

Abstract
Chronic low-grade inflammation in the pancreatic islets is observed in individuals with type 2 diabetes, and macrophage levels are elevated in the islets of these individuals. However, the molecular mechanisms underlying the interactions between the pancreatic β cells and macrophages and their involvement in inflammation are not fully understood. Here, we investigated the role of S100 calcium-binding protein A8 (S100A8), a member of the damage-associated molecular pattern molecules (DAMPs), in β-cell inflammation. Co-cultivation of pancreatic islets with unstimulated peritoneal macrophages in the presence of palmitate (to induce lipotoxicity) and high glucose (to induce glucotoxicity) synergistically increased the expression and release of islet-produced S100A8 in a Toll-like receptor 4 (TLR4)-independent manner. Consistently, a significant increase in the expression of the S100a8 gene was observed in the islets of diabetic db/db mice. Furthermore, the islet-derived S100A8 induced TLR4-mediated inflammatory cytokine production by migrating macrophages. When human islet cells were co-cultured with U937 human monocyte cells, the palmitate treatment up-regulated S100A8 expression. This S100A8-mediated interaction between islets and macrophages evoked β-cell apoptosis, which was ameliorated by TLR4 inhibition in the macrophages or S100A8 neutralization in the pancreatic islets. Of note, both glucotoxicity and lipotoxicity triggered S100A8 secretion from the pancreatic islets, which in turn promoted macrophage infiltration of the islets. Taken together, a positive feedback loop between islet-derived S100A8 and macrophages drives β-cell apoptosis and pancreatic islet inflammation. We conclude that developing therapeutic approaches to inhibit S100A8 may serve to prevent β-cell loss in patients with diabetes.
Keywords: apoptosis; diabetes; islet; macrophage; pancreatic islet; S100 proteins; glucolipotoxicity; islet inflammation; beta-cell apoptosis, S100A8
Introduction
Activation of the innate immune system and circulating levels of acute-phase inflammatory proteins play important roles in the onset and development of type 2 diabetes (1–3). Evidence of chronic inflammation has been demonstrated in the adipose tissue, liver, vascular endothelial cells, circulating leukocytes, and pancreatic islets in obese and/or diabetic humans (4–8). Chronic islet inflammation evokes a decline in the β-cell mass by promoting β-cell apoptosis, which is a hallmark of type 2 diabetes (9, 10).
It has been reported that macrophages are elevated in the pancreatic islets in patients with type 2 diabetes (11). Chronic hyperglycemia promotes amyloid formation in the islets by inducing the secretion of islet amyloid polypeptide (12), production of reactive oxygen species in β cells (13), and formation of advanced glycation end products (14, 15). These conditions lead to activation of the NLRP3 inflammasomes, IL-1β secretion, macrophage infiltration of the β cells, and pro-apoptotic processes (12, 16). Thus, islet inflammation is closely related to β-cell failure and apoptosis in diabetes. A previous study showed that the co-culture of MIN6 insulinoma cells with RAW264.7 macrophage cells in the presence of palmitate increased the expression of inflammatory genes in the MIN6 cells and decreased insulin secretion (17). However, the precise mechanisms involved in the mutual interaction between the pancreatic β cells and macrophages in diabetes remain unclear.
In this study, we identified S100a8 as an up-regulated gene after chronic glucose stimulation, which reflects a state of sustained hyperglycemia, in the pancreatic islets. S100A8 is a small calcium-binding protein that is found at high levels in the extracellular milieu under inflammatory conditions. Furthermore, the S100A8 protein is known to be associated with various chronic inflammatory diseases and both type 1 and type 2 diabetes (18, 19). S100A8 is thought to be a member of the damage-associated molecular pattern molecules and stimulates macrophages (20–23). Consequently, to test the hypothesis that S100A8 contributes to islet inflammation, we established a co-culture system with freshly isolated primary pancreatic islets and resident peritoneal macrophages to investigate the role(s) of S100A8 in the sustenance of islet inflammation.
Results
S100A8/A9 expression in the islets was up-regulated by chronic glucose stimulation
Chronic hyperglycemia induces β-cell apoptosis, in part, through continuous glucokinase activation (24). We previously identified the target genes of glucokinase by examining the gene expression profiles of glucokinase activator (GKA)5-treated isolated islets (NCBI GEO database GSE41248) (25). Among them, S100a8 and S100a9 (S100a8/a9) expression showed the greatest increase following chronic glucokinase activation of the islets (90- and 254-fold increase, respectively). We validated the gene expression changes in isolated islets stimulated with a glucokinase activator (Fig. 1A) and confirmed that glucose stimulation increased the expression of S100a8/a9 in the islets in a concentration-dependent manner (Fig. 1B). Thus, S100A8 expression was induced by high glucose in the islets without macrophages.
Figure 1.
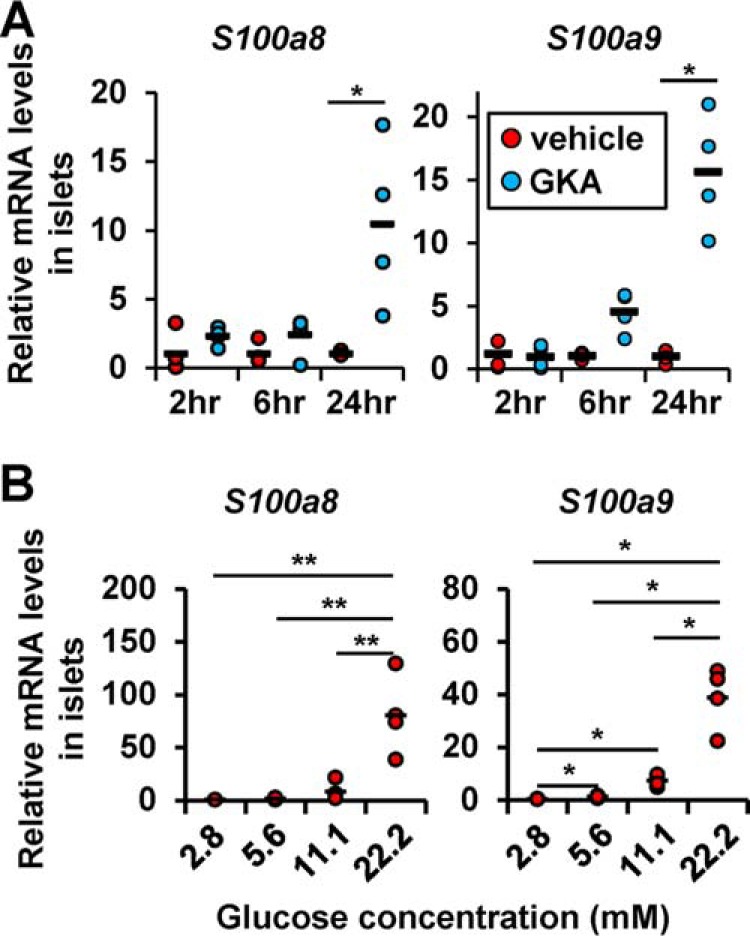
Glucose stimulation up-regulated S100A8/A9 expression in the islets. A, mRNA expression levels in the islets stimulated with GKA Cpd A (glucokinase activator; 30 μmol/liter) for 2, 6, or 24 h. Horizontal bars, mean values. *, p ≤ 0.05 versus other groups (n = 4/group). B, mRNA expression levels in islets cultured in the presence of 2.8, 5.6, 11.1, or 22.2 mmol/liter glucose for 24 h. Horizontal bars, mean values. *, p ≤ 0.05; **, p ≤ 0.01 (n = 4/group).
S100A8/A9 expression in the islets was enhanced by co-culture with macrophages in the presence of palmitate
We co-cultured islets with macrophages using co-culture inserts (Fig. 2A) and observed that the islet expression of S100a8, S100a9, Il-1b, Tnf-a, Il-6, and Ccl2 was increased in the presence of macrophages (Fig. 2B). The absence of elevated expression of the macrophage markers, Cd11b and F4/80, suggested that it was unlikely that there was contamination of the co-cultured islet samples with macrophages (Fig. 2C). The expression of S100a8 and S100a9, but not of Il-1b, Tnf-a, Il-6, or Ccl2, was enhanced in the islets co-cultured with macrophages in the presence of the saturated fatty acid palmitate (16:0) (Fig. 2B). We confirmed the secretion of S100A8, but not of S100A9, by ELISAs in the supernatant of islets co-cultured with macrophages in the presence of palmitate (Fig. 2D). Notably, secretion of S100A8 from the macrophages was not affected by the concentration of glucose or palmitate (Fig. 2E). S100A8 proteins were predominantly expressed in the mouse pancreatic islets, but not in acinar cells (Fig. 2F).
Figure 2.

Chronic stimulation with high concentrations of glucose, palmitate, and macrophages induced S100A8/A9 expression in the pancreatic islets. A, illustration of co-cultured islets and macrophages (right). Flow cytometry of the adherent peritoneal macrophages (left). B–D, isolated mouse pancreatic islets (50 islets) were co-cultured with peritoneal macrophages (1 × 105 cells) in the presence of BSA (0.5%) or palmitate (500 μmol/liter). B and C, mRNA expression levels in islets co-cultured with macrophages in the presence/absence of palmitate for 24 h. Horizontal bars, mean values. *, p ≤ 0.05; **, p ≤ 0.01 (n = 9). D, secreted S100A8 protein (left) and S100A9 protein (right) levels in the supernatant obtained from co-culture of islets with macrophages in the presence/absence of palmitate for 48 h. Horizontal bars, mean values. N.D., not detected; **, p ≤ 0.01 (n = 4). E, secreted S100A8 protein levels in the supernatant of macrophages cultured in the presence of 5.6 or 11.1 mmol/liter glucose with/without palmitate for 48 h. Horizontal bars, mean values (n = 3/group). F, pancreatic sections from 8-week-old male WT C57BL/6J mice were stained with antibodies to insulin (green), glucagon (red), and S100A8 (blue). Scale bar, 50 μm.
To test the possibility that the adipocyte-derived fatty acids contributed to the macrophage-mediated islet inflammation in vivo, we added isolated white adipocytes from the epididymal fat to the co-culture of islets with macrophages and examined islet gene expression. As expected, the adipocytes and macrophages synergistically increased the expression of S100a8 and S100a9 in the islets, and this was not associated with elevation of the expression of macrophage or adipocyte markers (Fig. 3A). Palmitate has been reported to induce islet inflammation through the TLR4/MyD88 pathway (17). Islets obtained from TLR4-knockout mice and co-cultured with WT macrophages in the presence of palmitate showed a significant increase in the expression of S100a8/a9 (Fig. 3B). These results suggest that TLR4-mediated signaling was not required for the S100A8 production induced by macrophage-derived factors and palmitate in the co-cultured islets.
Figure 3.

TLR4-independent S100A8 production induced by macrophage-derived factors and palmitate in the co-cultured islets. A, isolated pancreatic islets (50 islets) were co-cultured with white adipocytes in the presence of BSA (0.5%) or palmitate (500 μmol/liter) for 24 h. mRNA expression levels in co-cultured islets. Horizontal bars, mean values. *, p ≤ 0.05; **, p ≤ 0.01 (n = 9). B, mRNA expression levels in TLR4+/+ or TLR4−/− islets co-cultured with TLR4+/+ macrophages in the presence of BSA (0.5%) or palmitate (500 μmol/liter) for 24 h. Horizontal bars, mean values. *, p ≤ 0.05; **, p ≤ 0.01 (n = 6).
Glucotoxicity further enhanced the induction of S100A8/A9 in co-cultured islets
Chronic high ambient glucose concentration has been shown to accelerate inflammation in various tissues in diabetes (26, 27). We undertook experiments under normal glucose (5.6 mmol/liter) conditions and under high glucose (11.1 mmol/liter) conditions to mimic the environment in diabetes. The protein expression of S100A8/A9 in the co-cultured islets was enhanced following culture in the presence of 11.1 mmol/liter glucose (high concentration) (Fig. 4A). S100A8 secretion from islets co-cultured with macrophages in the presence of palmitate was also enhanced by glucose stimulation (Fig. 4B). High glucose enhanced palmitate-induced S100a8 and S100a9 gene expression, whereas expression of other inflammatory or macrophage markers in the co-cultured islets was not influenced by the glucose concentration (Fig. 4C). In addition, inflammatory gene expression in the macrophages was up-regulated by the ambient glucose level (Fig. 4D).
Figure 4.

Increased ambient glucose concentrations enhanced the expressions of S100A8/A9 in co-cultured islets with macrophages. Isolated pancreatic islets (200 islets for A and B, 50 islets for C and D) were co-cultured with peritoneal macrophages (5 × 105 cells for A and B, 1 × 105 cells for C and D) in medium containing BSA in the presence/absence of palmitate. A, isolated islets were co-cultured with macrophages in the presence of 5.6 or 11.1 mmol/liter of glucose with/without palmitate for 48 h. Total cell extracts from the islets were subjected to immunoblotting as indicated. B, secreted S100A8 protein levels in the supernatant from a co-culture of islets with macrophages in the presence/absence of palmitate and 5.6 or 11.1 mmol/liter glucose for 48 h. Horizontal bars, mean values. n.s., not significant; **, p ≤ 0.01 (n = 3). C, mRNA expression levels in islets co-cultured with macrophages in the presence of 5.6 or 11.1 mmol/liter glucose with/without palmitate for 24 h. Horizontal bars, mean values. **, p ≤ 0.01 (n = 6). D, mRNA expression levels in macrophages co-cultured with islets in the presence of 5.6 or 11.1 mmol/liter glucose with/without palmitate for 24 h. Horizontal bars, mean values. *, p ≤ 0.05; **, p ≤ 0.01 (n = 6).
We next examined the expression of S100a8/a9 in the islets of the db/db mouse, an established model of diabetes. Six- and 12-week-old db/db mice exhibited morbid obesity, severe hyperglycemia, and irregular α/β-cell distribution within the islets; however, the ratio of β to α cells and the proportion of apoptotic β cells were not altered in the db/db mice compared with control db/+ mice (Table S1 and Fig. 5 (A and B)). Isolated pancreatic islets from db/db mice, at both 6 and 12 weeks of age, showed higher levels of expression of S100a8/a9, of the inflammatory cytokine gene Il-6, and of the macrophage marker F4/80 compared with db/+ islets (Fig. 5C).
Figure 5.

Increased S100A8/A9 expressions in diabetic islets. A, quantification of β-cell mass as a proportion of the total α-cell mass in the islet. Horizontal bars, mean values (n = 6). B, the proportion of TUNEL-positive cells is shown as a percentage of the total number of insulin-positive cells in the sections. Horizontal bars, mean values (n = 5). Scale bar, 50 μm. C, mRNA expression levels in islets from 6- or 12-week-old db/db or db/+ mice. Horizontal bars, mean values. n.s., not significant; *, p ≤ 0.05; **, p ≤ 0.01 (n = 6).
Factors from the islets, but not from macrophages, activated the macrophages in co-culture via TLR4
In macrophages co-cultured with islets in the presence of palmitate, expression of Tnf-a, Ccl2, Il-1b, Il-6, Il-12, Il-22, Il-23, and Il-24 genes were elevated (Fig. 6A). IL-23 and IL-24 are potent inducers of oxidative and ER stress in β cells (28). No increase in the expression of S100a8/a9 genes was observed in the macrophages co-cultured with the islets in the presence of palmitate or in the presence of high ambient glucose (Fig. 6B). This implies that the production of S100A8 induced by co-culture with macrophages was predominantly derived from the islets. The aforementioned increase in the cytokine gene expression was blunted by the TLR4-inhibitory peptide VIPER (Fig. 6C). These results suggest that a humoral factor derived from the co-cultured islets stimulated the macrophages via TLR4.
Figure 6.

Activation of the macrophages in co-culture with islets via TLR4. Peritoneal macrophages (1 × 105 cells) were co-cultured with isolated pancreatic islets (50 islets) in a medium containing BSA (0.5%) in the presence/absence of palmitate. A and B, mRNA expression levels in macrophages co-cultured with islets for 24 h. Horizontal bars, mean values. **, p ≤ 0.01 (n = 5). C, mRNA expression levels in macrophages co-cultured with islets in the presence of the TLR4 inhibitory peptide VIPER or the control peptide CP7 for 24 h. Horizontal bars, mean values. *, p ≤ 0.05 (n = 5).
Because S100A8 is reported as a ligand of TLR4, we examined whether S100A8 induces inflammation in the co-cultured macrophages. Treatment with the recombinant S100A8-GST peptide increased the expression of the Tnf-a, Ccl2, Il-1b, Il-6, Il-12, Il-22, and Il-23 genes in the macrophages and prompted macrophage migration (Fig. 7, A and B). Neutralization of S100A8 using an antibody significantly reduced the migration of the macrophages induced by co-culture with islets (Fig. 7C). Neutralization of S100A8 with an antibody also reduced the cytokine expression of the macrophages induced by co-culture of the macrophages with islets in the presence of palmitate (Fig. 7D). Inhibition of TLR4 with VIPER or TAK-242 attenuated the migration of the macrophages and the cytokine expression in these cells induced by purified S100A8-GST peptide (Fig. 7, E and F). TLR4−/− macrophages exhibited a significant decrease of cytokine expression following co-culture with islets in the presence of palmitate as compared with TLR4+/+ macrophages (Fig. 7G). Taken together, S100A8 leads to the up-regulation of inflammation mediators in macrophages via TLR4.
Figure 7.

Islet-derived S100A8 activated macrophage migration and inflammation. A, mRNA expression levels in peritoneal macrophages (1 × 105 cells) stimulated with S100A8-GST peptide for 24 h. Horizontal bars, mean values. *, p ≤ 0.05; **, p ≤ 0.01 versus 0 ng/ml control (n = 3). B, fluorescence intensity of Calcein-AM–labeled macrophages migrating in response to S100A8-GST peptide stimulation for 24 h. Horizontal bars, mean values. *, p ≤ 0.05 versus 0 ng/ml control (n = 3). C, fluorescence intensity of migrated Calcein-AM–labeled macrophages migrating in response to co-culture with islets in the presence of anti-S100A8 neutralizing antibody (10 μg/ml) or IgG2B isotype control for 48 h. Horizontal bars, mean values. *, p ≤ 0.05; **, p ≤ 0.01 (n = 6). D, mRNA expression levels in macrophages co-cultured with the islets with/without palmitate in the presence of anti-S100A8 neutralizing antibody or IgG2B isotype control. Horizontal bars, mean values. **, p ≤ 0.01 (n = 4). E, fluorescence intensity of migrated Calcein-AM–labeled macrophages in response to co-culture with islets in the presence of recombinant S100A8 peptide and the TLR4-inhibitory peptide VIPER or TAK-242 (100 nmol/liter) for 24 h. Horizontal bars, mean values. *, p ≤ 0.05; **, p ≤ 0.01 (n = 6). F, mRNA expression levels in S100A8-stimulated macrophages cultured in the presence of the TLR4 inhibitory peptide VIPER or TAK-242 for 24 h. Horizontal bars, mean values. *, p ≤ 0.05; **, p ≤ 0.01 (n = 3). G, mRNA expression levels in TLR4+/+ or TLR4−/− macrophages co-cultured with TLR4+/+ islets in the presence of palmitate for 24 h. Horizontal bars, mean values. *, p ≤ 0.05; **, p ≤ 0.01 (n = 4).
Co-cultivation of islets with macrophages in the presence of palmitate coordinately promoted β-cell apoptosis via islet-derived S100A8 and macrophages
Exogenous S100a8/a9 expression induced by adenoviral transduction was also influenced by the ambient glucose levels (Fig. 8A), suggesting that glucose stimulation possibly enhanced translation or stabilized the S100A8/A9 mRNA or protein. Overexpression of S100A8/S100A9 exerted no effect on glucose-induced insulin secretion from the islets (Fig. 8B) or on the degree of apoptosis in the islets (Fig. 8C). S100A8 overexpression in the MIN6K8 β cells slightly decreased insulin secretion from the cells, and conversely, S100a8 knockdown was capable of restoring the insulin secretion, even in the absence of macrophages (Fig. 8D). In the presence of macrophages, however, S100A8 overexpression increased insulin secretion, whereas S100a8 knockdown tended to decrease insulin secretion in S100A8-overexpressing MIN6K8 β cells (Fig. 8E).
Figure 8.

Effects of S100A8 on insulin secretion from pancreatic islets. A, islets were treated with adenoviral LacZ or adenoviral S100A8/A9 for 48 h. The mRNA expression levels in the adenoviral S100A8/A9-transduced islets (50 islets) cultured in the presence of 5.6 or 11.1 mmol/liter glucose are shown. Horizontal bars, mean values. *, p ≤ 0.05 (n = 3). B, glucose-stimulated insulin secretion analysis from the adenoviral S100A8/A9-transduced islets (50 islets). Horizontal bars, mean values (n = 4). C, TUNEL assay in the adenoviral S100A8/A9-transduced islets (200 islets). Horizontal bars, mean values. D and E, glucose-stimulated insulin secretion analysis from shRNA-treated MIN6K8 cells. MIN6K8 cells were cultured for 24 h for adenoviral S100A8 transduction with/without peritoneal macrophages. D, without macrophages. Horizontal bars, mean values. **, p ≤ 0.01 (n = 8). E, with macrophages. Horizontal bars, mean values. **, p ≤ 0.01 (n = 4).
To assess the effects of the co-cultivation on the islets, β-cell apoptosis was evaluated. Co-culturing with macrophages increased the number of apoptotic β cells, and palmitate enhanced macrophage-induced β-cell apoptosis (Fig. 9A). The combination of glucose stimulation further induced apoptosis of β cells in the presence of macrophages (Fig. 9A). The apoptosis-associated Bax protein expression level, but not that of the necrosis-associated HMGB1 protein, increased in the islets co-cultured with macrophages in the presence of palmitate and ambient high glucose levels (Fig. 9B). The TLR4-inhibitory peptides VIPER and TAK-242 showed a tendency to reduce β-cell apoptosis caused by co-culturing of the islets with macrophages (Fig. 9, C and D). Furthermore, neutralization of S100A8 with an antibody specific to S100A8 significantly reduced the degree of β-cell apoptosis in the islets co-cultured with macrophages (Fig. 9, C and D).
Figure 9.

S100A8-stimulated macrophages induced β-cell apoptosis. Isolated pancreatic islets (200 islets) were co-cultured with peritoneal macrophages (5 × 105 cells) in medium containing BSA in the presence/absence of palmitate for 48 h. A and C, the islets were subjected to the TUNEL assay. The proportion of TUNEL-positive β cells is shown as a percentage of the co-cultured islets. Insulin is stained red, nuclei are stained blue, and TUNEL-positive nuclei are stained green. A, left, co-cultivation in the presence of 5.6 or 11.1 mmol/liter glucose. Horizontal bars, mean values. *, p ≤ 0.05; **, p ≤ 0.01 (n = 3). Right, representative images. B, isolated islets were co-cultured with macrophages in the presence of 5.6 or 11.1 mmol/liter glucose with/without palmitate for 48 h. Total cell extracts from the islets were subjected to immunoblotting as indicated. C, isolated islets were co-cultured with macrophages in the presence of 11.1 mmol/liter glucose with palmitate for 24 h. Co-cultivation in the presence of anti-S100A8 neutralizing antibody, IgG2B isotype control, the control peptide CP7, the TLR4 inhibitory peptide VIPER, or the TLR4 inhibitor TAK-242. Horizontal bars, mean values. *, p ≤ 0.05; **, p ≤ 0.01 (n = 6). D, representative images of C.
Expression of S100A8 in human islets
Next, investigation of human islets revealed that expression of S100A8 was significantly enhanced after stimulation with GKA in both nondiabetes and type 2 diabetes donors (Fig. 10A). Co-existence of human monocyte U937 cell line and palmitate significantly up-regulated the expression of S100A8 in human islets (Fig. 10B). We also explored the localization of S100A8 in human islets. Immunohistochemical staining for S100A8 was predominantly detected in β cells in the islets (Fig. 10C). The degree of β-cell apoptosis in the human islets co-cultured with U937 cells and palmitate tended to be decreased by the S100A8-specific neutralizing antibody (Fig. 10D). Further study is warranted to clarify the pathological significance of S100A8 in human islet inflammation.
Figure 10.

The expression of S100A8 was induced by glucose stimulation, monocytes, and palmitate in human islets. A, mRNA expression levels in human islets from nondiabetes (non-DM) donors after stimulation with GKA Cpd A (glucokinase activator; 30 μmol/liter) for 24 h. Horizontal bars, mean values. *, p ≤ 0.05; **, p ≤ 0.01 (n = 5). B, mRNA expression levels in human islets co-cultured with U937 human monocyte cell line (1 × 105 cells) in the presence of BSA (0.5%) or palmitate (500 μmol/liter) for 24 h. Horizontal bars, mean values. *, p ≤ 0.05; **, p ≤ 0.01 (n = 5). C, embedded human islets from nondiabetes donors were stained with antibodies to insulin (green), glucagon (red), and S100A8 (blue). Scale bar, 50 μm. D, human islets from nondiabetes donors were co-cultured with U937 cells in the presence of 11.1 mmol/liter glucose with palmitate for 24 h. They were co-cultivated in the presence of anti-S100A8 neutralizing antibody or IgG2B isotype control. The proportion of TUNEL-positive β cells is shown as a percentage of the co-cultured islets. Horizontal bars, mean values. *, p ≤ 0.05 (n = 5).
Discussion
The results of the present study identified S100A8 as an endogenous islet-derived secretory peptide that is induced by a combination of infiltrating macrophages, palmitate (lipotoxicity), and high glucose (glucotoxicity), resulting in the activation of macrophages and potentiation of islet inflammation and β-cell death through a positive feedback loop (Fig. 11). The current results are consistent with a recent report suggesting that the serum level of the S100A8/A9 complex is a sensitive marker of acute inflammation associated with islet transplant rejection (29).
Figure 11.

An illustrative model of islet inflammation–induced β-cell apoptosis via S100A8 in diabetes. A combination of infiltrating macrophages, saturated fatty acids (palmitate), and hyperglycemia augmented the production of S100A8. S100A8 secreted from the islets induced further macrophage migration and inflammation through TLR4. This positive feedback loop potentiates islet inflammation and β-cell death.
Several studies have shown that TLR4 signaling and MyD88 signaling in β cells play important roles in the development of islet inflammation (17, 30, 31). However, TLR4 stimulation did not induce S100A8/A9 production in the islets, whereas S100A8 protein from the islets acted as a ligand for the TLR4 expressed in macrophages. Because TLR4 and RAGE (receptor for advanced glycation end products) receptors are also expressed in the pancreatic β cells (17), it was suggested that S100A8 also exerts direct effects on the β cells. TLR4 stimulation by S100A8 triggered the release of the inflammatory cytokines Il-12, Il-23, and Il-24 from the macrophages, which resulted in β-cell apoptosis. Further production of S100A8 may evoke inflammation of the surrounding islets, neighboring tissues, or feeding vessels. TNFα, CCL2, and IL-1β are known to induce islet inflammation through the NF-κB pathway (3), and IL-23 and IL-24 have been shown to be potent inducers of oxidative and ER stress in β cells (28). The expression of these cytokines was also induced in the macrophages following stimulation with S100A8 in the present study.
Overexpression of S100A8/A9 had no inhibitory effect on insulin secretion from isolated islets (Fig. 8B). We also report that S100A8 per se impaired insulin secretion from MIN6K8 β cells in the absence of macrophages (Fig. 8D). However, overexpression of S100A8 enhanced insulin secretion from MIN6K8 β cells in the presence of macrophages (Fig. 8E). Overexpression of S100A8 has been reported in ductal adenocarcinoma of the pancreas, and it has been suggested that a peptide metabolite of S100A8 released from pancreatic cancer possibly suppresses insulin secretion to induce diabetes (32). Further research is required to clarify the effects of S100A8 on insulin secretion under similar pathophysiological conditions.
Our results suggested that S100A8 did not act directly to induce β-cell apoptosis, but via a mutual interaction with macrophages. S100A8 in β cells probably triggers destruction of the β-cell membrane, because S100A8 and S100A9 have the ability to form oligomers and induce amyloid deposition (33). Because S100A9 protein was detected in the islets but not in the culture medium, the contribution of S100A9 in our experimental conditions remains unclear. S100A9 protein, a heterodimerization partner of S100A8, possibly plays an essential role in the protein expression of S100A8 protein, as indicated by previous studies that found that S100A8 was not detectable in S100A9-KO peripheral tissues (20, 34).
Glucokinase-mediated induction of S100A8 production seems specific to pancreatic β cells, as deduced from the results of previous studies carried out in other organs (35–37). Glucokinase activation enhances adaptive β-cell proliferation and prevents β-cell apoptosis induced by glucotoxicity or ER stress (25, 38, 39). However, chronic glucokinase activation and high glucose act to trigger β-cell apoptosis (24, 39). There is considerable debate about the role of glucokinase in the protection of β cells from the S100A8-mediated positive feedback loop of islet inflammation under glucolipotoxicity. Additional studies are necessary to confirm the production of S100A8 in human β cells from obese or diabetic subjects.
In summary, our studies support the identification of S100A8 as a secretory protein to promote β-cell apoptosis and constitute an important step in the development of approaches to protect β cells in patients with diabetes.
Experimental procedures
Animals and animal care
Animal handling procedures were in accordance with institutional animal care and use committee protocols approved by Yokohama City University. Animals were housed in rooms maintained at a constant room temperature (25 °C) under a 12-h light (07:00 h)/12-h dark (19:00 h) cycle, and the animals were given free access to food and water. C57BL/6J mice were purchased from CLEA Japan (Tokyo, Japan). All of the mice used in this study belonged to the C57BL/6J background. db/db mice and db/+ mice (BKS.Cg-Leprdb/Leprdb and BKS.Cg-Leprdb/Dock7m) were purchased from Charles River Laboratories Japan (Yokohama, Japan). TLR4−/− mice were purchased from Oriental Bio Service (Kyoto, Japan).
Reagents, viruses, and cells
The S100A8 and S100A9 ELISA kits were purchased from Cloud-Clone Corp. (Houston, TX). GKA Cpd A was purchased from Merck (Darmstadt, Germany). Collagenase L was purchased from Nitta-Gelatin (Osaka, Japan). Thapsigargin was purchased from Sigma. Collagenase XI and LPS were purchased from Sigma-Aldrich. The TLR4 peptide inhibitor VIPER (40) was purchased from Novus Biologicals, LLC (Littleton, CO). TAK-242 was purchased from Chemscene, LLC (Monmouth Junction, NJ). d-Mannoheptulose was purchased from Carbosynth Ltd. (Compton, Berkshire, UK). Adenoviruses containing S100A8, S100A9, or LacZ were generated using the Virapower adenoviral expression system (Invitrogen). Five micrograms of adenoviral constructs were digested with PacI, and the linearized DNA was transfected into HEK293A cells. The adenovirus produced by these cells was then collected and subjected to three cycles of freezing and thawing to release the adenovirus. The resulting adenovirus was stored at −80 °C for later use. Viral titers were determined by plaque assays using cultured HEK293A cells, according to the manufacturer's instructions. Monoclonal rat IgG2B anti-mouse S100A8 antibody and monoclonal rat IgG2B isotype control were purchased from R&D Systems (Abingdon, UK). Calcein-AM was purchased from Dojindo Laboratories (Kumamoto, Japan). The MIN6K8 cell line was provided by Dr. Susumu Seino (Kobe University). Lentivirus particles expressing short hairpin RNA for S100A8 were purchased from Santa Cruz Biotechnology, Inc.
Isolation and co-culture of islets, resident peritoneal macrophages, and white adipocytes
Islets and peritoneal macrophages were isolated as described elsewhere (41, 42). The proportion of F4/80- and CD11b-positive macrophages was more than 90%, as confirmed by flow cytometry (Fig. 2A). Adipocytes were prepared by collagenase digestion (Nitta Gelatin) of epididymal fat tissue, as described previously (43). Glucose-stimulated insulin secretion from the islets was induced as described previously (44). The co-culture was performed at 37 °C in a Krebs–Ringer bicarbonate HEPES buffer, pH 7.4, containing 0.2% BSA. Isolated pancreatic islets and peritoneal macrophages were plated in a Netwell insert with a 74-μm mesh size polyester membrane (Corning, Inc.) and in the bottom wells, respectively, and the cultures were incubated for 24 or 48 h in RPMI1640 containing fatal bovine serum, Krebs–Ringer bicarbonate containing BSA, or 500 μmol/liter palmitate (Fig. 2A). Adipocytes (from 25 mg of epididymal fat) were co-cultured above the co-culture Netwell insert.
Real-time PCR
Total RNA isolation from pancreatic islets, cDNA synthesis, and quantitative PCR were performed as described previously (25, 45). Data were normalized according to the expression level of β-actin, 18S rRNA, or GAPDH. The primers used for the real-time PCR are listed in Table S2.
Immunohistochemical analysis
Pancreases and islets were fixed and immunostained as reported previously (25, 45). Pancreatic tissue sections were immunostained with antibodies to S100A8 (Santa Cruz Biotechnology, Abcam), S100A9 (Abcam), insulin (Santa Cruz Biotechnology), or glucagon (Abcam). Alexa Fluor 488-, 555-, and 647-conjugated secondary antibodies (Invitrogen) were used for the fluorescence microscopic analysis. β-cell apoptosis was evaluated using a TdT-mediated dUTP nick-end labeling (TUNEL) assay of the co-cultured β cells. Co-cultured islets (200 islets) were attached to poly-l-lysine–coated coverslips (Falcon) and subjected to a TUNEL assay using the ApopTag in situ detection kit (EMD Millipore, MA). All of the images were acquired using a FluoView FV1000-D confocal laser-scanning microscope (Olympus, Tokyo, Japan).
Immunoblotting
For immunoblotting, isolated islets (200 islets) were lysed in radioimmune precipitation buffer (Cell Signaling Technology, Danvers, MA) with complete protease inhibitor mixture (Roche Diagnostics). After centrifugation, the extracts were subjected to immunoblotting with antibodies. The primary antibodies used were Calgranulin A (S100A8), Calgranulin B (S100A9), Bcl-2–associated X protein (BAX), Bcl-2 (Santa Cruz Biotechnology), high-mobility group box 1 (HMGB1), and glyceraldehyde-3 phosphate dehydrogenase (GAPDH) (Abcam).
GST-fused proteins
GST-fused constructs comprising the mouse S100A8 and S100A9 proteins were generated in pGEX4T-1 and were received as kind gifts from Dr. Sachie Hiratsuka and Dr. Yoshihiro Maru (Department of Pharmacology, Tokyo Women's Medical University) (46). The proteins were expressed in Escherichia coli BL21 and purified on a GSH-Sepharose column.
Macrophage migration assay
The migrated macrophages were labeled with Calcein-AM (WAKO, Osaka, Japan) and measured using an ARVOTM MX plate reader (PerkinElmer Life Sciences) at an excitation wavelength of 485 nm and emission filter of 535 nm. The migration of macrophages was evaluated using 8-μm pore Falcon BD FluoroBlokTM inserts and plates (BD Biosciences) with isolated pancreatic islets (50 islets) or S100A8-GST in the presence or absence of anti-S100A8 antibody, isotype control IgG2b, VIPER/CP7, or TAK-242. Macrophages were seeded onto the insert mesh and incubated for 24 or 48 h at 37 °C under 5% CO2.
Human islets
Human islets were obtained from the Integrated Islet Distribution Program (National Institutes of Health). All studies and protocols used were approved by the Joslin Diabetes Center's Committee on Human Studies (approval CHS#5-05). Details of human islets are described in Table S3. Upon receipt, islets were cultured overnight in Miami Medium 1A (Cellgro). Co-culture was performed at 37 °C in final wash/culture medium (Cellgro) or RPMI1640 medium. Cadaveric human islets and U937 human monocyte cell line (ATCC) were plated in a Netwell insert with a 74-μm mesh size polyester membrane (Corning) and in the bottom wells, respectively.
Statistical analyses
All experiments were independently repeated at least three times. Horizontal bars indicate mean values. Statistical analyses were conducted using IBM SPSS Statistics version 19. Equality of variances was determined by using an F-test or Levene's test. Statistical comparisons between groups were analyzed for significance by an unpaired two-tailed Student's t test and one-way analysis of variance with post hoc Tukey tests for a parametric test or Welch's t test or Games–Howell test for a nonparametric test. Differences were considered significant at p < 0.05.
Author contributions
J. S. designed the research; H. I., J. S., Y. Togashi, K. T., T. O., M. K., Y. Tanaka, K. O., Y. S., and T. Y., performed the experiments; K. O., K. S., and R. N. K contributed to human islet studies. H. I., J. S., Y. Togashi, K. T., T. O., and Y. Terauchi analyzed the data; H. I., J. S., R. N. K., and Y. Terauchi wrote and edited the manuscript.
Supplementary Material
Acknowledgments
We thank Dr. Yoshihiro Maru and Dr. Sachie Hiratsuka (Tokyo Women's Medical University) for kindly gifting us the pGEX4T-1-S100A8 and -S100A9 plasmids and Dr. Susumu Seino (Kobe University) for providing the MIN6K8 cells. We also thank Mitsuyo Kaji, Eri Sakamoto (Yokohama City University), and Hiroko Madokoro (Keio University) for excellent technical assistance and Misa Katayama for excellent secretarial assistance.
This work was supported by Grant-in-Aid for Scientific Research (B) 16H05329 (to Y. Terauchi) from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT) of Japan, a grant-in-aid from the Japan Foundation for Applied Enzymology (to J. S.), and a Junior Scientist Development Grant supported by Novo Nordisk Pharma Ltd. (to J. S.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Tables S1–S3.
- GKA
- glucokinase activator
- TLR
- Toll-like receptor
- TUNEL
- TdT-mediated dUTP nick-end labeling
- ER
- endoplasmic reticulum.
References
- 1. Donath M. Y., Schumann D. M., Faulenbach M., Ellingsgaard H., Perren A., and Ehses J. A. (2008) Islet inflammation in type 2 diabetes: from metabolic stress to therapy. Diabetes Care 31, S161–S164 10.2337/dc08-s243 [DOI] [PubMed] [Google Scholar]
- 2. Pickup J. C., Mattock M. B., Chusney G. D., and Burt D. (1997) NIDDM as a disease of the innate immune system: association of acute-phase reactants and interleukin-6 with metabolic syndrome X. Diabetologia 40, 1286–1292 10.1007/s001250050822 [DOI] [PubMed] [Google Scholar]
- 3. Donath M. Y., and Shoelson S. E. (2011) Type 2 diabetes as an inflammatory disease. Nat. Rev. Immunol. 11, 98–107 10.1038/nri2925 [DOI] [PubMed] [Google Scholar]
- 4. Xu H., Barnes G. T., Yang Q., Tan G., Yang D., Chou C. J., Sole J., Nichols A., Ross J. S., Tartaglia L. A., and Chen H. (2003) Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Invest. 112, 1821–1830 10.1172/JCI200319451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cai D., Yuan M., Frantz D. F., Melendez P. A., Hansen L., Lee J., and Shoelson S. E. (2005) Local and systemic insulin resistance resulting from hepatic activation of IKK-β and NF-κB. Nat. Med. 11, 183–190 10.1038/nm1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brownlee M. (2001) Biochemistry and molecular cell biology of diabetic complications. Nature 414, 813–820 10.1038/414813a [DOI] [PubMed] [Google Scholar]
- 7. Morigi M., Angioletti S., Imberti B., Donadelli R., Micheletti G., Figliuzzi M., Remuzzi A., Zoja C., and Remuzzi G. (1998) Leukocyte-endothelial interaction is augmented by high glucose concentrations and hyperglycemia in a NF-κB-dependent fashion. J. Clin. Invest. 101, 1905–1915 10.1172/JCI656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Eguchi K., and Manabe I. (2013) Macrophages and islet inflammation in type 2 diabetes. Diabetes Obes. Metab. 15, 152–158 10.1111/dom.12168 [DOI] [PubMed] [Google Scholar]
- 9. Donath M. Y., and Halban P. A. (2004) Decreased beta-cell mass in diabetes: significance, mechanisms and therapeutic implications. Diabetologia 47, 581–589 10.1007/s00125-004-1336-4 [DOI] [PubMed] [Google Scholar]
- 10. Donath M. Y., Størling J., Maedler K., and Mandrup-Poulsen T. (2003) Inflammatory mediators and islet beta-cell failure: a link between type 1 and type 2 diabetes. J. Mol. Med. 81, 455–470 10.1007/s00109-003-0450-y [DOI] [PubMed] [Google Scholar]
- 11. Ehses J. A., Perren A., Eppler E., Ribaux P., Pospisilik J. A., Maor-Cahn R., Gueripel X., Ellingsgaard H., Schneider M. K., Biollaz G., Fontana A., Reinecke M., Homo-Delarche F., and Donath M. Y. (2007) Increased number of islet-associated macrophages in type 2 diabetes. Diabetes 56, 2356–2370 10.2337/db06-1650 [DOI] [PubMed] [Google Scholar]
- 12. Masters S. L., Dunne A., Subramanian S. L., Hull R. L., Tannahill G. M., Sharp F. A., Becker C., Franchi L., Yoshihara E., Chen Z., Mullooly N., Mielke L. A., Harris J., Coll R. C., Mills K. H., et al. (2010) Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1β in type 2 diabetes. Nat. Immunol. 11, 897–904 10.1038/ni.1935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhou R., Tardivel A., Thorens B., Choi I., and Tschopp J. (2010) Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 11, 136–140 10.1038/ni.1831 [DOI] [PubMed] [Google Scholar]
- 14. Vlassara H., and Palace M. R. (2002) Diabetes and advanced glycation endproducts. J. Int. Med. 251, 87–101 10.1046/j.1365-2796.2002.00932.x [DOI] [PubMed] [Google Scholar]
- 15. Zhu Y., Shu T., Lin Y., Wang H., Yang J., Shi Y., and Han X. (2011) Inhibition of the receptor for advanced glycation endproducts (RAGE) protects pancreatic beta-cells. Biochem. Biophys. Res. Commun. 404, 159–165 10.1016/j.bbrc.2010.11.085 [DOI] [PubMed] [Google Scholar]
- 16. Maedler K., Sergeev P., Ris F., Oberholzer J., Joller-Jemelka H. I., Spinas G. A., Kaiser N., Halban P. A., and Donath M. Y. (2002) Glucose-induced beta cell production of IL-1β contributes to glucotoxicity in human pancreatic islets. J. Clin. Invest. 110, 851–860 10.1172/JCI200215318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Eguchi K., Manabe I., Oishi-Tanaka Y., Ohsugi M., Kono N., Ogata F., Yagi N., Ohto U., Kimoto M., Miyake K., Tobe K., Arai H., Kadowaki T., and Nagai R. (2012) Saturated fatty acid and TLR signaling link beta cell dysfunction and islet inflammation. Cell Metab. 15, 518–533 10.1016/j.cmet.2012.01.023 [DOI] [PubMed] [Google Scholar]
- 18. Bouma G., Lam-Tse W. K., Wierenga-Wolf A. F., Drexhage H. A., and Versnel M. A. (2004) Increased serum levels of MRP-8/14 in type 1 diabetes induce an increased expression of CD11b and an enhanced adhesion of circulating monocytes to fibronectin. Diabetes 53, 1979–1986 10.2337/diabetes.53.8.1979 [DOI] [PubMed] [Google Scholar]
- 19. Burkhardt K., Schwarz S., Pan C., Stelter F., Kotliar K., Von Eynatten M., Sollinger D., Lanzl I., Heemann U., and Baumann M. (2009) Myeloid-related protein 8/14 complex describes microcirculatory alterations in patients with type 2 diabetes and nephropathy. Cardiovasc. Diabetol. 8, 10 10.1186/1475-2840-8-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vogl T., Tenbrock K., Ludwig S., Leukert N., Ehrhardt C., van Zoelen M. A., Nacken W., Foell D., van der Poll T., Sorg C., and Roth J. (2007) Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat. Med. 13, 1042–1049 10.1038/nm1638 [DOI] [PubMed] [Google Scholar]
- 21. Foell D., and Roth J. (2004) Proinflammatory S100 proteins in arthritis and autoimmune disease. Arthritis Rheum. 50, 3762–3771 10.1002/art.20631 [DOI] [PubMed] [Google Scholar]
- 22. Riva M., Källberg E., Björk P., Hancz D., Vogl T., Roth J., Ivars F., and Leanderson T. (2012) Induction of nuclear factor-κB responses by the S100A9 protein is Toll-like receptor-4-dependent. Immunology 137, 172–182 10.1111/j.1365-2567.2012.03619.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sunahori K., Yamamura M., Yamana J., Takasugi K., Kawashima M., Yamamoto H., Chazin W. J., Nakatani Y., Yui S., and Makino H. (2006) The S100A8/A9 heterodimer amplifies proinflammatory cytokine production by macrophages via activation of nuclear factor κB and p38 mitogen-activated protein kinase in rheumatoid arthritis. Arthritis Res. Ther. 8, R69 10.1186/ar1939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tornovsky-Babeay S., Dadon D., Ziv O., Tzipilevich E., Kadosh T., Schyr-Ben Haroush R., Hija A., Stolovich-Rain M., Furth-Lavi J., Granot Z., Porat S., Philipson L. H., Herold K. C., Bhatti T. R., Stanley C., et al. (2014) Type 2 diabetes and congenital hyperinsulinism cause DNA double-strand breaks and p53 activity in beta cells. Cell Metab. 19, 109–121 10.1016/j.cmet.2013.11.007 [DOI] [PubMed] [Google Scholar]
- 25. Shirakawa J., Togashi Y., Sakamoto E., Kaji M., Tajima K., Orime K., Inoue H., Kubota N., Kadowaki T., and Terauchi Y. (2013) Glucokinase activation ameliorates ER stress-induced apoptosis in pancreatic beta-cells. Diabetes 62, 3448–3458 10.2337/db13-0052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dasu M. R., and Jialal I. (2011) Free fatty acids in the presence of high glucose amplify monocyte inflammation via Toll-like receptors. Am. J. Physiol. Endocrinol. Metab. 300, E145–E154 10.1152/ajpendo.00490.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dasu M. R., Devaraj S., Zhao L., Hwang D. H., and Jialal I. (2008) High glucose induces toll-like receptor expression in human monocytes: mechanism of activation. Diabetes 57, 3090–3098 10.2337/db08-0564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hasnain S. Z., Borg D. J., Harcourt B. E., Tong H., Sheng Y. H., Ng C. P., Das I., Wang R., Chen A. C., Loudovaris T., Kay T. W., Thomas H. E., Whitehead J. P., Forbes J. M., Prins J. B., and McGuckin M. A. (2014) Glycemic control in diabetes is restored by therapeutic manipulation of cytokines that regulate beta cell stress. Nat. Med. 20, 1417–1426 10.1038/nm.3705 [DOI] [PubMed] [Google Scholar]
- 29. Ikemoto M., Matsumoto S., Egawa H., Okitsu T., Iwanaga Y., Umemoto S., Itoh H., Murayama H., and Fujita M. (2007) A case with transient increases in serum S100A8/A9 levels implying acute inflammatory responses after pancreatic islet transplantation. Ann. Clin. Biochem. 44, 570–572 10.1258/000456307782268156 [DOI] [PubMed] [Google Scholar]
- 30. Böni-Schnetzler M., Boller S., Debray S., Bouzakri K., Meier D. T., Prazak R., Kerr-Conte J., Pattou F., Ehses J. A., Schuit F. C., and Donath M. Y. (2009) Free fatty acids induce a proinflammatory response in islets via the abundantly expressed interleukin-1 receptor I. Endocrinology 150, 5218–5229 10.1210/en.2009-0543 [DOI] [PubMed] [Google Scholar]
- 31. Gao Q., Ma L. L., Gao X., Yan W., Williams P., and Yin D. P. (2010) TLR4 mediates early graft failure after intraportal islet transplantation. Am. J. Transplant. 10, 1588–1596 10.1111/j.1600-6143.2010.03151.x [DOI] [PubMed] [Google Scholar]
- 32. Basso D., Greco E., Padoan A., Fogar P., Scorzeto M., Fadi E., Bozzato D., Moz S., Navaglia F., Zambon C. F., Seraglia R., De Carlo E., Valerio A., Reggiani C., Pedrazzoli S., and Plebani M. (2011) Altered intracellular calcium fluxes in pancreatic cancer induced diabetes mellitus: Relevance of the S100A8 N-terminal peptide (NT-S100A8). J. Cell. Physiol. 226, 456–468 10.1002/jcp.22355 [DOI] [PubMed] [Google Scholar]
- 33. Vogl T., Gharibyan A. L., and Morozova-Roche L. A. (2012) Pro-inflammatory S100A8 and S100A9 proteins: self-assembly into multifunctional native and amyloid complexes. Int. J. Mol. Sci. 13, 2893–2917 10.3390/ijms13032893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Manitz M. P., Horst B., Seeliger S., Strey A., Skryabin B. V., Gunzer M., Frings W., Schönlau F., Roth J., Sorg C., and Nacken W. (2003) Loss of S100A9 (MRP14) results in reduced interleukin-8-induced CD11b surface expression, a polarized microfilament system, and diminished responsiveness to chemoattractants in vitro. Mol. Cell. Biol. 23, 1034–1043 10.1128/MCB.23.3.1034-1043.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sekimoto R., Fukuda S., Maeda N., Tsushima Y., Matsuda K., Mori T., Nakatsuji H., Nishizawa H., Kishida K., Kikuta J., Maijima Y., Funahashi T., Ishii M., and Shimomura I. (2015) Visualized macrophage dynamics and significance of S100A8 in obese fat. Proc. Natl. Acad. Sci. U.S.A. 112, E2058–E2066 10.1073/pnas.1409480112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Averill M. M., Kerkhoff C., and Bornfeldt K. E. (2012) S100A8 and S100A9 in cardiovascular biology and disease. Arterioscler. Thromb. Vasc. Biol. 32, 223–229 10.1161/ATVBAHA.111.236927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Perera C., McNeil H. P., and Geczy C. L. (2010) S100 Calgranulins in inflammatory arthritis. Immunol. Cell Biol. 88, 41–49 10.1038/icb.2009.88 [DOI] [PubMed] [Google Scholar]
- 38. Oh Y. S., Lee Y. J., Park K., Choi H. H., Yoo S., and Jun H. S. (2014) Treatment with glucokinase activator, YH-GKA, increases cell proliferation and decreases glucotoxic apoptosis in INS-1 cells. Eur. J. Pharm. Sci. 51, 137–145 10.1016/j.ejps.2013.09.005 [DOI] [PubMed] [Google Scholar]
- 39. Roma L. P., Duprez J., and Jonas J. C. (2015) Glucokinase activation is beneficial or toxic to cultured rat pancreatic islets depending on the prevailing glucose concentration. Am. J. Physiol. Endocrinol. Metab. 309, E632–E639 10.1152/ajpendo.00154.2015 [DOI] [PubMed] [Google Scholar]
- 40. Lysakova-Devine T., Keogh B., Harrington B., Nagpal K., Halle A., Golenbock D. T., Monie T., and Bowie A. G. (2010) Viral inhibitory peptide of TLR4, a peptide derived from vaccinia protein A46, specifically inhibits TLR4 by directly targeting MyD88 adaptor-like and TRIF-related adaptor molecule. J. Immunol. 185, 4261–4271 10.4049/jimmunol.1002013 [DOI] [PubMed] [Google Scholar]
- 41. Shirakawa J., Amo K., Ohminami H., Orime K., Togashi Y., Ito Y., Tajima K., Koganei M., Sasaki H., Takeda E., and Terauchi Y. (2011) Protective effects of dipeptidyl peptidase-4 (DPP-4) inhibitor against increased beta cell apoptosis induced by dietary sucrose and linoleic acid in mice with diabetes. J. Biol. Chem. 286, 25467–25476 10.1074/jbc.M110.217216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhang X., Goncalves R., and Mosser D. M. (2008) The isolation and characterization of murine macrophages. Curr. Protoc. Immunol. Chapter 14, Unit 14.11 10.1002/0471142735.im1401s83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shirakawa J., Fujii H., Ohnuma K., Sato K., Ito Y., Kaji M., Sakamoto E., Koganei M., Sasaki H., Nagashima Y., Amo K., Aoki K., Morimoto C., Takeda E., and Terauchi Y. (2011) Diet-induced adipose tissue inflammation and liver steatosis are prevented by DPP-4 inhibition in diabetic mice. Diabetes 60, 1246–1257 10.2337/db10-1338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shirakawa J., Okuyama T., Yoshida E., Shimizu M., Horigome Y., Tuno T., Hayasaka M., Abe S., Fuse M., Togashi Y., and Terauchi Y. (2014) Effects of the antitumor drug OSI-906, a dual inhibitor of IGF-1 receptor and insulin receptor, on the glycemic control, beta-cell functions, and beta-cell proliferation in male mice. Endocrinology 155, 2102–2111 10.1210/en.2013-2032 [DOI] [PubMed] [Google Scholar]
- 45. Shirakawa J., Fernandez M., Takatani T., El Ouaamari A., Jungtrakoon P., Okawa E. R., Zhang W., Yi P., Doria A., and Kulkarni R. N. (2017) Insulin signaling regulates the FoxM1/PLK1/CENP-A pathway to promote adaptive pancreatic beta cell proliferation. Cell Metab. 25, 868–882.e5 10.1016/j.cmet.2017.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hiratsuka S., Watanabe A., Aburatani H., and Maru Y. (2006) Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat. Cell Biol. 8, 1369–1375 10.1038/ncb1507 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.