirby N. M., Mudie S. T., Hawley A. M., Cookson D. J., Mertens H. D. T., Cowieson N., and Samardzic-Boban V. (2013) A low-background-intensity focusing small-angle X-ray scattering undulator beamline. J. Appl. Crystallogr. 46, 1670–1680 10.1107/S002188981302774X [DOI] [Google Scholar]
- 43. Mudie S. (2015) Scatterbrain—software for acquiring, processing and viewing SAXS/WAXS data at the Australian Synchrotron, Version 2.71, Australian Synchrotron, Clayton, Victoria, Australia [Google Scholar]
- 44. Whitten A. E., Cai S., and Trewhella J. (2008) MULCh: modules for the analysis of small-angle neutron contrast variation data from biomolecular assemblies. J. Appl. Crystallogr. 41, 222–226 10.1107/S0021889807055136 [DOI] [Google Scholar]
- 45. Konarev P. V., Volkov V. V., Sokolova A. V., Koch M. H. J., and Svergun D. I. (2003) PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 36, 1277–1282 10.1107/S0021889803012779 [DOI] [Google Scholar]
- 46. Petoukhov M. V., and Svergun D. I. (2005) Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophys. J. 89, 1237–1250 10.1529/biophysj.105.064154 [DOI] [PMC free article] [PubMed] [Google Scholar]