

Abstract
Germline-encoded receptors recognizing common pathogen-associated molecular patterns are a central element of the innate immune system and play an important role in shaping the host response to infection. Many of the innate immune molecules central to these signaling pathways are evolutionarily conserved. LysMD3 is a novel molecule containing a putative peptidoglycan-binding domain that has orthologs in humans, mice, zebrafish, flies, and worms. We found that the lysin motif (LysM) of LysMD3 is likely related to a previously described peptidoglycan-binding LysM found in bacteria. Mouse LysMD3 is a type II integral membrane protein that co-localizes with GM130+ structures, consistent with localization to the Golgi apparatus. We describe here two lines of mLysMD3-deficient mice for in vivo characterization of mLysMD3 function. We found that mLysMD3-deficient mice were born at Mendelian ratios and had no obvious pathological abnormalities. They also exhibited no obvious immune response deficiencies in a number of models of infection and inflammation. mLysMD3-deficient mice exhibited no signs of intestinal dysbiosis by 16S analysis or alterations in intestinal gene expression by RNA sequencing. We conclude that mLysMD3 contains a LysM with cytoplasmic orientation, but we were unable to define a physiological role for the molecule in vivo.
Keywords: immunology, mouse, mouse genetics, infection, inflammation, animal model, membrane protein, protein motif
Introduction
The innate immune response to infection relies heavily on signals transduced by germline-encoded receptors that recognize common pathogen-associated molecular patterns (PAMPs)4 such as bacterial and viral proteins, glycoproteins, and microbe-specific nucleic acids (1). These pattern recognition receptors tend to recognize microbial products that are absent in the host, thereby preventing self-reactivity, are often evolutionarily conserved, and may be members of a protein family that recognize similar but distinct microbial products.
Therefore, we were interested in characterizing the evolutionarily-conserved molecule, LysMD3, which has homologs in humans, mice, zebrafish, flies, and worms. LysMD3 is named for its N-terminal lysin motif (LysM) and, in mice and humans, is a member of a protein family that also includes LysMD1, LysMD2, and LysMD4. Recent studies of bacterial and plant LysMs suggest that LysMs bind the glycan backbone of peptidoglycan or the related molecule chitin (2–6). Although peptidoglycan is a ubiquitous bacterial component, relatively little is known about its interactions with the mammalian immune system. The intracellular receptors NOD1 and NOD2 have been shown to bind the peptidoglycan fragments Tri-DAP and muramyl-dipeptide, respectively (7), but it is unknown whether the mammalian immune system is capable of recognizing the ubiquitous polymeric glycan backbone of peptidoglycan. Furthermore, a recent study suggested that chitin oligomers may modulate mammalian angiogenesis through interactions with a yet unknown receptor (8). We hypothesized that the LysMD family may represent a novel family of pattern recognition receptors, potentially capable of recognizing important microbial ligands such as peptidoglycan or chitin.
Results from this study revealed that the LysM of LysMD3 is evolutionarily conserved, with orthologs in human, mice, zebrafish, flies, and worms. In mammalian cells, we found that LysMD3 was a type II integral membrane protein that co-localizes with the Golgi marker GM130 with a predicted cytoplasmic location of the LysM. To explore the function of LysMD3 in the immune response, we generated two lines of LysMD3-deficient mice and evaluated the role of LysMD3 in a number of infection and inflammation models. We found no evidence for a role for LysMD3 during the mammalian immune response in the models that we tested, nor did we find a role for LysMD3 in the control of the intestinal microbiota or intestinal gene expression. Additional studies will be required to determine a functional role for this evolutionarily-conserved molecule.
Results
LysMD3 is a predicted transmembrane protein containing an evolutionarily conserved LysM
Mouse LysMD3 (mLysMD3) is a 305-amino acid protein with an annotated N-terminal LysM and C-terminal transmembrane domain (Fig. 1A). Phylogenetic analysis of the minimal LysM sequence from multiple model organisms (Fig. 1, B and C) revealed that mLysMD1, mLysMD2, mLysMD3, and mLysMD4 each clustered with like sequences from other organisms, including Homo sapiens, Xenopus tropicalis, and Danio rerio. Furthermore, groups containing LysMD3 and LysMD4 sequences diverged from clusters containing LysMD1 and LysMD2 LysM sequences. Interestingly, the LysM from Caenorhabditis elegans protein F43G9 clustered with LysMD3 and LysMD4 sequences and apart from LysMD1 and LysMD2. Furthermore, whereas the exact relationship between the LysM of mLysMD3 and Drosophila melanogaster CG17985 is somewhat less defined, it is apparent that the D. melanogaster CG17985 clusters with sequences including mLysMD3/4 and not with mLysMD1/2. The other murine LysM-containing proteins Ncoa7 and Oxr1 cluster separately from CG17985 and the mLysMD family members and are more similar to the D. melanogaster protein Mtd (9).
Figure 1.

Phylogenetic analysis of LysMs. A, schematic of predicted domains of mouse LysMD3. TM, transmembrane. B, multiple sequence alignment of LysMs from the indicated organisms. Positions with a single, fully conserved residue are marked with an asterisk. Positions with conservation of residues with strongly similar properties (>0.5 in the Gonnet PAM 250 matrix) or weakly similar properties (≤0.5 in the Gonnet PAM 250 matrix) are marked with a “:” or “.”, respectively. Percent conservation of residues is indicated at the bottom. C, phylogenetic tree of LysMs based on the multiple sequence alignment in B. Where known, common protein names are listed. For E. coli protein MltD, two LysMs in the protein were evaluated (MltD-1 and MltD-2). D, sequence alignment of mLysMD3 and E. faecalis AltA. Secondary structural elements from E. faecalis AltA (PDB 2KMX) are indicated at the top. T, hydrogen-bonded turn. The figure was prepared using Espript3.
Although there was sequence diversity within LysMs across species, alignment of the deduced proteins demonstrated that several residues were remarkably conserved between prokaryotes and higher order organisms (Fig. 1B). In particular, an asparagine residue at position 31 is conserved in all the species analyzed. In addition, amino acids 11–13, with only a few exceptions, were conserved across LysMs from all species analyzed, including several bacterial proteins. A neighboring sequence of amino acids 16–19 had only conservative substitutions across all species.
The mLysMD3 LysM sequence was submitted to the Phyre2 Protein Fold Recognition Server (10) for protein structure prediction. The top hit from this analysis, in which 96% of the sequence was modeled with a confidence of 99.6%, was structure 2djp, a structure of the LysM of human LYSMD1 (data not shown). A model of the mLysMD3 LysM could also be generated based on the structure 2mkx, the LysM from Enterococcus faecalis protein AtlA, in which 85% of the sequence could be modeled with a confidence of 99.4% (Fig. 1D). Together, these data suggest that the LysM of mLysMD3 and those of mLysMD1, mLysMD2, and mLysMD4 are conserved across multiple divergent species.
LysMD3 co-localizes with GM130+ structures
We next sought to define the subcellular localization of LysMD3. Immunofluorescence staining of endogenous human LYSMD3 (hLYSMD3) in HeLa cells suggested that hLYSMD3 co-localizes with GM130+ structures, consistent with localization to the Golgi (Fig. 2A) (11). Importantly, this staining and the co-localization are not observed in HeLa cell lines in which hLYSMD3 expression has been eliminated through the use of CRISPR/Cas9-targeted genome editing. These data are further supported by similar staining patterns using a second commercially-available anti-hLYSMD3 antibody as well as transfection of HeLa cells with an mLysMD3-GFP expression construct (Fig. 2C) and subsequent staining for GFP (Fig. S1, A and B). Additionally, mouse embryonic fibroblast cells (MEFs) were stained for mLysMD3 expression using an affinity-purified polyclonal anti-mLysMD3 antibody raised against amino acids 1–205 of mLysMD3 (Fig. S1C). Although this antibody appears to nonspecifically stain the nucleus, LysMD3-specific signals were revealed by the absence of staining in MEFs deficient for mLysMD3 due to the insertion of a gene-trap cassette downstream of mLysMD3 exon 1 (Fig. S2A). GM130 co-localization was also observed for mLysMD3 in MEFs using this antibody (Fig. S1C). Taken together, these data suggest that human and mouse LysMD3 molecules are located on GM130+ structures, consistent with localization to the Golgi.
Figure 2.

LysMD3 is a type II integral membrane protein that co-localizes with GM130+ structures. A, WT and hLYSMD3-deficient ΔLYSMD3 HeLa cells were stained for LysMD3 using a polyclonal anti-hLYSMD3 antibody. Cells were co-stained for GM130, and phalloidin (blue) was used to visualize F-actin. B, WT BMMo were subjected to differential detergent fraction and immunoblot analysis. Control molecules were ERK1/2 (cytoplasmic fraction), transferrin receptor (TfR, membrane fraction), and histone H2AX (nuclear fraction). C, diagram of mLysMD3 constructs used in D and E. D, HeLa cells were transfected with a construct expressing FLAG-LysMD3-HA, then permeabilized with Triton X-100, digested with increasing doses of proteinase K, as indicated, and subjected to immunoblot analysis for FLAG, HA, and actin. E, HeLa cells were transfected with the indicated constructs. Cells were fixed and stained for GFP ± prior permeabilization with Triton X-100, as indicated. Following GFP staining, all samples were permeabilized, and phalloidin-594 was used to visualize F-actin.
Pfam analysis of the mLysMD3 amino acid sequence identified a putative transmembrane domain between amino acids 217 and 237 (Fig. 1A), suggesting that mLysMD3 is a single pass transmembrane molecule. To test this hypothesis, we first evaluated the cellular compartment with which mLysMD3 is associated. Differential detergent fractionation followed by immunoblotting suggested that mLysMD3 is associated with the membrane compartment of primary bone marrow macrophages (BMMo) and not the nucleus or cytoplasm (Fig. 2B). These data are consistent with the hypothesis that mLysMD3 is a transmembrane protein.
We next assessed mLysMD3 membrane topology in a protease protection assay using a recombinant mLysMD3 molecule with N-terminal FLAG and C-terminal HA tags (FLAG-LysMD3-HA, diagrammed in Fig. 2C). This construct was expressed in HeLa cells; the plasma membrane was selectively permeabilized using digitonin (12), and exposed epitopes were subjected to proteinase K digestion. Immunoblot analysis of the N-terminal FLAG epitope indicated that the FLAG tag was degraded upon permeabilization of the plasma membrane and addition of increasing concentrations of proteinase K, as shown by decreased detection of the FLAG-tagged full-length protein (Fig. 2D). In contrast, with increasing concentration of proteinase K, the HA tag was detected on a molecular species that shifted from a molecular mass of about 60 kDa to about 15 kDa, indicating that the C-terminal HA tag was within an intracellular vesicle not permeabilized by digitonin and therefore protected from proteinase K digestion.
This conclusion is further supported by immunofluorescence analysis using GFP-tagged mLysMD3 molecules (diagrammed in Fig. 2C). Transfection of HeLa cells with GFP-tagged molecules and anti-GFP staining demonstrated that an mLysMD3 construct with a C-terminal GFP tag could be immunostained at the cell surface without prior membrane permeabilization (Fig. 2E). This anti-GFP signal is not observed for an mLysMD3 construct with an N-terminal GFP tag without prior membrane permeabilization.
Overall, these data suggest that mLysMD3 is a type II integral membrane protein located in the Golgi, with the N-terminal LysM located in the cytoplasm.
Generation of LysMD3-deficient mice
To evaluate the role of mLysMD3 during the immune response in vivo, we generated a mouse line with gene trap (GT)–mediated disruption of mLysMD3 using commercially available ES cells. This 129P2Ola/Hsd background cell line contained a β-gal/neomycin resistance GT inserted between exons 1 and 2 of the mLysMD3 gene, upstream of the protein-coding region (Fig. S2A). The location of the GT cassette within mLysMD3 intron 1 was confirmed by PCR and Sanger sequencing across the cassette and its boundaries (data not shown). The mLysMD3 GT allele was backcrossed to a C57BL6/J (B6, WT) background with the assistance of microsatellite-based speed congenic analysis at each generation. mLysMD3GT/GT mice were viable and fertile, and crosses of mLysMD3+/GT mice generate offspring with genotypes at the expected Mendelian frequencies on both the 129P2Ola/Hsd and B6 backgrounds.
In knockout (KO) mice generated through gene-trap mutagenesis, it is possible that splicing over the gene trap may occur resulting in expression of the endogenous protein, although often at hypomorphic levels (13). Additionally, it is possible that backcrossing the mLysMD3 gene-trap allele from one background to another could select for a linked gene with differential immune responses between 129P2Ola/Hsd and B6.
We therefore also generated a KO mouse in which exon 2 of mLysMD3, containing the coding region for the LysM, was targeted using CRISPR/Cas9 genome editing in B6 embryos (Fig. S2B). Sequencing of the edited mLysMD3 allele revealed a deletion of ∼2 kb, corresponding to the genomic sequence between the guide gRNA sites (Fig. S2C). As observed for the mLysMD3 GT mouse line, mLysMD3EN/EN mice are viable and fertile, and crosses of heterozygous parents yield offspring at the expected Mendelian frequencies.
To evaluate expression of mLysMD3 in our two mouse lines, we used a qRT-PCR assay with primers spanning the exon 2–exon 3 junction of mLysMD3. This exon–exon junction was abolished in the LysMD3 EN mouse line and was downstream of the gene-trap cassette in the LysMD3 GT mouse line. In this assay, mLysMD3 transcripts are undetectable in littermates from both lines of mLysMD3-deficient mice (Fig. 3A) and are reduced in heterozygous mice, compared with WT.
Figure 3.

LysMD3-deficient mice lack LysMD3 RNA expression. A and B, qRT-PCR analysis of LysMD3 (A) and LysMD4 (B) expression in the indicated tissues from LysMD3 GT and LysMD3 EN mouse lines. All experimental mice were littermate-matched.
Genomic alterations have the potential to affect neighboring gene expression (14); therefore, we evaluated the expression of selected mLysMD3 neighboring genes. We found that expression of Polr3g was unchanged in both LysMD3 GT and LysMD3 EN mice (Fig. S2D), whereas expression of Adgrv1 was unchanged in LysMD3 EN mice and was marginally altered in LysMD3 GT mice in a gene-dose–dependent manner (Fig. S2D). Finally, given that LysMD3 is a member of a protein family, we evaluated the expression of LysMD4 in the absence of LysMD3. We found that LysMD4 expression was unaltered in both lines of LysMD3-deficient mice in the tissues evaluated (Fig. 3B).
These data suggest that alteration of mLysMD3 has minimal effect on expression of surrounding gene or LysMD4 expression, and that we have established two lines of mLysMD3-deficient mice, referred to as LysMD3 GT and LysMD3 EN hereinafter.
Lack of a role for mLysMD3 in the response to Citrobacter rodentium and Salmonella typhimurium infection
As our expression data suggested that mLysMD3 is expressed in the intestine, we first evaluated the response of mLysMD3-deficient mice to oral infection with the Gram-negative bacterium C. rodentium. In B6 background mice, oral C. rodentium infection causes the formation of lesions similar to those caused by the human enteric pathogen enteropathogenic Escherichia coli (15, 16). Over the course of infection with C. rodentium, minimal morbidity was observed in mLysMD3 WT or mLysMD3 KO littermate mice (Fig. 4A). Furthermore, no difference in C. rodentium fecal shedding was observed over 8 days of infection (Fig. 4B). Finally, no difference in colonic inflammation was observed 8 days post-infection (DPI) in the absence of mLysMD3 (Fig. 4, C and D).
Figure 4.

Lack of a role for LysMD3 in the control of C. rodentium and S. typhimurium infection. Littermate-matched male mice were infected with 1e9 CFU C. rodentium on day 0, and morbidity (A) and fecal shedding (B) were evaluated. At 8 DPI, colons were evaluated for inflammation (C and D). Each dot represents the histology score for one mouse. E and F, mice were infected with 1e8 CFU S. Typhimurium. At 2 DPI, colons were harvested and evaluated for inflammation. All experiments were performed in duplicate using littermate-matched male mice from the LysMD3 EN mouse line.
We next evaluated the response of mLysMD3-deficient mice to oral infection with the Gram-negative bacterium Salmonella enterica serotype Typhimurium (S. Typhimurium). B6 mice are highly susceptible to S. Typhimurium infection due to a mutation in Nramp (17), but they can be used to model intestinal inflammation during enteric salmonellosis (18). We found that mLysMD3 deficiency did not affect intestinal inflammation after S. Typhimurium infection in either the colon or cecum, as evaluated by clinical scoring of histological sections at 2 DPI (Fig. 4, E and F and data not shown).
Lack of a role for mLysMD3 in the response to intracellular bacteria
Given the cytoplasmic orientation of the LysMD3 LysM, we evaluated the role for LysMD3 in the sensing of intracellular bacteria. We evaluated the response of mice to infection with Listeria monocytogenes, an intracellular, cytoplasmic Gram-positive bacterium that is a major human pathogen (19). Mice were infected with L. monocytogenes and monitored for lethality. We observed no difference in lethality between mLysMD3 WT and mLysMD3 KO mice over the course of the experiment, with approximately 50% of each genotype succumbing to infection (Fig. 5A). Furthermore, we found no role for mLysMD3 in the ability of BMMo to activate T cells in response to L. monocytogenes infection in vitro (Fig. S3A).
Figure 5.

Lack of a role for LysMD3 in the control of multiple bacterial infections. A, age-matched mice from the LysMD3 GT mouse line were infected with 5e5 CFU L. monocytogenes strain EGD i.p. and monitored for lethality. Data were pooled from three experiments using male mice and one experiment using female mice. Nonlittermate B6 mice were used as controls. B, BMMo from the LysMD3 GT mouse line were infected with F. novicida strain U112 or isogenic mutant FPI at an m.o.i. of 10, and intracellular growth was evaluated. C and D, mice were infected by aerosol method with M. tuberculosis strain Erdman, and titers in the lungs were determined at 3 (C) and 13 (D) WPI. E–G, mice from the LysMD3 EN mouse line were infected with 1e6 CFU B. abortus i.p., and titers in the spleen (E) and liver (F) and serum IL6 levels (G) were determined at 3 DPI. Data were combined from two experimental replicates. H, BMMo from the LysMD3 EN mouse line were infected with L. pneumophila strains Lp02, ΔdotA, or ΔflaA ± IFNγ, and intracellular growth was evaluated. Data were combined from two experimental replicates. I–L, female littermate mice from the LysMD3 GT mouse line were inoculated transurethrally with 1e7 CFU of UPEC strain UTI89. At 24 HPI, urine was collected for CFU enumeration (I) and cytokine analysis (J), and serum was collected for cytokine analysis (K). L, MEFs from the LysMD3 GT mouse line were infected with C. trachomonatis at an m.o.i. of 10, and levels of secreted IFNb were evaluated by ELISA. M, MEFs from the LysMD3 GT mouse line were infected with S. flexneri at an m.o.i. of 1, and levels of secreted IFNb were evaluated by ELISA.
We next evaluated the ability of mLysMD3-deficient BMMo to respond to infection by a Gram-negative intracellular and cytoplasmic bacterium, Francisella novicida, a close relative of the human pathogen Francisella tularensis, the causative agent of tularemia (20). Two strains of F. novicida were used for these experiments: U112, a WT strain of F. novicida, and isogenic mutant ΔFPI, which is incapable of escaping the macrophage phagosome to enter the cytoplasm (21). We observed no difference in the ability of either F. novicida strain to grow intracellularly (Fig. 5B) or to stimulate cell death (Fig. S3B) or type I IFN production (Fig. S3C) in the absence of mLysMD3. Similarly, no difference in cell death or type I IFN production was seen at a high multiplicity of infection (MOI) of 100 (data not shown).
We next evaluated the ability of mLysMD3-deficient mice to control Mycobacterium tuberculosis infection in vivo (22–25). M. tuberculosis is an intracellular pathogen, classically thought to reside in an early endosomal compartment; however, recent studies have suggested that it may also grow in the cytoplasm (26). We found that there was no overall difference in M. tuberculosis titers in the lungs of B6, mLysMD3EN/EN, or mLysMD3GT/GT mice at 3 or 13 weeks post-infection (WPI) (Fig. 5, C and D). Although there was a slight reduction in bacterial titers in the spleens of mLysMD3GT/GT mice at three WPI compared with B6 controls (Fig. S3D), this reduction was not observed in mLysMD3EN/EN mice and was not seen at 13 WPI (Fig. S3E). Additionally, we did not observe differences in immune cell recruitment to the lung, spleen, or mediastinal lymph nodes of mLysMD3-deficient mice at either 3 or 13 WPI (data not shown).
We next evaluated the ability of mLysMD3-deficient mice to control Brucella abortus infection in vivo. Brucella species cause chronic granulomatous infection in both domestic animals and humans (27, 28). Littermate-matched mice were infected with B. abortus intraperitoneally (i.p.), and bacterial titers were determined at 3 DPI. We observed no difference in bacterial titers in the spleen or liver or serum IL6 levels in either LysMD3-deficient mouse line (Fig. 5, E–G, and Fig. S3, F–H).
We next evaluated the ability of mLysMD3-deficient BMMo to respond to infection with Legionella pneumophila, the causative agent of Legionnaire's disease (29). BMMo were infected with L. pneumophila strain LP02 or isogenic mutants ΔflaA or ΔdotA. ΔflaA mutants are nonflagellated, nonmotile, and grow to high titers in BMMo, although growth can be restricted by IFNγ priming of BMMo (30). Conversely, ΔdotA mutants lack the type IV secretion apparatus that is strictly required for intracellular growth (31). We found no difference in the ability of these bacterial strains to grow intracellularly or to induce cell death in the absence of mLysMD3 (Fig. 5H and Fig. S3I).
We next evaluated a possible role for mLysMD3 in the pathogenesis of urinary tract infections (UTIs) caused by the Gram-negative bacterium uropathogenic E. coli (UPEC), which occupies the luminal, intracellular cytoplasmic, and subcellular compartments during infection (32). Mice were transurethrally infected with the clinical UTI isolate, UTI89, and infection as well as the inflammatory response were evaluated 24 h later. We found no difference in urine bacterial titers or urine IL1β production at 24 h post-infection (HPI) in the absence of mLysMD3 (Fig. 5, I and J). Similarly, there was no difference in serum IL1β production at 24 HPI (Fig. 5K). Histological evaluation of bladders from UPEC-infected mice demonstrated no role for mLysMD3 in the severity of inflammation at 24 HPI (Fig. S3J). We also evaluated the ability of mLysMD3-deficient BMMo to respond to UPEC infection in vitro. We found there was no difference in IL1β production by mLysMD3-deficient BMMo at either 6 or 24 HPI (Fig. S3K).
Finally, we evaluated the ability of mLysMD3-deficient MEFs to respond to infection by clinically relevant intracellular bacteria species Chlamydia trachomatis and Shigella flexneri. We saw no difference between LysMD3 WT and KO cell lines in production of IFNβ in response to C. trachomatis (Fig. 5L) or S. flexneri (Fig. 5M). Similarly, no difference was seen in the production of IL6 in response to infection (Fig. S3, L and M).
Lack of a role for mLysMD3 in models of inflammation
Datasets from the Immunological Genome Project (Immgen) (33) suggest that mLysMD3 transcripts are expressed in cells of the immune system, as well as in intestinal tissues that could likely come into contact with pathogens. Therefore, we next evaluated a number of additional models of inflammation and infection.
Immgen datasets suggest that LysMD3 transcripts are highly expressed in neutrophils (33). Therefore, we hypothesized that mLysMD3 could regulate inflammation in the neutrophil-dependent KRN model of serum-induced arthritis (34, 35). Mice were intravenously injected with arthritogenic serum from KxB/N mice on day 0 and monitored for disease progression. In littermate-matched male mice, we found that there was no difference in arthritis induction, as assessed by weight loss (Fig. 6A), ankle swelling (Fig. 6B), and clinical scoring (data not shown) up to 7 days post-serum transfer. Furthermore, there were no observable histological differences in the affected joints at that time point (data not shown). Separate experiments with nonlittermate-matched mice suggested that resolution of swelling after arthritis induction was similarly unaffected by mLysMD3 deficiency (data not shown).
Figure 6.
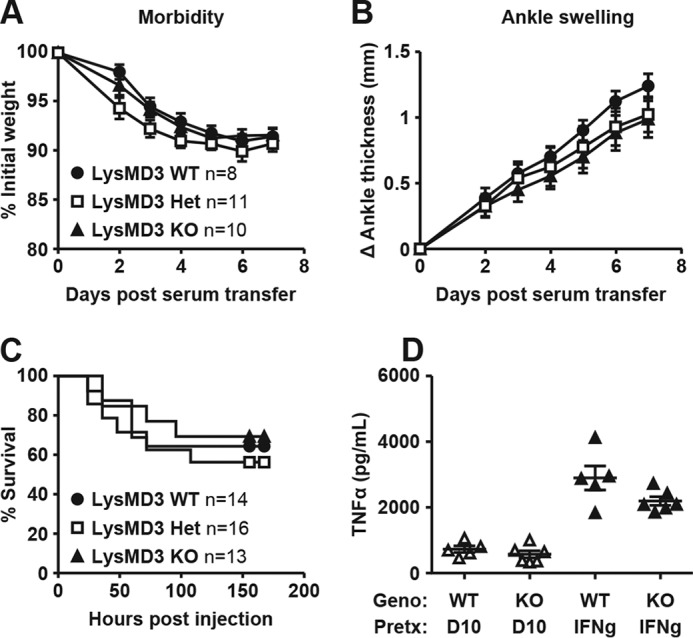
Lack of a role for LysMD3 in mouse models of inflammation. Mice were i.v. injected with KBN serum at day 0 and monitored for weight loss (A) and ankle swelling (B). Data were pooled from two experimental replicates using littermate male mice from the LysMD3 EN mouse line. C, mice were injected i.p. with 20 mg/kg LPS and monitored for death every 12 h. Data were pooled from three experimental replicates using littermate-matched female mice from the LysMD3 GT mouse line on a mixed B6/129P2Ola/Hsd background. D, PECs from the littermate-matched LysMD3 EN mouse line were stimulated with 10 ng/ml LPS ± IFNγ pretreatment. At 6 h post-stimulation, TNFα levels were measured in the supernatant fluids by ELISA. Data were pooled from two separate experimental replicates.
We next tested whether mLysMD3 played a role in the inflammatory response to bacterial products. To address this, we challenged mice with lipopolysaccharide (LPS) and monitored them for mortality (Fig. 6C). We found there was no difference in LPS-induced death in the absence of mLysMD3. Similarly, we found that peritoneal exudate cells (PECs) from littermate-matched mLysMD3-deficient mice produced similar levels of pro-inflammatory cytokines after ex vivo stimulation with LPS (Fig. 6D).
Lack of a role for mLysMD3 in response to viral infection
Given that mLysMD3 is highly expressed in the lung (Fig. 3A), we tested whether mLysMD3 played a role during respiratory viral infection. We first evaluated the ability of mLysMD3 to control influenza virus (IAV) infection in vivo. Mice were infected with mouse-adapted influenza virus H1N1 strain PR8 intranasally (i.n.) and monitored for morbidity and mortality. We found no difference in the susceptibility of mLysMD3-deficient mice to infection with IAV PR8 over the course of the experiment (Fig. 7A).
Figure 7.

Lack of a role for mLysMD3 in the control of influenza and gHV68 viral infection. A, mice were anesthetized and infected IN with H1N1 strain PR8 and monitored for weight loss. Data were pooled from two experiments using littermate-matched female mice from the LysMD3 EN line. B and C, mice were infected with MHV68 i.p. At 16 DPI, PECs (B) and splenocytes (C) were harvested, serially diluted, and plated on a monolayer of MEFs. At 3 weeks post-harvest, MEF monolayers were scored for cytopathic effect, indicating the present of latent virions. Data were pooled from three experiments using nonlittermate B6 mice as a control and the LysMD3 GT mouse line.
We also evaluated the role of mLysMD3 in the control of herpesvirus reactivation from latency. Mice were latently infected with the γ-herpesvirus MHV68 and the ability of MHV68 to reactivate from PECs was evaluated (36, 37). We found no difference in viral reactivation from latently infected LysMD3 WT or LysMD3-deficient PECs (Fig. 7, B and C).
Lack of a role for mLysMD3 in response to fungal infection
Given that LysM domains have been found capable of binding to chitin (5, 38), we evaluated the role of mLysMD3 in the host response to fungal infection. Mice were infected with Cryptococcus neoformans strain KN99α and monitored for lethality (Fig. 8A). In one of three experiments, LysMD3 KO mice were significantly more resistant to C. neoformans infection than their WT littermates. However, this finding was not replicated in two subsequent experiments. We also found no role for mLysMD3 in the response to Aspergillus fumigatus infection in vivo. Mice were infected with A. fumigatus i.n. and monitored for morbidity. We observed no difference in weight loss over the course of the experiment, and we saw no difference in cell recruitment to the lung at 4 DPI (Fig. 8, B and C). Evaluation of lung tissue from A. fumigatus-infected mice revealed no difference in lung inflammation or fungal hyphae growth in the absence of mLysMD3 (data not shown).
Figure 8.

Lack of a role for mLysMD3 in the response to C. neoformans and A. fumigatus infection. A, mice were infected i.n. with 5e4 CFU of C. neoformans and monitored for lethality. Aggregated results from three independent experiments are shown. Mice were sacrificed if the body weight dropped below 80% of initial starting weight. B and C, mice were infected i.n. with 1e7 A. fumigatus conidia and monitored for weight loss (B). At 4 DPI, lungs were lavaged, and BAL cell counts were determined (C).
Lack of a role for mLysMD3 in controlling intestinal bacterial populations or intestinal gene expression
Interactions between host and pathogen are likely to be distinct from interactions between host and commensal organisms. Given that publicly available datasets (39) and our own analysis (Fig. 3A) suggest that LysMD3 was expressed in the intestine, we assessed whether mLysMD3 regulated host interactions with the commensal microbiota. To test this hypothesis, we co-housed littermate WT and mLysMD3-deficient mice for 2 weeks to homogenize the microbiota between mice and to correct for variability due to differences in breeding cohorts. Mice were then singly housed for 2 weeks on a regimen of broad spectrum antibiotics in Kool Aid or Kool Aid alone. Fecal pellets were collected for 16S analysis, and ileal tissue samples were prepared for RNASeq. We found no difference between WT and mLysMD3-deficient mice in bacterial richness or diversity within treatment groups in either mLysMD3-deficient mouse line (Fig. 9, A and B). We did observe a significant drop in richness and diversity in the antibiotic-treated groups, as expected (Fig. 9, A and B). We also applied principal component analysis to these datasets and found that although there was a clear difference between Kool Aid-treated and antibiotic-treated mice, there was not a distinct separation of mice by genotype (Fig. 9C). Finally, RNASeq analysis of ileum samples from both the mLysMD3 GT and LysMD3 EN lines demonstrated no differentially expressed genes between WT and mLysMD3-deficient mice with or without antibiotic treatment that reached significance in both mouse lines (Fig. 9D).
Figure 9.

No microbiota or gene expression alterations in intestines of LysMD3-deficient mice. Littermate-matched WT and LysMD3-deficient (KO) mice from both mouse lines were co-housed for 2 weeks, then subjected to 2 weeks of oral broad-spectrum antibiotics in Kool Aid, or Kool Aid alone. Post-treatment fecal samples were subjected to 16S rRNA sequencing, and ileal RNA was subjected to RNA-Seq analysis. Based on 16S sequencing, bacterial species richness (A) and α diversity (B) were evaluated. C, principal component analysis was applied to weighted UniFrac distances based on 16S rRNA sequences. D, average expression values were calculated for each gene across replicates and plotted on a log10 scale by genotype. Genes with average expression values of <10 were omitted. Genes with at least 2-fold change in expression with adjusted p values of <0.1 appear as black with the remainder in gray. There were no genes with adjusted p values of <0.01.
Lack of a role for mLysMD3 in the innate immune response to C. rodentium infection
Finally, we tested whether mLysMD3 played a role in the innate immune response to infection, which may have been obscured by the presence of the adaptive immune system in previous experiments. To address this possibility, we crossed the LysMD3 EN mouse line to the Rag1 KO background (40). Littermate-matched LysMD3 WT and KO mice on the Rag1 KO background were infected with C. rodentium and monitored for lethality. We found no difference in lethality (Fig. 10A) or stool titers (Fig. 10B) over the course of C. rodentium infection.
Figure 10.
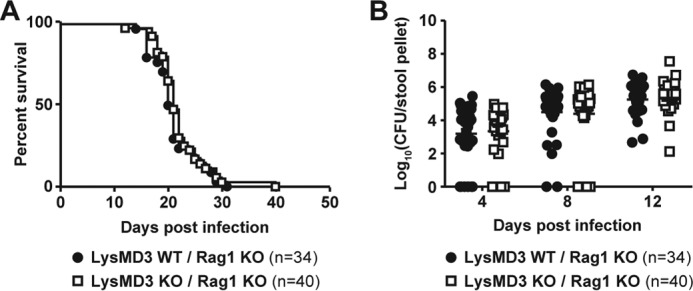
Lack of a role for LysMD3 in the innate immune response to C. rodentium infection. The LysMD3 EN mouse line was crossed onto the Rag1 KO background. Littermate mice were infected with 1e9 CFU C. rodentium i.g. Lethality (A) and C. rodentium fecal shedding (B) were monitored.
Discussion
The annotated LysM domain of mLysMD3 is evolutionarily related to similar domains in human, frog, and zebrafish with orthologs in flies and worms. We found that mouse and human LysMD3 proteins co-localize with GM130+ structures and that they are type II integral membrane proteins, with the LysM domain with a cytoplasmic orientation. In extensive studies of two independent lines of mLysMD3-deficient mice, we observed no evidence for a physiological role for this molecule in a range of pathogen infections, models of inflammation, or interactions with commensal bacterial. One explanation for these in vivo findings is that, while evolutionarily conserved, mLysMD3 has no physiological role. Alternatively, we simply failed to select the right model in which to detect a phenotype for mLysMD3 deficiency, or there is redundancy that obscures the function of this conserved molecule. The latter hypothesis is supported by the lack of an observable developmental phenotype in our mLysMD3-deficient mice.
Significance of mLysMD3 localization and immune functions of endoplasmic reticulum and Golgi proteins
We originally hypothesized that mLysMD3 was a candidate pattern recognition receptor due to homology between the LysMs in flies and plants. Furthermore, publicly available databases and datasets (39, 41, 42), as well as our own data, suggest that mLysMD3 is expressed in tissues where it could feasibly be involved in the sensing of invading pathogens or interactions with the commensal microbiota. Interestingly, however, we found that mLysMD3 co-localizes with GM130+ structures in human and mouse cells, consistent with localization to the Golgi (11), and furthermore, mLysMD3 is a type II integral membrane protein with LysM in the cytoplasm. Although traditional paradigms for PAMP-sensing focus on localization of pattern recognition receptors at the cell surface, it is now appreciated that intracellular organelles such as mitochondria, the endoplasmic reticulum, and the Golgi apparatus can serve innate immune signaling platforms (43). For instance, in primary cells isolated from Drosophila infected with Wolbachia pipientis bacteria, Wolbachia can be identified in a GM130+ cellular compartment or an immediately adjacent compartment (44). In mammals, S. Typhimurium establishes the Salmonella-containing vacuole near the Golgi apparatus (45). Furthermore, Salmonella enterica, B. abortus, C. trachomatis, and L. pneumophila have all been shown to co-opt intracellular trafficking and redirect Golgi-derived vesicles during infection (46). However, we were unable to define a role for mLysMD3 in models of these infections and multiple others. Furthermore, we found that mLysMD3-deficient mice have no evidence of dysbiosis or altered intestinal gene expression. It remains possible that mLysMD3 interacts specifically with a particular microbial product or ligand that we did not evaluate in our studies.
Evolutionary conservation
Despite the fact that mLysMD3 appears to be evolutionarily conserved with proteins found in flies, zebrafish, and worms, it is possible that its function is not conserved. Although Toll and Toll-like receptor family members are central to immune responses in flies and mammals (47, 48), the evolutionarily-conserved peptidoglycan recognition proteins (PGRP and PGLYRP), while essential for the Drosophila immune response, to date have been found to play a relatively minor role in mammalian immunity (49, 50). It is possible that LysMD3 is conserved for reasons completely unrelated to a role in immunity, despite its defining peptidoglycan-binding lysin motif, such as a role during organismal development. Indeed, the Drosophila molecule Toll plays an important role in dorsal-ventral polarity during embryogenesis (51), with secondary function as an important element of the immune response in adult flies (48, 52). Furthermore, LysMD family member expression in zebrafish has been shown to be up-regulated during embryogenesis, with localization to the central nervous system, but without alterations in expression in response to bacterial challenge (53).
mLysMD3 redundancy
LysMD3 is a member of a protein family, and LysMD4 is predicted to also contain a LysM and a transmembrane domain. It is possible that mLysMD4 may compensate for mLysMD3 deficiency in our mouse lines. However, we saw no evidence of mLysMD4 transcript up-regulation in mLysMD3-deficient mice, although it is possible that compensation may occur at the level of protein regulation or in the context of physiological levels of expression. It is also possible that the more distantly related family members mLysMD1 and mLysMD2 could compensate for mLysMD3 deficiency. However, obscuring of mLysMD3 function by mLysMD1 or mLysMD2 redundancy seems unlikely, given that the evolutionary distance between these molecules is significant (Fig. 1C), and mLysMD1 and mLysMD2 are not predicted to be membrane-bound proteins. Mice deficient for multiple family members will be required to evaluate a role for the LysMD protein family in mammalian biology.
Materials and methods
Mice
The embryonic stem cell line Lysmd3Gt(E201G10)Wrst (clone E201G10, 129P2Ola/Hsd background) was purchased from the German Gene Trap Consortium. A single male chimeric founder was bred to B6 mice and backcrossed to >99% B6J background with high-density (∼10 centimorgans) microsatellite–marker-based speed congenic analysis at each generation with the assistance of the Speed Congenics Facility of the Rheumatic Diseases Core Center at Washington University School of Medicine. LysMD3 gene trap mutant mice were genotyped using primers listed in Table 1, with GT mutant alleles identified by primers A + C and WT alleles identified by primers B + C. To generate CRISPR/Cas9-modified mLysMD3-deficient mice, B6 embryos were microinjected with mLysMD3 EN gRNA 1 and 2 (Table 1) and Cas9 mRNA and transferred to pseudopregnant recipient female mice, as described previously (54). Mice were screened for loss of exon 2 by Southern blotting, and targeted alleles were verified by Sanger sequencing. A single male founder was crossed to a B6 female, and F1 pups were intercrossed to generate subsequent breeding pairs and experimental animals. Mice were genotyped using the primers listed in Table 1, with WT alleles identified by primers A + B and EN mutant alleles identified by primers A + C. All mice were bred and housed in an enhanced barrier-specific pathogen-free facility at Washington University in St. Louis. All experiments were performed using age- and sex-matched mice between 8 and 12 weeks of age generated by intercrossing heterozygous mice (littermate mice), unless otherwise noted. B. abortus experiments were performed at the University of California Davis using age- and sex-matched littermate mice between 7 and 17 weeks of age. Experiments conducted at Washington University were approved by the Institutional Animal Care and Use Committee of Washington University. B. abortus experiments conducted at the University of California Davis were approved by the University of California Davis Institutional Animal Care and Use Committee.
Table 1.
Primers and gRNA
| Target | Primer sequence |
|---|---|
| mLysMD3 GT genotyping A | GAGCCCCCAAATGAAAGAC |
| mLysMD3 GT genotyping B | GTCTAGACCGGGTGGATGAA |
| mLysMD3 GT genotyping C | TCTTCGCGCATCTGTCTCTA |
| mLysMD3 EN genotyping A | TACAACAGGGTCTCAACCACG |
| mLysMD3 EN genotyping B | GCTGGCAGCAAGAACCTTTG |
| mLysMD3 EN genotyping C | AGGTCACCAGTAAAGGAATTTCA |
| Mouse actin qPCR F | GCTCCTTCGTTGCCGGTCCA |
| Mouse actin qPCR R | TTGCACATGCCGGAGCCGTT |
| Mouse actin qPCR probe | CACCAGTTCGCCATGGATGACGA |
| mLysMD3 EN gRNA 1 | GCCAGAGACACCCCGATACCTGG |
| mLysMD3 EN gRNA 2 | GCTTTGTAACCTGCCCGATAGGG |
| hLysMD3 gRNA | TGACCACTTGACTGAACTCCNGG |
Cell culture
MEFs were established from embryos derived from progeny of an LysMD3 GT heterozygous mouse cross, genotyped as above, and used before passage six. Unless otherwise stated, MEFs derived from WT littermates were used as controls. MEFs were grown in DMEM supplemented with 10% (v/v) FBS and 1% penicillin/streptomycin. All HeLa cell lines were maintained in DMEM with 10% FBS. hLYSMD3 HeLa KO cells (ΔLYSMD3) were generated using CRISPR/Cas9 genome editing at the Genome Engineering and iPSC Center at Washington University School of Medicine (St. Louis, MO), as described previously (55). Briefly, hLYSMD3 exon 2 was targeted using hLysMD3 gRNA (Table 1), and single cell clones were sequenced to confirm the complete absence of WT alleles and disruption of the ORF.
Plasmids
To generate an N-terminal FLAG and C-terminal HA expression vector, the Gateway cloning vector pTAG-attR-C1 (56), containing three N-terminal FLAG/CBP tags, was modified to encode three in-frame HA tags downstream of the Gateway cloning site. To generate an insert tagged with an N-terminal EGFP, the FLAG/CBP motif in pTAG-attR-C1 was replaced with EGFP. The cDNA IMAGE clone 3156298 containing mLysMD3 was purchased from ATCC. The mLysMD3 protein-coding region was PCR-amplified and cloned into the Gateway destination vectors described above. mLysMD3 was also cloned into pEGFP-N1 (Clontech) using Gibson assembly master mix (New England Biolabs). All expression plasmids were sequence-verified.
Antibodies
Rabbit polyclonal anti-LysMD3 antibody was raised against amino acids 1–205 of mouse LysMD3 (Cocalico). mLysMD3(1–205) was conjugated to AffiGel15 (Bio-Rad) and used to affinity-purify the resulting immune serum. Commercially available antibodies were as follows: anti-β-actin (Sigma A5316); anti-ERK1/2 (Cell Signaling 4695); anti-FLAG M2 (Sigma F1804); anti-GFP (Abcam ab6556); anti-GM130 (Pharmingen 610822); anti-H2AX (Millipore 07-627); anti-HA (Sigma H9658); anti-hLysMD3 (Sigma HPA018024 and Proteintech 24313-1-AP); and anti-transferrin receptor (Life Technologies, Inc., 13-6800). Secondary antibodies were as follows: donkey anti-rabbit AF488 (Invitrogen A21206); goat anti-mouse AF555 (Invitrogen A21425); goat anti-mouse AF633 (Invitrogen A21052); donkey anti-rabbit AF647 (Invitrogen A-31573); goat anti-mouse HRP (Jackson ImmunoResearch 115-035-146); and goat anti-rabbit HRP (Jackson ImmunoResearch 111-035-003).
Phylogenetic analysis
Phylogenetic analysis was performed using the maximum likelihood method in the MEGA6 package (57) with 500 bootstrap replicates. The tree with the highest log likelihood is shown. Phylogenetic trees were visualized using TreeView (58). Sequences used for the phylogenetic analysis were as follows: Caenorhabditis briggsae CBG12503, XP_002640031; C. elegans F43G9, NP_001122475; D. melanogaster CG12207, NP_650352; D. melanogaster CG15471, NP_572187; D. melanogaster CG17985, NP_610305; D. melanogaster Mtd, NP_652017; D. rerio LysMD1, NP_001070218; D. rerio LysMD2, NP_001003507; D. rerio LysMD3, NP_001002104; D. rerio LysMD4, NP_957144; Drosophila simulans GD10233, XP_002080462; E. coli MltD-1, NP_414747; E. coli MltD-2, NP_414747; E. faecalis AtlA, NP_814543; H. sapiens LysMD1, NP_997716; H. sapiens LysMD2, NP_699205; H. sapiens LysMD3, NP_938014; H. sapiens LysMD4, AAH84545; M. musculus LysMD1, NP_694761; M. musculus LysMD2, NP_081585; M. musculus LysMD3, NP_084533; M. musculus LysMD4, NP_780424; M. musculus Ncoa7, NP_766083; M. musculus Oxr1, NP_001123635; X. tropicalis LysMD1, NP_001096341; X. tropicalis LysMD2, NP_001037868; X. tropicalis LysMD3, NP_001017308; and X. tropicalis LysMD4, XP_012815036.
Cell fractionation and immunoblot
WT bone marrow macrophages were generated as described previously (59). Briefly, bone marrow cells were cultured in 10% FBS, 10% CMG14-12 supernatant fluids containing macrophage colony-stimulating factor (60). At day 7, cells were subjected to differential detergent fractionation using a QProteome kit (Qiagen), according to the manufacturer's instructions. Fractions were subjected to Western blot analysis with antibodies to the indicated proteins. All Western blotting experiments were repeated twice, and representative images are shown.
Protease protection assay
TransIT-LT1 (Mirus) was used to transfect an expression construct coding for FLAG-LysMD3-HA into HeLa cells. 24 h post-transfection, cells were treated with 80 μm digitonin to permeabilize the plasma membrane. Samples were treated with the indicated concentration of proteinase K on ice, and protease activity was inhibited by the addition of phenylmethylsulfonyl fluoride prior to Western blot analysis with the indicated antibodies. Experiments were repeated twice, and representative blots are shown.
Immunofluorescence
MEFs or HeLa cells were seeded on coverslips, fixed in 4% methanol-free paraformaldehyde, permeabilized using 0.5% Triton X-100, and blocked in 1% BSA and 10% serum corresponding to the secondary antibody species. Cells were stained using the indicated primary and secondary antibodies and phalloidin AF594 or AF647 (Invitrogen). Coverslips were mounted using Prolong Gold Antifade reagent (Invitrogen). For determination of membrane orientation by immunofluorescence staining, TransIT-LT1 (Mirus) was used to transfect HeLa cells with the indicated LysMD3 constructs. Cells were fixed, blocked, and stained with an anti-GFP antibody prior to permeabilization using 0.5% Triton X-100, re-blocking, and re-staining for GFP where appropriate. Phalloidin-Alexa594 (Invitrogen) was used to visualize actin. Immunofluorescence images were acquired using a Zeiss LSM510 confocal microscope or a Zeiss LSM880 confocal laser-scanning microscope. Unless otherwise noted, three separate experiments were performed, and representative images are shown.
qRT-PCR
RNA was extracted from tissues, and qRT-PCR was performed as described (61). RNA was extracted from tissues with TRIzol (Life Technologies, Inc.) or TRI Reagent (Sigma) according to the manufacturer's protocol. One μg of RNA was used as a template for random-primed cDNA synthesis with ImPromII reverse transcriptase (Promega). Transcripts were detected using predesigned TaqMan assays (IDT, Coralville, IA) listed in Table 2, and the absolute number of transcript copies was determined by comparison with target sequence-containing gBlocks (IDT) and normalized to actin (Table 1).
Table 2.
Predesigned qPCR assays
| Target | Assay |
|---|---|
| mLysMD3 | Mm.PT.58.10100260 |
| mLysMD4 | Mm.PT.58.12179173 |
| Adgrv1 | Mm.PT.58.43743756 |
| Polr3g | Mm.PT.58.10070464 |
C. rodentium
Kanamycin-resistant C. rodentium strain DBS120 (62, 63) cultures in log-phase growth were pelleted and resuspended in PBS with 3% bicarbonate. Mice were infected with ∼1e9 CFU intragastrically. Mice were subsequently weighed. Bacterial titers per stool pellet were determined by homogenizing a single stool pellet in 1 ml of PBS with 0.05% Triton X-100 using 1-mm silica beads (Biospec) on a mini-beadbeater 24 (Biospec) and plating serial dilutions on LB agar plates.
S. enterica serotype Typhimurium
S. enterica serotype Typhimurium SL1344 was used to inoculate streptomycin-pretreated mice, as described previously (18). Mice were fasted and pretreated with 200 μg of streptomycin 24 h before infection. On the day of infection, mice were fasted for 4 h, then gavaged with 1e8 CFU of SL1344 in PBS, and harvested in log-phase growth.
Histological preparation of tissues and colitis scoring
Colons and ceca were harvested and flushed with 10% neutral buffered formalin. Tissues were cut open, pinned flat, allowed to fix overnight at 4 °C, washed in 70% ethanol, and embedded in agar for processing. Sections were H&E-stained and scored for colitis severity using the following scoring system: 0, no acute inflammation; 1, acute inflammation in surface epithelium, lamina propria, or cryptitis; 2, crypt abscess; 3, acute inflammation present at muscularis mucosa or beyond; 4, ulcer or transmural inflammation. The extent of involved colon was also evaluated as an estimated percentage.
Listeria monocytogenes
L. monocytogenes strain EGD from a frozen glycerol stock was used to infect mice i.p. as described (64). L. monocytogenes experiments used B6 mice as controls and LysMD3-deficient mice were generated by intercrossing KO mice.
F. novicida
BMMo were differentiated in DMEM with 10% FBS and 20% macrophage colony-stimulating factor and infected with F. novicida U112 and ΔFPI as described previously (65, 66). Cytotoxicity was measured using lactate dehydrogenase release (Promega), and type I IFN levels were measured using ISRE-L929 reporter cells (67).
M. tuberculosis
M. tuberculosis Erdman strain bacteria were used to infect mice by the aerosol route, as described previously (64, 68). Approximately 100 CFU of M. tuberculosis was inoculated using an Inhalation Exposure System (Glass-Col, Terre Haute, IN). At 0 or 24 HPI, 1–2 mice were sacrificed, and the number of CFU delivered per mouse were quantitated to determine infection efficiency. Tissue bacterial titers were determined by plating dilutions of lung and spleen homogenate on 7H10 agar plates.
B. abortus
Littermate-matched mice between the ages of 7 and 17 weeks were bred and housed at Washington University and shipped to UC Davis, where they were housed in microisolator cages with sterile bedding and irradiated feed in a biosafety level 3 laboratory. Mice were infected intraperitoneally (i.p.) with 1e6 CFU of B. abortus 2308. Spleens and livers were collected aseptically. Spleens and livers were homogenized, and serial dilutions of the homogenate were plated on tryptic soy agar for enumeration of CFUs.
L. pneumophila
Primary BMMo were infected with L. pneumophila, as described previously (30, 69). Lactate dehydrogenase release was quantitated using the Cytotox assay (Promega) as described previously (30).
Uropathogenic E. coli
Anesthetized female mice were transurethrally inoculated with 1e7 CFU UPEC strain UTI89. At 24 HPI, urine was collected for CFU enumeration and cytokine analysis; serum was collected for cytokine analysis, and bladder tissues were removed for inflammation scoring (70, 71). IL1β concentrations in the urine and serum were determined by ELISA (R&D Systems). A semiquantitative scoring system, with a scale of 0 (normal) to 5 (necrosis with full-thickness inflammatory infiltration), was used to evaluate bladder inflammation at 24 HPI (71). UTI89 infection of BMMo was performed as described (72). Briefly, macrophages were differentiated from whole bone marrow in DMEM containing 15% FBS and 30% L929 conditioned media. Cells were re-plated on day 8 and challenged with UTI89 on day 9 at an MOI of 0.1. Supernatant fluids were collected at the indicated time points, and IL1β concentration was determined as above.
C. trachomatis and S. flexneri
Infections of BMMo with C. trachomatis serovar L2 434/Bu at an MOI of 10 or S. flexneri serovar 2a WT strain 2457T at an MOI of 1 were performed in triplicate as described (73, 74), and supernatant fluids were collected for cytokine analysis using L929-ISRE fibroblasts to measure IFNβ levels (73). Sandwich ELISA was used to determine IL6 levels (BD Biosciences).
Serum-induced arthritis
Serum was isolated from K/BxN mice as described previously (34). Serum was injected intravenously (i.v.) into littermate-matched male mice, and morbidity and ankle swelling were determined at the indicated times post-serum transfer (75).
LPS treatments
Mice were injected i.p. with 20 mg/kg LPS purified by phenol extraction (Sigma L-2880). PECs were isolated by flushing the peritoneal cavity with 10 ml of ice-cold DMEM containing 10% FBS. Cells were counted, plated on 96-well tissue culture-treated plates, and incubated for 3 days. Cells were stimulated with 100 ng/ml LPS purified by ion-exchange chromatography (Sigma L4524) ± 16 h IFNγ pretreatment. Supernatant fluids were collected 6 h post-stimulation, and TNFα was quantitated by ELISA (BD Biosciences).
Influenza
8–12-Week-old mice were anesthetized and inoculated with 50 TCID50 H1N1 influenza virus strain PR8 i.n., as described (76). Mice losing more than 30% of their initial body weight were sacrificed.
MHV68 ex vivo limiting dilution assay for reactivation from latency
MHV68 WUMS (ATCC VR1465) was used, and reactivation from latency was measured as described (36, 37, 77). Mice were infected with 1e6 PFU MHV68 i.p., and after 16 days, PECs or spleens were pooled from 3 to 5 mice, and serial dilutions of cells were plated on a MEF monolayer. After 3 weeks, wells were scored for cytopathic effect to detect reactivation. Data points represent the mean and standard error for three replicate experiments using three mice per genotype per experiment. MHV68 experiments used B6 mice as controls, and LysMD3-deficient mice were generated by intercrossing KO mice.
C. neoformans
C. neoformans strain KN99α was recovered from 15% glycerol stocks stored at −80 °C and maintained on YPD plates (1% yeast extract, 2% peptone, 2% dextrose, and 2% Bacto agar). A single colony was inoculated into YPD broth and grown for 16 h at 30 °C with shaking, collected by centrifugation, washed three times with sterile PBS, and counted on a Cellometer Auto M10 (Nexcelom Bioscience, Lawrence, MA). Female WT or LysMD3 KO littermate mice from the LysMD3 GT line were anesthetized by i.p. injection (of 150 μl of 2 mg/ml xylazine (VEDCO) and 10 mg/ml of ketaset (Fort Dodge Animal Health)) and i.n. inoculated with 5e4 CFU (verified by quantitative culture on YPD agar) in 50 μl of sterile PBS. Mice were euthanized when body weight fell below 80% of peak weight.
A. fumigatus
A. fumigatus CEA10 (CBS 144.89) was grown, and conidia were harvested similar to Ref. 78. Mice were infected i.n. with 1e7 conidia. Weight was monitored daily, and mice were sacrificed at 72 HPI. Lungs were lavaged with 1 ml of PBS with EDTA and fetal bovine serum. RBCs in the BAL were lysed, and cell counts were performed. Cytospin preparations were analyzed by light microscopy to determine differential counts. Lungs were fixed, paraffin-embedded, and stained with hematoxylin and eosin for assessment of pathological changes. Hyphae were stained using Grocott methamine silver.
Antibiotic treatment of mice for 16S analysis
Female WT and KO mice were co-housed for 2 weeks, at a ratio of 2 WT and 2 KO mice per cage. After 2 weeks, mice were individually housed and were administered grape Kool-Aid ± a broad-spectrum antibiotic mixture in their drinking water (79) for another 2 weeks. The antibiotic mixture consisted of 1 g/liter ampicillin, 1 g/liter metronidazole, 1 g/liter neomycin, 0.5 g/liter vancomycin (Sigma) in 20 mg/ml grape Kool-Aid (Kraft Foods, Northfield, IL). After antibiotic treatment, stool pellets were collected for 16S analysis (79).
16S rRNA Illumina sequencing and analysis
Preparation of fecal pellets for 16S analysis was as described previously (79, 80). Briefly, DNA was phenol/chloroform-extracted and amplified in triplicate with Golay-barcoded primers specific for the V4 region of the 16S rRNA. Amplicons were pooled and purified with 0.6× Agencourt Ampure XP beads (Beckman-Coulter) prior to sequencing at the Center for Genome Sciences, Washington University School of Medicine, by the 2 × 250-bp protocol on the Illumina MiSeq platform. 16S rRNA gene sequences were resolved using dada2 (81). Taxonomy was assigned to resolved sequences according to the Greengenes database (version 13.8) (82). All subsequent analyses were performed using PhyloSeq (version 1.16.2) (83) to calculate richness, per sample Shannon diversity, and both weighted and unweighted UniFrac distances among samples (84). Differential abundance of bacterial taxa between experimental groups was determined using the PhyloSeq DESeq2 extension using the Wald significance test and a local fit type (version 1.6.3) (83, 85).
RNA-Seq
RNA from the distal ileum was purified; an Illumina sequencing library was generated, and libraries were sequenced (HiSeq platform), as described previously (37). Five to six mice were included in each group. DESeq2 was used to identify differentially expressed genes (85).
Sequencing data
RNA-seq and 16S sequencing data were deposited to the European nucleotide archive under the accession numbers PRJEB23707 and PRJEB25196, respectively.
Antigen presentation
Antigen presentation by BMMo was assessed as described (86, 87). BMMo were generated in 10% FBS and 10% CMG14–12 supernatant fluids (60) and infected with L. monocytogenes strain 10403S or pulsed with LLO peptide ± IFNγ priming. At 1 HPI, infected BMMo were washed with Dulbecco's PBS, and gentamycin was added. Splenic CD4 T cells isolated from LLO56tg mice (86), bearing a T cell receptor specific for LLO190-225, were isolated using a CD4-negative selection kit (Miltenyi Biotech, Bergisch Gladbach, Germany) and added to the macrophage cultures. After 24 h, cells were stained for CD4 (eFluor450, Clone RM4-5, eBioscience) and CD69 (PECy7, Clone H1.2F3, BioLegend) to evaluate activation status.
Author contributions
C. C. Y., M. T. B., D. W. L., J. E. D., G. K. A., C. A. M., and H. W. V. conceptualization; C. C. Y., G. Z., C. D., and S. A. H. data curation; C. C. Y. validation; C. C. Y., M. T. B., D. W. L., G. Z., T.-C. L., V. E. D.-O., J. P. H., J. M. K., E. L. S., C. R. H., R. A. I., S. P., K. M. S., C. W., A. V. M., E. C., and T. K. investigation; C. C. Y. visualization; C. C. Y., D. W. L., G. Z., C. D., T.-C. L., V. E. D.-O., J. P. H., J. M. K., E. L. S., C. R. H., R. A. I., S. P., K. M. S., C. W., S. H., A. V. M., E. C., T. K., M. N. S., I. U. M., P. M. A., D. M. M., M.C. D., T. L. D., R. M. T., C. L. S., and G. K. A. methodology; C. C. Y. writing-original draft; C. C. Y., M. T. B., D. W. L., G. Z., C. D., T.-C. L., V. E. D.-O., J. P. H., J. M. K., E. L. S., C. R. H., R. A. I., S. P., K. M. S., C. W., S. H., A. V. M., E. C., T. K., M. N. S., S. A. H., I. U. M., P. M. A., D. M. M., M.C. D., T. L. D., R. M. T., J. E. D., C. L. S., G. K. A., C. A. M., and H. W. V. writing-review and editing; M. T. B., D. W. L., M. N. S., S. A. H., I. U. M., P. M. A., D. M. M., M.C. D., T. L. D., R. M. T., C. L. S., G. K. A., C. A. M., and H. W. V. supervision; G. Z., C. D., T.-C. L., and S. A. H. formal analysis; C. D. software; S. H. resources; C. A. M. and H. W. V. project administration; H. W. V. funding acquisition.
Supplementary Material
Acknowledgments
We thank Darren Kreamalmeyer for assistance with animal husbandry; Andrea Bredemeyer, Barry Sleckman, and W. Tim Schaiff for technical assistance; and Broc T. McCune, Thomas Burke, and Daniel Portnoy for providing reagents. Experimental support was provided by the Genome Engineering Center of the Alvin J. Siteman Cancer Center, the Speed Congenics Facility, the Microinjection Core of the Rheumatic Diseases Core Center, and the Molecular Microbiology Imaging Facility at Washington University School of Medicine and Barnes-Jewish Hospital in St. Louis, MO. The Siteman Cancer Center is supported by National Institutes of Health Grant P30CA091842. The Rheumatic Diseases Core Center is supported by National Institutes of Health Grant P30AR048335.
This work was supported in part by the Children's Discovery Institute of Washington University and St. Louis Children's Hospital (to M. C. D.), Grant 274415 from the Crohn's and Colitis Foundation Genetics Initiative (to H. W. V.), National Science Foundation Fellowships DGE-1143954 (to J. P. H.) and DGE-1143954 (to J. M. K.), and National Institutes of Health Grants T32AI007163 (to C. C. Y.), T32GM007067 (to J. M. K.), T32AI007172 (to C. R. H.), F32AI108089 (to K. M. S.), R01DK100644 (to I. U. M.), R01AI24157 (to P. M. A.), R01AI095396 (to D. M. M.), R01AI102882 (to T. L. D.), and U19AI109725 (to H. W. V.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S3.
- PAMP
- pathogen-associated molecular pattern
- qRT
- quantitative RT
- CFU
- colony-forming unit
- CBP
- calmodulin-binding peptide
- DPI
- days post-infection
- MOI
- multiplicity of infection
- WPI
- weeks post-infection
- IFN
- interferon
- HPI
- hours post-infection
- MEF
- mouse embryonic fibroblast
- LPS
- lipopolysaccharide
- UPEC
- uropathogenic E. coli
- PEC
- peritoneal exudate cell
- TNF
- tumor necrosis factor
- KO
- knockout
- RNASeq
- RNA sequencing
- EGFP
- enhanced green fluorescent protein
- i.n.
- intranasally
- i.g.
- intragastrically
- i.p.
- intraperitoneally
- LysM
- lysin motif
- DMEM
- Dulbecco's modified Eagle's medium
- FBS
- fetal bovine serum
- BMMo
- bone marrow macrophage
- UPEC
- uropathogenic E. coli
- IAV
- influenza virus
- h
- human
- GT
- gene trap
- UTI
- urinary tract infection.
References
- 1. Newton K., and Dixit V. M. (2012) Signaling in innate immunity and inflammation. Cold Spring Harb. Perspect. Biol. 4, a006049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Eckert C., Lecerf M., Dubost L., Arthur M., and Mesnage S. (2006) Functional analysis of AtlA, the major N-acetylglucosaminidase of Enterococcus faecalis. J. Bacteriol. 188, 8513–8519 10.1128/JB.01145-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Andre G., Leenhouts K., Hols P., and Dufrêne Y. F. (2008) Detection and localization of single LysM-peptidoglycan interactions. J. Bacteriol. 190, 7079–7086 10.1128/JB.00519-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Frankel M. B., and Schneewind O. (2012) Determinants of murein hydrolase targeting to cross-wall of Staphylococcus aureus peptidoglycan. J. Biol. Chem. 287, 10460–10471 10.1074/jbc.M111.336404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mesnage S., Dellarole M., Baxter N. J., Rouget J.-B., Dimitrov J. D., Wang N., Fujimoto Y., Hounslow A. M., Lacroix-Desmazes S., Fukase K., Foster S. J., and Williamson M. P. (2014) Molecular basis for bacterial peptidoglycan recognition by LysM domains. Nat. Commun. 5, 4269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wong J. E., Midtgaard S. R., Gysel K., Thygesen M. B., Sørensen K. K., Jensen K. J., Stougaard J., Thirup S., and Blaise M. (2015) An intermolecular binding mechanism involving multiple LysM domains mediates carbohydrate recognition by an endopeptidase. Acta Crystallogr. D Biol. Crystallogr. 71, 592–605 10.1107/S139900471402793X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Caruso R., Warner N., Inohara N., and Núñez G. (2014) NOD1 and NOD2: signaling, host defense, and inflammatory disease. Immunity 41, 898–908 10.1016/j.immuni.2014.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Djordjevic M. A., Bezos A., Susanti Marmuse L., Driguez H., Samain E., Vauzeilles B., Beau J.-M., Kordbacheh F., Rolfe B. G., Schwörer R., Daines A. M., Gresshoff P. M., and Parish C. R. (2014) Lipo-chitin oligosaccharides, plant symbiosis signalling molecules that modulate mammalian angiogenesis in vitro. PLoS ONE 9, e112635 10.1371/journal.pone.0112635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang Z., Berkey C. D., and Watnick P. I. (2012) The Drosophila protein mustard tailors the innate immune response activated by the immune deficiency pathway. J. Immunol. 188, 3993–4000 10.4049/jimmunol.1103301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kelley L. A., Mezulis S., Yates C. M., Wass M. N., and Sternberg M. J. (2015) The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858 10.1038/nprot.2015.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nakamura N., Rabouille C., Watson R., Nilsson T., Hui N., Slusarewicz P., Kreis T. E., and Warren G. (1995) Characterization of a cis-Golgi matrix protein, GM130. J. Cell Biol. 131, 1715–1726 10.1083/jcb.131.6.1715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Plutner H., Davidson H. W., Saraste J., and Balch W. E. (1992) Morphological analysis of protein transport from the ER to Golgi membranes in digitonin-permeabilized cells: role of the P58 containing compartment. J. Cell Biol. 119, 1097–1116 10.1083/jcb.119.5.1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cadwell K., Liu J. Y., Brown S. L., Miyoshi H., Loh J., Lennerz J. K., Kishi C., Kc W., Carrero J. A., Hunt S., Stone C. D., Brunt E. M., Xavier R. J., Sleckman B. P., Li E., et al. (2008) A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 456, 259–263 10.1038/nature07416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pham C. T., MacIvor D. M., Hug B. A., Heusel J. W., and Ley T. J. (1996) Long-range disruption of gene expression by a selectable marker cassette. Proc. Natl. Acad. Sci. U.S.A. 93, 13090–13095 10.1073/pnas.93.23.13090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Eckmann L. (2006) Animal models of inflammatory bowel disease: lessons from enteric infections. Ann. N.Y. Acad. Sci. 1072, 28–38 10.1196/annals.1326.008 [DOI] [PubMed] [Google Scholar]
- 16. Mundy R., MacDonald T. T., Dougan G., Frankel G., and Wiles S. (2005) Citrobacter rodentium of mice and man. Cell. Microbiol. 7, 1697–1706 10.1111/j.1462-5822.2005.00625.x [DOI] [PubMed] [Google Scholar]
- 17. Vidal S., Gros P., and Skamene E. (1995) Natural resistance to infection with intracellular parasites: molecular genetics identifies Nramp1 as the Bcg/Ity/Lsh locus. J. Leukocyte Biol. 58, 382–390 10.1002/jlb.58.4.382 [DOI] [PubMed] [Google Scholar]
- 18. Barthel M., Hapfelmeier S., Quintanilla-Martínez L., Kremer M., Rohde M., Hogardt M., Pfeffer K., Rüssmann H., and Hardt W.-D. (2003) Pretreatment of mice with streptomycin provides a Salmonella enterica serovar Typhimurium colitis model that allows analysis of both pathogen and host. Infect. Immun. 71, 2839–2858 10.1128/IAI.71.5.2839-2858.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pamer E. G. (2004) Immune responses to Listeria monocytogenes. Nat. Rev. Immunol. 4, 812–823 10.1038/nri1461 [DOI] [PubMed] [Google Scholar]
- 20. Oyston P. C., Sjostedt A., and Titball R. W. (2004) Tularaemia: bioterrorism defence renews interest in Francisella tularensis. Nat. Rev. Microbiol. 2, 967–978 10.1038/nrmicro1045 [DOI] [PubMed] [Google Scholar]
- 21. Weiss D. S., Brotcke A., Henry T., Margolis J. J., Chan K., and Monack D. M. (2007) In vivo negative selection screen identifies genes required for Francisella virulence. Proc. Natl. Acad. Sci. U.S.A. 104, 6037–6042 10.1073/pnas.0609675104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Frieden T. R., Sterling T. R., Munsiff S. S., Watt C. J., and Dye C. (2003) Tuberculosis. Lancet 362, 887–899 [DOI] [PubMed] [Google Scholar]
- 23. Zumla A., Raviglione M., Hafner R., and von Reyn C. F. (2013) Tuberculosis. N. Engl. J. Med. 368, 745–755 10.1056/NEJMra1200894 [DOI] [PubMed] [Google Scholar]
- 24. O'Garra A., Redford P. S., McNab F. W., Bloom C. I., Wilkinson R. J., and Berry M. P. (2013) The immune response in tuberculosis. Annu. Rev. Immunol. 31, 475–527 10.1146/annurev-immunol-032712-095939 [DOI] [PubMed] [Google Scholar]
- 25. Flynn J. L., and Chan J. (2001) Immunology of tuberculosis. Annu. Rev. Immunol. 19, 93–129 10.1146/annurev.immunol.19.1.93 [DOI] [PubMed] [Google Scholar]
- 26. Philips J. A., and Ernst J. D. (2012) Tuberculosis pathogenesis and immunity. Annu. Rev. Pathol. 7, 353–384 10.1146/annurev-pathol-011811-132458 [DOI] [PubMed] [Google Scholar]
- 27. Pappas G., Akritidis N., Bosilkovski M., and Tsianos E. (2005) Brucellosis. N. Engl. J. Med. 352, 2325–2336 10.1056/NEJMra050570 [DOI] [PubMed] [Google Scholar]
- 28. Byndloss M. X., and Tsolis R. M. (2016) Brucella spp. virulence factors and immunity. Annu. Rev. Anim. Biosci. 4, 111–127 10.1146/annurev-animal-021815-111326 [DOI] [PubMed] [Google Scholar]
- 29. Fields B. S., Benson R. F., and Besser R. E. (2002) Legionella and Legionnaires' disease: 25 years of investigation. Clin. Microbiol. Rev. 15, 506–526 10.1128/CMR.15.3.506-526.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ren T., Zamboni D. S., Roy C. R., Dietrich W. F., and Vance R. E. (2006) Flagellin-deficient Legionella mutants evade caspase-1- and Naip5-mediated macrophage immunity. PLoS Pathog. 2, e18 10.1371/journal.ppat.0020018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vance R. E. (2010) Immunology taught by bacteria. J. Clin. Immunol. 30, 507–511 10.1007/s10875-010-9389-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Terlizzi M. E., Gribaudo G., and Maffei M. E. (2017) UroPathogenic Escherichia coli (UPEC) infections: virulence factors, bladder responses, antibiotic, and non-antibiotic antimicrobial strategies. Front. Microbiol. 8, 1566 10.3389/fmicb.2017.01566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Heng T. S., Painter M. W., and Immunological Genome Project Consortium. (2008) The Immunological Genome Project: networks of gene expression in immune cells. Nat. Immunol. 9, 1091–1094 10.1038/ni1008-1091 [DOI] [PubMed] [Google Scholar]
- 34. Wipke B. T., and Allen P. M. (2001) Essential role of neutrophils in the initiation and progression of a murine model of rheumatoid arthritis. J. Immunol. 167, 1601–1608 10.4049/jimmunol.167.3.1601 [DOI] [PubMed] [Google Scholar]
- 35. Sadik C. D., Kim N. D., Iwakura Y., and Luster A. D. (2012) Neutrophils orchestrate their own recruitment in murine arthritis through C5aR and FcγR signaling. Proc. Natl. Acad. Sci. U.S.A. 109, E3177–E3185 10.1073/pnas.1213797109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tibbetts S. A., Loh J., Van Berkel V., McClellan J. S., Jacoby M. A., Kapadia S. B., Speck S. H., and Virgin H. W. 4th. (2003) Establishment and maintenance of gammaherpesvirus latency are independent of infective dose and route of infection. J. Virol. 77, 7696–7701 10.1128/JVI.77.13.7696-7701.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Park S., Buck M. D., Desai C., Zhang X., Loginicheva E., Martinez J., Freeman M. L., Saitoh T., Akira S., Guan J.-L., He Y.-W., Blackman M. A., Handley S. A., Levine B., Green D. R., et al. (2016) Autophagy genes enhance murine gammaherpesvirus 68 reactivation from latency by preventing virus-induced systemic inflammation. Cell Host Microbe 19, 91–101 10.1016/j.chom.2015.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tanaka K., Nguyen C. T., Liang Y., Cao Y., and Stacey G. (2013) Role of LysM receptors in chitin-triggered plant innate immunity. Plant Signal. Behav. 8, e22598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wu C., Orozco C., Boyer J., Leglise M., Goodale J., Batalov S., Hodge C. L., Haase J., Janes J., Huss J. W. 3rd., and Su A. I. (2009) BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 10, R130 10.1186/gb-2009-10-11-r130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mombaerts P., Iacomini J., Johnson R. S., Herrup K., Tonegawa S., and Papaioannou V. E. (1992) RAG-1-deficient mice have no mature B and T lymphocytes. Cell 68, 869–877 10.1016/0092-8674(92)90030-G [DOI] [PubMed] [Google Scholar]
- 41. Lattin J. E., Schroder K., Su A. I., Walker J. R., Zhang J., Wiltshire T., Saijo K., Glass C. K., Hume D. A., Kellie S., and Sweet M. J. (2008) Expression analysis of G protein-coupled receptors in mouse macrophages. Immunome Res. 4, 5 10.1186/1745-7580-4-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Su A. I., Wiltshire T., Batalov S., Lapp H., Ching K. A., Block D., Zhang J., Soden R., Hayakawa M., Kreiman G., Cooke M. P., Walker J. R., and Hogenesch J. B. (2004) A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl. Acad. Sci. U.S.A. 101, 6062–6067 10.1073/pnas.0400782101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Weinberg S. E., Sena L. A., and Chandel N. S. (2015) Mitochondria in the regulation of innate and adaptive immunity. Immunity 42, 406–417 10.1016/j.immuni.2015.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cho K.-O., Kim G.-W., and Lee O.-K. (2011) Wolbachia bacteria reside in host Golgi-related vesicles whose position is regulated by polarity proteins. PLoS ONE 6, e22703 10.1371/journal.pone.0022703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hilbi H., and Haas A. (2012) Secretive bacterial pathogens and the secretory pathway. Traffic 13, 1187–1197 10.1111/j.1600-0854.2012.01344.x [DOI] [PubMed] [Google Scholar]
- 46. Dolat L., and Valdivia R. H. (2017) Bacterial subversion of COG-dependent membrane traffic. Trends Cell Biol. 27, 877–878 10.1016/j.tcb.2017.09.004 [DOI] [PubMed] [Google Scholar]
- 47. Medzhitov R., Preston-Hurlburt P., and Janeway C. A. (1997) A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 388, 394–397 10.1038/41131 [DOI] [PubMed] [Google Scholar]
- 48. Lemaitre B., Nicolas E., Michaut L., Reichhart J. M., and Hoffmann J. A. (1996) The dorsoventral regulatory gene cassette spätzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell 86, 973–983 10.1016/S0092-8674(00)80172-5 [DOI] [PubMed] [Google Scholar]
- 49. Dziarski R., and Gupta D. (2010) Review: Mammalian peptidoglycan recognition proteins (PGRPs) in innate immunity. Innate Immun. 16, 168–174 10.1177/1753425910366059 [DOI] [PubMed] [Google Scholar]
- 50. Xu M., Wang Z., and Locksley R. M. (2004) Innate immune responses in peptidoglycan recognition protein L-deficient mice. Mol. Cell. Biol. 24, 7949–7957 10.1128/MCB.24.18.7949-7957.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Anderson K. V., Jürgens G., and Nüsslein-Volhard C. (1985) Establishment of dorsal-ventral polarity in the Drosophila embryo: genetic studies on the role of the Toll gene product. Cell 42, 779–789 10.1016/0092-8674(85)90274-0 [DOI] [PubMed] [Google Scholar]
- 52. Lemaitre B., and Hoffmann J. (2007) The host defense of Drosophila melanogaster. Annu. Rev. Immunol. 25, 697–743 10.1146/annurev.immunol.25.022106.141615 [DOI] [PubMed] [Google Scholar]
- 53. Laroche F. J., Tulotta C., Lamers G. E., Meijer A. H., Yang P., Verbeek F. J., Blaise M., Stougaard J., and Spaink H. P. (2013) The embryonic expression patterns of zebrafish genes encoding LysM-domains. Gene Expr. Patterns 13, 212–224 10.1016/j.gep.2013.02.007 [DOI] [PubMed] [Google Scholar]
- 54. Parikh B. A., Beckman D. L., Patel S. J., White J. M., and Yokoyama W. M. (2015) Detailed phenotypic and molecular analyses of genetically modified mice generated by CRISPR-Cas9-mediated editing. PLoS ONE 10, e0116484 10.1371/journal.pone.0116484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Selleck E. M., Orchard R. C., Lassen K. G., Beatty W. L., Xavier R. J., Levine B., Virgin H. W., and Sibley L. D. (2015) A noncanonical autophagy pathway restricts Toxoplasma gondii growth in a strain-specific manner in IFN-γ-activated human cells. mBio. 6, e01157–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lee S., Salwinski L., Zhang C., Chu D., Sampankanpanich C., Reyes N. A., Vangeloff A., Xing F., Li X., Wu T.-T., Sahasrabudhe S., Deng H., Lacount D. J., and Sun R. (2011) An integrated approach to elucidate the intra-viral and viral-cellular protein interaction networks of a gamma-herpesvirus. PLoS Pathog. 7, e1002297 10.1371/journal.ppat.1002297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tamura K., Stecher G., Peterson D., Filipski A., and Kumar S. (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729 10.1093/molbev/mst197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Page R. D. (1996) TreeView: an application to display phylogenetic trees on personal computers. Comput. Appl. Biosci. 12, 357–358 [DOI] [PubMed] [Google Scholar]
- 59. Hwang S., Maloney N. S., Bruinsma M. W., Goel G., Duan E., Zhang L., Shrestha B., Diamond M. S., Dani A., Sosnovtsev S. V., Green K. Y., Lopez-Otín C., Xavier R. J., Thackray L. B., and Virgin H. W. (2012) Nondegradative role of Atg5-Atg12/ Atg16L1 autophagy protein complex in antiviral activity of interferon γ. Cell Host Microbe 11, 397–409 10.1016/j.chom.2012.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Takeshita S., Kaji K., and Kudo A. (2000) Identification and characterization of the new osteoclast progenitor with macrophage phenotypes being able to differentiate into mature osteoclasts. J. Bone Miner. Res. 15, 1477–1488 10.1359/jbmr.2000.15.8.1477 [DOI] [PubMed] [Google Scholar]
- 61. Yokoyama C. C., Loh J., Zhao G., Stappenbeck T. S., Wang D., Huang H. V., Virgin H. W., and Thackray L. B. (2012) Adaptive immunity restricts replication of novel murine astroviruses. J. Virol. 86, 12262–12270 10.1128/JVI.02018-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Schauer D. B., and Falkow S. (1993) The eae gene of Citrobacter freundii biotype 4280 is necessary for colonization in transmissible murine colonic hyperplasia. Infect. Immun. 61, 4654–4661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kim Y.-G., Kamada N., Shaw M. H., Warner N., Chen G. Y., Franchi L., and Núñez G. (2011) The Nod2 sensor promotes intestinal pathogen eradication via the chemokine CCL2-dependent recruitment of inflammatory monocytes. Immunity 34, 769–780 10.1016/j.immuni.2011.04.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. MacDuff D. A., Reese T. A., Kimmey J. M., Weiss L. A., Song C., Zhang X., Kambal A., Duan E., Carrero J. A., Boisson B., Laplantine E., Israel A., Picard C., Colonna M., Edelson B. T., Sibley L. D., Stallings C. L., Casanova J.-L., Iwai K., and Virgin H. W. (2015) Phenotypic complementation of genetic immunodeficiency by chronic herpesvirus infection. Elife 4, e04494 10.7554/eLife.04494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Peng K., Broz P., Jones J., Joubert L.-M., and Monack D. (2011) Elevated AIM2-mediated pyroptosis triggered by hypercytotoxic Francisella mutant strains is attributed to increased intracellular bacteriolysis. Cell. Microbiol. 13, 1586–1600 10.1111/j.1462-5822.2011.01643.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Storek K. M., Gertsvolf N. A., Ohlson M. B., and Monack D. M. (2015) cGAS and Ifi204 cooperate to produce type I IFNs in response to Francisella infection. J. Immunol. 194, 3236–3245 10.4049/jimmunol.1402764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Jiang Z., Georgel P., Du X., Shamel L., Sovath S., Mudd S., Huber M., Kalis C., Keck S., Galanos C., Freudenberg M., and Beutler B. (2005) CD14 is required for MyD88-independent LPS signaling. Nat. Immunol. 6, 565–570 10.1038/ni1207 [DOI] [PubMed] [Google Scholar]
- 68. Kimmey J. M., Huynh J. P., Weiss L. A., Park S., Kambal A., Debnath J., Virgin H. W., and Stallings C. L. (2015) Unique role for ATG5 in neutrophil-mediated immunopathology during M. tuberculosis infection. Nature 528, 565–569 10.1038/nature16451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wright E. K., Goodart S. A., Growney J. D., Hadinoto V., Endrizzi M. G., Long E. M., Sadigh K., Abney A. L., Bernstein-Hanley I., and Dietrich W. F. (2003) Naip5 affects host susceptibility to the intracellular pathogen Legionella pneumophila. Curr. Biol. 13, 27–36 10.1016/S0960-9822(02)01359-3 [DOI] [PubMed] [Google Scholar]
- 70. Mysorekar I. U., and Hultgren S. J. (2006) Mechanisms of uropathogenic Escherichia coli persistence and eradication from the urinary tract. Proc. Natl. Acad. Sci. U.S.A. 103, 14170–14175 10.1073/pnas.0602136103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Stemler K. M., Crock L. W., Lai H. H., Mills J. C., Gereau R. W. 4th., and Mysorekar I. U. (2013) Protamine sulfate induced bladder injury protects from distention induced bladder pain. J. Urol. 189, 343–351 10.1016/j.juro.2012.08.189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Symington J. W., Wang C., Twentyman J., Owusu-Boaitey N., Schwendener R., Núñez G., Schilling J. D., and Mysorekar I. U. (2015) ATG16L1 deficiency in macrophages drives clearance of uropathogenic E. coli in an IL-1β-dependent manner. Mucosal Immunol. 8, 1388–1399 10.1038/mi.2015.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Jehl S. P., Nogueira C. V., Zhang X., and Starnbach M. N. (2012) IFNγ inhibits the cytosolic replication of Shigella flexneri via the cytoplasmic RNA sensor RIG-I. PLoS Pathog. 8, e1002809 10.1371/journal.ppat.1002809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Gondek D. C., Olive A. J., Stary G., and Starnbach M. N. (2012) CD4+ T cells are necessary and sufficient to confer protection against Chlamydia trachomatis infection in the murine upper genital tract. J. Immunol. 189, 2441–2449 10.4049/jimmunol.1103032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Monach P., Hattori K., Huang H., Hyatt E., Morse J., Nguyen L., Ortiz-Lopez A., Wu H.-J., Mathis D., and Benoist C. (2007) The K/BxN mouse model of inflammatory arthritis, theory and practice. Methods Mol. Med. 136, 269–282 10.1007/978-1-59745-402-5_20 [DOI] [PubMed] [Google Scholar]
- 76. Lu Q., Yokoyama C. C., Williams J. W., Baldridge M. T., Jin X., DesRochers B., Bricker T., Wilen C. B., Bagaitkar J., Loginicheva E., Sergushichev A., Kreamalmeyer D., Keller B. C., Zhao Y., Kambal A., et al. (2016) Homeostatic control of innate lung inflammation by Vici syndrome gene Epg5 and additional autophagy genes promotes influenza pathogenesis. Cell Host Microbe 19, 102–113 10.1016/j.chom.2015.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Weck K. E., Barkon M. L., Yoo L. I., Speck S. H., Virgin H. W. (1996) Mature B cells are required for acute splenic infection, but not for establishment of latency, by murine gammaherpesvirus 68. J. Virol. 70, 6775–6780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Morgenstern D. E., Gifford M. A., Li L. L., Doerschuk C. M., and Dinauer M. C. (1997) Absence of respiratory burst in X-linked chronic granulomatous disease mice leads to abnormalities in both host defense and inflammatory response to Aspergillus fumigatus. J. Exp. Med. 185, 207–218 10.1084/jem.185.2.207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Baldridge M. T., Nice T. J., McCune B. T., Yokoyama C. C., Kambal A., Wheadon M., Diamond M. S., Ivanova Y., Artyomov M., and Virgin H. W. (2015) Commensal microbes and interferon-λ determine persistence of enteric murine norovirus infection. Science 347, 266–269 10.1126/science.1258025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Caporaso J. G., Lauber C. L., Walters W. A., Berg-Lyons D., Lozupone C. A., Turnbaugh P. J., Fierer N., and Knight R. (2011) Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. U.S.A. 108, Suppl. 1, 4516–4522 10.1073/pnas.1000080107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Callahan B. J., McMurdie P. J., Rosen M. J., Han A. W., Johnson A. J., and Holmes S. P. (2016) DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583 10.1038/nmeth.3869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. McDonald D., Price M. N., Goodrich J., Nawrocki E. P., DeSantis T. Z., Probst A., Andersen G. L., Knight R., and Hugenholtz P. (2012) An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 6, 610–618 10.1038/ismej.2011.139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. McMurdie P. J., and Holmes S. (2012) Phyloseq: A bioconductor package for handling and analysis of high-throughput phylogenetic sequence data. Pac. Symp. Biocomput. 2012, 235–246 [PMC free article] [PubMed] [Google Scholar]
- 84. Lozupone C., and Knight R. (2005) UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 71, 8228–8235 10.1128/AEM.71.12.8228-8235.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Love M. I., Huber W., and Anders S. (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Weber K. S., Li Q. J., Persaud S. P., Campbell J. D., Davis M. M., and Allen P. M. (2012) Distinct CD4+ helper T cells involved in primary and secondary responses to infection. Proc. Natl. Acad. Sci. U.S.A. 109, 9511–9516 10.1073/pnas.1202408109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Redmann V., Lamb C. A., Hwang S., Orchard R. C., Kim S., Razi M., Milam A., Park S., Yokoyama C. C., Kambal A., Kreamalmeyer D., Bosch M. K., Xiao M., Green K., Kim J., et al. (2016) Clec16a is critical for autolysosome function and Purkinje cell survival. Sci. Rep. 6, 23326 10.1038/srep23326 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.