

Abstract
HIV-1 is spread by cell-free virions and by cell–cell viral transfer. We asked whether the structure and function of a broad neutralizing antibody (bNAb) epitope, the membrane-proximal ectodomain region (MPER) of the viral gp41 transmembrane glycoprotein, differ in cell-free and cell–cell-transmitted viruses and whether this difference could be related to Ab neutralization sensitivity. Whereas cell-free viruses bearing W666A and I675A substitutions in the MPER lacked infectivity, cell-associated mutant viruses were able to initiate robust spreading infection. Infectivity was restored to cell-free viruses by additional substitutions in the cytoplasmic tail (CT) of gp41 known to disrupt interactions with the viral matrix protein. We observed contrasting effects on cell-free virus infectivity when W666A was introduced to two transmitted/founder isolates, but both mutants could still mediate cell–cell spread. Domain swapping indicated that the disparate W666A phenotypes of the cell-free transmitted/founder viruses are controlled by sequences in variable regions 1, 2, and 4 of gp120. The sequential passaging of an MPER mutant (W672A) in peripheral blood mononuclear cells enabled selection of viral revertants with loss-of-glycan suppressor mutations in variable region 1, suggesting a functional interaction between variable region 1 and the MPER. An MPER-directed bNAb neutralized cell-free virus but not cell–cell viral spread. Our results suggest that the MPER of cell–cell-transmitted virions has a malleable structure that tolerates mutagenic disruption but is not accessible to bNAbs. In cell-free virions, interactions mediated by the CT impose an alternative MPER structure that is less tolerant of mutagenic alteration and is efficiently targeted by bNAbs.
Keywords: human immunodeficiency virus (HIV), glycoprotein, virus entry, viral replication, mutant, membrane fusion, cell–cell transmission, neutralization
Introduction
HIV-1 assembles at and buds from polarized cholesterol- and GM1-rich domains within the plasma membrane of infected T cells following the accumulation of the virion core protein precursor, Gag, and the mature envelope glycoprotein (Env)4 complex, gp120–gp41, at these sites (1–6). Cell–cell contact can also trigger the rapid recruitment of Gag and Env to GM1-rich polarized caps on the HIV-1–infected cell surface, whereas CD4 and the chemokine coreceptor (CCR5 or CXCR4) are recruited to the contact point on the uninfected cell, forming the virological synapse (VS) (7–9). Virological synapse formation is triggered by Env–CD4 interaction and involves cytoskeletal remodeling that polarizes Gag, Env, CD4, and chemokine receptors, as well as secretory organelles and mitochondria to the cell–cell adhesions (10–12). The cytoplasmic tail (CT) of gp120–gp41 plays a key role in signaling the recruitment of Gag to the VS, a process that depends on residues within the matrix protein (MA) domain. The functional interaction between the CT and MA regulates the duration and stability of interactions between infected and uninfected target cells (13). Cell-associated virus can be transferred directly from an infected cell to an uninfected cell via the VS, and this cell-to-cell mode of viral spread has been observed for CD4+ T cell–CD4+ T cell (7, 14), dendritic cell–CD4+ T cell (15, 16), and monocyte-derived macrophage (MDM)–CD4+ T cell (17) conjugates.
Cell–cell viral spread between T cells in vitro is at least 10-fold more efficient than the cell-free spread (18), whereas VS-mediated transmission by MDM is 10–100-fold more efficient than cell-free infection (19), correlating with higher multiplicities of infection within VSs (19–21). Cell-to-cell HIV-1 transmission may contribute significantly to viral spread in vivo. Mathematical modeling suggests that a hybrid cell-free and cell-to-cell spreading mode is the most consistent with the major phases of infection, with hybrid spreading being critical to seed and establish infection, whereas the cell-to-cell spread is important for HIV-1 progression, becoming increasingly effective as infection progresses (22). Cell-to-cell infection may account for ∼60% of viral infection (23). An examination of the dynamic behavior of HIV-infected T cells in the lymph nodes of humanized mice revealed motile small syncytia that establish tethering interactions that may facilitate cell-to-cell transmission through VSs (24). Such small HIV-1-induced syncytia have been observed in the lymph nodes of HIV-1–infected individuals (25, 26) and can be recapitulated in vitro in 3D extracellular matrix hydrogels (27). In this latter context, the syncytia transiently interact with uninfected cells, leading to rapid virus transfer. Further support for cell–cell viral transmission in vivo was provided by the observation that the inoculation of humanized mice with cells coinfected with two viral genotypes leads to high levels of co-transmission to target cells in highly localized microanatomical clusters within lymphoid tissue. Within these clusters, the HIV-infected cells induced arrest of interacting uninfected CD4+ T cells to form Env-dependent cell–cell conjugates (28). These observations indicate that cell-to-cell viral spread is likely to be a significant mode of transmission in vivo and that its blockade should be a consideration in drug therapy and vaccination strategies.
Virological synapse-mediated HIV-1 transmission can confer replicative advantages to virus such that it overcomes exogenous barriers to transmission. For example, VS-mediated viral transmission is less sensitive to commonly used nucleoside reverse transcription inhibitors such as nevirapine, zidovudine, and tenofovir (29–32). Importantly, VS-mediated HIV-1 transmission between CD4+ T cells and between HIV-1–infected MDMs and uninfected CD4+ T cells is less sensitive to neutralization by bNAbs, when compared with cell-free virus infections, indicating that this mode of spread may represent an obstacle to successful vaccine development and neutralizing antibody therapy (19, 33–36). Although these differences between cell-to-cell and cell-free virus transmission can be explained in part by a higher local multiplicity of infection at the VS, it is also plausible that cell-free and cell-associated viruses possess structural differences that confer distinct functional advantages to the two viral forms.
To examine this idea, we assessed the role of the MPER of the HIV-1 transmembrane glycoprotein, gp41, in cell-free and cell-to-cell HIV-1 transmission. The MPER is a conserved 23-residue amphipathic sequence at the C terminus of the gp41 ectodomain and is a critical determinant of membrane fusion and infectivity. Spectroscopic studies of the MPER indicate that it forms a kinked α-helix in the interfacial region of the viral envelope lying parallel to the membrane plane. It includes a tilted N-terminal helix, linked via a hinge to a near-flat C-terminal helix. Conserved aromatic and hydrophobic residues penetrate into the hydrophobic phase of the membrane (37–39). Mutational studies revealed that the conserved W666-W670-W672-W678-W680 motif of the MPER functions cooperatively in the membrane fusion process (40, 41) and that hydrophobic and aromatic MPER residues participate in forming a clasp that stabilizes the membrane-interactive end of the 6-helix bundle conformation of gp41 to initiate membrane fusion (42, 43).
The MPER is of interest to the HIV-1 vaccine research field because it represents the major epitope in gp41 that is recognized by potent human bNAbs such as 2F5, 4E10, 10E8, and Z13 (44–46), some of which can confer complete protection against mucosal cell-free simian-HIV challenge of macaques following passive immunization (47). Distinct modes of MPER binding have been identified for 2F5, 4E10, 10E8, and Z13. 2F5 and 4E10 induce conformational changes in the MPER relative to membrane, 2F5 lifting, and inducing a helix-to-turn transition in the N-helix (37, 48), whereas 4E10 binds to the hinge by extracting Trp672 and Phe673 (37, 38, 49). The secondary structures of the 10E8 and 4E10 epitopes are similar, but 4E10 contacts a substantially larger area of the helical face due to a less acute binding angle relative to the C-helix (44, 50). Cryo-EM studies of HIV-1 particles indicate that the trimeric ectodomain of Env is lifted from the envelope when 10E8 is bound, consistent with a conformational change in the MPER (50). Z13 binds to and immobilizes the MPER hinge with little change in membrane orientation or conformation (37, 51). bNAbs directed to the MPER have low efficacy against VS-mediated viral transmission, even though they potently neutralize cell-free virus (19, 33–36). An understanding of the basis of the differential efficacy of MPER bNAbs in neutralizing cell-free versus cell-to-cell viral transmission is needed to inform approaches for developing vaccines that target this highly conserved epitope.
Here, we show that mutations in the MPER, which block cell-free virus infection, are tolerated in the context of cell-to-cell transmission and that the block to cell-free virus infectivity can be partially reversed by mutations in the CT of gp41. We also observed that an MPER mutation causes contrasting effects in two transmitted/founder (T/F) Envs as follows: blockade of cell-free virus infectivity for isolate SC45, versus infectivity enhancement for isolate PRB958. These inter-strain phenotypic differences were modulated by the first and fourth variable regions (V1 and V4) of the receptor-binding glycoprotein, gp120, and correlated with the relative accessibility of the epitope recognized by bNAb 10E8 within the MPER. Our data indicate that the MPER adopts distinct structural/functional forms during cell-free and cell-to-cell viral spread, with the latter form being unavailable for interaction with bNAbs.
Results
Cell-associated MPER mutant viruses retain the ability to initiate spreading infection but the corresponding cell-free viral mutants do not
bNAbs directed to the MPER have low efficacy against VS-mediated viral transmission, even though they potently neutralize cell-free virus (19, 33–36). To examine the role of the MPER in cell-free infection and cell–cell viral spread, an assay was developed to compare the spread of two well-characterized HIV-1AD8 MPER mutants, W666A and I675A (Fig. 1, A and B; Fig. S1A) (52), by cell-free and cell-to-cell modes. The U87.CD4.CCR5 glioblastoma cell line (53, 54), which expresses receptors necessary for cell–cell spread, including CD4, CCR5, and ICAM-1 (8, 54–57), was used as the infection target. VSV G-pseudotyping in trans was used to facilitate entry of W666A- and I675A-mutated (and WT) AD8 viruses into the U87.CD4.CCR5 cells to enable subsequent reverse transcription, integration, and production of cell-associated (and cell-free) viruses. Nonpseudotyped virions were used as control cell-free virus-only inocula. Nonpseudotyped and VSV-G-pseudotyped inocula were normalized according to reverse transcriptase (RT) activity prior to their addition to the U87.CD4.CCR5 cells. At 24 h post-inoculation, the cells were trypsinized and washed to remove surface-adsorbed virus and then re-plated and cultured for 10 days; the RT activity present in culture supernatants was used as a measure of virus production. The results indicate that both nonpseudotyped and VSV G-pseudotyped WT viruses are able to initiate infection of U87.CD4.CCR5 cells and virus egress into culture supernatants (Fig. 1, C and D). By contrast, very little virus production was observed for the W596L/K601D (WLKD) mutant that lacks gp120 association (58), indicating that a fusion-competent glycoprotein complex is required for virus transmission in cultures initiated by cell-free and cell-associated virus. Because VSV G-pseudotyped WLKD virions are fully competent to initiate infection (see Fig. 2D), whereas the nonpseudotyped WLKD viruses are not (Fig. 1C), the RT activity produced by VSV G-pseudotyped WLKD indicates the amount of virus that is released following a single round of infection. We therefore infer that the accumulation of virus in supernatants during the culture is due to spreading infection.
Figure 1.

Viral replication initiated by cell-free and cell-associated MPER mutant viruses in U87.CD4.CCR5 cells. A, NMR structure of MPER peptide (amino acids 662–683; PDB code 2PV6) in dodecylphosphocholine micelle (38). The MPER peptide forms a kinked helix in the interfacial region (bordered by arcs) with aromatic and hydrophobic side chains penetrating into the hydrophobic phase (stippled). The side chains of amino acids mutated to alanine in this study are shown. B, functional characteristics of W666A, W672A, and I675A AD8 Env mutants described in Bellamy-McIntyre et al. (52). +++, >90% of WT activity, +/−, <2% of WT activity. U87.CD4.CCR5 cells were inoculated with unpseudotyped (C) or VSV G-pseudotyped (D) HIV-1AD8 particles (50,000 cpm of RT activity per inoculum) and then trypsinized 24 h later to remove residual adsorbed virus. The cells were then replated and cultured for a further 10 days. Mean RT activity ± S.D. of triplicate samples is shown. Representative of two independent experiments. E, day-7 and day-10 culture supernatants obtained from D were filtered through 0.45-μm nitrocellulose filters and then used to infect naive U87.CD4.CCR5 cells. Mean RT activity ± S.D. of triplicate samples is shown. Representative of two independent experiments. F and G, PHA-stimulated PBMCs were inoculated with nonpseudotyped (left panels) or VSV G-pseudotyped (right panels) HIV-1 particles and then washed extensively 24 h later. The cells were replated and cultured for a further 10 days. The p24 content of viral supernatants was determined at the indicated time points by ELISA. Data are representative of two independent experiments. H, infectivity of WT and mutated viruses for TZM-bl cells. Serially diluted virus-containing supernatants were incubated with TZM-bl cells for 2 days prior to lysis and luciferase assay. The mean relative light units (RLU) ± S.E. was obtained from three independent experiments shown. I, nonpseudotyped and VSV G-pseudotyped HIV-1AD8 virions fail to infect a CD4-negative cell line, Huh7.5. The experiment was conducted as for C and D. Mean RT activity ± S.D. of triplicate samples is shown. J, infection of Huh7.5 cells by VSV G-pseudotyped NL4.3lucR−E− luciferase reporter virus. Mean RLU ± S.D. of triplicate samples is shown. No Env, nonpseudotyped reporter particles.
Figure 2.

L8S/S9R matrix mutation blocks viral spread initiated by cell-associated virus. A, L8S/S9R decreases viral infectivity for TZM-bl cells. Serially diluted virus-containing supernatants were incubated with TZM-bl cells for 2 days prior to lysis and luciferase assay. The mean RLU ± S.E. obtained from two independent experiments is shown. B, cell-surface–expressed Env glycoproteins expressed from WT and mutated pAD8 infectious clones retain fusion competence. 293T cells that had been cotransfected with WT or mutated pAD8 infectious clones and pCAG-T7 were cocultured with BHK21 cells that had been cotransfected with pCCR5 and pT4luc. Luciferase activity was measured 16 h later. The mean RLU ± S.E. obtained from three independent experiments is shown. *, p < 0.05 WT versus mutant; two-tailed t test assuming unequal variances. C, L8S/S9R blocks spreading infection initiated by cell-associated virus in U87.CD4.CCR5 cells. As for Fig. 1D, except that the virus content of culture supernatants was measured by p24 ELISA. Data are representative of three independent experiments. D, VSV G-pseudotyped L8S/S9R-containing viruses retain infectivity for TZM-bl cells. Serially diluted virus-containing supernatants were incubated with TZM-bl cells for 2 days prior to lysis and luciferase assay. The mean RLU ± S.E. obtained from two independent experiments is shown.
W666A and I675A viruses lacked replication competence in U87.CD4.CCR5 cells when the infection was initiated with nonpseudotyped cell-free viruses (Fig. 1C). By contrast, the mutated cell-associated viruses gave rise to substantial RT activity in the culture supernatants consistent with spreading infection (Fig. 1D). In this context, W666A and I675A gave rise to more virus than WT at day 10. Day-7 and day-10 supernatants that had been obtained from cultures initiated with VSV G-pseudotyped W666A and I675A viruses were unable to efficiently infect naive U87.CD4.CCR5 cells (Fig. 1E). These data indicate that cell-free W666A and I675A viruses produced in cultures initiated by VSV G-pseudotyped viruses remain entry-defective; therefore, W666A and I675A viral spread occurs via the cell-to-cell route. The data also suggest that the W666A and I675A virus production observed in Fig. 1D is not simply due to mutant virus reversion or due to infection by cell-free VSV G-pseudotyped viruses persisting after the trypsin treatment.
We next examined the infectivity of W666A and I675A mutants in phytohemagglutinin (PHA)-stimulated PBMCs pooled from three normal donors (Fig. S1B). Spreading infection did not occur in the stimulated PBMCs when cell-free AD8-W666A and AD8-I675 viruses were used to initiate infection (Fig. 1, F and G, left panels), consistent with the results obtained with U87.CD4.CCR5 cells (see Fig. 1C). By contrast, spreading infection was observed when the first round of infection was initiated by VSV G-pseudotyped AD8-W666A and AD8-I675 viruses (Fig. 1, F and G, right panels), again consistent with that observed with U87.CD4.CCR5 cells (see Fig. 1D). The fusion-incompetent gp120-shedding mutant, AD8-WLKD, did not spread in the PBMCs. Because cell-free AD8-W666A and AD8-I675 viruses are not infectious for stimulated PBMCs and U87.CD4.CCR5 cells, we infer that the spreading infection observed in cultures initiated with VSV G-pseudotyped MPER mutants occurs via the cell-to-cell route only. A difference observed between the two systems is that cell–cell spread by W666A and I675A viruses is less efficient in stimulated PBMCs relative to U87.CD4.CCR5 cells (compare Fig. 1D with F and G, right panels).
The cell-free entry ability of the mutants was further examined in TZM-bl cells, which express ∼4-fold higher levels of CD4 relative to U87.CD4.CCR5 cells but about the same amount of CCR5 (Fig. S1C). In this assay, infection is allowed to proceed for 48 h, which corresponds to 1–2 infection cycles, suggesting that the infectivity readout is largely due to cell-free virus infection. A low level of infectivity was observed for the two mutants at a 1/10 dilution of inocula (Fig. 1H), which corresponds to ∼5-fold higher virus content than that used with U87.CD4.CCR5 and stimulated PBMCs. These data suggest that at high CD4 concentrations, residual infectivity can be detected for concentrated W666A and I675A virus inocula.
Inoculation of the nonpermissive cell line, Huh7.5, with VSV G-pseudotyped WT virus did not give rise to RT activity in culture supernatants (Fig. 1I) even though VSV G pseudotyping of pNL4.3R-E-luc reporter viruses enabled efficient infection of these cells (Fig. 1J). Thus cell–cell spread requires CD4 and CCR5. The data indicate that the W666A and I675A mutations are tolerated in viruses transmitted via the cell–cell route, although the mutations block the infectivity of cell-free virions.
Viral spread initiated by cell-associated MPER mutants is blocked by a mutation in MA that reduces Env incorporation
We next asked whether the apparent ability of MPER mutants to mediate cell–cell spread in U87.CD4.CCR5 cells depends on the assembly of Env-containing virions. The introduction of L8S/S9R mutations into MA has been shown previously to inhibit Env incorporation into virions (59) and to block cell–cell transmission (60). We first determined that the L8S/S9R MA mutation severely limited the infectivity of cell-free WT AD8 virus for TZM-bl reporter cells and reduced the residual infectivity of W666A and I675A viruses to WLKD background levels (Fig. 2A). By contrast, the fusion activities of 293T cell-surface–expressed WT, W666A, and I675A Env derived from MA-L8S/S9R-containing proviral constructs were similar to their counterparts derived from proviruses with an intact MA domain (Fig. 2B). These data confirm that MA-L8S/S9R blocks cell-free virus infectivity but does not affect the intrinsic fusion function of the cell-surface Env containing W666A and I675A. We next tested the effects of the MA-L8S/S9R mutation on cell–cell spread in U87.CD4.CCR5 cells. The cells were inoculated with VSV G-pseudotyped viruses with and without the MA-L8S/S9R mutation, trypsinized 24 h later, and then cultured for a further 14 days. The p24 content of the cell-free culture supernatants was used as a measure of virus production. Fig. 2C confirms that WT, W666A, and I675A but not WLKD viruses can mediate cell–cell spread in U87.CD4.CCR5 cultures and that this spreading infection is completely blocked by the MA-L8S/S9R mutation. Finally, we used the TZM-bl reporter system to confirm that VSV G-pseudotyped viruses bearing the MA-L8S/S9R mutation were competent to initiate infection (Fig. 2D), thereby ruling out the possibility that the MA-L8S/S9R-mediated block to cell–cell spread in U87.CD4.CCR5 cultures was simply due to a block in the initial VSV G-mediated entry step. These data indicate that viral spread initiated by cell-associated WT, W666A, and I675A viruses depends on the assembly of Env-containing virions.
Mutations in the CT of gp41 mitigate the cell-free virus infectivity defect associated with W666A and I675A
The W666A and I675A mutations diminished the infectivity of cell-free virions, whereas previous studies have shown that membrane fusion mediated by these Env mutants expressed at the cell surface in the absence of other viral proteins is not affected (52). It may be that the MPER mutations adversely affect Env function in the context of assembled cell-free virions, where interactions between MA and the CT of gp41 (61–63) can modulate the structure and function of the ectodomain (64–66). To investigate the influence of the CT on the function of the MPER mutants, stop codons were introduced after positions 712, 727, and 752 in the WT, W666A, and I675A pcDNA3.1-AD8env expression vectors to give the ΔCT144, ΔCT129, ΔCT104 mutants, respectively (Fig. 3A). These truncations remove various CT subdomains. For example, ΔCT104 removes a C-terminal dileucine endocytosis motif (67–70) and the so-called lentiviral lytic helical peptides (LLP)-1, LLP-3, and LLP-2 (71–73), as well as two intracellular sorting motifs, Tyr795–Trp/Leu, and Tyr802–Trp (74–77), present within LLP-3; ΔCT129 removes the former motifs plus the highly immunogenic “Kennedy sequence” (78, 79), whereas ΔCT144 removes the former motifs plus the dominant tyrosine-based sorting signal, Tyr712–Xaa–Xaa–Leu (80–83).
Figure 3.

Mutations in the CT of gp41 restore infectivity to cell-free MPER mutant virions. A, schematic representation of gp41. HR1, HR2 are heptad repeats 1 and 2 respectively; TMD, transmembrane domain; LLP, lentiviral lytic peptides; Y712SPL, dominant tyrosine-based sorting signal; YW, intracellular sorting motifs; LL, C-terminal dileucine endocytosis motif. B, expression of truncated AD8 glycoproteins in 293T cells transfected with WT and mutated Env expression vectors. Cell lysates were subjected to reducing SDS-PAGE and Western blotting with DV-012 polyclonal anti-gp120 serum. C, cell–cell fusion activities of truncated AD8 Env glycoproteins. 293T cells that had been cotransfected with WT or mutated pcDNA3.1AD8env vectors and pCAG-T7 were cocultured with BHK21 cells that had been cotransfected with pCCR5 and pT4luc. Luciferase activity was measured 18 h later. The mean RLU ± S.E. obtained from at least three independent experiments is shown. D and E, single-cycle infectivity of NL4.3LucR−E− luciferase reporter viruses pseudotyped with WT and mutated AD8 Env glycoproteins for U87.CD4.CCR5 cells. The cells were assayed for luciferase activity at 52 h post-transfection. Mean RLU ± S.E. from at least three independent experiments is shown. *, p < 0.05; **, p < 0.01; glycoprotein construct with mutated CT versus corresponding construct with unmutated CT; two-tailed t test assuming unequal variances. F, infectivity of WT and mutated HIV-1AD8 viruses for TZM-bl cells. Serially diluted virus-containing supernatants were incubated with TZM-bl cells for 2 days prior to lysis and luciferase assay. The mean RLU ± S.D. obtained from a representative experiment is shown. G, gp120 content of virions. Virions were pelleted through a sucrose cushion and then subjected to reducing SDS-PAGE and Western blotting with DV012 (upper panel) and HIVIG (lower panel). gp120-incorporation indices ((gp120mutant pixels ÷ p24mutant pixels) × (p24WT pixels ÷ gp120WT pixels)) are shown below. The image was obtained from a single gel with the splice point indicated.
The expression and processing of the truncation mutants in 293T cells were first confirmed by Western blotting with DV-102 anti-gp120 antibody (Fig. 3B). The apparent molecular weights of gp160 were progressively smaller for ΔCT104, ΔCT129, and ΔCT144, respectively, consistent with the removal of gp41 CT sequences, whereas the apparent molecular weight of gp120 remained constant. The W666A and I675A mutations did not significantly affect the cell–cell fusion activities of the corresponding full-length and C-terminally truncated Envs, indicating that the CT does not influence the membrane fusion abilities of the MPER mutant glycoproteins (Fig. 3C).
We next tested the effects of the CT truncations on the infectivity of Env-pseudotyped luciferase reporter viruses for U87.CD4.CCR5 cells in a single-cycle assay (Fig. 3D and Fig. S1D). The ΔCT144 truncation restored the infectivity of the W666A and I675A mutants to within 1 log10 of WT, whereas the ΔCT129 and ΔCT104 did not significantly change the entry activities of the MPER mutants. These data locate an inhibitory determinant within the 713–727 sequence of CT that operates in conjunction with W666A and I675A.
Several lines of evidence indicate that the gp41 CT interacts with the MA protein shell beneath the inner leaflet of the viral envelope. In this context, the MA shell may include a hexameric array of MA trimers, the central aperture of the hexamer potentially providing a docking site for the CT (61–63, 84). Mutations in MA such as L49D can destabilize the interaction between gp120 and gp41 on the virion surface; however, Env can be uncoupled from the L49D MA defect by ΔCT144, and this effect can be reproduced by the Y712S substitution in the gp41 CT (64). We therefore asked whether Y712S in the context of a full-length CT mimics the restorative effects of ΔCT144 for W666A and I675A in the U87.CD4.CCR5 single-cycle assay (Fig. S1D). Fig. 3E indicates that combining either ΔCT144 or Y712S with W666A or I675A increases single-cycle entry function by ∼10-fold with respect to the corresponding MPER mutants with a WT CT. Next, we asked whether the Y712S-dependent increase in W666A and I675A pseudotype infectivity could be reproduced with HIV-1 particles derived from full-length proviruses. The Y712S mutation was therefore introduced to WT, W666A, and I675A AD8 infectious clones, and the infectivity of corresponding cell-free viruses was determined using TZM-bl reporter cells (Fig. S1C). In agreement with the single-cycle entry data, W666A and I675A reduced cell-free virus infectivity by ∼2.5 log10, whereas the introduction of Y712S to these MPER mutants restored their infectivity to within 1 log10 of WT (Fig. 3F). Because Tyr712–Xaa–Xaa–Leu represents a dominant endocytosis motif and Tyr712 modulates CT-MA interactions, we examined whether Y712S affected Env incorporation into virions. Virions produced in HeLa cells, which are permissive for CT mutant viruses, were pelleted through a sucrose cushion, and their gp120 and p24 content was analyzed by reducing SDS-PAGE and Western blotting. Slightly lower gp120 relative to p24 incorporation was observed for W666A and I675A when compared with WT; however, no major changes to relative gp120 incorporation of the mutants with the introduction of Y712S were observed (Fig. 3G, upper panels). (Attempts were made to examine the effects of Y712S on cell-associated virus transmission in U87.CD4.CCR5 cells; however, even WT virus spread was blocked by the mutation (data not shown) suggesting that this cell line is not permissive for viruses bearing mutations in the CT.) Thus, the cell-free virus infectivity defects associated with W666A and I675A may in part be due to a subtle gp120–gp41 association defect. However, the restorative effect of Y712S was not due to increased glycoprotein incorporation, indicating that a substantial component of the cell-free virus infectivity defect is independently mediated through an interaction between the CT and MA.
Mutations in the MPER do not alter the kinetics of membrane fusion
We employed a real-time fluorescence-based assay (85) to examine the kinetics of membrane fusion mediated by the W666A and I675A Env mutants expressed at the cell surface. This assay dissects membrane fusion into two kinetic phases as follows: the lag time that reflects the ability of Env to recruit and be activated by receptors, and the fusion rate that correlates with chemokine receptor affinity and infectivity (85–88). 293T cells expressing Env and β-lactamase were cocultured with JC53 targets (HeLa cells stably expressing CD4 and CCR5) loaded with the green fluorescent substrate, CCF2-AM; upon formation of a fusion pore, small molecule transfer between the cocultured cells enables β-lactamase to cleave CCF2-AM, resulting in blue fluorescence. The ratio of blue/green fluorescence was used as a measure of fusion. Fig. 4A indicates an ∼30-min lag time and an exponential phase of ∼120 min prior to the final extent of fusion being reached for WT, consistent with the Env fusion curves originally generated by Lineberger et al. (85). By contrast, a fusion curve was not generated by G597A, a fusion- and entry-defective gp41 mutant (89). To compare fusion mediated by the MPER mutants, the ratio of blue/green fluorescence over time was normalized against the final extent of fusion. The lag times and rates of fusion for W666A and I675A were not significantly different from those generated by WT, indicating that the mutations did not affect the kinetics of Env-mediated membrane fusion (Fig. 4B). The data confirm that W666A and I675A mutations do not alter the intrinsic fusion function of Env when expressed in the absence of other viral proteins.
Figure 4.
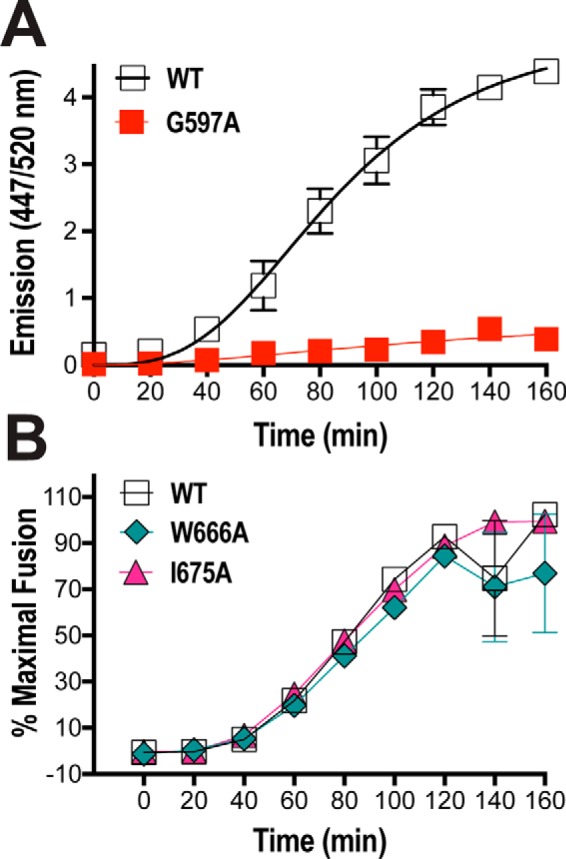
Real-time fusion kinetics of MPER mutant Envs. A, 293T effector cells cotransfected with expression vectors encoding β-lactamase plus WT Env or a fusion-incompetent Env mutant (G597A) were cocultured with CCF2-AM–loaded JC53 targets in 96-well plates at 37 °C for 180 min. Individual wells were read for green and blue fluorescence (excitation at 409 nm; emission at 520 and 447 nm, respectively) every 20 min in a FLUOstar plate reader (BMG). The ratio of blue/green fluorescence is shown. B, fusion kinetics of MPER mutants. The assay conditions were as per C. The ratio of blue/green fluorescence was calculated and then normalized to the final extent of fusion. Mean ± S.E. of four independent experiments is shown.
Differential functional effects of the W666A MPER mutation in the context of two T/F clones
We next examined whether the distinct effects of the MPER mutations on cell-free versus cell-associated viral transmission are a general feature of HIV-1 isolates by introducing W666A into two clade B T/F Envs, SC45.4B5.2631 (SC45) and PRB958_06.TB1.4305 (PRB958), which share 82.7% amino acid identity in the Env ectodomain (Fig. S2) (90). Transmitted/founder viruses establish infection in naive hosts and are the targets of future prophylactic vaccines; therefore, it is important to examine the structure and function of this key bNAb epitope in this context. The WT and mutated T/F env regions were used to replace the corresponding region of pNL4.3 to generate the chimeric infectious clones pNL.SC45 and pNL.PRB958. The data presented in Fig. 5A (left panel) show that W666A strongly inhibited the infectivity of cell-free NL.SC45 virus for U87.CD4.CCR5 cells, and the W666A-associated block to infectivity was negated when viral transmission was initiated with cell-associated NL.SC45 virus with increased levels of p24 produced in mutant viral cultures at days 7 and 10 relative to WT (Fig. 5A, right panel). These data are consistent with those obtained with AD8 virus (see Fig. 1, C and D). In marked contrast, the infectivity of cell-free NL.PRB958 virus was enhanced by W666A with 5-fold greater amounts of p24 being produced at days 7 and 10 relative to WT; a similar ratio of p24 production was observed for NL.PRB958-W666A and WT in cultures initiated with cell-associated virus (Fig. 5A). Thus, the W666A mutation has opposing effects on NL.SC45 and NL.PRB958 cell-free virus infectivity.
Figure 5.
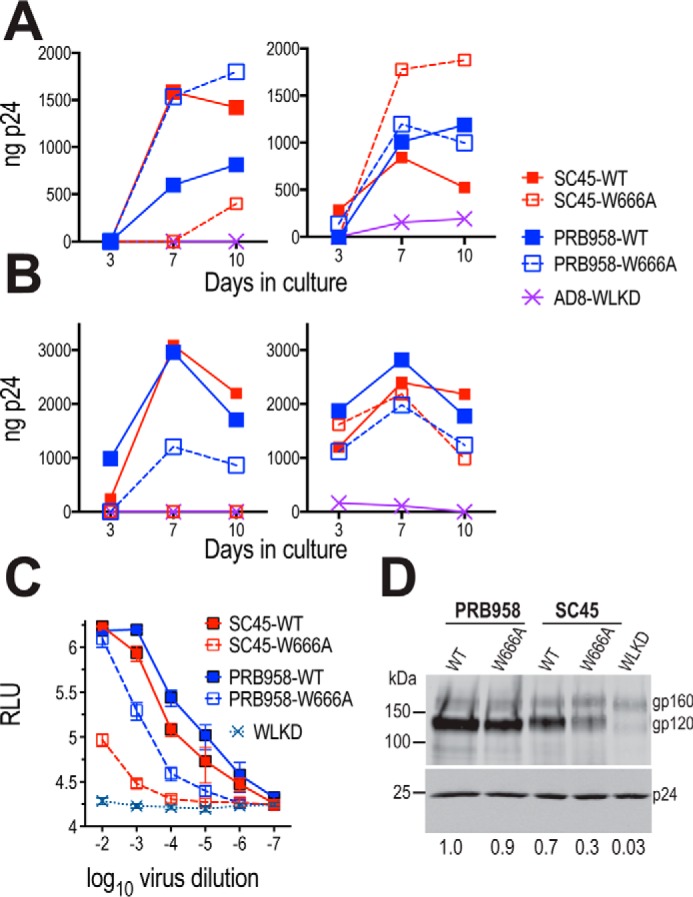
Distinct effects of W666A in Envs from two T/F isolates. A, U87.CD4.CCR5 cells were inoculated with unpseudotyped (left panel) or VSV G-pseudotyped (right panel) HIV-1 particles and then trypsinized 24 h later to remove residual adsorbed virus. The cells were then replated and cultured for a further 10 days. The p24 content of viral supernatants was determined at the indicated time points by ELISA. Representative of three independent experiments. B, PHA-stimulated PBMCs were inoculated with nonpseudotyped (left panel) or VSV G-pseudotyped (right panel) HIV-1 particles and then washed extensively 24 h later. The cells were replated and cultured for a further 10 days. The p24 content of viral supernatants was determined at the indicated time points by ELISA. Data are representative of two independent experiments. C, infectivity of WT and mutated viruses for TZM-bl cells. Serially diluted virus-containing supernatants were incubated with TZM-bl cells for 2 days prior to lysis and luciferase assay. The mean RLU ± S.E. obtained from six independent experiments is shown. D, gp120 content of virions. Virions were pelleted through a sucrose cushion and then subjected to reducing SDS-PAGE and Western blotting with DV-012 (upper panel) and HIVIG (lower panel). gp120-incorporation indices relative to PRB958-WT are shown below.
We next compared the effects of W666A on the infectivity of cell-free and cell-associated NL.SC45 and NL.PRB958 viruses in stimulated PBMCs. W666A blocked the infectivity of cell-free NL.SC45 virus for the primary cells, consistent with the data obtained with U87.CD4.CCR5 cells (Fig. 5B, left panel). Cell-free NL.PRB958-W666A retained substantial infectivity for PBMCs; however, the enhancing effect of the MPER mutation observed in U87.CD4.CCR5 cells was not evident, with ∼3-fold lower levels of mutant being produced relative to WT at the peak of virus production by the primary cells. All mutants were able to mediate spreading infection in PBMCs when the culture was initiated with cell-associated viruses (Fig. 5B, right panel). Again, the enhancing effects of the mutations observed in the U87.CD4.CCR5 cells were not evident.
The effects of W666A on cell-free NL.SC45 and NL.PRB958 virus infectivity were further investigated using TZM-bl target cells. In this assay, the MPER mutation caused an ∼150-fold reduction in NL.SC45 infectivity, whereas an ∼10-fold infectivity reduction was observed for NL.PRB958-W666A (Fig. 5C). These infectivity reductions observed for cell-free NL.SC45-W666A and NL.PRB958-W666A viruses in the TZM-bl assay are consistent with the data obtained with cell-free virus infection of PBMC targets, suggesting that cell-free virus is an important component of spreading infection in the PBMC cultures. Interestingly, cell-free NL.PRB958-W666A showed enhanced infectivity in the U87.CD4.CCR5 culture. Cell–cell transmission may account for the majority of spreading infection in the 10-day U87.CD4.CCR5 cultures, provided that a threshold of infection is achieved in the first round. The data indicate that the functional consequences of MPER modification can vary in a strain-dependent manner.
The effect of the W666A mutation on Env incorporation into virions was examined by Western blotting. Fig. 5D reveals that W666A leads to markedly reduced gp120 in SC45 virions, whereas WT levels are present in PRB958. The data indicate that a component of the infectivity loss observed for cell-free SC45-W666A virus is due to decreased gp120 in virions, whereas the infectivity phenotype of cell-free PRB958-W666A is likely to be a subtle change in the Env complex. As for AD8, these functional defects appeared to be mitigated in virions spread via the cell–cell mode.
Molecular basis of the SC45 and PRB958 W666A phenotypes
A domain-swapping approach, whereby SC45 env sequences were swapped into the pNL.PRB958 vector backbone (PR.SC chimeras), was employed to identify the molecular determinants controlling the distinct infectivity phenotypes of NL.SC45-W666A and NL.PRB958-W666A viruses. The specific aim was to identify which structural elements of SC45 Env confer an SC45-W666A-like phenotype to NL.PRB958. The TZM-bl infectivity assay (Fig. S1C) was used to screen a panel of domain-swapped NL.PR.SC chimeras (Fig. 6A). In this assay, W666A caused an ∼150-fold reduction in NL.SC45 infectivity, whereas an ∼10-fold infectivity reduction was observed for NL.PRB958-W666A (Fig. 6B, parental; see also Fig. 5C). The introduction of the SC45 gp120 domain into the NL.PRB958 backbone (Fig. 6B, PR.SC.gp120) resulted in NL.SC45-like infectivity for PR.SC.gp120-WT, whereas an intermediate infectivity phenotype was observed for PR.SC.gp120-W666A. The MPERs of PRB958 and SC45 differ at three positions: S671N, E674N, and N677Q (PRB958 → SC45, Fig. S2). These mutations were introduced to the MPER of PR.SC.gp120-W666A to give PR.SC.gp120Mx-W666A. This was done to determine whether the SC45-W666A phenotype required the SC45 gp120 domain to be matched with the cognate MPER. This was not the case because PR.SC.gp120Mx-W666A exhibited the intermediate infectivity phenotype of PR.SC.gp120-W666A, wherein the gp120 and MPER domains are mismatched (Fig. 6B, PR.SC.gp120 panel).
Figure 6.

Phenotypic analysis of PRB958-SC45 chimeric Env glycoproteins. A, schematic representation of PRB958-SC45 chimeric Env glycoproteins. The chimeras contain NL4.3 flanking sequences 5′ and 3′ to the indicated KpnI and BamHI sites, respectively. B, infectivity of WT and mutated viruses for TZM-bl cells. Serially diluted virus-containing supernatants were incubated with TZM-bl cells for 2 days prior to lysis and luciferase assay. The mean RLU ± S.E. obtained from at least three independent experiments is shown. C, gp120 content of virions. Virions were pelleted through a sucrose cushion and then subjected to reducing SDS-PAGE and Western blotting with DV012 (upper panel) and HIVIG (lower panel). gp120-incorporation indices are shown below. The data were obtained from three gels, as indicated. The arrow indicates the position of the 150-kDa marker. D, spreading infection in U87.CD4.CCR5 cells initiated by cell-free and cell-associated virus. Data are representative of two independent experiments. E, spreading infection in PHA-stimulated PBMCs initiated by cell-free and cell-associated virus. Data are representative of two independent experiments.
To further map the determinants in gp120 responsible for the SC45 phenotype, gp120 subdomains were chimerized to the NL.PRB958 backbone. PR.SC.V1V2 exhibited NL.PRB958-like infectivity for WT and intermediate infectivity for W666A, whereas PR.SC.V3 exhibited NL.PRB958-like infectivity for W666A. The combination of SC45 V4 and V5 with V1V2 (PR.SC.V1245) led to greatly diminished infectivity for the WT version, indicating that this SC45 subdomain combination is functionally incompatible with a PRB958 backbone (Fig. 6B). Chimeras containing the V1V2C2V3 and V1V4V5 regions of SC45 on a PRB958-WT background exhibited 2–3 log10 decreases in cell-free virus infectivity relative to PRB958-WT (data not shown) and were not pursued further. The most informative chimeras were PR.SC.V12V4 and PR.SC.V1V4, which largely recapitulated the NL.SC45 infectivity phenotype (Fig. 6B), indicating that the external V1 and V4 loops of gp120 modulate the function of the W666A-mutated MPER in cell-free virus.
The ability of the chimeric W666A mutants to incorporate gp120 into virions was determined by Western blotting. The data confirm the gp120-incorporation defect of SC45-W666A (Fig. 6C). Interestingly, the PR.SC.gp120 and PR.SC.V1V2 W666A mutants exhibited similar gp120-incorporation defects to SC45-W666A but were more infectious than the parental mutant, suggesting that reduced gp120 incorporation does not completely account for defective cell-free virus infectivity. This idea was supported by the observation that the two chimeras that most closely approximated SC45-W666A infectivity had distinct gp120-incorporation phenotypes; W666A reduced PR.SC.V12V4 virion gp120 content by ∼50% with respect to PR.SC.V12V4-WT, whereas the gp120 content of PR.SC.V1V4 was affected to a lesser degree by W666A. The data suggest that V1 and V4 modulate the cell-free infectivity phenotype of SC45-W666A, whereas V2 contributes to the gp120-incorporation phenotype. Interestingly, PR.SC.gp120-W666A exhibited a less attenuated infectivity phenotype than PR.SC.V1V2V4 in the TZM-bl assay, despite the two chimeras exhibiting similar gp120-incorporation defects. The SC45-W666A infectivity phenotype appears to be partially suppressed when SC45 gp120 sequences outside of V1, V2, and V4 are included in the PRB958 Env backbone.
We next examined the abilities of cell-free and cell-associated PRB.SC.V1V4-WT and W666A viruses to mediate spreading infection in U87.CD4.CCR5 cells and PHA-stimulated PBMCs. In U87.CD4.CCR5 cultures initiated with cell-free viruses, W666A had an enhancing effect on PRB958 virus production, whereas SC45 was inhibited by the mutation, consistent with the previous findings (Fig. 6D, left panels). In contrast, PR.SC.V14-WT and W666A appeared to replicate at similar levels, i.e. neither enhancement nor inhibition of virus production was observed with W666A. This result was not expected, given that cell-free PR.SC.V14-W666A exhibited a highly attenuated phenotype in the TZM-bl single-cycle assay. WT and W666A-mutated SC45, PRB958, and PR.SC.V1V4 viruses were competent to mediate cell-to-cell transmission in U87.CD4.CCR5 cells (Fig. 6D, right panels). The abilities of cell-free and cell-associated PR.SC.V1V4-WT and -W666A chimeras to mediate spreading infection in PHA-stimulated PBMCs were next tested. Nonpseudotyped and VSV G-pseudotyped inocula were employed to initiate the first round of PBMC infection after which the cells were washed extensively prior to culture for 14 days. The data indicate that W666A blocks the infectivity of cell-free PRB.SC.V1V4 viruses, whereas cell-associated PRB.SC.V1V4-W666A is able to efficiently mediate spreading infection in PBMCs (Fig. 6E). Overall, the data indicate that the V1 and V4 domains of SC45 act in concert to confer the attenuated infectivity phenotype observed with cell-free SC45-W666A in TZM-bl cells and PBMCs. Whereas the enhancing effect of W666A for cell-free PRB958 virus infectivity in the U87.CD4.CCR5 system was negated by the introduction of SC45 V1 and V4, the transmission block seen with SC45-W666A was not recapitulated. It is possible that the residual infectivity of cell-free PR.SC.V14-W666A (Fig. 6B) enables seeding of the U87.CD4.CCR5 cells to enable subsequent rounds of highly efficient cell–cell transmission.
Cell-free PRB958 and SC45 pseudovirions exhibit differential sensitivities to bNAbs directed to the MPER
The structural context of SC45 and MPER sequences was probed in neutralization assays with the 2F5 and 10E8 bNAbs, which bind to different segments of the MPER (Fig. 7, A and B), as well as bNAbs directed to the gp120–gp41 complex (PGT151) and the gp120 domain (VRC01, PGT121, and PG9) (Fig. 7C). 2F5 recognizes an N-terminal epitope 656NEQELLELDK665WASLW670, which adopts an extended conformation when bound to antibody (48), whereas the more C-terminal 10E8 epitope, 670WNWFDI675TNWLW680YIK683, maintains a helical conformation when bound (44, 50). Serially diluted bNAbs were preincubated with luciferase reporter viruses pseudotyped with PRB958 or SC45 Envs, and the mixtures were incubated with U87.CD4.CCR5 cells for 48 h prior to lysis and luciferase assay. Whereas the two Envs exhibited similar neutralization sensitivities to 2F5, PRB958 Env exhibited greater sensitivity to 10E8 neutralization than SC45 Env, the neutralization IC50 being 4.3-fold higher for the latter (Fig. 7D). PRB958 and SC45 Env pseudotypes were neutralized to the same degree by bNAb PGT151, which is directed to a complex glycan-dependent epitope contributed by gp120 and gp41, and specifically recognizes the cleaved, trimeric form of Env (91) VRC01, which is directed to the CD4-binding site (92), and PGT121, which is directed to an epitope involving the V3 glycan at Asn332 (93). In contrast, the two Envs exhibited differential sensitivities to PG9, which is directed to a V1V2 Asn156/Asn160-glycan-dependent epitope (94). In this case, PRB958 was completely neutralized by PG9, whereas the neutralization of SC45 was incomplete. As most of the key PG9 contact residues (Asn156, Asn160, Arg168, and Phe176) (95) are present in both isolates, this suggests that the conformation of this region differs. Overall, the data suggest that the 10E8 epitope is more accessible to antibody binding in PRB958 Env than in SC45 indicative of alternative structural contexts in the two Envs. This is unlikely to be due to gross differences in the global organization of the gp120–gp41 trimer, as the two Envs exhibited similar sensitivities to the cleaved gp120–gp41 trimer, CD4bs and Asn332 glycan-dependent bNAbs, but may be linked to differences in the sequence and or structure of V1V2 to which PG9 binds.
Figure 7.

Neutralization sensitivity of PRB958 and SC45 Envs. A, alignment of MPER sequences. The asterisks indicate contact residues for bNAbs 10E8 (red) and 2F5 (blue). B, 2F5 (blue) and 10E8 (red) contact residues in the context of the lipid micelle-associated MPER peptide (38). C, location of bNAb epitopes in the gp120–gp41 SOSIP.664 trimer (PDB code 5FYJ) (120). The MPER is absent from the structure but would be located at the base of the trimer (50) as indicated by arrow. D, neutralization of cell-free viruses by various bNAbs. U87.CD4.CCR5 cells were incubated with pseudovirus-IgG mixtures for 2 days prior to lysis and assay for luciferase activity. Neutralizing activities were measured in triplicate, and the average percent luciferase activity was determined for each assay. The data are the means ± S.E. obtained from at least two independent experiments. E, neutralization of cell-associated viruses. U87.CD4.CCR5 cells were infected with VSV G-pseudotyped viruses and then trypsinized 24 h later. The cells were seeded in triplicate into 96-well tissue culture plates in the presence of serially diluted bNAbs. Three days later, 50% of the culture supernatant was replaced with fresh medium containing the appropriate dilution of bNAb. The p24 content of viral supernatants was determined at day-7 post-seeding by ELISA. Neutralizing activities were measured in triplicate and reported as the average percent p24 content. The data are representative of three independent experiments. HC84-1 is a control human monoclonal bNAb directed to the E2 glycoprotein of HCV (139).
We next examined whether the MPER within cell-associated PRB958 and SC45 viruses is accessible to neutralizing antibody. To this end U87.CD4.CCR5 cells were infected with VSV G-pseudotyped NL.PRB958 and NL.SC45 viruses for 24 h, after which they were trypsinized and replated into 96-well tissue culture plates in the presence of serially diluted bNAbs 10E8 and PGT121. An HCV-specific bNAb HC84-1 was included as a negative control. At day-3 postinfection, one-half of the tissue culture medium was replaced with complete medium containing serially diluted bNAbs. The tissue culture medium was then assayed for p24 content at day 7. Both NL.PRB958 and NL.SC45 viruses resisted neutralization by the MPER-directed bNAb 10E8, although they were efficiently neutralized by the V3 glycan-directed bNAb, PGT121 (Fig. 7E). In this context, PRB958 was more sensitive to PGT121, which had a 3.6-fold lower IC50 for PRB958 relative to SC45. These data suggest that the structural context and/or conformation of the MPER in cell-associated virus precludes its interaction with 10E8, in contrast to the case observed with cell-free virus. Furthermore, because 10E8 was able to completely block cell-free virus infection at the concentrations used in the cell–cell transmission assay, the data also suggest that viral spread in U87.CD4.CCR5 cells predominantly occurs via the cell-to-cell route.
Sequential passaging of a W672A mutant suggests a functional link between glycans in V1 and the MPER
An alternative approach was taken to identify determinants within Env that modulate the function of the MPER. The W666A and I675A HIV-1AD8 MPER mutants as well as a third well-characterized mutant, W672A (see Fig. 1, A and B), which all exhibit diminished replication capacity, were subjected to serial passaging in PBMCs to force the emergence of 2nd-site suppressor mutations. The location of the suppressor potentially indicates an element within gp120–gp41 that is linked to MPER function. W666A and I675A largely ablated the infectivity of cell-free AD8 for stimulated PBMCs, whereas the presence of a low level of RT activity in day-14 W672A culture supernatant indicates that this mutant retains a low level of replication competence (Fig. 8A). The U87.CD4.CCR5 assay revealed that W672A was competent to spread via the cell–cell route (Fig. 8B, right panel). The mutant cell-free viruses were subjected to six serial passages in PHA-stimulated PBMCs. Replication competence was not restored to the W666A mutant in two independent long-term cultures, consistent with the highly attenuated replication phenotype of this mutant (Fig. 8C, data not shown). By contrast, replication-competent viruses appeared in the W672A and I675A cultures during the second passage, and infectivity was maintained in subsequent passages. The env region was PCR-amplified from proviral DNA obtained from the W672A and I675A long-term culture at days 30 and 60 and ligated into the pcDNA3.1AD8env vector. The entire env region of individual clones was subjected to DNA sequencing. The W672A mutation was maintained in all clones obtained at both time points; however, 2nd site mutations leading to the deletion of either one of two potential PNGSs at Asn136 or Asn142 in V1 were observed in 10 of 13 day-60 clones (Fig. 8D). The Asn136 PNGS was lost either by deletion of 136NVTNI (ΔNVTNI) or the T138I substitution, whereas the adjacent Asn142 site was lost through deletion of 139NIN (ΔNIN). The 136 PNGS is well conserved across multiple clades of HIV-1, whereas the 142 PNGS is less conserved (Fig. 8E), and their deletion has been shown to affect fusion and infectivity in a strain-dependent manner (96–98). In the case of I675A, 50% of day-30 clones and 100% of day-60 clones contained an A675V pseudoreversion with no dominant second site mutations found (data not shown). Both Ile and Val have branched aliphatic side chains that differ by a single methyl group. As the I675V replacement is likely to be functionally neutral, this reversion mechanism was not examined further.
Figure 8.

In vitro evolution of pAD8-W672A. A, 14-day replication kinetics of WT and mutated AD8 viruses. Virus stocks produced in 293T cells were normalized according to RT activity and used to infect PHA-stimulated PBMCs. RT activity was measured in culture supernatants obtained at days 3, 7, 10, and 14 postinfection. The mean RT activity ± S.D. of duplicate samples is shown. B, U87.CD4.CCR5 cells were inoculated with unpseudotyped (left panel) or VSV G-pseudotyped (right panel) HIV-1AD8 particles (50,000 cpm of RT activity per inoculum) and then trypsinized 24 h later to remove residual adsorbed virus. The cells were then replated and cultured for a further 10 days. Mean RT activity ± S.D. of triplicate samples is shown. Data are representative of two independent experiments. C, long-term PBMC culture of WT and mutated AD8 viruses. Viruses produced by transfected 293T cells were normalized according to RT activity prior to infection of PHA-stimulated PBMCs. The PBMCs used in each passage were obtained from different donors. Cell-free virus collected at day 10 of each passage was normalized for RT activity and used to infect fresh PHA-stimulated PBMCs. The mean RT activity of duplicate samples is shown. D, frequency of genotypes observed in env clones obtained at days 30 and 60 from the W672A culture. E, alignment of V1 sequences, with potential N-linked glycosylation sites highlighted in green. F, migration of immunoprecipitated 35S-labeled gp120 molecules containing second site mutations in SDS-PAGE. G, 14-day replication kinetics of HIV-1AD8-W672A viruses ± 2nd site mutations in V1. Virus stocks produced in 293T cells were normalized according to RT activity and used to infect PHA-stimulated PBMCs. RT activity was measured in culture supernatants obtained at days 3, 7, 10, and 14 postinfection. The mean RT activity of duplicate samples is shown.
SDS-PAGE analysis of immunoprecipitated radiolabeled gp120 constructs containing the ΔNVTNI, T138I, and ΔNIN mutations indicated that the mutants migrated to lower molecular weight positions compared with WT, which is consistent with the loss of glycosylation (Fig. 8F). The ΔNVTNI, T138I, and ΔNIN mutations improved the ability of W672A-containing AD8 viruses to replicate in PBMCs (Fig. 8G), indicating that the loss of N-linked glycosylation in V1 partially suppresses the W672A infectivity defect and suggesting that this gp120 region is functionally linked to the MPER.
Discussion
Mutations in the MPER that inhibited cell-free virus infectivity were tolerated in virions transmitted via the cell-to-cell route and in cell–cell fusion mediated by the Env glycoproteins expressed at the cell surface. The data point to distinct structural and/or functional roles for the MPER in the two modes of viral spread and in cell–cell fusion. Substantial infectivity was restored to the cell-free viral MPER mutants by additional mutations in the CT (truncation to 712 and Y712S) that have been shown to functionally decouple Env from MA (62, 64, 65). Recent biochemical and electron microscopic studies suggest a direct interaction between CT and MA within virions where a hexameric assembly of MA trimers provides a docking site for the CT (61–63, 84). Thus, the effects of the MPER mutations on cell-free virus infectivity are to a degree controlled by the CT, most likely via interactions with MA. The Y712S CT mutation did not reverse a subtle gp120-incorporation defect seen with W666A and I675A mutant AD8 virions, suggesting that the restoration of infectivity to the mutants by Y712S does not involve changes in virion Env complex stability. Within cell-free virions, CT-mediated interactions control a number of functional characteristics of gp120–gp41, including the clustering of Env spikes (99), and their acquisition of fusion competence following Gag maturation (100), as well as Gag maturation-dependent antigenic changes in the Env ectodomain, including within the MPER (66). CT may modulate the role of the MPER present in cell-free virions in one or more of these functions.
In association with membrane mimics, the MPER includes a metastable tilted N-helix (amino acids 666–672) linked via a Phe673–Asp/Asn674 hinge to a stable near-flat C-helix (amino acids 675–683) (37–39). Cryo-EM of HIV-1 particles in the presence and absence of 10E8 Fab locates the MPER to the base of the Env trimer, embedded in the envelope in its ground state (50). Trp666, Trp672, and Ile675 are located on the apolar face of the MPER helices and penetrate into the acyl chain region by 4.5, 5, and 11 Å, respectively (37). The replacement of Trp and Ile with the helix-forming residue, Ala (101), potentially removes up to 148 and 74 Å2 of buried surface, respectively (102), which may diminish stabilizing protein–membrane interactions. In integral membrane proteins, Trp tends to locate at the membrane headgroup region, whereas Ile and Ala have a greater propensity to interact with the acyl chain region (103–105). The effects of W666A, W672A, and I675A on the binding free energy of the MPER to the phosphatidylcholine interface were therefore estimated using the Totalizer feature of MPEx (106). This analysis (Fig. S3) suggested that W666A and W672A cause increases in binding free energy. Furthermore, W666A was predicted to decrease the hydrophobic moment or amphiphilicity (107) of the N-helix, whereas, by contrast, W672A was predicted to increase this property. Thus, W666A and W672A may decrease the depth of penetration and alter the tilt and/or orientation of the N-helical segment in the interfacial region of the cell-free viral envelope, thereby blocking infectivity. By contrast, subtle changes in interfacial binding energy and hydrophobic moment were predicted for I675A. This mutation may therefore block the infectivity of cell-free virions by diminishing stabilizing interactions with the acyl chain region of the envelope.
Recent data suggest that the functional role of the CT in VS-transmitted viruses is distinct from that of cell-free viruses as small truncations and point mutations within the CT that are largely tolerated by viruses transmitted between Jurkat donors and MT4 targets impair the infectivity of cell-free virions (108). (Larger CT truncations were observed to block cell–cell transmission (108); however, this could be due to the inability of virions produced in Jurkat cells to incorporate Envs with large CT truncations (109).) Thus, the mode of interaction between the CT and MA within cell-free virions may impose an MPER structure that is perturbed by W666A and I675A
Fusion between infected T cells and uninfected T cells or macrophages and the formation of multinucleated syncytia may be associated with efficient cell-to-cell transfer of HIV-1 (24–28, 110). We observed that the W666A and I675A mutations did not affect the lag prior to fusion pore formation or the rate of fusion mediated by cell-surface–expressed Env. The intrinsic fusogenicity of Env expressed at the cell surface in the absence of other viral proteins is therefore not affected by ablation of lipid-interacting side chains at 666 and 675. It therefore appears that Env complexes present in virions transmitted via the cell-to-cell route resemble cell-surface–expressed glycoprotein forms with respect to the MPER being resistant to mutagenic alteration. Assuming that this MPER structure represents a native conformer, the replacement of lipid-buried residues such as Trp666, Trp672, and Ile675 with Ala may be functionally tolerated if there is sufficient flexibility to allow alternative side chains to mediate functional interactions with the viral envelope. A model summarizing the potential effects of the MPER mutations is presented in Fig. 9.
Figure 9.

Structural modulation of the MPER, a model. Left, depiction of the Env glycoprotein in a cell-free virion. The trimeric gp120–gp41 ectodomain was drawn using the coordinates from PDB code 5FYJ. The MPER (a monomer is shown for clarity) is embedded as a kinked helix in the polar headgroup (light blue)–acyl chain (white) interfacial region of the viral envelope as indicated by NMR studies of MPER peptides associated with membrane mimics (37, 38) and cryo-EM of HIV-1 particles (50). The hydrophobic acyl-chain–interactive face of the MPER helices is colored teal. The N-terminal segment of the CT (707–752) is unstructured in the absence of MA interactions (143) but becomes organized when engaged by MA trimers in hexameric arrays (62–64, 84). In this context, the MPER is in a conformation that is sensitive to mutations such as W666A, W672A, and I675A that potentially alter its interaction with the envelope. The effects of these mutations are mitigated by truncation of the CT or by Y712S, which uncouple Env from Gag due in part to disruption of the YXXL motif. In the SC54 isolate, the bridge formed between the V1-Asn142 and V4-Asn386 glycans and the V2–β-hairpin–stabilizing effects of the Pro183–Tyr191 interaction at the trimer apex confer a mutation-sensitive phenotype to the MPER via allosteric mechanisms. Right, in VS-transmitted virions, the MPER is resistant to mutations and neutralization by bNAb 10E8 perhaps due to an alternative CT conformation (108) and interaction with the MA domain. The various protein domains were drawn with PyMOL using the following coordinates: gp120–gp41 ectodomain, PDB code 5FYJ (120); MPER, PDB code 2PV6 (38); membrane-spanning sequence, PDB code 5JYN (144); CT, PDB code 5VWL (143); MA trimer, PDB code 1HIW (145).
A feature that potentially distinguishes the two modes of viral spread is the timing of virion maturation. Evidence has been provided for CD4-dependent transfer of immature viruses across the VS to the target cell, where they enter endocytic compartments and undergo Gag maturation and activation for virus–endosome fusion (57, 111, 112). In immature particles, interactions between the CT and the MA domain of the Gag precursor trap Env in a fusion-inactive state, Gag processing being required to release this trap (100, 113–115). Therefore, although cell-free virions encounter receptors in a mature, fusion-competent state, VS-transferred virions may be fusion-incompetent when they bind to CD4, requiring maturation within endosomes for the activation of fusion (57). As the MPER adopts distinct epitope conformations in immature and mature virions (66), it is plausible that the structure and function of the MPER are sensitive to disruption by W666A and I675A in a setting where Env–CD4 interactions occur in the context of mature Gag and fusion-competent Env, whereas these mutations are tolerated in cell–cell transmission where CD4 binds to Env prior to Gag maturation and virus–cell fusion.
Spreading infection in U87.CD4.CCR5 cells initiated by cell-associated AD8, SC45, and PRB958 viruses bearing W666A was more efficient than in PBMCs. Our observation that the 10E8 bNAb could not block cell–cell transmission between U87.CD4.CCR5 cells even though cell-free virus infection was completely neutralized by this bNAb suggests that the major mode of viral spread in these cells is via the cell-to-cell route. cryo-EM has shown HIV-1-budding sites of glioblastoma-derived cells such as U87 to be associated with lamellipodia-like areas and filopodial structures (116, 117), bearing some resemblance to those involved in VS-mediated viral transmission between immune cells (16, 118). It may be that VSs formed by such structures in adherent U87.CD4.CCR5 cells are sustained for long periods throughout the culture period leading to highly efficient cell–cell transmission, whereas T cell–T cell and monocytic cell–T cell VSs (as would form in PBMC cultures) are transient (8, 17, 18, 119). Using Quantibrite beads in flow cytometry, we estimated ∼10- and ∼4-fold higher CD4 and CCR5 expression levels, respectively, on U87.CD4.CCR5 cells versus PBMCs (Fig. S1). As CD4-Env interactions play a critical role in initiating VSs (8), the markedly higher CD4 numbers on U87.CD4.CCR5 cells is also consistent with more efficient viral transmission in this cell type. Cell-free virus infection may therefore make a greater contribution to spreading infection in PBMCs than in U87.CD4.CCR5 cells. This may explain why the slightly attenuated TZM-bl cell infectivity phenotype of cell-free PRB958-W666A correlated with that seen in PBMCs, whereas its infectivity was enhanced relative to WT in U87.CD4.CCR5 cells.
The analysis of SC45 and PRB958 T/F Envs revealed that the effects of W666A on cell-free virus infectivity are isolate-specific. The most striking difference was in U87.CD4.CCR5 cells where the infectivity of cell-free SC45-W666A was largely lost, but the infectivity of PRB958-W666A was enhanced. However, when infections were initiated with cell-associated SC45- and PRB958-W666A viruses, efficient viral spread in U87.CD4.CCR5 cells and PBMCs was observed for both mutants. Thus, the resilience of the MPER in cell–cell-transmitted virus to mutational disruption appears to be a conserved feature, whereas the MPER in cell-free virus responds to W666A in an isolate-specific manner. Domain swapping indicated that the SC45 V1, V2, and V4 domains in combination transferred most aspects of the attenuated cell-free virus infectivity phenotype of SC45-W666A to PRB958. For example, the gp120-shedding phenotype of SC45-W666A was replicated in PR.SC.V124-W666A, whereas PR.SC.V14-W666A exhibited a moderate shedding phenotype but a similarly low level of residual infectivity for TZM-bl cells. V1 and V4 may therefore modulate the function of the MPER as it relates to cell-free virus infectivity, whereas V2 influences gp120–gp41 association in this context. The defective infectivity phenotype of cell-free PR.SC.V14-W666A for TZM-bl cells was recapitulated in PBMCs, again suggesting that the cell-free mode of viral transmission is a major contributor to viral spread in this system.
To gain a better understanding of how V1 and V4 may synergize in modulating the structure and function of the MPER, two recently described crystal structures of disulfide-linked gp120–gp41 trimers (SOSIP.664) (120) were used as homology models of SC45 and PRB958. Like SC45, X1193.c1 SOSIP.664 contains a relatively long V1 loop with four PNGSs, whereas V4 contains five PNGSs. In contrast, the BG505 SOSIP.664 and PRB958 Env contain short V1 loops with two PNGSs but longer V4 loops containing five PNGSs (Fig. S4). X1193.c1 V1 forms an extended loop that points outwards from the 3-fold axis and overlays the V4 loop, allowing the Asn142-linked GlcNAc group to hydrogen-bond with the terminal mannose-9 moiety of the Asn386 glycan in V4 (Fig. S5). These PNGSs are conserved in SC45, suggesting that they could be a point of interaction between V1 and V4. In BG505, glycan–glycan interactions are mediated by the mannose-7 and mannose-5 moieties of the V1-Asn137 and V4-Asn386 glycans, respectively (Fig. S5), which are conserved in PRB958. These glycan-mediated interactions between V1 and V4 may provide a physical link between the two variable domains such that they act in tandem to modulate virus infectivity (Fig. 9). In contrast, the SC45 and PRB958 V3 sequences are highly conserved (Fig. S2) (81% identity, 100% homology), perhaps explaining why this loop is functionally interchangeable between the two isolates. The V2 loop of SC45 was found to largely contribute the gp120-incorporation defect of SC45-W666A to PRB958 when V1 or V1 plus V4 of the former were also present. The most obvious differences between the SC45 and PRB958 V2 sequences are the P183Q consensus-to-nonconsensus residue substitution and deletion of Asn186–Asn187 (Fig. S4). The X1193.c1 SOSIP.664 crystal structure suggests that P183Q would alter a stacking interaction with the conserved Tyr191 within a short β-sandwich that constrains a hairpin loop; the Asp186–Asn187 deletion would occur at the apex of this short hairpin. Interestingly, the loop is disordered in BG505 SOSIP.664, which contains Gln183 (120), suggesting that the Pro183–Tyr191 interaction in SC45 Env stabilizes this V2 segment (Fig. S5). Intermonomer V2–V3 interactions are believed to modulate the stability of the trimer apex (50). It is plausible that rigidity in the V2 hairpin of SC45 stabilizes the trimer apex against accommodating subtle structural perturbations associated with mutations that destabilize the Env complex. In common with SC45, AD8 also contains the Pro183–Ile–Asp–Asp186–Asn187 sequence in V2, and its gp120–gp41 complex is sensitive to W666A.
Taken together, the data suggest that amino acid changes in V1, V2, and V4 at the top of the trimer apex can allosterically modulate the structure and function of the MPER at the base. We did not detect differences in the global conformation of SC45 and PRB958 Envs in neutralization assays with the PGT151, VRC01, and PGT121 bNAbs, suggesting that the allosteric modulation of the MPER by the variable domains may involve subtle structural changes in the Env ectodomain. We have previously shown that loss of the V1 glycans at Asn136 or Asn141/Asn142 brings about changes in the gp120–gp41 association site (96). We have also found that restoration of infectivity to a defective DSR double mutant (W596L and K601D) involves a D674E mutation in the MPER (58). These results suggest that the 136/142 glycans in V1 are allosterically linked to the function of the DSR, which is in turn linked to the function of the MPER. There is also evidence that the central helical region 1 (HR1) coiled coil of gp41 may link CD4-induced conformational changes and the MPER as T569A mutation in HR1 is associated with increased exposure of epitopes in both the MPER and CD4bs (121). Thus, the functional link between the variable loops of gp120 and the MPER is likely to involve multiple structural elements within Env.
Independent evidence for a functional linkage between V1 and the MPER was provided by the finding that the cell-free virus replication defect of AD8-W672A could be suppressed by mutations in V1 that ablate N-linked glycosylation at Asn136 (ΔN136VTNI or T138I) or Asn141 (ΔN139IN). However, in contrast to the SC45-PRB958 domain swapping data, changes in V4 did not accompany the V1 changes in the context of AD8 revertants. Although this could be an isolate-specific phenomenon, we note that the W672A clones containing V1 modifications exhibited delayed replication kinetics relative to WT, suggesting that other suppressor mutations are required to restore full infectivity to W672A. We previously observed that loss of the Asn136 and Asn141/142 glycans in AD8 Env do not affect cell-free virus neutralization by 2F5 (96) nor binding by 10E8 to HIV-like particles incorporating the Env mutants (data not shown). However, the glycan deletions restored gp120–gp41 association and membrane fusion function to Envs bearing mutations in the disulfide-bonded region of gp41 (96), which mediates contacts with gp120. These data suggest that the glycan changes in V1 restore function to W672A by an indirect mechanism involving the gp120–gp41 association site.
MPER-directed bNAbs have broad neutralizing activity in vitro (44, 122), and their passive transfer to macaques contributes to protection from mucosal challenge with cell-free pathogenic chimeric simian-HIV (123, 124), stimulating intense study of this region as a potential vaccine target. Our findings indicate that the MPER may have distinct conformations and functions in cell-free versus cell–cell viral spread that correlate with neutralization resistance in the latter. One potential mechanism for resistance to MPER-directed bNAbs is an Env trimer conformation in VS-associated virions that denies antibody access to the MPER. This idea is illustrated by the finding that the Asn88 and Asn625 N-linked glycans of gp120 can sterically hinder antibody–MPER interactions (125). Ruprecht et al. (126) showed that the neutralization activity of MPER-directed bNAbs is associated with the induction of gp120 shedding. It is possible that VS-associated virions can resist gp120 shedding in response to perturbations of MPER structure that are known to be induced by antibody binding (37, 38, 44, 48–51).
Overall, these findings have important implications for MPER-targeted vaccine design, particularly if the cell-to-cell mode of viral spread represents an important transmission mode in vivo (22–24, 27, 28). The findings that the cell-to-cell HIV-1 spread between primary T cells (35), primary MDMs and T cells (19), and U87.CD4.CCR5 cells is relatively resistant to neutralization by MPER-directed bNAbs compared with gp120-directed bNAbs (19, 34, 35) suggest that vaccine design should focus on bNAb epitopes expressed in other regions of the gp120–gp41 complex.
Experimental procedures
Env expression vectors and proviral clones
The construction of WT and W666A-, W672A-, and I675A-mutated pCDNA3.1-AD8env vectors is described elsewhere (52, 89). Termination codons and the Y712S mutation were introduced to the gp41 CT using overlap extension PCR. The env mutations were introduced to the pAD8 infectious clone (obtained from K. Peden (127), NIAID, National Institutes of Health) by transferring the EcoRI–BspMI env-containing fragment from pCDNA3.1-AD8env vectors into pAD8. The L8S/S9R mutation (59, 60) was introduced to the MA domain of pAD8 by the QuikChange II XL protocol (Agilent). The sequences of the mutated open reading frames were confirmed using BigDye terminator version 3.1 (ABI). Vectors encoding the env region of the clade B T/F isolates, SC45.4B5.2631 and PRB958_06.TB1.4305, were obtained through the AIDS Reagent Program, Division of AIDS, NIAID, National Institutes of Health, from Drs. Beatrice H. Hahn, Brandon F. Keele, and George M. Shaw (90). Chimeric infectious clones containing the SC45 and PRB958 env regions in a pNL4.3 background (AIDS Reagent Program, Division of AIDS, NIAID, National Institutes of Health, from Dr. Malcolm Martin (128)) were prepared by replacing the KpnI–BamHI (nucleotides 6343–8465) or KpnI–PpuMI (nucleotides 6343–8389) regions of pNL4.3 with the corresponding SC45 and PRB958 regions, respectively.
Infection of U87.CD4.CCR5 cells
Infection of U87.CD4.CCR5 cells (from H. Deng and D. Littman (54), AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, National Institutes of Health) was carried out as described (58). Briefly, virus stocks were prepared by transfecting 293T cell (American Type Culture Collection) monolayers with infectious clones using FuGENE HD (Roche Applied Science). Virus-containing transfection supernatants were normalized according to RT activity (129) or p24 content (National Cancer Institute-Frederick) and used to infect U87.CD4.CCR5 monolayers in 25-cm2 culture flasks. The supernatants were assayed for RT activity or p24 content at various time points. To assess cell-to-cell transmission, HIV-1 particles were pseudotyped with VSV G by cotransfection of 293T cells with pAD8 infectious clones and pHEF-VSV G (from Dr. L.-J. Chang (130), AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, National Institutes of Health). VSV G-pseudotyped virus-containing transfection supernatants were normalized according to RT activity (129) or p24 content as for nonpseudotyped viruses. U87.CD4.CCR5 monolayers in 25-cm2 culture flasks were inoculated with the HIV-VSV G pseudotypes, and then, at 24-h postinfection, trypsinized to remove surface-adsorbed virions. The cells were washed with PBS, replated, and then cultured for 10–14 days. The culture supernatants were assayed for RT activity or p24 content at various time points.
Infection of PBMCs
Phytohemagglutinin-stimulated PBMCs (106) (prepared from buffy packs obtained from the Australian Red Cross Blood Bank, as described previously (123)) were plated in 12-well culture dishes in RPMI 1640 medium, 10% FCS, and 2 units of IL-2 and incubated at 37 °C for 3 h in a CO2 incubator. Phytohemagglutinin and IL-2 were obtained from Sigma and Genscript, respectively. To examine cell-associated virus transmission, the cells were infected with VSV G-pseudotyped viruses (inocula normalized according to p24 content) and incubated for 24 h at 37 °C in a CO2 incubator. The infected cells were washed three times with PBS and then cocultured with 5 × 106 naive PHA-stimulated PBMC targets in 2 ml of RPMI 1640 medium, 10% FCS/IL-2 for 10 days at 37 °C in a CO2 incubator. To examine the infectivity of cell-free viruses, 106 PHA-stimulated PBMCs were infected with nonpseudotyped viruses (inocula normalized according to p24 content) and cultured in 2 ml of RPMI 1640 medium, 10% FCS/IL-2 for 10 days at 37 °C in a CO2 incubator. The supernatants were assayed for p24 content at days 3, 7, and 10. In some cases, the infectivity of cell-free HIV-1 for PBMCs was determined as described previously (129). Equal amounts of virus (normalized according to RT activity) were serially diluted (10-fold) in a 96-well plate containing RPMI 1640 medium, 10% FCS and IL-2. PHA-stimulated PBMCs (105) were added to each well in a final volume of 200 μl. The plate was incubated at 37 °C, and at days 3, 7, 10, and 14, the supernatants were assayed for RT activity.
Infection of TZM-bl cells
TZM-bl cells, a HeLa cell line expressing CD4 and CCR5 and harboring integrated copies of the luciferase and β-gal genes under control of the HIV-1 promoter, were obtained from J. C. Kappes, X. Wu, and Tranzyme Inc., AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, National Institutes of Health (131–133). Serially diluted nonpseudotyped and VSV G-pseudotyped virus stocks were used to inoculate the TZM-bl cells in triplicate (104 cells in 100 μl per well of a 96-well tissue culture plate). Two days later, the cells were lysed and assayed for luciferase activity (Promega).
Single cycle infectivity assays
Single-cycle infectivity assays were conducted as described previously (52). Env-pseudotyped luciferase reporter viruses were produced by cotransfecting 293T cells with Env expression vectors plus the luciferase reporter virus vector, pNL4.3.Luc.R−E− (N. Landau, AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, National Institutes of Health (134)). The infectivity of pseudotyped viruses was determined in U87.CD4.CCR5 cells using the Promega luciferase assay system at 48 h postinfection.
Real-time cell–cell fusion assay employing β-lactamase
Real-time fusion kinetics were determined using a modification of the assay originally described by Lineberger et al. (85). 293T effector cells (2.5 × 105 cells/well of a 12-well culture plate) were cotransfected with pCDNA3.1-AD8env and pCDNA3.1-β-lactamase plasmids. At 30 h post-transfection, JC53 target cells (from D. Kabat, Oregon Health and Science University (135)) were seeded at a density of 4 × 104 cells/well in a clear-bottom black-sided 96-well plate (BMG). At 48 h post-transfection, the JC53 targets were labeled with 5 μm CCF2-AM (Invitrogen) for 60 min at room temperature and then washed twice in PBS. 293T effector cells (2 × 104, Hanks'-buffered saline containing 2 mm glutamine, 2 μm HEPES, and 2 mm probenicid) were then added to the JC53 targets in triplicate. Fluorescence was quantified using a BMG FLUOstar fluorimeter, using a 405-nm excitation filter, and 460-nm (blue fluorescence) and 538-nm (green fluorescence) emission filters. The cells were maintained at 37 °C, and readings were taken every 20 min for 180 min. The ratio of blue-green fluorescence was normalized against the final extent of fusion to determine the lag time and rate of fusion.
Neutralization assays
Cell-free virus neutralization assays were conducted, as described previously (96, 136). Briefly, Env-pseudotyped NL4.3.Luc.R−E− luciferase reporter viruses were incubated with an equal volume of serially diluted IgG for 1 h at 37 °C. The virus/IgG mixtures were then added to U87.CD4.CCR5 cells (104 cells per well of a 96-well tissue culture plate) and incubated for 2 days prior to lysis and assay for luciferase activity (Promega, Madison, WI). For cell-associated virus neutralization assays, U87.CD4.CCR5 cells were infected with VSV G-pseudotyped HIV-1 particles and then treated with trypsin prior to washing and replating in 96-well tissue culture plates in the presence of serially diluted bNAbs. At day 3, 50% of the culture supernatant was replaced with fresh medium containing the appropriate dilution of bNAb. The p24 content of each well was determined at 7 days postinfection using p24 ELISA. Purified IgG of bNAbs PG9 and PGT121 (93, 94) was obtained from the IAVI Neutralizing Antibody Consortium; 2F5 and 4E10 were obtained from the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, National Institutes of Health, from H. Katinger; and VRC01 (137), 10E8 (44), and PGT151 (91, 138) and the hepatitis C virus E2 glycoprotein bNAb HC84.1 (139) were produced in Freestyle 293 cells (Invitrogen) following cotransfection with pCDNA3-based IgG1 heavy and light chain expression vectors containing the VRC01, 10E8, PGT151, and HC84.1 variable regions that were produced in-house (Fig. S6).
Western blotting
Supernatants from 293T or HeLa cells (American Type Culture Collection) transfected with infectious clones were filtered through 0.45-μm nitrocellulose filters and then centrifuged over 1.5 ml of 25% w/v sucrose/PBS cushions (Beckman SW41 Ti rotor, 25,000 rpm, 2.5 h, 4 °C) prior to reducing SDS-PAGE and Western blotting with DV-012 (AIDS Reagent Program, Division of AIDS, NIAID, National Institutes of Health, from Dr. Michael Phelan) to detect gp120 and pooled IgG from HIV-1–infected individuals to detect Gag proteins. To detect viral proteins in cell lysates, transfected 293T cells were washed with PBS and then lysed in PBS, 1% Triton X-100 containing 1 mm EDTA, 1 mm PMSF, leupeptin, and aprotinin (Sigma). The lysates were clarified by centrifugation and then subjected to reducing SDS-PAGE and Western blotting with DV-012 to detect gp120.
Luciferase assay of cell–cell fusion
The ability of cell-surface-expressed Env proteins derived from pAD8 proviral vectors to mediate cell–cell fusion was determined as described previously (52). Briefly, 293T cells were cotransfected with pAD8 proviral vectors and the bacteriophage T7 RNA polymerase expression vector, pCAG-T7 (140). Twenty four hours later, BHK21 target cells (American Type Culture Collection) were cotransfected with pc.CCR5 (AIDS Reagent Program, Division of AIDS, NIAID, National Institutes of Health, from N. Landau (141)) and pT4luc, a bicistronic vector that expresses human CD4 from a CMV promoter and firefly luciferase from a T7 promoter (142), and incubated for a further 24 h. Targets and effectors were then cocultured in triplicate in a 96-well plate (18 h, 37 °C) and assayed for luciferase activity (SteadyGlo, Promega).
Immunoprecipitation
gp120 proteins were expressed in 293T cells and biosynthetically labeled with 35S-Met/Cys (PerkinElmer Life Sciences) for 40 min and then chased with unlabeled media for 6 h. The glycoproteins were immunoprecipitated with IgG from a HIV-positive individual and protein G-Sepharose and then subjected to reducing SDS-PAGE and scanning in a phosphorimager.
Author contributions
V. G. S. N., A. K. B.-M., D. N. H., H. A. D. K., H. E. D., and P. P. formal analysis; V. G. S. N., A. K. B.-M., C.-S. L., D. N. H., H. A. D. K., and P. P. validation; V. G. S. N., A. K. B.-M., A. E. L., C.-S. L., D. N. H., H. A. D. K., and P. P. investigation; V. G. S. N., A. K. B.-M., A. E. L., C.-S. L., D. N. H., H. A. D. K., and P. P. methodology; V. G. S. N., A. K. B.-M., A. E. L., C.-S. L., D. N. H., H. A. D. K., H. E. D., and P. P. writing-review and editing; A. K. B.-M., H. E. D., and P. P. conceptualization; A. K. B.-M. and P. P. writing-original draft; H. E. D. and P. P. supervision; P. P. data curation; P. P. funding acquisition; P. P. project administration.
Supplementary Material
This work was supported by National Health and Medical Research Council of Australia Grants 1070890 and 1125822 (to P. P.) and 1041897 (to H. E. D.) and the Australian Centre for HIV and Hepatitis Virology Research (to P. P.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S6 and supporting Refs. 1–5.
- Env
- envelope glycoprotein
- MPER
- membrane proximal ectodomain region
- PBMC
- peripheral blood mononuclear cell
- bNAb
- broad neutralizing antibody
- CT
- cytoplasmic tail
- MA
- matrix protein
- T/F
- transmitted/founder
- VS
- virological synapse
- MDM
- monocyte-derived macrophage
- RT
- reverse transcriptase
- WLKD
- W596L/K601D
- RLU
- relative light units
- LLP
- lentiviral lytic helical peptide
- PHA
- phytohemagglutinin
- SC45
- SC45.4B5.2631
- PRB958
- PRB958_06.TB1.4305
- PDB
- Protein Data Bank.
References
- 1. Bhattacharya J., Repik A., and Clapham P. R. (2006) Gag regulates association of human immunodeficiency virus type 1 envelope with detergent-resistant membranes. J. Virol. 80, 5292–5300 10.1128/JVI.01469-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Leung K., Kim J. O., Ganesh L., Kabat J., Schwartz O., and Nabel G. J. (2008) HIV-1 assembly: viral glycoproteins segregate quantally to lipid rafts that associate individually with HIV-1 capsids and virions. Cell Host Microbe 3, 285–292 10.1016/j.chom.2008.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lindwasser O. W., and Resh M. D. (2001) Multimerization of human immunodeficiency virus type 1 Gag promotes its localization to barges, raft-like membrane microdomains. J. Virol. 75, 7913–7924 10.1128/JVI.75.17.7913-7924.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Muranyi W., Malkusch S., Müller B., Heilemann M., and Kräusslich H. G. (2013) Super-resolution microscopy reveals specific recruitment of HIV-1 envelope proteins to viral assembly sites dependent on the envelope C-terminal tail. PLoS Pathog. 9, e1003198 10.1371/journal.ppat.1003198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ono A., and Freed E. O. (2001) Plasma membrane rafts play a critical role in HIV-1 assembly and release. Proc. Natl. Acad. Sci. U.S.A. 98, 13925–13930 10.1073/pnas.241320298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nguyen D. H., and Hildreth J. E. (2000) Evidence for budding of human immunodeficiency virus type 1 selectively from glycolipid-enriched membrane lipid rafts. J. Virol. 74, 3264–3272 10.1128/JVI.74.7.3264-3272.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hübner W., McNerney G. P., Chen P., Dale B. M., Gordon R. E., Chuang F. Y., Li X. D., Asmuth D. M., Huser T., and Chen B. K. (2009) Quantitative 3D video microscopy of HIV transfer across T cell virological synapses. Science 323, 1743–1747 10.1126/science.1167525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jolly C., Kashefi K., Hollinshead M., and Sattentau Q. J. (2004) HIV-1 cell to cell transfer across an Env-induced, actin-dependent synapse. J. Exp. Med. 199, 283–293 10.1084/jem.20030648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jolly C., and Sattentau Q. J. (2005) Human immunodeficiency virus type 1 virological synapse formation in T cells requires lipid raft integrity. J. Virol. 79, 12088–12094 10.1128/JVI.79.18.12088-12094.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Groppelli E., Starling S., and Jolly C. (2015) Contact-induced mitochondrial polarization supports HIV-1 virological synapse formation. J. Virol. 89, 14–24 10.1128/JVI.02425-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jolly C., Mitar I., and Sattentau Q. J. (2007) Requirement for an intact T-cell actin and tubulin cytoskeleton for efficient assembly and spread of human immunodeficiency virus type 1. J. Virol. 81, 5547–5560 10.1128/JVI.01469-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jolly C., Welsch S., Michor S., and Sattentau Q. J. (2011) The regulated secretory pathway in CD4(+) T cells contributes to human immunodeficiency virus type-1 cell-to-cell spread at the virological synapse. PLoS Pathog. 7, e1002226 10.1371/journal.ppat.1002226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gardiner J. C., Mauer E. J., and Sherer N. M. (2016) HIV-1 Gag, envelope, and extracellular determinants cooperate to regulate the stability and turnover of virological synapses. J. Virol. 90, 6583–6597 10.1128/JVI.00600-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Martin N., and Sattentau Q. (2009) Cell-to-cell HIV-1 spread and its implications for immune evasion. Curr. Opin. HIV AIDS 4, 143–149 10.1097/COH.0b013e328322f94a [DOI] [PubMed] [Google Scholar]
- 15. Blanchet F., Moris A., Mitchell J. P., and Piguet V. (2011) A look at HIV journey: from dendritic cells to infection spread in CD4(+) T cells. Curr. Opin. HIV AIDS 6, 391–397 10.1097/COH.0b013e328349b0a0 [DOI] [PubMed] [Google Scholar]
- 16. Felts R. L., Narayan K., Estes J. D., Shi D., Trubey C. M., Fu J., Hartnell L. M., Ruthel G. T., Schneider D. K., Nagashima K., Bess J. W. Jr., Bavari S., Lowekamp B. C., Bliss D., Lifson J. D., and Subramaniam S. (2010) 3D visualization of HIV transfer at the virological synapse between dendritic cells and T cells. Proc. Natl. Acad. Sci. U.S.A. 107, 13336–13341 10.1073/pnas.1003040107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Groot F., Welsch S., and Sattentau Q. J. (2008) Efficient HIV-1 transmission from macrophages to T cells across transient virological synapses. Blood 111, 4660–4663 10.1182/blood-2007-12-130070 [DOI] [PubMed] [Google Scholar]
- 18. Martin N., Welsch S., Jolly C., Briggs J. A., Vaux D., and Sattentau Q. J. (2010) Virological synapse-mediated spread of human immunodeficiency virus type 1 between T cells is sensitive to entry inhibition. J. Virol. 84, 3516–3527 10.1128/JVI.02651-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Duncan C. J., Williams J. P., Schiffner T., Gärtner K., Ochsenbauer C., Kappes J., Russell R. A., Frater J., and Sattentau Q. J. (2014) High-multiplicity HIV-1 infection and neutralizing antibody evasion mediated by the macrophage-T cell virological synapse. J. Virol. 88, 2025–2034 10.1128/JVI.03245-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Del Portillo A., Tripodi J., Najfeld V., Wodarz D., Levy D. N., and Chen B. K. (2011) Multiploid inheritance of HIV-1 during cell-to-cell infection. J. Virol. 85, 7169–7176 10.1128/JVI.00231-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Russell R. A., Martin N., Mitar I., Jones E., and Sattentau Q. J. (2013) Multiple proviral integration events after virological synapse-mediated HIV-1 spread. Virology 443, 143–149 10.1016/j.virol.2013.05.005 [DOI] [PubMed] [Google Scholar]
- 22. Zhang C., Zhou S., Groppelli E., Pellegrino P., Williams I., Borrow P., Chain B. M., and Jolly C. (2015) Hybrid spreading mechanisms and T cell activation shape the dynamics of HIV-1 infection. PLoS Comput. Biol. 11, e1004179 10.1371/journal.pcbi.1004179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Iwami S., Takeuchi J. S., Nakaoka S., Mammano F., Clavel F., Inaba H., Kobayashi T., Misawa N., Aihara K., Koyanagi Y., and Sato K. (2015) Cell-to-cell infection by HIV contributes over half of virus infection. eLife 4, e08150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Murooka T. T., Deruaz M., Marangoni F., Vrbanac V. D., Seung E., von Andrian U. H., Tager A. M., Luster A. D., and Mempel T. R. (2012) HIV-infected T cells are migratory vehicles for viral dissemination. Nature 490, 283–287 10.1038/nature11398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Orenstein J. M. (2000) In vivo cytolysis and fusion of human immunodeficiency virus type 1-infected lymphocytes in lymphoid tissue. J. Infect. Dis. 182, 338–342 10.1086/315640 [DOI] [PubMed] [Google Scholar]
- 26. Orenstein J. M. (2008) HIV expression in surgical specimens. AIDS Res. Hum. Retroviruses 24, 947–955 10.1089/aid.2008.0265 [DOI] [PubMed] [Google Scholar]
- 27. Symeonides M., Murooka T. T., Bellfy L. N., Roy N. H., Mempel T. R., and Thali M. (2015) HIV-1-induced small T cell syncytia can transfer virus particles to target cells through transient contacts. Viruses 7, 6590–6603 10.3390/v7122959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Law K. M., Komarova N. L., Yewdall A. W., Lee R. K., Herrera O. L., Wodarz D., and Chen B. K. (2016) In vivo HIV-1 cell-to-cell transmission promotes multicopy micro-compartmentalized infection. Cell Rep. 15, 2771–2783 10.1016/j.celrep.2016.05.059 [DOI] [PubMed] [Google Scholar]
- 29. Agosto L. M., Zhong P., Munro J., and Mothes W. (2014) Highly active antiretroviral therapies are effective against HIV-1 cell-to-cell transmission. PLoS Pathog. 10, e1003982 10.1371/journal.ppat.1003982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Duncan C. J., Russell R. A., and Sattentau Q. J. (2013) High multiplicity HIV-1 cell-to-cell transmission from macrophages to CD4+ T cells limits antiretroviral efficacy. AIDS 27, 2201–2206 10.1097/QAD.0b013e3283632ec4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sigal A., Kim J. T., Balazs A. B., Dekel E., Mayo A., Milo R., and Baltimore D. (2011) Cell-to-cell spread of HIV permits ongoing replication despite antiretroviral therapy. Nature 477, 95–98 10.1038/nature10347 [DOI] [PubMed] [Google Scholar]
- 32. Titanji B. K., Aasa-Chapman M., Pillay D., and Jolly C. (2013) Protease inhibitors effectively block cell-to-cell spread of HIV-1 between T cells. Retrovirology 10, 161 10.1186/1742-4690-10-161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Durham N. D., Yewdall A. W., Chen P., Lee R., Zony C., Robinson J. E., and Chen B. K. (2012) Neutralization resistance of virological synapse-mediated HIV-1 Infection is regulated by the gp41 cytoplasmic tail. J. Virol. 86, 7484–7495 10.1128/JVI.00230-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gombos R. B., Kolodkin-Gal D., Eslamizar L., Owuor J. O., Mazzola E., Gonzalez A. M., Korioth-Schmitz B., Gelman R. S., Montefiori D. C., Haynes B. F., and Schmitz J. E. (2015) Inhibitory effect of individual or combinations of broadly neutralizing antibodies and antiviral reagents against cell-free and cell-to-cell HIV-1 transmission. J. Virol. 89, 7813–7828 10.1128/JVI.00783-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Malbec M., Porrot F., Rua R., Horwitz J., Klein F., Halper-Stromberg A., Scheid J. F., Eden C., Mouquet H., Nussenzweig M. C., and Schwartz O. (2013) Broadly neutralizing antibodies that inhibit HIV-1 cell to cell transmission. J. Exp. Med. 210, 2813–2821 10.1084/jem.20131244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Reh L., Magnus C., Schanz M., Weber J., Uhr T., Rusert P., and Trkola A. (2015) Capacity of broadly neutralizing antibodies to inhibit HIV-1 cell-cell transmission is strain- and epitope-dependent. PLoS Pathog. 11, e1004966 10.1371/journal.ppat.1004966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Song L., Sun Z. Y., Coleman K. E., Zwick M. B., Gach J. S., Wang J. H., Reinherz E. L., Wagner G., and Kim M. (2009) Broadly neutralizing anti-HIV-1 antibodies disrupt a hinge-related function of gp41 at the membrane interface. Proc. Natl. Acad. Sci. U.S.A. 106, 9057–9062 10.1073/pnas.0901474106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sun Z. Y., Oh K. J., Kim M., Yu J., Brusic V., Song L., Qiao Z., Wang J. H., Wagner G., and Reinherz E. L. (2008) HIV-1 broadly neutralizing antibody extracts its epitope from a kinked gp41 ectodomain region on the viral membrane. Immunity 28, 52–63 10.1016/j.immuni.2007.11.018 [DOI] [PubMed] [Google Scholar]
- 39. Sun Z. Y., Cheng Y., Kim M., Song L., Choi J., Kudahl U. J., Brusic V., Chowdhury B., Yu L., Seaman M. S., Bellot G., Shih W. M., Wagner G., and Reinherz E. L. (2014) Disruption of helix-capping residues 671 and 674 reveals a role in HIV-1 entry for a specialized hinge segment of the membrane proximal external region of gp41. J. Mol. Biol. 426, 1095–1108 10.1016/j.jmb.2013.09.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Muñoz-Barroso I., Salzwedel K., Hunter E., and Blumenthal R. (1999) Role of the membrane-proximal domain in the initial stages of human immunodeficiency virus type 1 envelope glycoprotein-mediated membrane fusion. J. Virol. 73, 6089–6092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Salzwedel K., West J. T., and Hunter E. (1999) A conserved tryptophan-rich motif in the membrane-proximal region of the human immunodeficiency virus type 1 gp41 ectodomain is important for Env-mediated fusion and virus infectivity. J. Virol. 73, 2469–2480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Buzon V., Natrajan G., Schibli D., Campelo F., Kozlov M. M., and Weissenhorn W. (2010) Crystal structure of HIV-1 gp41 including both fusion peptide and membrane proximal external regions. PLoS Pathog. 6, e1000880 10.1371/journal.ppat.1000880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lay C. S., Ludlow L. E., Stapleton D., Bellamy-McIntyre A. K., Ramsland P. A., Drummer H. E., and Poumbourios P. (2011) Role for the terminal clasp of HIV-1 gp41 glycoprotein in the initiation of membrane fusion. J. Biol. Chem. 286, 41331–41343 10.1074/jbc.M111.299826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Huang J., Ofek G., Laub L., Louder M. K., Doria-Rose N. A., Longo N. S., Imamichi H., Bailer R. T., Chakrabarti B., Sharma S. K., Alam S. M., Wang T., Yang Y., Zhang B., Migueles S. A., et al. (2012) Broad and potent neutralization of HIV-1 by a gp41-specific human antibody. Nature 491, 406–412 10.1038/nature11544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Muster T., Steindl F., Purtscher M., Trkola A., Klima A., Himmler G., Rüker F., and Katinger H. (1993) A conserved neutralizing epitope on gp41 of human immunodeficiency virus type 1. J. Virol. 67, 6642–6647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zwick M. B., Labrijn A. F., Wang M., Spenlehauer C., Saphire E. O., Binley J. M., Moore J. P., Stiegler G., Katinger H., Burton D. R., and Parren P. W. (2001) Broadly neutralizing antibodies targeted to the membrane-proximal external region of human immunodeficiency virus type 1 glycoprotein gp41. J. Virol. 75, 10892–10905 10.1128/JVI.75.22.10892-10905.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hessell A. J., Rakasz E. G., Tehrani D. M., Huber M., Weisgrau K. L., Landucci G., Forthal D. N., Koff W. C., Poignard P., Watkins D. I., and Burton D. R. (2010) Broadly neutralizing monoclonal antibodies 2F5 and 4E10 directed against the human immunodeficiency virus type 1 gp41 membrane-proximal external region protect against mucosal challenge by simian-human immunodeficiency virus SHIVBa-L. J. Virol. 84, 1302–1313 10.1128/JVI.01272-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ofek G., Tang M., Sambor A., Katinger H., Mascola J. R., Wyatt R., and Kwong P. D. (2004) Structure and mechanistic analysis of the anti-human immunodeficiency virus type 1 antibody 2F5 in complex with its gp41 epitope. J. Virol. 78, 10724–10737 10.1128/JVI.78.19.10724-10737.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cardoso R. M., Zwick M. B., Stanfield R. L., Kunert R., Binley J. M., Katinger H., Burton D. R., and Wilson I. A. (2005) Broadly neutralizing anti-HIV antibody 4E10 recognizes a helical conformation of a highly conserved fusion-associated motif in gp41. Immunity 22, 163–173 10.1016/j.immuni.2004.12.011 [DOI] [PubMed] [Google Scholar]
- 50. Lee J. H., Ozorowski G., and Ward A. B. (2016) Cryo-EM structure of a native, fully glycosylated, cleaved HIV-1 envelope trimer. Science 351, 1043–1048 10.1126/science.aad2450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pejchal R., Gach J. S., Brunel F. M., Cardoso R. M., Stanfield R. L., Dawson P. E., Burton D. R., Zwick M. B., and Wilson I. A. (2009) A conformational switch in human immunodeficiency virus gp41 revealed by the structures of overlapping epitopes recognized by neutralizing antibodies. J. Virol. 83, 8451–8462 10.1128/JVI.00685-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bellamy-McIntyre A. K., Lay C. S., Baär S., Maerz A. L., Talbo G. H., Drummer H. E., and Poumbourios P. (2007) Functional links between the fusion peptide-proximal polar segment and membrane-proximal region of human immunodeficiency virus gp41 in distinct phases of membrane fusion. J. Biol. Chem. 282, 23104–23116 10.1074/jbc.M703485200 [DOI] [PubMed] [Google Scholar]
- 53. Allen M., Bjerke M., Edlund H., Nelander S., and Westermark B. (2016) Origin of the U87MG glioma cell line: Good news and bad news. Sci. Transl. Med. 8, 354re3 [DOI] [PubMed] [Google Scholar]
- 54. Björndal A., Deng H., Jansson M., Fiore J. R., Colognesi C., Karlsson A., Albert J., Scarlatti G., Littman D. R., and Fenyö E. M. (1997) Coreceptor usage of primary human immunodeficiency virus type 1 isolates varies according to biological phenotype. J. Virol. 71, 7478–7487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jolly C., Mitar I., and Sattentau Q. J. (2007) Adhesion molecule interactions facilitate human immunodeficiency virus type 1-induced virological synapse formation between T cells. J. Virol. 81, 13916–13921 10.1128/JVI.01585-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kondo S., Yin D., Takeuchi J., Morimura T., Miyatake S. I., Nakatsu S., Oda Y., and Kikuchi H. (1994) Tumour necrosis factor-α induces an increase in susceptibility of human glioblastoma U87-MG cells to natural killer cell-mediated lysis. Br. J. Cancer 69, 627–632 10.1038/bjc.1994.123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Dale B. M., McNerney G. P., Thompson D. L., Hubner W., de Los Reyes K., Chuang F. Y., Huser T., and Chen B. K. (2011) Cell-to-cell transfer of HIV-1 via virological synapses leads to endosomal virion maturation that activates viral membrane fusion. Cell Host Microbe 10, 551–562 10.1016/j.chom.2011.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Khasawneh A. I., Laumaea A., Harrison D. N., Bellamy-McIntyre A. K., Drummer H. E., and Poumbourios P. (2013) Forced virus evolution reveals functional crosstalk between the disulfide bonded region and membrane proximal ectodomain region of HIV-1 gp41. Retrovirology 10, 44 10.1186/1742-4690-10-44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Dorfman T., Mammano F., Haseltine W. A., and Göttlinger H. G. (1994) Role of the matrix protein in the virion association of the human immunodeficiency virus type 1 envelope glycoprotein. J. Virol. 68, 1689–1696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Monel B., Beaumont E., Vendrame D., Schwartz O., Brand D., and Mammano F. (2012) HIV cell-to-cell transmission requires the production of infectious virus particles and does not proceed through env-mediated fusion pores. J. Virol. 86, 3924–3933 10.1128/JVI.06478-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Alfadhli A., Mack A., Ritchie C., Cylinder I., Harper L., Tedbury P. R., Freed E. O., and Barklis E. (2016) Trimer enhancement mutation effects on HIV-1 matrix protein binding activities. J. Virol. 90, 5657–5664 10.1128/JVI.00509-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tedbury P. R., Ablan S. D., and Freed E. O. (2013) Global rescue of defects in HIV-1 envelope glycoprotein incorporation: implications for matrix structure. PLoS Pathog. 9, e1003739 10.1371/journal.ppat.1003739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Tedbury P. R., Novikova M., Ablan S. D., and Freed E. O. (2016) Biochemical evidence of a role for matrix trimerization in HIV-1 envelope glycoprotein incorporation. Proc. Natl. Acad. Sci. U.S.A. 113, E182–E190 10.1073/pnas.1516618113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Davis M. R., Jiang J., Zhou J., Freed E. O., and Aiken C. (2006) A mutation in the human immunodeficiency virus type 1 Gag protein destabilizes the interaction of the envelope protein subunits gp120 and gp41. J. Virol. 80, 2405–2417 10.1128/JVI.80.5.2405-2417.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Edwards T. G., Wyss S., Reeves J. D., Zolla-Pazner S., Hoxie J. A., Doms R. W., and Baribaud F. (2002) Truncation of the cytoplasmic domain induces exposure of conserved regions in the ectodomain of human immunodeficiency virus type 1 envelope protein. J. Virol. 76, 2683–2691 10.1128/JVI.76.6.2683-2691.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Joyner A. S., Willis J. R., Crowe J. E. Jr., and Aiken C. (2011) Maturation-induced cloaking of neutralization epitopes on HIV-1 particles. PLoS Pathog. 7, e1002234 10.1371/journal.ppat.1002234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Byland R., Vance P. J., Hoxie J. A., and Marsh M. (2007) A conserved dileucine motif mediates clathrin and AP-2-dependent endocytosis of the HIV-1 envelope protein. Mol. Biol. Cell 18, 414–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Miller M. A., Garry R. F., Jaynes J. M., and Montelaro R. C. (1991) A structural correlation between lentivirus transmembrane proteins and natural cytolytic peptides. AIDS Res. Hum. Retroviruses 7, 511–519 10.1089/aid.1991.7.511 [DOI] [PubMed] [Google Scholar]
- 69. Tencza S. B., Douglass J. P., Creighton D. J. Jr., Montelaro R. C., and Mietzner T. A. (1997) Novel antimicrobial peptides derived from human immunodeficiency virus type 1 and other lentivirus transmembrane proteins. Antimicrob. Agents Chemother. 41, 2394–2398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wyss S., Berlioz-Torrent C., Boge M., Blot G., Höning S., Benarous R., and Thali M. (2001) The highly conserved C-terminal dileucine motif in the cytosolic domain of the human immunodeficiency virus type 1 envelope glycoprotein is critical for its association with the AP-1 clathrin adaptor (correction of adapter). J. Virol. 75, 2982–2992 10.1128/JVI.75.6.2982-2992.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Eisenberg D., and Wesson M. (1990) The most highly amphiphilic α-helices include two amino acid segments in human immunodeficiency virus glycoprotein 41. Biopolymers 29, 171–177 10.1002/bip.360290122 [DOI] [PubMed] [Google Scholar]
- 72. Kliger Y., and Shai Y. (1997) A leucine zipper-like sequence from the cytoplasmic tail of the HIV-1 envelope glycoprotein binds and perturbs lipid bilayers. Biochemistry 36, 5157–5169 10.1021/bi962935r [DOI] [PubMed] [Google Scholar]
- 73. Venable R. M., Pastor R. W., Brooks B. R., and Carson F. W. (1989) Theoretically determined three-dimensional structures for amphipathic segments of the HIV-1 gp41 envelope protein. AIDS Res. Hum. Retroviruses 5, 7–22 10.1089/aid.1989.5.7 [DOI] [PubMed] [Google Scholar]
- 74. Bauby H., Lopez-Vergès S., Hoeffel G., Delcroix-Genête D., Janvier K., Mammano F., Hosmalin A., and Berlioz-Torrent C. (2010) TIP47 is required for the production of infectious HIV-1 particles from primary macrophages. Traffic 11, 455–467 10.1111/j.1600-0854.2010.01036.x [DOI] [PubMed] [Google Scholar]
- 75. Checkley M. A., Luttge B. G., Mercredi P. Y., Kyere S. K., Donlan J., Murakami T., Summers M. F., Cocklin S., and Freed E. O. (2013) Reevaluation of the requirement for TIP47 in human immunodeficiency virus type 1 envelope glycoprotein incorporation. J. Virol. 87, 3561–3570 10.1128/JVI.03299-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Lopez-Vergès S., Camus G., Blot G., Beauvoir R., Benarous R., and Berlioz-Torrent C. (2006) Tail-interacting protein TIP47 is a connector between Gag and Env and is required for Env incorporation into HIV-1 virions. Proc. Natl. Acad. Sci. U.S.A. 103, 14947–14952 10.1073/pnas.0602941103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Qi M., Chu H., Chen X., Choi J., Wen X., Hammonds J., Ding L., Hunter E., and Spearman P. (2015) A tyrosine-based motif in the HIV-1 envelope glycoprotein tail mediates cell-type- and Rab11-FIP1C-dependent incorporation into virions. Proc. Natl. Acad. Sci. U.S.A. 112, 7575–7580 10.1073/pnas.1504174112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kennedy R. C., Henkel R. D., Pauletti D., Allan J. S., Lee T. H., Essex M., and Dreesman G. R. (1986) Antiserum to a synthetic peptide recognizes the HTLV-III envelope glycoprotein. Science 231, 1556–1559 10.1126/science.3006246 [DOI] [PubMed] [Google Scholar]
- 79. Postler T. S., Martinez-Navio J. M., Yuste E., and Desrosiers R. C. (2012) Evidence against extracellular exposure of a highly immunogenic region in the C-terminal domain of the simian immunodeficiency virus gp41 transmembrane protein. J. Virol. 86, 1145–1157 10.1128/JVI.06463-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lodge R., Lalonde J. P., Lemay G., and Cohen E. A. (1997) The membrane-proximal intracytoplasmic tyrosine residue of HIV-1 envelope glycoprotein is critical for basolateral targeting of viral budding in MDCK cells. EMBO J. 16, 695–705 10.1093/emboj/16.4.695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ohno H., Aguilar R. C., Fournier M. C., Hennecke S., Cosson P., and Bonifacino J. S. (1997) Interaction of endocytic signals from the HIV-1 envelope glycoprotein complex with members of the adaptor medium chain family. Virology 238, 305–315 10.1006/viro.1997.8839 [DOI] [PubMed] [Google Scholar]
- 82. Rowell J. F., Stanhope P. E., and Siliciano R. F. (1995) Endocytosis of endogenously synthesized HIV-1 envelope protein. Mechanism and role in processing for association with class II MHC. J. Immunol. 155, 473–488 [PubMed] [Google Scholar]
- 83. West J. T., Weldon S. K., Wyss S., Lin X., Yu Q., Thali M., and Hunter E. (2002) Mutation of the dominant endocytosis motif in human immunodeficiency virus type 1 gp41 can complement matrix mutations without increasing Env incorporation. J. Virol. 76, 3338–3349 10.1128/JVI.76.7.3338-3349.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Alfadhli A., Barklis R. L., and Barklis E. (2009) HIV-1 matrix organizes as a hexamer of trimers on membranes containing phosphatidylinositol-(4,5)-bisphosphate. Virology 387, 466–472 10.1016/j.virol.2009.02.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Lineberger J. E., Danzeisen R., Hazuda D. J., Simon A. J., and Miller M. D. (2002) Altering expression levels of human immunodeficiency virus type 1 gp120–gp41 affects efficiency but not kinetics of cell–cell fusion. J. Virol. 76, 3522–3533 10.1128/JVI.76.7.3522-3533.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Platt E. J., Durnin J. P., and Kabat D. (2005) Kinetic factors control efficiencies of cell entry, efficacies of entry inhibitors, and mechanisms of adaptation of human immunodeficiency virus. J. Virol. 79, 4347–4356 10.1128/JVI.79.7.4347-4356.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Putcharoen O., Lee S. H., Henrich T. J., Hu Z., Vanichanan J., Coakley E., Greaves W., Gulick R. M., Kuritzkes D. R., and Tsibris A. M. (2012) HIV-1 clinical isolates resistant to CCR5 antagonists exhibit delayed entry kinetics that are corrected in the presence of drug. J. Virol. 86, 1119–1128 10.1128/JVI.06421-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Reeves J. D., Miamidian J. L., Biscone M. J., Lee F. H., Ahmad N., Pierson T. C., and Doms R. W. (2004) Impact of mutations in the coreceptor binding site on human immunodeficiency virus type 1 fusion, infection, and entry inhibitor sensitivity. J. Virol. 78, 5476–5485 10.1128/JVI.78.10.5476-5485.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Poumbourios P., Maerz A. L., and Drummer H. E. (2003) Functional evolution of the HIV-1 envelope glycoprotein 120 association site of glycoprotein 41. J. Biol. Chem. 278, 42149–42160 10.1074/jbc.M305223200 [DOI] [PubMed] [Google Scholar]
- 90. Keele B. F., Giorgi E. E., Salazar-Gonzalez J. F., Decker J. M., Pham K. T., Salazar M. G., Sun C., Grayson T., Wang S., Li H., Wei X., Jiang C., Kirchherr J. L., Gao F., Anderson J. A., et al. (2008) Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc. Natl. Acad. Sci. U.S.A. 105, 7552–7557 10.1073/pnas.0802203105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Blattner C., Lee J. H., Sliepen K., Derking R., Falkowska E., de la Peña A. T., Cupo A., Julien J. P., van Gils M., Lee P. S., Peng W., Paulson J. C., Poignard P., Burton D. R., Moore J. P., et al. (2014) Structural delineation of a quaternary, cleavage-dependent epitope at the gp41-gp120 interface on intact HIV-1 Env trimers. Immunity 40, 669–680 10.1016/j.immuni.2014.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Zhou T., Georgiev I., Wu X., Yang Z. Y., Dai K., Finzi A., Kwon Y. D., Scheid J. F., Shi W., Xu L., Yang Y., Zhu J., Nussenzweig M. C., Sodroski J., Shapiro L., et al. (2010) Structural basis for broad and potent neutralization of HIV-1 by antibody VRC01. Science 329, 811–817 10.1126/science.1192819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Walker L. M., Huber M., Doores K. J., Falkowska E., Pejchal R., Julien J. P., Wang S. K., Ramos A., Chan-Hui P. Y., Moyle M., Mitcham J. L., Hammond P. W., Olsen O. A., Phung P., Fling S., et al. (2011) Broad neutralization coverage of HIV by multiple highly potent antibodies. Nature 477, 466–470 10.1038/nature10373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Walker L. M., Phogat S. K., Chan-Hui P. Y., Wagner D., Phung P., Goss J. L., Wrin T., Simek M. D., Fling S., Mitcham J. L., Lehrman J. K., Priddy F. H., Olsen O. A., Frey S. M., Hammond P. W., et al. (2009) Broad and potent neutralizing antibodies from an African donor reveal a new HIV-1 vaccine target. Science 326, 285–289 10.1126/science.1178746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. McLellan J. S., Pancera M., Carrico C., Gorman J., Julien J. P., Khayat R., Louder R., Pejchal R., Sastry M., Dai K., O'Dell S., Patel N., Shahzad-ul-Hussan S., Yang Y., Zhang B., et al. (2011) Structure of HIV-1 gp120 V1/V2 domain with broadly neutralizing antibody PG9. Nature 480, 336–343 10.1038/nature10696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Drummer H. E., Hill M. K., Maerz A. L., Wood S., Ramsland P. A., Mak J., and Poumbourios P. (2013) Allosteric modulation of the HIV-1 gp120–gp41 association site by adjacent gp120 variable region 1 (V1) N-glycans linked to neutralization sensitivity. PLoS Pathog. 9, e1003218 10.1371/journal.ppat.1003218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Ogert R. A., Lee M. K., Ross W., Buckler-White A., Martin M. A., and Cho M. W. (2001) N-Linked glycosylation sites adjacent to and within the V1/V2 and the V3 loops of dualtropic human immunodeficiency virus type 1 isolate DH12 gp120 affect coreceptor usage and cellular tropism. J. Virol. 75, 5998–6006 10.1128/JVI.75.13.5998-6006.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Wolk T., and Schreiber M. (2006) N-Glycans in the gp120 V1/V2 domain of the HIV-1 strain NL4–3 are indispensable for viral infectivity and resistance against antibody neutralization. Med. Microbiol. Immunol. 195, 165–172 10.1007/s00430-006-0016-z [DOI] [PubMed] [Google Scholar]
- 99. Chojnacki J., Staudt T., Glass B., Bingen P., Engelhardt J., Anders M., Schneider J., Müller B., Hell S. W., and Kräusslich H. G. (2012) Maturation-dependent HIV-1 surface protein redistribution revealed by fluorescence nanoscopy. Science 338, 524–528 10.1126/science.1226359 [DOI] [PubMed] [Google Scholar]
- 100. Wyma D. J., Jiang J., Shi J., Zhou J., Lineberger J. E., Miller M. D., and Aiken C. (2004) Coupling of human immunodeficiency virus type 1 fusion to virion maturation: a novel role of the gp41 cytoplasmic tail. J. Virol. 78, 3429–3435 10.1128/JVI.78.7.3429-3435.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Myers J. K., Pace C. N., and Scholtz J. M. (1997) A direct comparison of helix propensity in proteins and peptides. Proc. Natl. Acad. Sci. U.S.A. 94, 2833–2837 10.1073/pnas.94.7.2833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Tien M. Z., Meyer A. G., Sydykova D. K., Spielman S. J., and Wilke C. O. (2013) Maximum allowed solvent accessibilities of residues in proteins. PLoS ONE 8, e80635 10.1371/journal.pone.0080635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Killian J. A., and von Heijne G. (2000) How proteins adapt to a membrane-water interface. Trends Biochem. Sci. 25, 429–434 10.1016/S0968-0004(00)01626-1 [DOI] [PubMed] [Google Scholar]
- 104. Senes A., Chadi D. C., Law P. B., Walters R. F., Nanda V., and Degrado W. F. (2007) E(z), a depth-dependent potential for assessing the energies of insertion of amino acid side-chains into membranes: derivation and applications to determining the orientation of transmembrane and interfacial helices. J. Mol. Biol. 366, 436–448 10.1016/j.jmb.2006.09.020 [DOI] [PubMed] [Google Scholar]
- 105. Ulmschneider M. B., and Sansom M. S. (2001) Amino acid distributions in integral membrane protein structures. Biochim. Biophys. Acta 1512, 1–14 10.1016/S0005-2736(01)00299-1 [DOI] [PubMed] [Google Scholar]
- 106. Snider C., Jayasinghe S., Hristova K., and White S. H. (2009) MPEx: a tool for exploring membrane proteins. Protein Sci. 18, 2624–2628 10.1002/pro.256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Eisenberg D., Weiss R. M., and Terwilliger T. C. (1982) The helical hydrophobic moment: a measure of the amphiphilicity of a helix. Nature 299, 371–374 10.1038/299371a0 [DOI] [PubMed] [Google Scholar]
- 108. Durham N. D., and Chen B. K. (2015) HIV-1 cell-free and cell-to-cell infections are differentially regulated by distinct determinants in the Env gp41 cytoplasmic tail. J. Virol. 89, 9324–9337 10.1128/JVI.00655-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Murakami T., and Freed E. O. (2000) The long cytoplasmic tail of gp41 is required in a cell type-dependent manner for HIV-1 envelope glycoprotein incorporation into virions. Proc. Natl. Acad. Sci. U.S.A. 97, 343–348 10.1073/pnas.97.1.343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Bracq L., Xie M., Lambele M., Vu L. T., Matz J., Schmitt A., Delon J., Zhou P., Randriamampita C., Bouchet J., and Benichou S. (2017) T cell-macrophage fusion triggers multinucleated giant cell formation for HIV-1 spreading. J. Virol. 2017 JVI.01237–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Chen P., Hübner W., Spinelli M. A., and Chen B. K. (2007) Predominant mode of human immunodeficiency virus transfer between T cells is mediated by sustained Env-dependent neutralization-resistant virological synapses. J. Virol. 81, 12582–12595 10.1128/JVI.00381-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Wang L., Eng E. T., Law K., Gordon R. E., Rice W. J., and Chen B. K. (2017) Visualization of HIV T cell virological synapses and virus-containing compartments by three-dimensional correlative light and electron microscopy. J. Virol. 91, e01605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Jiang J., and Aiken C. (2007) Maturation-dependent human immunodeficiency virus type 1 particle fusion requires a carboxyl-terminal region of the gp41 cytoplasmic tail. J. Virol. 81, 9999–10008 10.1128/JVI.00592-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Murakami T., Ablan S., Freed E. O., and Tanaka Y. (2004) Regulation of human immunodeficiency virus type 1 Env-mediated membrane fusion by viral protease activity. J. Virol. 78, 1026–1031 10.1128/JVI.78.2.1026-1031.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Wyma D. J., Kotov A., and Aiken C. (2000) Evidence for a stable interaction of gp41 with Pr55(Gag) in immature human immunodeficiency virus type 1 particles. J. Virol. 74, 9381–9387 10.1128/JVI.74.20.9381-9387.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Carlson L. A., de Marco A., Oberwinkler H., Habermann A., Briggs J. A., Kräusslich H. G., and Grünewald K. (2010) Cryo electron tomography of native HIV-1 budding sites. PLoS Pathog. 6, e1001173 10.1371/journal.ppat.1001173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Stauffer S., Rahman S. A., de Marco A., Carlson L. A., Glass B., Oberwinkler H., Herold N., Briggs J. A., Müller B., Grünewald K., and Kräusslich H. G. (2014) The nucleocapsid domain of Gag is dispensable for actin incorporation into HIV-1 and for association of viral budding sites with cortical F-actin. J. Virol. 88, 7893–7903 10.1128/JVI.00428-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Do T., Murphy G., Earl L. A., Del Prete G. Q., Grandinetti G., Li G. H., Estes J. D., Rao P., Trubey C. M., Thomas J., Spector J., Bliss D., Nath A., Lifson J. D., and Subramaniam S. (2014) Three-dimensional imaging of HIV-1 virological synapses reveals membrane architectures involved in virus transmission. J. Virol. 88, 10327–10339 10.1128/JVI.00788-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Sowinski S., Jolly C., Berninghausen O., Purbhoo M. A., Chauveau A., Köhler K., Oddos S., Eissmann P., Brodsky F. M., Hopkins C., Onfelt B., Sattentau Q., and Davis D. M. (2008) Membrane nanotubes physically connect T cells over long distances presenting a novel route for HIV-1 transmission. Nat. Cell Biol. 10, 211–219 10.1038/ncb1682 [DOI] [PubMed] [Google Scholar]
- 120. Stewart-Jones G. B., Soto C., Lemmin T., Chuang G. Y., Druz A., Kong R., Thomas P. V., Wagh K., Zhou T., Behrens A. J., Bylund T., Choi C. W., Davison J. R., Georgiev I. S., Joyce M. G., et al. (2016) Trimeric HIV-1-Env structures define glycan shields from Clades A, B, and G. Cell 165, 813–826 10.1016/j.cell.2016.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Blish C. A., Nguyen M. A., and Overbaugh J. (2008) Enhancing exposure of HIV-1 neutralization epitopes through mutations in gp41. PLoS Med. 5, e9 10.1371/journal.pmed.0050009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Binley J. M., Wrin T., Korber B., Zwick M. B., Wang M., Chappey C., Stiegler G., Kunert R., Zolla-Pazner S., Katinger H., Petropoulos C. J., and Burton D. R. (2004) Comprehensive cross-clade neutralization analysis of a panel of anti-human immunodeficiency virus type 1 monoclonal antibodies. J. Virol. 78, 13232–13252 10.1128/JVI.78.23.13232-13252.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Ferrantelli F., Hofmann-Lehmann R., Rasmussen R. A., Wang T., Xu W., Li P. L., Montefiori D. C., Cavacini L. A., Katinger H., Stiegler G., Anderson D. C., McClure H. M., and Ruprecht R. M. (2003) Post-exposure prophylaxis with human monoclonal antibodies prevented SHIV89.6P infection or disease in neonatal macaques. AIDS 17, 301–309 10.1097/00002030-200302140-00003 [DOI] [PubMed] [Google Scholar]
- 124. Mascola J. R., Stiegler G., VanCott T. C., Katinger H., Carpenter C. B., Hanson C. E., Beary H., Hayes D., Frankel S. S., Birx D. L., and Lewis M. G. (2000) Protection of macaques against vaginal transmission of a pathogenic HIV-1/SIV chimeric virus by passive infusion of neutralizing antibodies. Nat. Med. 6, 207–210 10.1038/72318 [DOI] [PubMed] [Google Scholar]
- 125. Lee B., Sharron M., Montaner L. J., Weissman D., and Doms R. W. (1999) Quantification of CD4, CCR5, and CXCR4 levels on lymphocyte subsets, dendritic cells, and differentially conditioned monocyte-derived macrophages. Proc. Natl. Acad. Sci. U.S.A. 96, 5215–5220 10.1073/pnas.96.9.5215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Ruprecht C. R., Krarup A., Reynell L., Mann A. M., Brandenberg O. F., Berlinger L., Abela I. A., Regoes R. R., Günthard H. F., Rusert P., and Trkola A. (2011) MPER-specific antibodies induce gp120 shedding and irreversibly neutralize HIV-1. J. Exp. Med. 208, 439–454 10.1084/jem.20101907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Theodore T. S., Englund G., Buckler-White A., Buckler C. E., Martin M. A., and Peden K. W. (1996) Construction and characterization of a stable full-length macrophage-tropic HIV type 1 molecular clone that directs the production of high titers of progeny virions. AIDS Res. Hum. Retroviruses 12, 191–194 10.1089/aid.1996.12.191 [DOI] [PubMed] [Google Scholar]
- 128. Adachi A., Gendelman H. E., Koenig S., Folks T., Willey R., Rabson A., and Martin M. A. (1986) Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol. 59, 284–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Hill M. K., Shehu-Xhilaga M., Campbell S. M., Poumbourios P., Crowe S. M., and Mak J. (2003) The dimer initiation sequence stem-loop of human immunodeficiency virus type 1 is dispensable for viral replication in peripheral blood mononuclear cells. J. Virol. 77, 8329–8335 10.1128/JVI.77.15.8329-8335.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Chang L. J., Urlacher V., Iwakuma T., Cui Y., and Zucali J. (1999) Efficacy and safety analyses of a recombinant human immunodeficiency virus type 1 derived vector system. Gene Ther. 6, 715–728 10.1038/sj.gt.3300895 [DOI] [PubMed] [Google Scholar]
- 131. Derdeyn C. A., Decker J. M., Sfakianos J. N., Wu X., O'Brien W. A., Ratner L., Kappes J. C., Shaw G. M., and Hunter E. (2000) Sensitivity of human immunodeficiency virus type 1 to the fusion inhibitor T-20 is modulated by coreceptor specificity defined by the V3 loop of gp120. J. Virol. 74, 8358–8367 10.1128/JVI.74.18.8358-8367.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Platt E. J., Bilska M., Kozak S. L., Kabat D., and Montefiori D. C. (2009) Evidence that ecotropic murine leukemia virus contamination in TZM-bl cells does not affect the outcome of neutralizing antibody assays with human immunodeficiency virus type 1. J. Virol. 83, 8289–8292 10.1128/JVI.00709-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Wei X., Decker J. M., Liu H., Zhang Z., Arani R. B., Kilby J. M., Saag M. S., Wu X., Shaw G. M., and Kappes J. C. (2002) Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob. Agents Chemother. 46, 1896–1905 10.1128/AAC.46.6.1896-1905.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Connor R. I., Chen B. K., Choe S., and Landau N. R. (1995) Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology 206, 935–944 10.1006/viro.1995.1016 [DOI] [PubMed] [Google Scholar]
- 135. Platt E. J., Wehrly K., Kuhmann S. E., Chesebro B., and Kabat D. (1998) Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. J. Virol. 72, 2855–2864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Dhillon A. K., Donners H., Pantophlet R., Johnson W. E., Decker J. M., Shaw G. M., Lee F. H., Richman D. D., Doms R. W., Vanham G., and Burton D. R. (2007) Dissecting the neutralizing antibody specificities of broadly neutralizing sera from human immunodeficiency virus type 1-infected donors. J. Virol. 81, 6548–6562 10.1128/JVI.02749-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Wu X., Yang Z. Y., Li Y., Hogerkorp C. M., Schief W. R., Seaman M. S., Zhou T., Schmidt S. D., Wu L., Xu L., Longo N. S., McKee K., O'Dell S., Louder M. K., Wycuff D. L., et al. (2010) Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to HIV-1. Science 329, 856–861 10.1126/science.1187659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Falkowska E., Le K. M., Ramos A., Doores K. J., Lee J. H., Blattner C., Ramirez A., Derking R., van Gils M. J., Liang C. H., Mcbride R., von Bredow B., Shivatare S. S., Wu C. Y., Chan-Hui P. Y., et al. (2014) Broadly neutralizing HIV antibodies define a glycan-dependent epitope on the prefusion conformation of gp41 on cleaved envelope trimers. Immunity 40, 657–668 10.1016/j.immuni.2014.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Krey T., Meola A., Keck Z. Y., Damier-Piolle L., Foung S. K., and Rey F. A. (2013) Structural basis of HCV neutralization by human monoclonal antibodies resistant to viral neutralization escape. PLoS Pathog. 9, e1003364 10.1371/journal.ppat.1003364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Takikawa S., Ishii K., Aizaki H., Suzuki T., Asakura H., Matsuura Y., and Miyamura T. (2000) Cell fusion activity of hepatitis C virus envelope proteins. J. Virol. 74, 5066–5074 10.1128/JVI.74.11.5066-5074.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Deng H., Liu R., Ellmeier W., Choe S., Unutmaz D., Burkhart M., Di Marzio P., Marmon S., Sutton R. E., Hill C. M., Davis C. B., Peiper S. C., Schall T. J., Littman D. R., and Landau N. R. (1996) Identification of a major co-receptor for primary isolates of HIV-1. Nature 381, 661–666 10.1038/381661a0 [DOI] [PubMed] [Google Scholar]
- 142. Maerz A. L., Drummer H. E., Wilson K. A., and Poumbourios P. (2001) Functional analysis of the disulfide-bonded loop/chain reversal region of human immunodeficiency virus type 1 gp41 reveals a critical role in gp120–gp41 association. J. Virol. 75, 6635–6644 10.1128/JVI.75.14.6635-6644.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Murphy R. E., Samal A. B., Vlach J., and Saad J. S. (2017) Solution structure and membrane interaction of the cytoplasmic tail of HIV-1 gp41 protein. Structure 25, 1708–1718 10.1016/j.str.2017.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Dev J., Park D., Fu Q., Chen J., Ha H. J., Ghantous F., Herrmann T., Chang W., Liu Z., Frey G., Seaman M. S., Chen B., and Chou J. J. (2016) Structural basis for membrane anchoring of HIV-1 envelope spike. Science 353, 172–175 10.1126/science.aaf7066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Hill C. P., Worthylake D., Bancroft D. P., Christensen A. M., and Sundquist W. I. (1996) Crystal structures of the trimeric human immunodeficiency virus type 1 matrix protein: implications for membrane association and assembly. Proc. Natl. Acad. Sci. U.S.A. 93, 3099–3104 10.1073/pnas.93.7.3099 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.