

Abstract
Utilization of energy-rich carbon sources such as glucose is fundamental to the evolutionary success of bacteria. Glucose can be catabolized via glycolysis for feeding the intermediary metabolism. The methylglyoxal synthase MgsA produces methylglyoxal from the glycolytic intermediate dihydroxyacetone phosphate. Methylglyoxal is toxic, requiring stringent regulation of MgsA activity. In the Gram-positive bacterium Bacillus subtilis, an interaction with the phosphoprotein Crh controls MgsA activity. In the absence of preferred carbon sources, Crh is present in the nonphosphorylated state and binds to and thereby inhibits MgsA. To better understand the mechanism of regulation of MgsA, here we performed biochemical and structural analyses of B. subtilis MgsA and of its interaction with Crh. Our results indicated that MgsA forms a hexamer (i.e. a trimer of dimers) in the crystal structure, whereas it seems to exist in an equilibrium between a dimer and hexamer in solution. In the hexamer, two alternative dimers could be distinguished, but only one appeared to prevail in solution. Further analysis strongly suggested that the hexamer is the biologically active form. In vitro cross-linking studies revealed that Crh interacts with the N-terminal helices of MgsA and that the Crh–MgsA binding inactivates MgsA by distorting and thereby blocking its active site. In summary, our results indicate that dimeric and hexameric MgsA species exist in an equilibrium in solution, that the hexameric species is the active form, and that binding to Crh deforms and blocks the active site in MgsA.
Keywords: prokaryotic signal-transduction, protein cross-linking, protein structure, protein-protein interaction, Bacillus, bacteria, bacterial genetics, bacterial metabolism, metabolic regulation, crystal structure, catabolite regulation, Crh, glycolysis, methylglyoxal synthase, phosphoprotein, methylglyoxal toxicity, glucose catabolism, Crh, HPr
Introduction
The rapid and selective utilization of energy-rich carbon sources is essential for the evolutionary success of bacteria. For many bacteria, including the Gram-positive model organism Bacillus subtilis, glucose is the preferred source of carbon and energy (1). Glucose can be catabolized via glycolysis and thus feed into the intermediary metabolism. The availability of glucose results in a variety of regulatory events to transport and metabolize this sugar efficiently as well as to prevent the simultaneous utilization of secondary carbon sources. In B. subtilis, several regulators are involved in these processes. The RNA-binding transcription antiterminator GlcT is required for the expression of the glucose-specific transporter of the phosphotransferase system (PTS)5 (2). Two regulators, CggR and CcpN, control the expression of enzymes involved in the lower part of glycolysis and gluconeogenesis, respectively (3, 4). Finally, the global transcription factor CcpA represses the expression of genes involved in the utilization of less preferred carbon sources and activates genes for overflow metabolism and amino acid biosynthesis (5–8). The DNA-binding activity of CcpA is controlled by an interaction with a regulatory protein that acts as cofactor. This protein, the HPr protein of the PTS, can be phosphorylated on a serine residue (Ser46) in response to the availability of preferred carbon sources by a dedicated kinase, HPrK. In the presence of glucose, the intracellular pools of ATP and fructose 1,6-bisphosphate are high, and these metabolites activate the kinase activity of HPrK (9–12). The resulting HPr-pSer acts as the cofactor for CcpA (13, 14). The CcpA-HPr complex is central to the global process of carbon catabolite repression in Gram-positive bacteria with a low GC content (Firmicutes) (1, 15–17).
B. subtilis and closely related bacilli contain a paralog of HPr, Crh (18). In contrast to HPr, which has its main function as a phosphotransferase in the PTS, Crh does not participate in PTS-mediated sugar transport (18). The role of this protein has long been enigmatic. The HPrK also phosphorylates Crh in a carbon source–dependent manner (18, 19), and it had been proposed that it acts as another cofactor for CcpA (20). However, both the weak expression of Crh and its low affinity to CcpA (21, 22) suggest that the physiological role of the protein might be different. To gain more insight into the function of this protein, we have searched for Crh interaction partners and found that it binds the methylglyoxal synthase MgsA (23).
MgsA is a key protein of a bypass of glycolysis, the formation of methylglyoxal from dihydroxyacetone phosphate. Methylglyoxal has been shown to regulate cell elongation (24), and this bypass acts as an essential overflow mechanism if phosphorylated glycolytic intermediates accumulate in the cell. These phosphorylated intermediates are toxic for the cell (25, 26); however, methylglyoxal at elevated concentrations is toxic as well (27). Thus, the bacteria have a choice that is comparable with the one between Scylla and Charybdis. Therefore, the formation of methylglyoxal has to be tightly controlled, and the bacterium needs efficient mechanisms for its disposal. Indeed, three pathways for the degradation of methylglyoxal to lactate or acetol have been discovered in B. subtilis (28).
The mgsA gene encoding the methylglyoxal synthase is part of a seven-gene operon in B. subtilis. In addition to MgsA, this operon encodes enzymes for bacillithiol production, which are required for the major pathway of methylglyoxal degradation, as well as essential enzymes for cell wall biosynthesis and protein biotinylation. This operon including the mgsA gene is constitutively expressed, but expression is increased by thiol depletion (29, 30). Thus, regulation of MgsA enzymatic activity seems to be the major mechanism to control the production of the toxic metabolite methylglyoxal. In both Escherichia coli and B. subtilis, this enzyme is inhibited by Pi, which is generated by the conversion of dihydroxyacetone phosphate to methylglyoxal (23, 31, 32). In addition, nonphosphorylated Crh inactivates MgsA by direct protein-protein interaction in B. subtilis (23). This form of Crh is present if B. subtilis grows with poor carbon sources (23). Thus, MgsA activity is inhibited if B. subtilis faces nutrient limitation.
It is interesting to note that MgsA interacts specifically with nonphosphorylated Crh, but not with the phosphorylated form of the protein. Moreover, HPr, which exhibits 45% identity with Crh, is also unable to bind MgsA. In a previous study, we have shown that the N-terminal α-helix of Crh is essential for the interaction of the two proteins. Moreover, replacement of the four differing amino acids in HPr in this helix with those found in Crh allowed efficient binding of HPr to MgsA (23).
In this work, we have studied the molecular details of MgsA multimerization and the interaction between Crh and MgsA. Our findings suggest that MgsA is active only as a hexamer and, upon binding of Crh, the active site is deformed and thus blocked. Crh binding might result in decreased flexibility of the active center assemblies of MgsA hexamers and, thus, in loss of MgsA activity.
Results
Effect of Crh on the status of MgsA
To gain insight into the molecular mechanism by which Crh controls MgsA activity, we performed a size-exclusion chromatography and multiangle light scattering (SEC-MALS) analysis of recombinantly expressed and freshly purified proteins. The results using MgsA identified exclusively dimers when a low concentration of MgsA (13 μm) was used (see Fig. 1A). This concentration is still about 10-fold increased compared with the cellular concentration (about 1 μm as calculated from Ref. 33). This is in contrast to the known oligomeric states of MgsA from other organisms showing a multimer exhibiting predominantly a tetrameric state in solution (32, 34–36). Moreover, in MgsA from Thermus sp. GH5, only a hexameric state has been found in solution and in all the crystal structures available to date (see also Fig. S1) (37–39). To mimic the conditions in the crystallization experiments, the concentration was increased by a factor of about 10 (135 versus 13 μm), resulting in MgsA eluting as a dimer, and a broader peak width was observed for MgsA when the protein was injected at a higher concentration, indicating a higher-number oligomerization state (Fig. 1B). These results, using MgsA alone, strongly suggest an equilibrium of dimers (corresponding to 38 kDa as determined by MALS) and multimers (by calculation using the MALS data a tetramer, which would correspond roughly to the calculated 62 kDa). Interpretation of the MALS data and the shape of the elution profile from the gel-filtration experiments argue in favor of a highly dynamic assembly and disassembly of MgsA between a stable dimeric and an instable hexameric state, resulting in an average multimeric state of about 4. This would best explain the observed differences between the in-solution and in crystallo MgsA oligomerization states.
Figure 1.
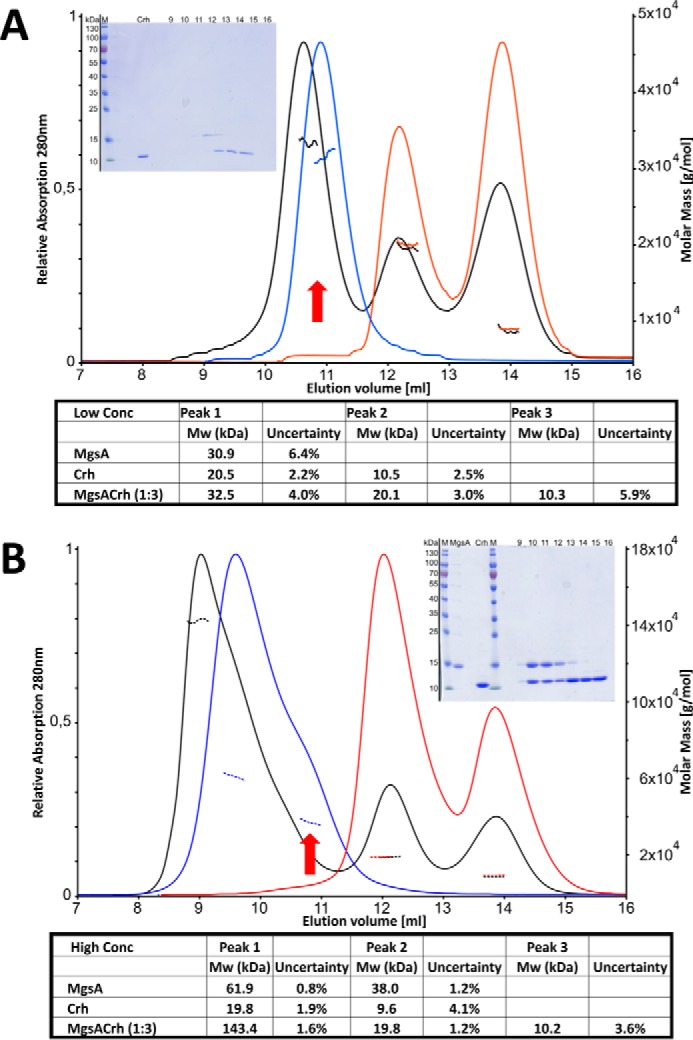
The MgsA binds to Crh only if MgsA is present in hexamers. SEC-MALS experiments suggest that only the MgsA hexamer binds Crh, resulting in a 1:1 stoichiometry. A, low MgsA concentration (13 μm). B, high MgsA concentration (135 μm). In each case, Crh was used in a 3-fold molar excess. Insets, Coomassie-stained gel of the respective fractions (equivalent to the ml of elution volume) obtained in the SEC run of the MgsA/Crh mixture. Red arrows, elution volume of dimeric MgsA. Below each chromatogram, the calculated molar masses determined by MALS are listed. Blue lines, MgsA; red lines, Crh; black lines, mixture of MgsA and Crh (concentrations as indicated). Dotted line, molecular masses as determined by MALS.
Moreover, our experiments with MgsA revealed that the stability of the hexameric complex strongly depends on the surrounding medium, as, for example, phosphate or glycerol strongly increases the percentage of MgsA in a hexameric state (Fig. S2; also see “Discussion”).
The purified regulator of MgsA, Crh, elutes in two peaks corresponding to the dimer and monomer form in a varying ratio, as has been already observed earlier (40) (Fig. 1, peaks corresponding to a calculated mass of 20 and 10 kDa, respectively).
In the presence of a 3-fold excess of Crh over MgsA, the formation of larger complexes was observed for MgsA at higher concentration (Fig. 1B). The elution volume of this corresponds to a mass of 143 kDa, which can be best explained by a hexamer of MgsA (105 kDa) bound to six Crh molecules (9.6 kDa each). This calculation is supported by the SDS-PAGE analysis of the respective fractions, which show the presence of equal amounts of both proteins (see Fig. 1, inset). Interestingly, at low concentration, only a slight shift of the first peak was observed in the elution volume (Fig. 1A), which, together with mass determined by MALS, could be interpreted as either a weak interaction between MgsA and Crh or even as a MgsA dimer and as a lack of interaction with Crh. The results from the SDS-PAGE support this conclusion (Fig. 1A, inset).
The crystal structure of MgsA
To obtain structural information on B. subtilis MgsA, purified MgsA was crystallized, and X-ray diffraction data were collected. The crystals diffracted to a resolution of 2.34 Å, and the phase problem was solved by means of molecular replacement using the methylglyoxal synthase from Thermus sp. (PDB code 2XW6) as a search model. The crystal structure was refined at 2.34 Å resolution (see Table 1 for data statistics).
Table 1.
Data collection and refinement statistics
The numbers in parentheses refer to the highest-resolution shell.
| Data collection | |
| Wavelength (Å) | 0.7732 |
| Resolution range (Å) | 48.99–2.34 (2.423–2.34) |
| Space group | P 21 21 21 |
| Unit cell parameters | |
| a, b, c (Å) | 108.93, 109.71, 199.95 |
| α = β = γ (°) | 90 |
| Total reflections | 478,161 (48,123) |
| Unique reflections | 101,094 (10,027) |
| Multiplicity | 4.7 (4.8) |
| Completeness (%) | 99.41 (99.72) |
| Mean I/σ(I) | 15.94 (2.41) |
| Wilson B-factor (Å2) | 46.98 |
| Rmerge | 0.06155 (0.6581) |
| Rmeas | 0.06938 |
| CC1/2 | 0.999 (0.739) |
| CC | 1 (0.922) |
| Refinement | |
| Rwork | 0.170 (0.252) |
| Rfree | 0.205 (0.277) |
| No. of non-hydrogen atoms | 11,898 |
| No. of macromolecules | 11,278 |
| No. of ligands | 77 |
| No. of waters | 526 |
| No. of protein residues | 1464 |
| RMSD (bonds) (Å) | 0.007 |
| RMSD (angles) (degrees) | 1.079 |
| Ramachandran favored (%) | 99.2 |
| Ramachandran allowed (%) | 0.28 |
| Ramachandran outliers (%) | 0 |
| Clashscore | 2.00 |
| Average B-factor (Å2) | 54.7 |
| B-factor for macromolecules | 54.6 |
| B-factor for ligands | 71.8 |
| B-factor for solvent | 53.8 |
| PDB code | 6F2C |
The core of the MgsA monomer is composed of the prototypic five β-strand/α-helix repeats, resulting in a protein with a centered all parallel β-sheet and the arrangement flanked by three α-helices on one side and two on the other (Fig. 2A). Short helical turns (denoted η2 and η1) precede helix α4 or connect directly to helix α1, respectively. In contrast to the canonical motif, helix α1 is followed by a short α-helix, numbered as α2 before the next β-strand (β2) (Fig. 2A). Surprisingly, the ∼17 C-terminal residues, which are thought to be involved in interaction with neighboring MgsA molecules and allosteric regulation of the active site (37, 39), are missing; most likely, they are disordered due to an increased flexibility (see below, and also see “Discussion”).
Figure 2.
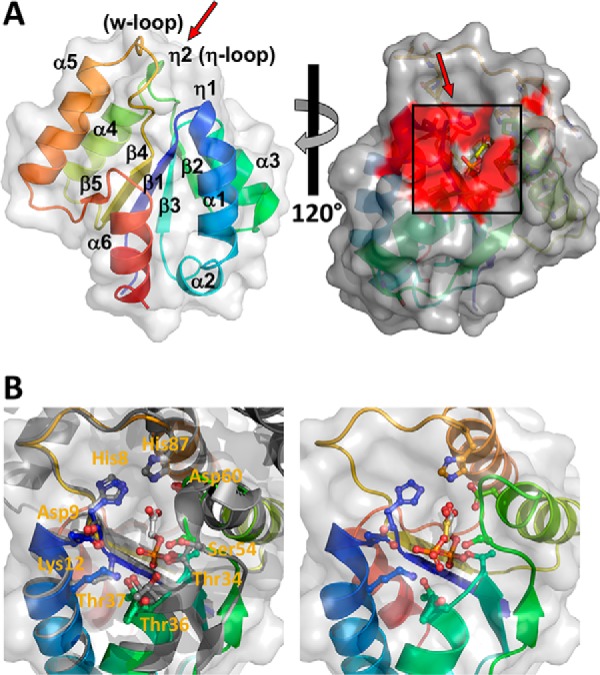
The structure of B. subtilis MgsA exhibits an overall globular shape. A, the central five-stranded β-sheet is flanked by six α-helices. The coloring is in a rainbow from the N terminus (blue) to the C terminus (red). The surface structure is depicted in gray; the individual secondary structure motifs are indicated and numbered as α-helices (α) or β-strand (β) from the N to the C terminus. The panel on the right shows a surface representation of an MgsA monomer in gray with the surface region of important active site residues highlighted in red. B, left, magnification of the active site as deduced from an overlay with the E. coli MgsA (gray) with 2-phosphoglycolic acid (ball-and-stick mode, PDB code 1EGH) bound, serving as reference ligand. Cavity conformation and arrangement of active site residues are highly conserved (see also Figs. S3 and S5). Identical residues of B. subtilis MgsA within the Mgs family involved in ligand binding are depicted with their side chains in ball-and-stick mode with the carbon atoms labeled according to A, with oxygen in red and nitrogen in blue. Residue numbering is according to B. subtilis MgsA. Right, result of a docking experiment with the substrate dihydroxyacetone phosphate (carbons in yellow, phosphorus in orange) and phosphoglycolohydroxamic acid (PDB code 1IK4) as reference.
The search for homologous proteins performed using the DALI server (41) identified other methylglyoxal synthases deposited in the PDB as having the highest degree of similarity to MgsA (Z-score of 24 to 19 and a root mean square deviation (RMSD) ranging between 0.7 and 1.7 Å over 120–150 residues) (Fig. S3). The superposition reveals that the central cores of all enzymes are highly similar, with the structures of the methylgyloxal synthases from E. coli and Thermotoga maritima bearing an additional domain formed by N- and C-terminal regions visible in the crystal structures (Fig. S3). The second-best match are the large subunits of carbamoyl phosphate synthetases (Z-score of 14 to 10) with a root mean square deviation of 2.3–2.9 Å over 108–114 residues in the C-terminal region. The most prominent difference from the large subunit of the carbamoyl phosphate synthetase from E. coli is a major structural rearrangement in the region of the transition from strand β3 to helix α3.
The structure-based sequence alignment of MgsA from B. subtilis and the other four organisms from which structures have been deposited in the Protein Data Bank helped to identify structurally conserved regions in MgsA (Fig. S4) in all of these structures. In general, they are mostly localized in the region of the C-terminal ends of the β-strands, the adjacent loop regions, or the beginning of the following α-helices (Fig. S4).
The superimposition of MgsA and the methylglyoxal synthase from E. coli (50% identity and 73% similarity (PDB code 1B93)) (37) as well as the structures from Thermus thermophilus (PDB code 1WO8) and Thermus sp. (PDB code 2XW6) allowed us to deduce the localization of the active-site residues in MgsA. The other proteins have been crystallized in the presence of phosphate (PDB code 2XW6), sulfate (PDB code 1WO8), formate ions (PDB code 1B93) (37), malonate (PDB code 2X8W), phosphoglycolohydroxamic acid (PDB code 1IK4) (42), or the competitive inhibitor 2-phosphoglycolate (PDB code 1EGH) (43). The active-site region of MgsA is formed by the residues located in the loop regions subsequent to the C termini of β-strands 1- 4, harboring also most of the highly conserved residues (Fig. 2B (left); see also Fig. S4). The residues involved in the active-site formation almost perfectly match in position in the B. subtilis and E. coli enzymes, in particular residues His8, Asp9, Lys12, Thr34, Thr36, Thr37, Ser54, Asp60, and His87 (according to the numbering of B. subtilis MgsA). Their arrangement results in the formation of a well-defined deep cavity that is blocked by residues of the loop connecting strand β4 with helix α5 (termed the w-loop), which harbors the conserved His87 at its N-terminal end (Fig. 2 and Fig. S4). Two histidines (i.e. His8 and His87) are located at the base of this steep and prolonged wall of the cavity formed by the loop connecting strand β3 with helix α4 (termed the η-loop, as it harbors the η2-helix). On the opposing side, a shallow saddle is formed by a patch of three threonines (residues 34, 36, and 37) and Gly35. This quasi-contained region harbors the active center of the enzyme formed by residues Asp9 and Lys12 on one side as well as Ser54 and Asp60 on the other. As the active site has been identified by localization of products and an inhibitor in it, the binding mode of the educt was investigated by in silico docking experiments. Using a relaxed MgsA molecule and having the educt 1,3-dihydroxyacetonephosphate docked into it, the obtained model exhibits an arrangement of the ligand similar to that observed for 2-phosphoglycolate in E. coli MgsA (Fig. 2B, right). Surprisingly, docking failed completely if the obtained crystal structure was used, omitting the relaxation step before the docking experiment (data not shown), suggesting that the minute structural changes in the active site arrangement caused by the relaxation have a dramatic effect on binding and perhaps also activity.
As described for the E. coli protein, the B. subtilis MgsA forms hexamers in the crystal structure (Fig. 3). Overall, the hexamer can be described as a trimer of dimers (chains AB, CD, and EF; Fig. 3). The resulting alternating and antiparallel arrangement of the individual subunits in the hexamer results in six active centers, with three on each side (Fig. 3) and a high connectivity between the individual molecules. Each subunit interacts with four of the five other subunits of the hexamer. Subunit A forms a “triangle” with two other subunits (C and E; as defined by orientation of their active centers) with a rotation of 120°, resulting in interaction of the w-loop and η-loop with the identical loops of the other two subunits, covering about 150 Å2 of surface area each, resulting in 300 Å2 of surface area per subunit. Moreover, both the w- and the η-loop regions interact with the central scaffold of the subunits from the second triangle (subunits B, D, and F) (i.e. helices α4 and α5). Interestingly, both loops harbor highly conserved residues that take part in the active-site formation and function. As loops are usually quite flexible, their interaction with this rigid core of the neighboring molecule entity might positively or negatively influence active-site pocket accessibility and function.
Figure 3.
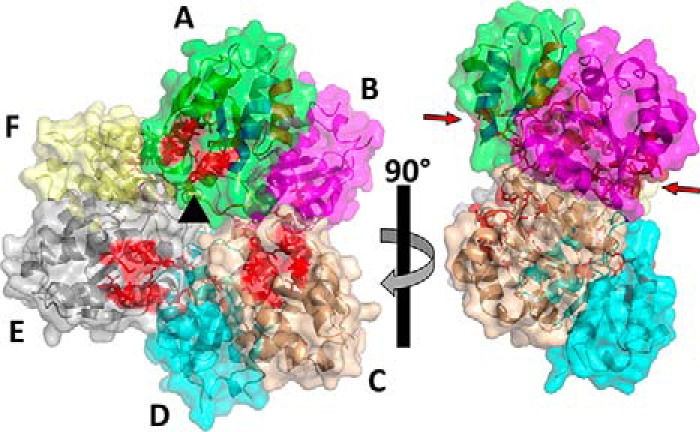
Structure of the B. subtilis MgsA hexamer. The hexamer is composed of three dimers of MgsA, which arrange in the hexamer with three active sites (active-site residues indicated in red) arranged by 120° rotations (indicated by the black triangle in the center) on either side of the hexamer. The individual molecules are indicated in different colors and labeled A–F (top right panel). Bottom right panel, side view of the hexamer. The arrows indicate the active sites of molecules A and B.
The majority of interactions within the hexamer are formed by two directly neighboring subunits within the hexamer by surface areas of ∼850 and ∼750 Å2 (Fig. 4; see below for details). The smaller of the two surfaces (e.g. molecule A and F, denoted AF) is formed by helices α4 and α5 of both molecules that are oriented in opposing directions with an almost 45° tilt with respect to each other (Fig. 4). The C-terminal ends of these helices are followed by the two loop regions, the η-loop and the w-loop. The larger surface (denoted AB) is formed by helices α5 and α6 as well as β5 of both molecules. Strands β5 are oriented antiparallel and thus form the continuation of the distorted central β sheet from one subunit to the β sheet of the other. The two strands are in the center of a rhombus with the four helices forming their sides (Fig. 4B). Taken together, in the crystal structure, the hexamer of MgsA is stabilized by an intricate interaction pattern involving contacts between almost all subunits present in the molecular assembly.
Figure 4.
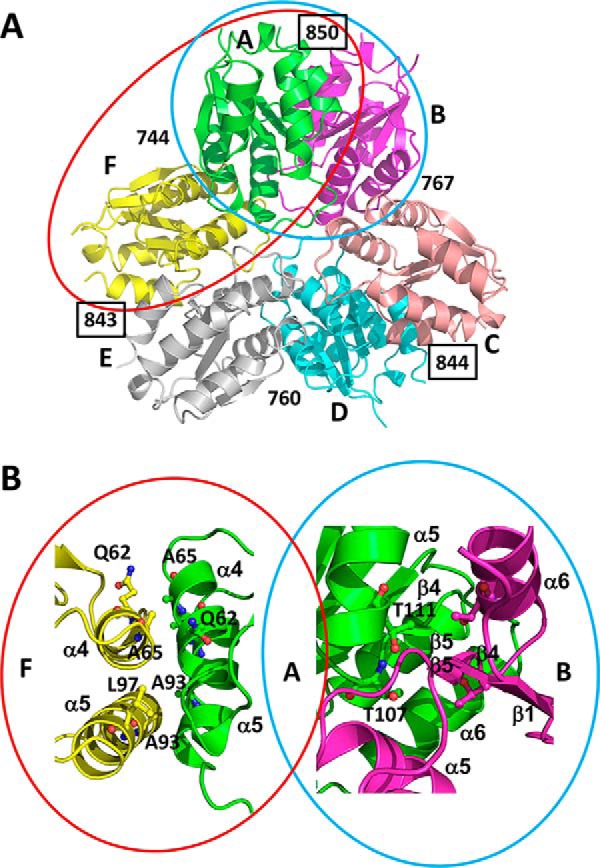
Mutational and structural analysis suggests hexamer disassembly into MgsA dimers. A, the disassembly of the hexamer (molecules colored and labeled as in Fig. 3) into dimers could result in two different dimers, one leaving the active sites of one subunit in contact with the neighboring molecule (AF, indicated by a red ellipsoid) or disrupting the active site connecting surface (AB, indicated by a cyan ellipsoid). The numbers depicted indicate the average surface area (in Å2) of the individual molecules involved in interaction with the neighboring molecule. See “The functional MgsA dimer” for details. B, magnification of the interfaces AF and AB. The point mutations tested for interference with hexamer formation of MgsA are indicated, and only those located at the interface formed by the MgsA molecules A and F prevent hexamerization. Thus, the monomers that interact to form the stable dimer require the other dimer interface (e.g. AB).
The functional MgsA dimer
Based on the fact that MgsA at low concentrations forms dimers in solution but hexamers in the crystal structure, we asked which of the two dimer interfaces observed in the crystal structure is physiologically relevant in dimer formation. One possible dimer might be formed by interaction of helices α4 and α5 (dimer AF in Fig. 4). Alternatively, MgsA may be present as an AB-type dimer. The latter potential dimer exhibits the larger interface by interaction of helices α5 and α6 and strand β5. Both potential dimers would lack specific interactions present in the hexamer thought to affect accessibility of the active site and potentially inhibit their function.
To understand which of the two dimer assemblies is more likely to be present in the cell, we performed an analysis of the properties of the two alternative interfaces. The analysis revealed that the contact surface area AF is around 757.5 ± 8.4 Å2, whereas the other interaction surface (AB) is around 849.1 ± 6.1 Å2. The AB dimer interface is formed by 16 hydrogen bonds (13–17 for the other interfaces in the complexes) and only four salt bridges (3–4), whereas the AF dimer interface is formed by eight hydrogen bonds (8–10 for the other interfaces in the complexes) and eight salt bridges (eight for all), also indicating a weaker interaction for the latter. Moreover, the calculated solvation energies derived from the respective interaction surfaces (−6.6 ± 0.1 for the smaller AF interface and −10.2 ± 0.4 for the larger AB interface) support the idea that the larger interaction surface is physiologically relevant. Taken together, this analysis suggests that MgsA forms dimers of the AB type.
To discriminate experimentally between the two possibilities of MgsA dimer arrangements, various mutant forms of MgsA were generated and tested for their ability to form multimeric assemblies (see Table 2). One of them, MgsA-T107R/T111R, is thought to interfere with the AB complex formation without having a major impact on the overall folding of MgsA. Interestingly, the corresponding protein variants formed aggregates, suggestive of either misfolding or a severe defect in dimer formation. A possible explanation would be that the amino acid substitutions cause improper interactions, resulting in large aggregates. Three other mutant proteins (i.e. MgsA-Q62R and the double mutant forms MgsA-Q62R/A65R and MgsA-A93W/L97W) are predicted to interfere with the formation of the AF dimer. In contrast to the MgsA variants described above, these proteins were soluble. According to the elution volume determined by gel filtration and mass determination by SEC-MALS using concentrations where WT MgsA still assembles in the multimeric form (see Fig. 5 and Table 2), they formed predominantly dimers. The location of these substitutions on the surface of MgsA reveals that the corresponding residues of the two subunits are arranged opposite each other (Fig. 4, presented as sticks). Thus, these results indicate that the mutations do not interfere with the formation of functional AB dimers. To assess whether this dimer arrangement is enzymatically active, the activity of purified MgsA-Q62R was determined and compared with WT MgsA. A specific activity of 0.15 mol min−1 g−1 was determined for WT MgsA. In contrast, no activity was observed for MgsA-Q62R (activity below 0.005 mol min−1 g−1). Taken together, these results support the idea that the stable dimer formed is of the AB type and confirm that the hexameric state is required for activity.
Table 2.
Calculated and predicted molecular mass of the various (partially) soluble MgsA forms tested in SEC-MALS
| Experimental mass | Predicted massa | |
|---|---|---|
| kDa | kDa | |
| MgsA | ||
| WT (peak) | 62 | 69.9 (4-mer) |
| 104.8 (6-mer) | ||
| WT (shoulder) | 38 | 34,9 (2-mer) |
| T107R/T111R | Aggregate | 35.1 (2-mer) |
| Q62R/A65R | 34.1 | 35.1 (2-mer) |
| Q62R | 41.3 | 34.9 (2-mer) |
| A93W/L97W | 34.4 | 35.3 (2-mer) |
| Crh | ||
| WT | 20.5/19.8 | 21.0 (2-mer) |
| 10.3/9.6 | 10.5 (1-mer) | |
a All masses were calculated via ExPASy ProtParam as expressed, including tags.
Figure 5.
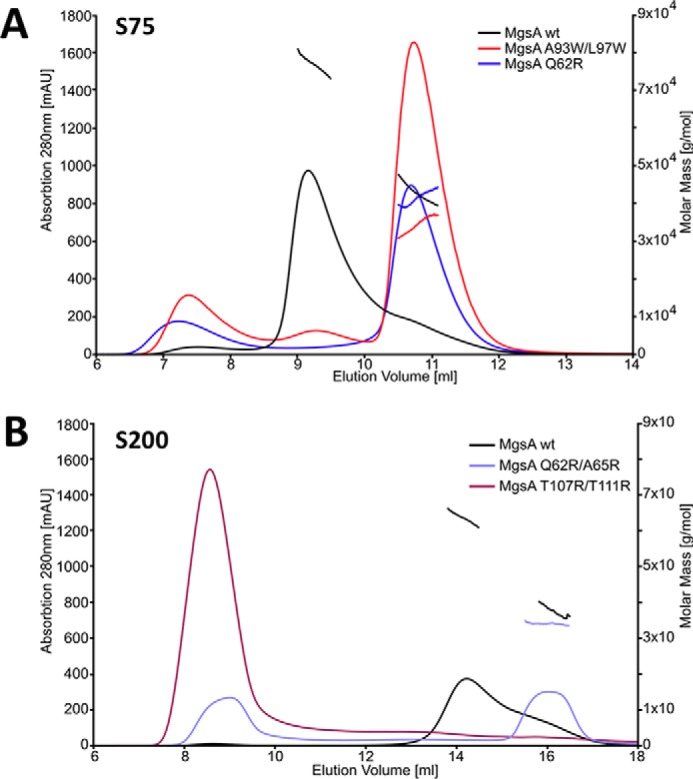
The analysis of the different MgsA variants suggests that the AB-type dimer is the one that exists in solution not the AF-type dimer. SEC-MALS experiments suggest that only the WT MgsA is folded correctly and capable of forming hexamers. The solid lines indicate the absorption profile of the SEC run, whereas the stray lines indicate the molecular mass observed in the MALS measurement at the respective elution volume. A, in contrast to WT MgsA, the point mutants A93W/L97W and Q69R form predominantly soluble dimers. B, opposing the WT MgsA, the point mutant Q62R/A65R forms mostly dimers and T107R/T111R aggregates. See “The functional MgsA dimer” for details.
Identification of the site of interaction between Crh and MgsA
To get a more precise idea of the structural basis for the regulation of MgsA by Crh, we performed an in vitro chemical cross-linking analysis. The WT proteins were purified and incubated with the cross-linker bis-sulfosuccinimidyl suberate (BS3), either individually or in a 1:1 ratio of MgsA to Crh. The resulting complexes were analyzed by SDS-PAGE (Fig. 6A). In the presence of cross-linker, MgsA and Crh homodimers and multimers as well as the putative complex of MgsA and Crh were detected (Fig. 6A, red box). Subsequently, the cross-linked protein complexes were digested with trypsin and cross-linked peptides identified by MS. This analysis revealed the presence of intra- and intermolecular cross-links. Intramolecular cross-links derived from SDS-PAGE bands containing exclusively cross-linked MgsA or Crh may result from subunit interactions of oligomeric proteins, but they can also simply reflect the spatial proximity of two amino acids within one protein molecule.
Figure 6.
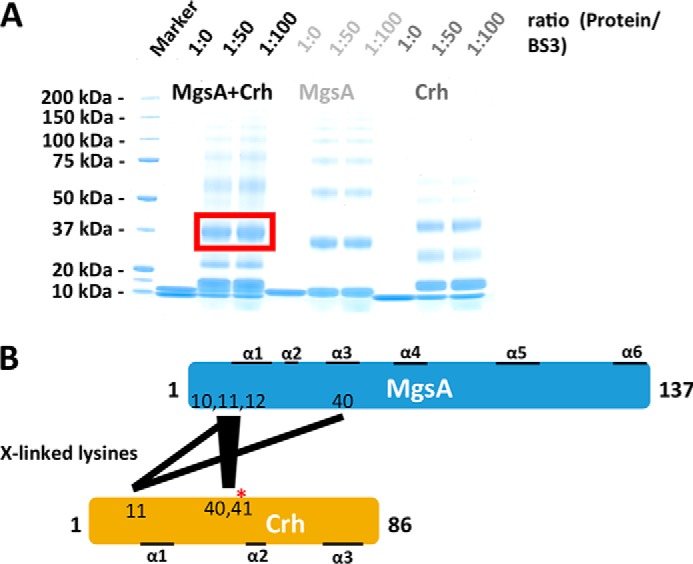
Determination of Crh cross-links to MgsA. A, an in vitro chemical cross-linking of MgsA and Crh either alone or in a complex formed in a 1:1 ratio. The samples were treated with the predetermined (Fig. S8) optimal amounts of BS3 using a 1:50 and 1:100 protein/cross-linker ratio. According to the size, bands that represent the complex of MgsA and/or MgsA-Crh were analyzed by LC-MS. B, schematic representation of the intermolecular cross-links determined (see also Table 3 and Figs. S5 and S6).
For Crh cross-linked alone (Fig. 6A), the lysine residue 5 forms intramolecular cross-links with Lys11, Lys37, Lys40, and Lys41. Moreover, Lys11 is arranged close to Lys37, Lys40, and Lys41. Finally, Lys41 cross-links to Lys45. These intramolecular cross-links are in good agreement with the observed proximity of these residues in the Crh structure and thus represent intracross-links in monomeric Crh (Fig. S5A) (PDB code 1K1C) (69). Additional cross-links of Lys11 to Lys45 and of Lys11 to Lys11 reflect cross-links between two molecules of Crh dimers (Fig. S5B) (PDB code 2AK7).
In contrast, for MgsA alone (Fig. 6A), we observed only a few intramolecular cross-links within the monomer, predominantly located in the η1-loop with Lys10 cross-linked to Lys12. In MgsA dimers, Lys10–Lys10 and Lys12–Lys12 cross-links were detected. These cross-links are in excellent agreement with the structure of MgsA (Fig. S6).
For the MgsA-Crh complex, we observed several additional intra- and intermolecular cross-links (Table 3). For MgsA, intramolecular cross-links between Lys40 and Lys10, Lys11, and Lys12 were detected that have not been observed in the analysis of the monomeric or dimeric forms of MgsA (Fig. 6). This suggests that the region of helices α1 and α3 is arranged quite differently upon interaction of MgsA with Crh. Both helices participate in the formation of the active site cavity, and their altered positioning would also add to the explanation of why the dimeric form of MgsA is inactive (see above). Importantly, we observed several intermolecular cross-links (Table 3). The lysine residues 10, 11, and 12 of the η1-loop in MgsA were found to be cross-linked predominantly to the lysine residues 11, 40, and 41 of Crh (Fig. 6B). In addition to the stretch of these three consecutive lysine residues located in one region of the complex, Lys40 located in the neighboring helix α3 of MgsA was linked to Crh Lys11. These findings highlight (i) the importance of the N-terminal region of MgsA for the interaction with Crh and (ii) the relevance of the region around the phosphorylation site Ser46 in Crh. It should be noted that MgsA contains six lysine residues that are all located in the N-terminal part of the protein. The other two lysines, Lys2 and Lys27, are most likely too distant to be accessible for cross-links after complex formation. This suggests that the stretch of the three consecutive lysine residues and the Lys40 are all located close to the interaction surface of the two molecules and thus were linked to Crh. Taken together, the biochemical cross-linking experiments indicate that the N-terminal region of MgsA is required for the interaction with Crh (Fig. 6B).
Table 3.
Intermolecular cross-links between MgsA and Crh
*, cross-linked lysine residues.
| MgsA residue | Crh residue | MgsA peptide | Crh peptide |
|---|---|---|---|
| Lys10 | Lys40 | IALIAHDK*KK | DGK*K |
| Lys10 | Lys41 | IALIAHDK*K | K*VNAK |
| Lys11 | Lys40 | IALIAHDKK*K | DGK*K |
| Lys12 | Lys41 | K*QDMVQFTTAYR | K*VNAK |
| Lys10 | Lys11 | IALIAHDK*K | LK*TGLQAR |
| Lys12 | Lys11 | KK*QDMVQFTTAYR | LK*TGLQAR |
| Lys40 | Lys11 | NHDLYATGTTGLK*IHEATGLQIER | LK*TGLQAR |
In a complementary approach to understand MgsA regulation, we used computational modeling to predict where Crh might bind onto MgsA (23, 44). Blind docking was performed as described under “Experimental procedures.” The only constraint used is based on the observation that interaction of Crh requires a narrow region formed by helices α1 and α2, which contains a residue, Ala20, shown to be important for interaction to MgsA (23). In the solution obtained, Crh is positioned in the vicinity of a hydrophobic patch formed by helices α1 and α6 of MgsA with Crh helices α1 and α2 oriented in a perpendicular fashion, also generating a conserved hydrophobic patch on Crh (44) (Fig. 7 and Fig. S7A). The obtained arrangement positions the region encompassing residue Ala20 at a prominent position in close proximity to MgsA (Fig. 7A). Any increase in side-chain size or charge would strongly interfere with the interaction properties of Crh and thus explain the results obtained (23).
Figure 7.
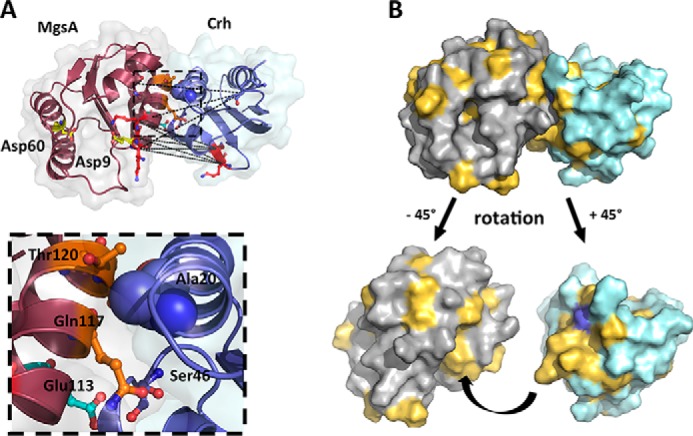
Model of the MgsA-Crh interaction. A, the model of MgsA-Crh complex obtained by blind docking experiments fulfills the described criteria. The MgsA molecule (gray surface and molecule colored dark red) is in close contact with Crh (surface in light cyan and the molecule colored in purple-blue) in the vicinity of the MgsA active site. For reference, the catalytically active site residues of MgsA, Asp9 and Asp60, are depicted in ball-and-stick mode. The lysines involved in MgsA-Crh cross-links (positions 11, 12, and 13 in MgsA and 40 and 41 in Crh) are depicted in ball-and-stick mode (red), and the distances between the respective Cα atoms are indicated by black dashed lines, which range from 22.8 to 25.1 Å (from Cα to Cα). The Ala20 used as the single constraint in the initial docking step is depicted in purple-blue spheres, whereas the phosphorylation-sensitive inhibitory site Ser46 is indicated in ball-and-stick mode colored with the carbon atoms in purple. A nearby positioned negatively charged glutamate (position 113) interfering with binding of phosphorylated Crh is indicted in cyan (located in helix 2). B, analysis of the hydrophobic patches on both MgsA and Crh, indicated in light orange on the gray (MgsA) or light cyan (Crh) surfaces. Below the interaction, hydrophobic patches of the two molecules are visualized by a rotation of the individual molecules as indicated, omitting the partner molecule. The surface area of Ala20 used as the initial criterion for docking is highlighted in purple-blue (see “Identification of the site of interaction between Crh and MgsA” for details).
The following criteria were used to confirm, and nicely fit to, the obtained model. First, the cross-linking results, in particular the distances of the lysines cross-linked between MgsA and Crh, agree with the docking model within the required limits (Fig. 7A). In agreement with the model derived from the docking, the interaction interface between Crh and MgsA is defined by intramolecular cross-links on two distant regions of the interaction surface (Fig. 7A). Second, a region with negative charge should be located on MgsA in the vicinity of Ser46 to allow for inhibition of interaction if Crh is phosphorylated (23). In the model, Glu113 is located at a perfect spatial distance such that a phosphorylation of Ser46 in Crh would strongly interfere with this interaction (Fig. 7A). Third, a remarkable cluster of highly conserved residues in helices α1 and α2 in Crh is found (23). Similarly, on MgsA, Ala112 followed by an acidic residue is conserved, and two small patches of hydrophobic residues located in helices α1 and α6, respectively, are involved in the formation of the two opposing hydrophobic surfaces (Fig. 7B and Fig. S3).
In sum, the docking model of Crh to MgsA seems quite reasonable, as it fulfills the multiple criteria described in this work and in the literature. The possible impact of Crh binding on MgsA activity will be discussed below.
Discussion
In this work, we have unraveled the molecular mechanism that links carbon source availability to the activity of B. subtilis methylglyoxal synthase.
Surprisingly, the results of the SEC-MALS analysis and crystal structure determination provided ambiguous results concerning the oligomeric state of MgsA. In agreement with the crystal structures of E. coli (37), T. thermophilus, and Thermus sp. MgsA, only a hexameric state was observed in crystals. In contrast, dimers and multimers (tetramers/hexamers) were found in solution, and at low concentrations, only dimers were detected. This discrepancy is not unprecedented for MgsA; for the E. coli protein, elution volumes corresponding to tetramers rather than hexamers were observed (32, 34–36). Given the low concentration of MgsA in B. subtilis (about 1,000 molecules/cell, about 1 μm) (33), the dimeric form of the AB type is also likely to be present in the cell, and the conversion into the hexameric form depends on the cellular environment. Moreover, our data suggest that the hexamer is the functional form, and the formation of hexamers is a prerequisite for MgsA activity. The hexamer stabilization is increased if phosphate ions (and presumably also if substrate levels) are increased. Mechanistically, this could be explained by structural alterations in the C-terminal residues present in MgsA, forming an arm that reaches from one subunit to a neighboring subunit and interacts in the region of and in some cases with the active site (Fig. S3) (37, 43). In the latter case, an arginine is required for the proposed interaction with a phosphate in the active site, which is also present in the C-terminal region of B. subtilis MgsA (Fig. S3). Moreover, the residues that have been shown to be important and highly conserved for another allosteric pathway are conserved in MgsA as well (Fig. S3) (37, 45). Interestingly, Thermus sp. GH5 MgsA bears a shortened C-terminal extension that lacks this C-terminal Arg residue but has been shown to still form a stable hexamer in solution independent of Pi (39).
Under conditions of nutrient limitation, MgsA activity is inhibited by a regulatory interaction with the carbon flux regulator, Crh. In contrast, under conditions of good nutrient supply, Crh becomes phosphorylated on Ser46 by the HPr kinase (19). The phosphorylated form of Crh is unable to bind MgsA (23), and thus the methylglyoxal bypass of glycolysis is operative under these conditions. This is of physiological relevance to prevent the accumulation of toxic phosphorylated glycolytic intermediates.
Protein-protein interactions are a common theme in the regulation of a variety of biological processes, such as nutrient transport, gene expression, or metabolism. Usually, the interactions affect the activity of the target protein by preventing access of a molecule or by modifying the local stability of important domains. In bacterial signaling, these paradigms have been well established for the control of ammonium uptake by the regulatory PII protein, as well as for the global control of carbon catabolite repression (14, 46). In its phosphorylated state, Crh has been shown to interact with the global transcription factor CcpA, and here the phosphate moiety on Ser46 is charge-compensated by two basic residues (Arg303 and Lys307) on CcpA (47). Our docking studies suggest that the charge of the hydroxyl group of Ser46 forms a strong hydrogen bond with Glu113 of MgsA.
The in silico protein-protein docking studies, supported by the cross-linking data and the positioning of Ser46 opposite a negatively charged residue (Glu113), suggest that Crh binding to MgsA interferes with the structural properties of the active site of MgsA and reduces or shuts down its activity. This may result from allosteric effects upon binding, which influence the active-site structure, especially by interaction with helices α1, α3, and α6, and hereby allosterically delocalize the active-site loops toward the center of the active site and thus prevent activity (Fig. S7B). Only minute movements seem to be sufficient, as suggested by the ligand-docking experiments. As mentioned, docking was successful only in case of a model relaxation procedure before docking, which resulted only in slight structural alterations in the active site.
The bacterial phosphotransferase system is a key component in the control of carbon metabolism in bacteria. In E. coli, the phosphorylation state of the Crr protein determines the activity of adenylate cyclase and of a variety of sugar transporters and catabolic enzymes. In the presence of preferred carbon sources, nonphosphorylated Crr binds to several sugar permeases as well as to the glycerol kinase to inhibit the activity of these proteins. In contrast, phosphorylated Crr, which indicates the absence of preferred carbon sources, binds the adenylate cyclase and triggers the synthesis of the second messenger cAMP and thus the expression of catabolic genes and operons. In Gram-positive bacteria, the HPr protein has multiple functions in sugar transport, control of general catabolite repression via interaction with the global transcription factor CcpA, and the regulation of individual pathways through regulatory interactions with operon-specific regulators. All of these interactions are controlled by the phosphorylation state of HPr and may involve the phosphorylation of the target proteins, as observed for the glycerol kinase and multiple transcriptional regulators. Alternatively, HPr(pSer) binds to transcription factors, such as CcpA and RbsR, to trigger their DNA-binding activity, and HPr(pHis) binds to the transcription activator RhgR to stimulate its activity (15, 16).
With this work, we have added another facet to the control of carbon catabolism by the PTS; the Crh protein, a regulatory paralog of HPr, binds the metabolic enzyme methylglyoxal synthase in the presence of preferred carbon sources to prevent the accumulation of toxic intermediates. Binding of Crh results in inhibition of MgsA via the deformation of the active site, which in consequence is no longer accessible for the substrate molecules. Taking into account the cellular concentrations of both interaction partners, the control of MgsA activity is likely to be the major function of Crh.
Experimental procedures
Bacterial strains and growth conditions
E. coli strains XL1-Blue (48) and BL21(DE3) (Stratagene) were used for cloning experiments and expression of recombinant proteins, respectively. E. coli was routinely grown in lysogeny broth (LB) at 37 °C. Ampicillin (100 μg/ml) was used for selection of recombinant E. coli strains. LB plates were prepared by the addition of 17 g of Bacto agar/liter (Difco) to LB medium.
DNA manipulation and transformation
Transformation of E. coli and plasmid DNA extraction were performed using standard procedures (49). Restriction enzymes, T4 DNA ligase, and DNA polymerases were used as recommended by the manufacturers. DNA fragments were purified by using the QIAquick PCR purification kit (Qiagen, Germany). Phusion DNA polymerase was used for the PCR as recommended by the manufacturer. DNA sequences were determined using the dideoxy chain termination method (49). All plasmid inserts derived from PCR products were verified by DNA sequencing.
Protein purification
His-tagged Crh was purified from E. coli BL21 (DE3) carrying plasmid pAG1 (18) as described previously (23).
For expression and subsequent purification of Strep-MgsA and its variants, E. coli BL21(DE3) carrying plasmid pGP1301 (WT) (23) and its mutant derivatives were used. Plasmids expressing the mutant mgsA alleles were obtained as follows. The mgsA gene was amplified using primers SK59 and SK60 (23) and the appropriate mutagenic primers by PCR (50, 51). The PCR fragments were inserted between the SacI and BamHI restriction sites of the expression vector pGP172 (52). Cleared lysates of cells expressing MgsA were prepared as described (23) and subsequently passed over columns containing 1 ml of Strep-Tactin matrix (IBA, Göttingen, Germany). The columns were washed with 2.5 ml of buffer W (100 mm Tris/HCl, pH 8.0, 150 mm NaCl, 1 mm EDTA), and subsequently Strep-MgsA was eluted using 1 ml of buffer W containing 2.5 mm desthiobiotin. An S75 Sepharose gel-filtration column (GE Healthcare) equilibrated in 50 mm Tris/Cl, pH 7.5, 150 mm sodium-chloride was used for final purification.
If required for particular experiments, HEPES buffer (150 mm NaCl, 50 mm HEPES, pH 7.5) was used for the purification procedure. The Bio-Rad dye-binding assay was used to determine protein concentrations. BSA was used as a standard.
Methylglyoxal synthase activity assay
The activity of MgsA was determined spectrophotometrically by a direct assay as described previously (32). Briefly, the reaction mixture (total volume of 0.5 ml) contained 40 mm imidazole buffer, pH 7.5, 3 mm dihydroxyacetone phosphate, and 1 μg (1.12 μm) of purified Strep-MgsA. After incubation at 30 °C for 10 min, 100 μl of each sample was removed and added to a solution containing 0.9 ml of H2O and 0.33 ml of 2,4-dinitrophenylhydrazine (0.1% 2,4-dinitrophenylhydrazine in 2 m HCl). After incubation at 30 °C for 15 min, 1.67 ml of 10% (w/v) NaOH was added, and the A555 was determined after an additional incubation at 30 °C for 15 min. The readings were converted into μmol of methylglyoxal using a molar extinction coefficient of 4.48 × 104.
Protein-protein cross-linking
To determine interacting regions of the proteins, we cross-linked the proteins either alone or as complexes using BS3, digested the cross-linked complexes (53), and identified the linked peptides by MS. The optimal cross-linker to protein ratio was determined by using 2.5-μg aliquots of the MgsA-Crh complex and a series of cross-linker molar excesses of 5-, 10-, 25-, 50-, 100-, and 200-fold as well as a control without cross-linker (see Fig. S8A). Samples were allowed to react with freshly prepared BS3 (20 mg/ml in DMSO) for 30 min at room temperature. The cross-linked samples were analyzed by SDS-PAGE on a 4–12% gel (Invitrogen) or on a freshly prepared 15% SDS gel. The gels were analyzed by Coomassie staining. The identity of the proteins in the complexes was verified by Western blot analysis using antibodies against Crh and the Strep-Tag (for the detection of Strep-MgsA) (data not shown).
For MS analysis, MgsA and Crh were cross-linked with freshly prepared BS3 in a 50:1 and 100:1 cross-linker/protein ratio, allowed to react, and separated as described above. Bands corresponding to the monomeric protein, the homooligomers of various states, and the cross-linked complex were excised from the gel (see Fig. S8B). In-gel digestion and extraction of peptides was achieved as described elsewhere (53). The solution of extracted peptides was concentrated on a vacuum evaporator and redissolved in 20 μl of injection buffer (5% (v/v) acetonitrile, 0.1% (v/v) trifluoroacetic acid), of which 6 μl were loaded in technical duplicate onto an in-house packed C18 column (75 μm × 300 mm; Reprosil-Pur 120C18-AQ, 1.9 μm, Dr. Maisch GmbH). Chromatographic separation was achieved on an UltiMate 3000 RSLC nanosystem (Thermo Fisher Scientific) from 9 to 42% buffer B (80% (v/v) acetonitrile, 0.08% (v/v) formic acid), 300-nl/min flow rate, and 38-min duration. Eluting peptides were sprayed into a QExactive HF-X (Thermo Fisher Scientific) mass spectrometer. MS1 scans were performed with a scan range from m/z 350 to 1600, a resolution of 120,000, 1e6 AGC target, and 50-ms maximum injection time. Each survey scan was followed by MS2 scans of the 20 most abundant precursors fragmented with a normalized collision energy of 30, a resolution of 30,000, 1e5 AGC target, and 128-ms maximum injection time. Only charge states from 3 to 8 were considered, and no dynamic exclusion was set. Raw files were converted to mgf format with Proteome Discoverer version 2.1 (Thermo Fisher Scientific) and submitted to a cross-link database search with Plink version 1.23 (54). Briefly, spectra were searched against the sequences of MgsA and Crh considering three missed cleavages, cysteine carbamidomethylation as fixed, and methionine oxidation as a variable modification, 10-ppm precursor mass deviation, and 1% false discovery rate cut-off. Subsequently, identified cross-link matches were examined manually.
Western blotting
For Western blot analysis, proteins were separated by 12% SDS-PAGE and transferred onto polyvinylidene difluoride membranes (Bio-Rad) by electroblotting. Rabbit anti-Crh (19) or anti-Strep polyclonal antibodies (PromoCell, Heidelberg, Germany; 1:10,000) served as primary antibodies. The antibodies were visualized by using anti-rabbit immunoglobulin alkaline phosphatase secondary antibodies (Promega) and the CDP-Star detection system (Roche Diagnostics), as described previously (55).
Determination of the oligomerization state of MgsA and the MgsA-Crh complex
The oligomerization status of MgsA and the MgsA-Crh complex was determined by SEC-MALS. For this purpose, purified proteins were applied onto the column either alone or as a premixed and preincubated sample in gel-filtration buffer (50 mm Tris/Cl, pH 7.5, 150 mm sodium-chloride, if not indicated otherwise). The buffer was filtered (0.1-μm filters) and degassed in line (Model 2003, Biotech AB, Onsala, Sweden) before protein separation on a S75/S200 Superdex 10/300GL column on an Äkta Purifier (both from GE Healthcare). Subsequently, the eluate was analyzed in line with a miniDawn Treos multiangle light scattering system followed by an Optilab T-rEX refractive index detector (both from Wyatt Technology Europe GmbH, Dernbach, Germany) before fractionation. Data analysis was performed using ASTRA version 6.1 software (Wyatt Technology) and also compared with a gel-filtration standard (Bio-Rad).
Crystallization and data collection
Purified MgsA at a concentration of 11.5 mg/ml was crystallized in SwissCI 96-well plates using the sitting-drop vapor diffusion method. Initial screening of protein crystallization conditions in a nanoliter-scale droplet array was performed automatically using a pipetting robot (PhoenixRE, Rigaku) for several commercially available crystallization screens. The best-diffracting crystals grew in a condition of the Morpheus screen (56) composed of 10% (w/v) PEG 4000, 0.1 m M-buffer, 20% (w/v) glycerol, and 30 mm M-Divalent Cations buffer as an additive (M-buffer: 0.1 m Bicine/Trizma base, pH 8.5; M-Divalent Cations buffer: 0.3 m magnesium chloride, 0.3 m calcium chloride). Before diffraction experiments, crystals were transferred into a cryosolution composed of reservoir solution supplemented with 10% (v/v) glycerol. Oscillation images were collected at PETRA III (EMBL, Hamburg, Germany) and processed with XDS (57, 58). The scaling process revealed an orthorhombic lattice with unit cell parameters of a = 108.93 Å, b = 109.71 Å, c = 199.95 Å, α = β = γ = 90.00°. Systematic absences indicated the presence of screw axes along a, b, and c, resulting in the P2(1)2(1)2(1) space group. The Matthews coefficient (Vm = 3.5 Å3/Da) suggested 12 molecules in the asymmetric unit corresponding to a solvent content of 65%. The data collection statistics are summarized below (see Table 1).
Structure determination and refinement
The phase problem was solved by means of molecular replacement (MR) using PHASER and the structure of the methylglyoxal synthase from Thermus sp. (PDB code 2XW6) as a search model. The search model was identified based on an HHPRED (59) search for homologous proteins and trimmed to the last common atom using the sequence alignment and tools from the ROSETTA package (60) before the MR search. The initial MR solution was first rebuilt using Rosetta model completion and relaxation (60), followed by alternating cycles of refinement using Phenix (61) and manual rebuilding with Coot (62). Standard parameters, including weights optimization, TLS parameterization, and building of solvent molecules, were used during refinement. A random set of 5% of reflections, selected in thin resolution shells, was excluded from refinement to monitor Rfree (63). The final model consisting of two MgsA hexamers, 526 water molecules, 9 glycerol molecules and 23 Cl ions has been refined at a resolution of 2.34 Å to R and Rfree factors of 17.0 and 20.49%, respectively. Each of the 12 MgsA monomers is made up of 121 residues, of which two or three result from the N-terminally placed Strep-tag (sequence Val-Ser-Ser). Seventeen residues at the C terminus as well as 20 residues of the Strep-tag could not be localized in the electron density map and are most likely disordered. The refinement statistics are summarized in Table 1. The quality of the model was assessed using MOLPROBITY, as implemented in Phenix (61). Secondary structure predictions were performed using DSSP (64). Coordinates were superimposed with LSQKAB (65) from the CCP4 program suite (66) or as implemented in PyMOL (version 1.8.4., Schrödinger, LLC, New York). Exploration of macromolecular interfaces was performed using “Protein interfaces, surfaces and assemblies” (PISA) services at the European Bioinformatics Institute (http://www.ebi.ac.uk/pdbe/prot_int/pistart.html)6 using standard settings (67). Structure-based sequence alignments were performed using Expresso (68, 69), and the respective graphs were prepared using ESPript (70).
Computational modeling
In silico docking experiments of the substrate dihydroxyacetone phosphate into MgsA was performed using Autodock Vina (71). The MgsA active site was identified based on superposition with the structure of the E. coli methylglyoxal synthase complexed with phosphoglycolo-hydroxamic acid (PDB code 1IK4), serving also as the reference for ligand-binding mode. The substrate molecule as well as side chains forming the active site were kept flexible during the docking trials: Ile6, His8, Asp9, Lys12, Thr34, Thr36, Thr37, Ser54, Asp60, Phe77, His87, and Val91. Interestingly, successful docking results were obtained only for the receptor model that was relaxed using the Rosetta application (72). The relax protocol in Rosetta performs a simple all-atom model refinement in the Rosetta force field that searches the local conformational space around the starting structure (RMSD calculated for Cα atoms forming the MgsA hexamer amounted to 0.06 Å). This indicates that the receptor molecule was slightly too compressed, and very small model-alterations were necessary to make the narrow active site capable of accommodating the ligand molecule. The final docking model, representing the lowest-energy decoy, reveals a very similar position and orientation of the docked dihydroxyacetone phosphate to the reference phosphoglycolohydroxamic acid (distances between the carbonyl oxygen and the phosphate atoms are 0.5 and 2.0 Å, respectively. Blind protein-protein docking experiments of Crh molecules (PDB code 1ZVV, chain W) to a hexameric form of MgsA have been performed using Rosetta (73) in a two-stage protocol. The first stage, where aggressive sampling is done, was performed in the centroid (low-resolution) mode with a single site constraint defined based on our previous study (23): Crh residue Ala20, located in the middle of helix 1, should be in contact with the receptor (MgsA). The 10,000 decoys were sorted by Rosetta score to select the 300 best preliminary docking models, which were clustered by pairwise RMSD calculation. The central decoy from the cluster with the lowest score was subjected to an all-atom refinement stage (high resolution) that optimizes both rigid-body orientations and side-chain conformations of the docked molecules. The 1,000 docked models were scored based on the Rosetta total energy score as well as the interface score I_sc, which represents the energy of the interactions across the interface. The best-scoring decoys were clustered, and the central model from the best cluster was chosen as the final docking model. This model was subsequently used for the further analysis and visual inspection.
Author contributions
A. D., B. G., and J. S. conceptualization; A. D., C. P. Z., J. A., J. G., R. H., I. P., and P. N. data curation; A. D., H. U., B. G., R. F., and J. S. supervision; A. D., C. P. Z., J. A., J. G., R. H., P. N., H. U., I. P., and B. G. validation; A. D., C. P. Z., J. A., I. P., J. G., R. H., P. N., H. U., B. G., and R. F. investigation; A. D. and J. A. visualization; A. D., C. P. Z., J. A., I. P., J. G., R. H., P. N., H. U., and B. G. methodology; A. D. and J. S. writing-original draft; A. D., C. P. Z., P. N., H. U., B. G., R. F., and J. S. writing-review and editing; P. N. software; H. U., R. F., and J. S. funding acquisition; B. G. and J. S. resources; B. G., R. F., and J. S. project administration.
Supplementary Material
Acknowledgments
We are grateful to the staff of the European Synchrotron Radiation Facility beam line (IS23-1, Grenoble, France) for support during data collection. We thank Bastian Behrens for help with some experiments.
This work was supported in part by Deutsche Forschungsgemeinschaft (DFG) Grant HI 291/13-1 and SFB860. The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (code 6F2C) have been deposited in the Protein Data Bank (http://wwpdb.org/).
This article contains Figs. S1–S8.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- PTS
- phosphotransferase system
- SEC
- size-exclusion chromatography
- MALS
- multiangle light scattering
- PDB
- Protein Data Bank
- RMSD
- root mean square deviation
- BS3
- bis-sulfosuccinimidyl suberate
- LB
- lysogeny broth
- MR
- molecular replacement
- I_sc
- interface score
- Bicine
- N,N-bis(2-hydroxyethyl)glycine.
References
- 1. Fujita Y. (2009) Carbon catabolite control of the metabolic network in Bacillus subtilis. Biosci. Biotechnol. Biochem. 73, 245–259 10.1271/bbb.80479 [DOI] [PubMed] [Google Scholar]
- 2. Bachem S., and Stülke J. (1998) Regulation of the Bacillus subtilis GlcT antiterminator protein by components of the phosphotransferase system. J. Bacteriol. 180, 5319–5326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fillinger S., Boschi-Muller S., Azza S., Dervyn E., Branlant G., and Aymerich S. (2000) Two glyceraldehyde-3-phosphate dehydrogenases with opposite physiological roles in a nonphotosynthetic bacterium. J. Biol. Chem. 275, 14031–14037 10.1074/jbc.275.19.14031 [DOI] [PubMed] [Google Scholar]
- 4. Servant P., Le Coq D., and Aymerich S. (2005) CcpN (YqzB), a novel regulator for CcpA-independent catabolite repression of Bacillus subtilis gluconeogenic genes. Mol. Microbiol. 55, 1435–1451 10.1111/j.1365-2958.2005.04473.x [DOI] [PubMed] [Google Scholar]
- 5. Moreno M. S., Schneider B. L., Maile R. R., Weyler W., and Saier M. H. (2001) Catabolite repression mediated by the CcpA protein in Bacillus subtilis: novel modes of regulation revealed by whole-genome analysis. Mol. Microbiol. 39, 1366–1381 10.1111/j.1365-2958.2001.02328.x [DOI] [PubMed] [Google Scholar]
- 6. Ludwig H., Meinken C., Matin A., and Stülke J. (2002) Insufficient expression of the ilv-leu operon encoding enzymes of branched-chain amino acid biosynthesis limits growth of a Bacillus subtilis ccpA mutant. J. Bacteriol. 184, 5174–5178 10.1128/JB.184.18.5174-5178.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wacker I., Ludwig H., Reif I., Blencke H. M., Detsch C., and Stülke J. (2003) The regulatory link between carbon and nitrogen metabolism in Bacillus subtilis: regulation of the gltAB operon by the carbon catabolite protein CcpA. Microbiology 149, 3001–3009 10.1099/mic.0.26479-0 [DOI] [PubMed] [Google Scholar]
- 8. Shivers R. P., and Sonenshein A. L. (2005) Bacillus subtilis ilvB operon: an intersection of global regulons. Mol. Microbiol. 56, 1549–1559 10.1111/j.1365-2958.2005.04634.x [DOI] [PubMed] [Google Scholar]
- 9. Jault J. M., Fieulaine S., Nessler S., Gonzalo P., Di Pietro A., Deutscher J., and Galinier A. (2000) The HPr kinase from Bacillus subtilis is a homo-oligomeric enzyme which exhibits strong positive cooperativity for nucleotide and fructose 1,6-bisphosphate binding. J. Biol. Chem. 275, 1773–1780 10.1074/jbc.275.3.1773 [DOI] [PubMed] [Google Scholar]
- 10. Hanson K. G., Steinhauer K., Reizer J., Hillen W., and Stülke J. (2002) HPr kinase/phosphatase of Bacillus subtilis: expression of the gene and effects of mutations on enzyme activity, growth and carbon catabolite repression. Microbiology 148, 1805–1811 10.1099/00221287-148-6-1805 [DOI] [PubMed] [Google Scholar]
- 11. Singh K. D., Schmalisch M. H., Stülke J., and Görke B. (2008) Carbon catabolite repression in Bacillus subtilis: quantitative analysis of repression exerted by different carbon sources. J. Bacteriol. 190, 7275–7284 10.1128/JB.00848-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Meyer F. M., Jules M., Mehne F. M. P., Le Coq D., Landmann J. J., Görke B., Aymerich S., and Stülke J. (2011) Malate-mediated carbon catabolite repression in Bacillus subtilis involves the HprK/ CcpA pathway. J. Bacteriol. 193, 6939–6949 10.1128/JB.06197-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Deutscher J., Küster E., Bergstedt U., Charrier V., and Hillen W. (1995) Protein kinase-dependent HPr/CcpA interaction links glycolytic activity to carbon catabolite repression in Gram-positive bacteria. Mol. Microbiol. 15, 1049–1053 10.1111/j.1365-2958.1995.tb02280.x [DOI] [PubMed] [Google Scholar]
- 14. Schumacher M. A., Allen G. S., Diel M., Seidel G., Hillen W., and Brennan R. G. (2004) Structural basis for allosteric control of the transcription regulator CcpA by the phosphoprotein HPr-Ser46-P. Cell 118, 731–741 10.1016/j.cell.2004.08.027 [DOI] [PubMed] [Google Scholar]
- 15. Görke B., and Stülke J. (2008) Carbon catabolite repression in bacteria: many ways to make the most out of nutrients. Nat. Rev. Microbiol. 6, 613–624 10.1038/nrmicro1932 [DOI] [PubMed] [Google Scholar]
- 16. Deutscher J., Aké F. M., Derkaoui M., Zébré A. C., Cao T. N., Bouraoui H., Kentache T., Mokhtari A., Milohanic E., and Joyet P. (2014) The bacterial phosphoenolpyruvate:carbohydrate phosphotransferase system: regulation by protein phosphorylation and phosphorylation-dependent protein-protein interactions. Microbiol. Mol. Biol. Rev. 78, 231–256 10.1128/MMBR.00001-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Warner J. B., and Lolkema J. S. (2003) CcpA-dependent carbon catabolite repression in bacteria. Microbiol. Mol. Biol. Rev. 67, 475–490 10.1128/MMBR.67.4.475-490.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Galinier A., Haiech J., Kilhoffer M. C., Jaquinod M., Stülke J., Deutscher J., and Martin-Verstraete I. (1997) The Bacillus subtilis crh gene encodes a HPr-like protein involved in carbon catabolite repression. Proc. Natl. Acad. Sci. U.S.A. 94, 8439–8444 10.1073/pnas.94.16.8439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Landmann J. J., Werner S., Hillen W., Stülke J., and Görke B. (2012) Carbon source control of the phosphorylation state of the Bacillus subtilis carbon-flux regulator Crh in vivo. FEMS Microbiol. Lett. 327, 47–53 10.1111/j.1574-6968.2011.02456.x [DOI] [PubMed] [Google Scholar]
- 20. Galinier A., Deutscher J., and Martin-Verstraete I. (1999) Phosphorylation of either Crh or HPr mediates binding of CcpA to the Bacillus subtilis xyn cre and catabolite repression of the xyn operon. J. Mol. Biol. 286, 307–314 10.1006/jmbi.1998.2492 [DOI] [PubMed] [Google Scholar]
- 21. Görke B., Fraysse L., and Galinier A. (2004) Drastic differences in Crh and HPr synthesis levels reflect their different impacts on catabolite repression in Bacillus subtilis. J. Bacteriol. 186, 2992–2995 10.1128/JB.186.10.2992-2995.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Seidel G., Diel M., Fuchsbauer N., and Hillen W. (2005) Quantitative interdependence of coeffectors, CcpA, and cre in carbon catabolite regulation of Bacillus subtilis. FEBS J. 272, 2566–2577 10.1111/j.1742-4658.2005.04682.x [DOI] [PubMed] [Google Scholar]
- 23. Landmann J. J., Busse R. A., Latz J. H., Singh K. D., Stülke J., and Görke B. (2011) Crh, the paralogue of the phosphocarrier protein HPr, controls the methylglyoxal bypass of glycolysis in Bacillus subtilis. Mol. Microbiol. 82, 770–787 10.1111/j.1365-2958.2011.07857.x [DOI] [PubMed] [Google Scholar]
- 24. Shin S. M., Song S. H., Lee J. W., Kwak M. K., and Kang S. O. (2017) Methylglyoxal synthase regulates cell elongation via alterations of cellular methylglyoxal and spermidine content in Bacillus subtilis. Int. J. Biochem. Cell Biol. 91, 14–28 10.1016/j.biocel.2017.08.005 [DOI] [PubMed] [Google Scholar]
- 25. Böck A., and Neidhardt F. C. (1966) Properties of a mutant of Escherichia coli with a temperature-sensitive fructose-1,6-diphosphate aldolase. J. Bacteriol. 92, 470–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kadner R. J., Murphy G. P., and Stephens C. M. (1992) Two mechanisms for growth inhibition by elevated transport of sugar phosphates in Escherichia coli. J. Gen. Microbiol. 138, 2007–2014 10.1099/00221287-138-10-2007 [DOI] [PubMed] [Google Scholar]
- 27. Tötemeyer S., Booth N. A., Nichols W. W., Dunbar B., and Booth I. R. (1998) From famine to feast: the role of methylglyoxal production in Escherichia coli. Mol. Microbiol. 27, 553–562 10.1046/j.1365-2958.1998.00700.x [DOI] [PubMed] [Google Scholar]
- 28. Chandrangsu P., Dusi R., Hamilton C. J., and Helmann J. D. (2014) Methylglyoxal resistance in Bacillus subtilis: contributions of bacillithiol-dependent and independent pathways. Mol. Microbiol. 91, 706–715 10.1111/mmi.12489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gaballa A., Newton G. L., Antelmann H., Parsonage D., Upton H., Rawat M., Claiborne A., Fahey R. C., and Helmann J. D. (2010) Biosynthesis and functions of bacillithiol, a major low-molecular weight thiol in Bacilli. Proc. Natl. Acad. Sci. U.S.A. 107, 6482–6486 10.1073/pnas.1000928107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gaballa A., Antelmann H., Hamilton C. J., and Helmann J. D. (2013) Regulation of Bacillus subtilis bacillithiol biosynthesis operons by Spx. Microbiology 159, 2025–2035 10.1099/mic.0.070482-0 [DOI] [PubMed] [Google Scholar]
- 31. Cooper R. A. (1984) Metabolism of methylglyoxal in microorganisms. Annu. Rev. Microbiol. 38, 49–68 10.1146/annurev.mi.38.100184.000405 [DOI] [PubMed] [Google Scholar]
- 32. Hopper D. J., and Cooper R. A. (1972) The purification and properties of Escherichia coli methylglyoxal synthase. Biochem. J. 128, 321–329 10.1042/bj1280321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Maass S., Wachlin G., Bernhardt J., Eymann C., Fromion V., Riedel K., Becher D., and Hecker M. (2014) Highly precise quantification of protein molecules per cell during stress and starvation responses in Bacillus subtilis. Mol. Cell. Proteomics 13, 2260–2276 10.1074/mcp.M113.035741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cooper R. A. (1974) Methylglyoxal formation during glucose catabolism by Pseudomonas saccharophila: identification of methylglyoxal synthase. Eur. J. Biochem. 44, 81–86 10.1111/j.1432-1033.1974.tb03459.x [DOI] [PubMed] [Google Scholar]
- 35. Saadat D., and Harrison D. H. (1998) Identification of catalytic bases in the active site of Escherichia coli methylglyoxal synthase: cloning expression, and functional characterization of conserved aspartic acid residues. Biochemistry 37, 10074–10086 10.1021/bi980409p [DOI] [PubMed] [Google Scholar]
- 36. Huang K., Rudolph F. B., and Bennett G. N. (1999) Characterization of methylglyoxal synthase from Clostridium acetobutylicum ATCC824 and its use in the formation of 1,2-propanediol. Appl. Environ. Microbiol. 65, 3244–3247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Saadat D., and Harrison D. H. (1999) The crystal structure of methylglyoxal synthase from Escherichia coli. Structure 7, 309–317 10.1016/S0969-2126(99)80041-0 [DOI] [PubMed] [Google Scholar]
- 38. Hatti K., Biswas A., Chaudhary S., Dadireddy V., Sekar K., Srinivasan N., and Murthy M. R. N. (2017) Structure determination of contaminant proteins using the MarathonMR procedure. J. Struct. Biol 197, 372–378 10.1016/j.jsb.2017.01.005 [DOI] [PubMed] [Google Scholar]
- 39. Pazhang M., Khajeh K., Asghari S. M., Falahati H., and Naderi-Manesh H. (2010) Cloning, expression, and characterization of a novel methylglyoxal synthase from Thermus sp. strain GH5. Appl. Biochem. Biotechnol. 162, 1519–1528 10.1007/s12010-010-8933-0 [DOI] [PubMed] [Google Scholar]
- 40. Penin F., Favier A., Montserret R., Brutscher B., Deutscher J., Marion D., and Galinier D. (2001) Evidence for a dimerisation state of the Bacillus subtilis catabolite repression HPr-like protein, Crh. J. Mol. Microbiol. Biotechnol. 3, 429–432 [PubMed] [Google Scholar]
- 41. Holm L., and Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 10.1093/nar/gkq366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Marks G. T., Harris T. K., Massiah M. A., Mildvan A. S., and Harrison D. H. (2001) Mechanistic implications of methylglyoxal synthase complexed with phosphoglycolohydroxamic acid as observed by X-ray crystallography and NMR spectroscopy. Biochemistry 40, 6805–6818 10.1021/bi0028237 [DOI] [PubMed] [Google Scholar]
- 43. Saadat D., and Harrison D. H. (2000) Mirroring perfection: the structure of methylglyoxal synthase complexed with the competitive inhibitor 2-phosphoglycolate. Biochemistry 39, 2950–2960 10.1021/bi992666f [DOI] [PubMed] [Google Scholar]
- 44. Favier A., Brutscher B., Blackledge M., Galinier A., Deutscher J., Penin F., and Marion D. (2002) Solution structure and dynamics of Crh, the Bacillus subtilis catabolite repression HPr. J. Mol. Biol. 317, 131–144 10.1006/jmbi.2002.5397 [DOI] [PubMed] [Google Scholar]
- 45. Falahati H., Pazhang M., Zareian S., Ghaemi N., Rofougaran R., Hofer A., Rezaie A. R., and Khajeh K. (2013) Transmitting the allosteric signal in methylglyoxal synthase. Protein Eng. Des. Sel. 26, 445–452 10.1093/protein/gzt014 [DOI] [PubMed] [Google Scholar]
- 46. Coutts G., Thomas G., Blakey D., and Merrick M. (2002) Membrane sequestration of the signal transduction protein GlnK by the ammonium transporter AmtB. EMBO J. 21, 536–545 10.1093/emboj/21.4.536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Schumacher M. A., Seidel G., Hillen W., and Brennan R. G. (2006) Phosphoprotein Crh-Ser46-P displays altered binding to CcpA to effect carbon catabolite regulation. J. Biol. Chem. 281, 6793–6800 10.1074/jbc.M509977200 [DOI] [PubMed] [Google Scholar]
- 48. Woodcock D. M., Crowther P. J., Doherty J., Jefferson S., DeCruz E., Noyer-Weidner M., Smith S. S., Michael M. Z., and Graham M. W. (1989) Quantitative evaluation of Escherichia coli host strains for tolerance to cytosine methylation in plasmid and phage recombinants. Nucleic Acids Res. 17, 3469–3478 10.1093/nar/17.9.3469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sambrook J., and Russell D. (2001) Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 50. Bi W., and Stambrook P. J. (1998) Site-directed mutagenesis by combined chain reaction. Anal. Biochem. 256, 137–140 10.1006/abio.1997.2516 [DOI] [PubMed] [Google Scholar]
- 51. Hames C., Halbedel S., Schilling O., and Stülke J. (2005) Multiple-mutation reaction: a method for simultaneous introduction of multiple mutations into the glpK gene of Mycoplasma pneumoniae. Appl. Environ. Microbiol. 71, 4097–4100 10.1128/AEM.71.7.4097-4100.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Merzbacher M., Detsch C., Hillen W., and Stülke J. (2004) Mycoplasma pneumoniae HPr kinase/phosphorylase: assigning functional roles to the P-loop and the HPrK/P signature sequence motif. Eur. J. Biochem. 271, 367–374 10.1046/j.1432-1033.2003.03935.x [DOI] [PubMed] [Google Scholar]
- 53. Christian H., Hofele R. V., Urlaub H., and Ficner R. (2014) Insights into the activation of the helicase Prp43 by biochemical studies and structural mass spectrometry. Nucleic Acids Res. 42, 1162–1179 10.1093/nar/gkt985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yang B., Wu Y. J., Zhu M., Fan S. B., Lin J., Zhang K., Li S., Chi H., Li Y. X., Chen H. F., Luo S. K., Ding Y. H., Wang L. H., Hao Z., Xiu L. Y., et al. (2012) Identification of cross-linked peptides from complex samples. Nat. Methods 9, 904–906 10.1038/nmeth.2099 [DOI] [PubMed] [Google Scholar]
- 55. Schmalisch M. H., Bachem S., and Stülke J. (2003) Control of the Bacillus subtilis antiterminator protein GlcT by phosphorylation. J. Biol. Chem. 278, 51108–51115 10.1074/jbc.M309972200 [DOI] [PubMed] [Google Scholar]
- 56. Gorrec F. (2009) The MORPHEUS protein crystallization screen. J. Appl. Crystallogr. 42, 1035–1042 10.1107/S0021889809042022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kabsch W. (2010) Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. D Biol. Crystallogr. 66, 133–144 10.1107/S0907444909047374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Söding J., Biegert A., and Lupas A. N. (2005) The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 33, W244–W248 10.1093/nar/gki408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. DiMaio F., Terwilliger T. C., Read R. J., Wlodawer A., Oberdorfer G., Wagner U., Valkov E., Alon A., Fass D., Axelrod H. L., Das D., Vorobiev S. M., Iwaï H., Pokkuluri P. R., and Baker D. (2011) Improved molecular replacement by density- and energy-guided protein structure optimization. Nature 473, 540–543 10.1038/nature09964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Brünger A. T. (1993) Assessment of phase accuracy by cross validation: the free R value: methods and applications. Acta Crystallogr. D Biol. Crystallogr. 49, 24–36 10.1107/S0907444992007352 [DOI] [PubMed] [Google Scholar]
- 64. Kabsch W., and Sander C. (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637 10.1002/bip.360221211 [DOI] [PubMed] [Google Scholar]
- 65. Kabsch W., Kabsch H., and Eisenberg D. (1976) Packing in a new crystalline form of glutamine synthetase from Escherichia coli. J. Mol. Biol. 100, 283–291 10.1016/S0022-2836(76)80064-2 [DOI] [PubMed] [Google Scholar]
- 66. Collaborative Computational Project, Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D 50, 760–763 10.1107/S0907444994003112 [DOI] [PubMed] [Google Scholar]
- 67. Krissinel E., and Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 10.1016/j.jmb.2007.05.022 [DOI] [PubMed] [Google Scholar]
- 68. Notredame C., Higgins D. G., and Heringa J. (2000) T-coffee: a novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302, 205–217 10.1006/jmbi.2000.4042 [DOI] [PubMed] [Google Scholar]
- 69. Armougom F., Moretti S., Poirot O., Audic S., Dumas P., Schaeli B., Keduas V., and Notredame C. (2006) Expresso: automatic incorporation of structural information in multiple sequence alignments using 3D-Coffee. Nucleic Acids Res. 34, W604–W608 10.1093/nar/gkl092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Robert X., and Gouet P. (2014) Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324 10.1093/nar/gku316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Trott O., and Olson A. J. (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 31, 455–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Nivón L. G., Moretti R., and Baker D. (2013) A pareto-optimal refinement method for protein design scaffolds. PLoS One 8, e59004 10.1371/journal.pone.0059004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Leaver-Fay A., Tyka M., Lewis S. M., Lange O. F., Thompson J., Jacak R., Kaufman K., Renfrew P. D., Smith C. A., Sheffler W., Davis I. W., Cooper S., Treuille A., Mandell D. J., Richter F., Ban Y. E., et al. (2011) ROSETTA3: an object-oriented soft-ware suite for the simulation and design of macromolecules. Methods Enzymol. 487, 545–574 10.1016/B978-0-12-381270-4.00019-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.