

Abstract
Arsenic trioxide (ATO, As2O3) is currently used to treat acute promyelocytic leukemia. However, expanding its use to include high-dose treatment of other cancers is severely hampered by serious side effects on healthy organs. To address these limitations, we loaded ATO onto folate (FA)-labeled human serum albumin (HSA) pretreated with glutathione (GSH) based on the low pH- and GSH-sensitive arsenic-sulfur bond, and we termed the resulting smart nanodrug as FA-HSA-ATO. FA-HSA-ATO could specifically recognize folate receptor-β-positive (FRβ+) chronic myeloid leukemia (CML) cells, resulting in more intracellular accumulation of ATO. Furthermore, the nanodrug could upregulate FRβ expression in CML cancer cells and xenograft tumor model, facilitating even more recruitment and uptake of FRβ-targeting drugs. In vitro and in vivo experiments indicate that the nanodrug significantly alleviates side effects and improves therapeutic efficacy of ATO on CML and xenograft tumor model.
Keywords: arsenic trioxide, human serum albumin, smart nanodrug, chronic myeloid leukemia, folate receptor-β
COMMUNICATION
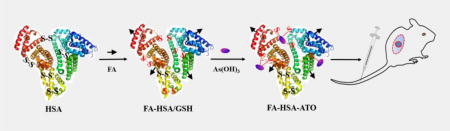
As2O3 (ATO) was loaded onto folate (FA)-labeled human serum albumin (HSA) pretreated with glutathione based on low pH- and GSH-sensitive arsenic-sulfur bond formation to produce smart nanodrug FA-HSA-ATO. The nanodrug could upregulate FRβ expression in CML, facilitating even more recruitment and uptake of FRβ-targeting drugs. In vitro and in vivo experiments indicate the nanodrug significantly improves therapeutic efficacy of ATO in CML treatment.
Arsenic trioxide (ATO, As2O3) is a front-line antineoplastic agent approved by the U.S. Food and Drug Administration (FDA) for treatment of acute promyelocytic leukemia (APL) with a complete remission rate of 83–95%[1]. ATO affects numerous intracellular signal transduction pathways and alters cellular function, resulting in cellular apoptosis, inhibition of angiogenesis and the promotion of differentiation[2]. Thus, with the success of ATO in APL treatment, the use of ATO for treatment of other hematologic cancers and solid tumors has been aggressively investigated. However, compared to APL cells, ATO is known to be poorly uptaken by chronic myeloid leukemia (CML) and solid tumor cells, making it necessary to use higher doses to treat these cancers[3]. Yet, such strategy presents the limitations of toxic side effects and poor biodistribution in the body at high doses[3,4]. To address these limitations, various delivery systems have been conceived to improve the therapeutic index of ATO[5]. Typical examples are ATO-loading liposome systems, in which ATO is encapsulated through the formation of insoluble arsenite compounds with transition metal ions or platinum-based drugs[5b,5c]. Such strategies open new avenues to improve the therapeutic index of ATO. However, the encapsulated ATO shows attenuated cytotoxicity compared to free ATO, resulting from incomplete release from the insoluble arsenic compounds[5b]. Therefore, we contemplated the rational design of a delivery platform that would expand the therapeutic applications of arsenic in CML, but without the limitations noted above.
Human serum albumin (HSA) is the most abundant endogenous protein in plasma. It has been found that HSA has extraordinary binding affinity not only for endogenous solutes in plasma, including metal ions, fatty acids, amino acids and metabolites[6], but also for many exogenous drugs, for example, Paclitaxel (PTX), platinum-based drugs and Chlorin e6[7]. HSA has a long plasma half-life of 19 days and can be preferentially uptaken by tumor cells[8]. In addition, HSA is low in cost, fully biocompatible, and easy to manipulate by genetic engineering[9]. All of these properties make HSA an ideal material for drug delivery. Usually, drug molecules are adsorbed on the hydrophobic domains of HSA and then encapsulated in HSA nanoparticles by desolvation[10]. The resulting drug-HSA complex significantly improves therapeutic efficacy. The most outstanding example is an HSA-PTX nanodrug (trade name: Abraxane), which has been approved by the FDA for the treatment of metastatic breast cancer and non-small cell lung cancer[11]. These successful reports have encouraged us to develop HSA as an effective carrier to expand the applications of ATO.
Inspired by previous reports that the arsenic-sulfur (AsIII-S) bond could be easily formed between arsenic and free sulfhydryl groups[12], we developed a smart nanodrug by conjugating ATO molecules with folate-labeled HSA molecules (FA-HSA, Table S1) through AsIII-S bond formation. The nanodrug, termed FA-HSA-ATO, expands the application of ATO beyond the treatment of APL to other cancers, for example, CML. HSA contains one sulfhydryl group and 17 pairs of disulfide bonds. Thus, FA-HSA provides an efficient carrier to load ATO after reduction with GSH (Figure 1a). The loading of ATO onto the reduced HSA was monitored with 4,4'-dithiodipyridine[13]. As seen in Figure S1, the ratio of free sulfhydryl group to FA-HSA, reduced FA-HSA and FA-HSA-ATO were 0.48, 7.59 and 0.63, respectively, indicating that ATO was successfully loaded on FA-HSA through the formation of AsIII-S bonds. The average loaded amounts of ATO on FA-HSA, as determined by Inductively Coupled Plasma Optical Emission Spectrometry (ICP-OES), were 3.8 ± 0.76 ATO molecules per FA-HSA.
Figure 1.

(a) Cartoon depictions of FA-HSA-ATO synthesis. (b) UV-vis spectra of HSA, HSA-ATO and FA-HSA-ATO. (c) Dynamic light scattering (DLS) of various HSA formulations.
The as-prepared FA-HSA-ATO in Dulbecco's phosphate buffered saline without Ca2+ and Mg2+ (D-PBS, pH 7.4) was a transparent yellow solution with two UV-vis absorption peaks centered at 280 nm and 363 nm (Figure 1b and Figure S2), representing the characteristic absorbances of HSA and FA, respectively. Dynamic light scattering (DLS) analysis showed that the hydrodynamic diameter of FA-HSA-ATO was 43 ± 5.1 nm, larger than that of HSA alone (12 ± 2.6 nm) or HSA-ATO (39 ± 4.7 nm) (Figure 1c), indicating that the formation of the AsIII-S bond increases the size of HSA. This phenomenon of size increase by ATO was also demonstrated by transmission electron microscopy (TEM) (Figure S3). However, the circular dichroism (CD) spectra showed that FA-HSA-ATO retains the secondary structure of HSA (Figure S4).
The AsIII-S bond is susceptible to an acidic or reductive environment[5e,12a]. The release of arsenic from FA-HSA-ATO in a series of buffer solutions with different pH values (pH 7.4, 6.5, 6.0 and 5.5) or in PBS with GSH at different concentrations (0.01, 1, 5 or 10 mM) was investigated by ICP-OES. It could be seen that ATO release from FA-HSA-ATO was slow in PBS, even in RPMI 1640 cell medium with 10% FBS or human serum at pH 7.4, with a release rate less than of 15% after 24-h incubation (Figure S5 and S6). However, when the pH of the buffer decreased, or the concentration of GSH increased, the release of ATO from the nanodrug became rapid and gradually complete. For example, when nanodrug was incubated with buffer at pH 6.0 or containing 5 mM GSH, the release half-life of ATO was ~3 h and ~2 h, respectively. Therefore, it is expected that FA-HSA-ATO will unload its cargo in the endo/lysosomes and cytosol after internalization. It was also observed that the release of arsenic was accompanied by a size decrease of FA-HSA-ATO (Figure S5c, S5d, and Table S2).
To demonstrate the in vitro folate receptor β (FRβ)-targeting ability of FA-HSA-ATO, FITC-labeled FA-HSA-ATO (FA/FITC-HSA-ATO) was prepared (Table S1). Then, its binding on CML FRβ-positive (FRβ+) K562 cells was analyzed by confocal laser scanning microscopy (CLSM) and flow cytometry, and compared to FITC-HSA-ATO. As shown in Figure 2 and Figure S7, K562 cells incubated with FA/FITC-HSA-ATO presented more fluorescence signals than cells incubated with FITC-HSA-ATO. When K562 cells were incubated with FA/FITC-HSA-ATO in the presence of 2 mM of FA, the excess FA competitively blocked the binding of the nanodrug to K562 cells, resulting in a significant decrease in the fluorescence signal (Figure 2). These results suggest that it is the recognition between FA and FRβ that mediates the binding of FA-HSA-ATO to K562 cells.
Figure 2.

Flow cytometric analysis of FRβ-targeting ability of FA-HSA-ATO.
It has been reported that ATO can prompt the differentiation of primary leukemia cells from patients with CML[2,14] and FRβ is the terminal marker of differentiation for neutrophils during hematopoiesis[15]. Thus, the expression of FRβ in K562 cells with or without treatment of ATO and FA-HSA-ATO was investigated by Western Blotting. As shown in Figure 3a, K562 cells treated with FA-HSA-ATO or ATO presented 2-fold higher expression of FRβ than K562 cells. The induced expression of FRβ resulted in more binding of FITC-PEG-FA on K562 cells, which could be completely blocked by excess FA (Figure 3b). Thus, these results prove that FA-HSA-ATO can upregulate the expression of FRβ in K562 cells. The upregulation of FRβ expression in K562 cells strongly depends on drug dosage and induction time, as shown in Figure 3c and 3d. More importantly, the level of FRβ expression in K562 xenograft tumor could also be upregulated by FA-HSA-ATO. As shown in Figure 3e, tumor tissues from mice treated with FA-HSA-ATO presented higher green fluorescence signals than those from untreated mice after staining with FITC-PEG-FA. This phenomenon could also be observed in AML U937 cells and HL-60 cells, but not in human lymphoblastic leukemia Ramos cells and CCRF-CEM cells (Figure S8). Therefore, the ability of FA-HSA-ATO to induce the upregulation of FRβ expression should afford the nanodrug with self-amplified FRβ-targeting ability for CML and, hence, improve the therapeutic index of ATO.
Figure 3.

Upregulation of FR expression in primary mononuclear CML K562 cells and K562 mouse xenograft tumor tissue, as induced by FA-HSA-ATO. (a) FR-β expression detected in K562 cells treated with 1 µM of FA-HSA-ATO for 72 h by Western blot using anti-FR-β antibody. (b) Binding analysis of FITC-PEG-FA on K562 cells with or without drug induction. (c) and (d) Binding analysis of FITC-PEG-FA on K562 cells pretreated with FA-HSA-ATO at different concentrations or different times (2 µM). (e) Effect of FA-HSA-ATO on FRβ expression in K562 xenograft tumor 10 days post-treatment. The tissue section was stained with DAPI (blue) and FRβ-recognizing FITC-PEG-FA (green).
The amounts of intracellular arsenic, extracellular free arsenic and arsenic in extracellular nanodrug were quantitatively assayed by ICP-OES after K562 cells, six other FRβ+ cell lines, and two FR-negative (FRβ−) cell lines were treated with three ATO formulations for 6 hours and 24 hours, respectively. Table S3 shows that FA-HSA-ATO can cause more arsenic accumulation in the 7 FRβ+ cancer cell lines and less free arsenic in cell medium than HSA-ATO and ATO after 6- or 24-h incubation. It also reveals that the intracellular arsenic accumulation strongly depended on the incubation time of drugs. These data revealed that FA-HSA-ATO can improve the intracellular accumulation of arsenic, while reducing the extracellular residue.
The enhanced intracellular accumulation caused more cytotoxicity. As shown in Table S4 and Figure S9, FA-HSA-ATO exerted more cytotoxicity against the 7 FRβ+ cancer cell lines than HSA-ATO and ATO, but had cytotoxicity similar to that of HAS-ATO on the 2 FRβ− cancer cell lines. These results clearly demonstrate that the active targeting ability of FA-HSA-ATO could enhance cytotoxicity of arsenic on targeted cells. When FRβ on K562 cells were occupied by excess FA, the cytotoxicity of FA-HSA-ATO on K562 cells decreased distinctly. The competition experiment pinpoints the FRβ-targeting ability of FA-HSA-ATO as the decisive factor in improving the cytotoxicity of ATO in FRβ+ cancer cell lines. Meanwhile, FA-HSA-ATO showed better cytotoxicity than folate-targeted liposomal arsenic f-Lip(Ni, As)[5c] in K562 cells (Figure S10).
It has been reported that FRβ is overexpressed in CML and acute myelogenous leukemia (AML), but not in ALL and normal mononuclear cells[15]. Therefore, we further evaluated the therapeutic efficacy of FA-HSA-ATO on leukemia mononuclear cells from peripheral blood of 16 CML, 15 AML and 8 acute lymphoblastic leukemia (ALL) patients, as well as 9 healthy donors. As seen in Figure S11, FA-HSA-ATO exhibited more cytotoxicity than ATO on the primary mononuclear cells from CML and AML patients, but showed cytotoxicity similar to that of ATO on cells from ALL patients. It was noteworthy that FA-HSA-ATO also caused less cytotoxicity on the primary mononuclear cells from healthy donors compared to free ATO. These results strongly support that FRβ-targeting ability of the nanodrug enhances the cytotoxicity of ATO on targeted cells and alleviates its side effects on normal cells.
In their study on all-trans retinoic acid, Pan et al. stated that the upregulated expression of FRβ in AML induced by all-trans retinoic acid could improve the therapeutic outcome of FRβ-targeted liposomal doxorubicin[16]. Similarly, to study whether FA-HSA-ATO can improve the cytotoxicity of other FRβ-targeting drugs, the cytotoxicity of FA-HSA-PTX was evaluated on K562 cells and leukemia mononuclear cells from CML patients treated with or without FA-HSA-ATO for 72 h. As shown in Figure S12a, FA-HSA-PTX exerted more cytotoxicity against K562 cells pretreated with a low concentration of FA-HSA-ATO (1 µM) compared to K562 cells without treatment, with an IC50 of 1.61 µM vs. 5.96 µM (calculation of IC50 was based on concentration of arsenic). After treatment with the low concentration of FA-HSA-ATO (1 µM), the leukemia mononuclear cells from 8 CML patients were also more sensitive toward FA-HSA-PTX (Figure S12b), with an apoptosis rate of 47.12% vs. 22.94% for these cells without treatment (P < 0.0001; t tests). Collectively, these data indicate that FA-HSA-ATO can upregulate FR expression, recruit more FR-targeting drugs to cancer cells, and improve the therapeutic efficacy of itself and a second FRβ-targeting drug.
To test the in vivo tumor-targeting capability of FA-HSA-ATO, mice bearing the K562 tumor were imaged after i.v. injection of FA/FITC-HSA-ATO or FITC-HSA-ATO, respectively. As shown in Figure 4a, active FRβ-targeting of FA/FITC-HSA-ATO caused more accumulation of nanodrug than passive targeting of HSA-ATO nanodrug. The in vivo targeting ability of FA-HSA-ATO was further tested by investigating the distribution of arsenic in the main organs of mice bearing K562 tumors at 8 h after i.v. administration of FA-HSA-ATO compared with that in mice bearing K562 tumors using the same dose of free ATO and HSA-ATO. As shown in Figure 4b, the intratumoral arsenic concentration of the group treated with FA-HSA-ATO showed about a 6-fold and 1.5-fold increase compared to that of groups treated with free ATO and HSA-ATO, respectively. Pharmacokinetics data showed that the blood circulation half-life of FA-HSA-ATO (t1/2α = 0.85 h and t1/2β = 9.71 h) and HSA-ATO (t1/2α = 0.82 h and t1/2β = 6.54 h) was significantly prolonged compared to free ATO (t1/2α = 0.48 h and t1/2β = 4.27 h) (Figure 4c). Thus, the dramatically enhanced arsenic accumulation of the FA-HSA-ATO in tumor may be attributed to its tumor-targeting ability and the prolonged circulation time. More importantly, FA-HSA-ATO arsenic accumulation was noticeably reduced in heart, liver, spleen, lung and kidney compared with tumor. These results suggest that FA-HSA-ATO can deliver the loaded ATO to tumors in vivo, while reducing the accumulation of ATO in healthy organs.
Figure 4.

In vivo targeting ability, blood circulation and tissue biodistribution of FA-HSA-ATO. (a) In vivo targeting ability of FITC-HSA-ATO injected i.v. in K562 tumor-bearing mouse. (b) Biodistribution of ATO, HSA-ATO and FA-HSA-ATO in main mouse organs, as determined by ICP-OES. Results are expressed as a percentage of the total injected dose per tissue mass (%ID/g). Data are shown as mean ± SD (n = 3). (c) Blood circulation curves of ATO, HSA-ATO and FA-HSA-ATO in mice, as determined by ICP-OES. Data are shown as mean ± SD (n = 3).
The in vivo anticancer therapeutic efficacy of FA-HSA-ATO was evaluated in a murine K562 xenograft model. The i.p. administration of ATO, HSA-ATO and FA-HSA-ATO at 5 mg/kg/day ATO or ATO equivalents, which was chosen based on a preliminary experiment (Figure S13), significantly reduced tumor growth (Figure 5a, Figure S14). The tissue sections from each group after treatment were then stained with H&E. It was found that the three ATO formulations significantly caused tumor cell apoptosis, with signs of cellular nucleus enlargement, large vesicular nuclei, or concentrated nuclei (Figure 5b). The tumors treated with three arsenic formulations also presented lower expression of proliferation marker Ki67 compared to those treated with HSA (Figure S15). Free ATO clearly inhibited tumor growth, resulting in a median survival time of 28 d in the group. However, a significant drop of body weight was observed at 24 d (Figure 5c, 5d). These results indicate that free ATO causes severe side effects in mice. The median survival times of mice treated with HSA-ATO increased to 36 d, whereas treatment with FA-HSA-ATO resulted in 80% survival of mice with a survival time of 45 d. Thus, FA-HSA-ATO was more effective than either free ATO (P=0.0005; log-rank test) or HSA-ATO (P=0.0059; log-rank test) in prolonging animal survival. Moreover, no significant drop of body weight was observed in mice treated with FA-HSA-ATO (Figure 5c, 5d), and no obvious damage to major organs was observed in the H&E-stained slices of main organs from these mice (Figure S16). Therefore, our results demonstrate that FA-HSA-ATO improves the therapeutic index and alleviates side effects during treatment of CML.
Figure 5.

Antitumor activity and toxicity of FA-HSA-ATO. K562 tumor cells were implanted s.c. into mice on day 0. (a) Tumor growth curves for mouse groups are indicated. (b) H&E staining for pathological changes in tumor sections from each group to determine the effectiveness of FA-HSA-ATO. (c) Animal survival curves in different groups. Asterisks indicate P = 0.0005 and 0.0059 for FA-HSA-ATO compared with ATO and HSA-ATO, respectively (log-rank test). (d) The effect of different treatments on mouse body weight (bars represent means ± SD, n = 5).
In summary, we have shown a smart FA-HSA-ATO nanodrug that expands the use of ATO from APL treatment to CML treatment. The nanodrug was simply prepared by loading ATO on folate-modified HSA via low pH- and GSH-sensitive AsIII-S bonds. In vitro and in vivo experiments proved that FA-HSA-ATO with self-amplified FRβ-targeting ability could significantly enhance intracellular arsenic accumulation and prolong the circulation time of ATO, resulting in improved therapeutic index and alleviation of side effects in the treatment of CML K562 tumors. Based on the appealing performance demonstrated in the present study, the overexpression of FRβ in many cancers and their metastatic stroma (e.g., liver cancer and lung cancer)[17], as well as the nature of the carrier moiety and target moiety, FA-HSA-ATO is expected to be a promising candidate to extend the application of ATO from APL to CML, as well as other cancers.
Supplementary Material
Acknowledgments
This work is supported by NSFC grants (21521063, 21327009, 21405041, and 61527806), Science and Technology Project of Hunan Province (2015RS4019) and China Postdoctoral Science Foundation Funded Project (2016T90751, 2015M570679 and 2013T60828), and by the US National Institutes of Health (GM079359 and CA133086).
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Contributor Information
Dr. Yongbo Peng, Molecular Science and Biomedicine Laboratory, State Key Laboratory for Chemo/Bio-Sensing and Chemometrics, College of Chemistry and Chemical Engineering, College of Biology and Collaborative Research Center of Molecular Engineering for Theranostics, Hunan University, Changsha 410082 (China)
Prof. Dr. Zilong Zhao, Molecular Science and Biomedicine Laboratory, State Key Laboratory for Chemo/Bio-Sensing and Chemometrics, College of Chemistry and Chemical Engineering, College of Biology and Collaborative Research Center of Molecular Engineering for Theranostics, Hunan University, Changsha 410082 (China)
Teng Liu, Molecular Science and Biomedicine Laboratory, State Key Laboratory for Chemo/Bio-Sensing and Chemometrics, College of Chemistry and Chemical Engineering, College of Biology and Collaborative Research Center of Molecular Engineering for Theranostics, Hunan University, Changsha 410082 (China); Department of Infectious Diseases, Xiangya Hospital, Central South University, Changsha, 410008 (China).
Prof. Xiong Li, Department of Infectious Diseases, Xiangya Hospital, Central South University, Changsha, 410008 (China)
Dr. Xiaoxiao Hu, Molecular Science and Biomedicine Laboratory, State Key Laboratory for Chemo/Bio-Sensing and Chemometrics, College of Chemistry and Chemical Engineering, College of Biology and Collaborative Research Center of Molecular Engineering for Theranostics, Hunan University, Changsha 410082 (China)
Dr. Xiaoping Wei, Center for Clinical Molecular Medicine, Ministry of Education Key Laboratory of Child Development and Dis-orders, Children's Hospital of Chongqing Medical University, Chongqing 400014 (China)
Prof. Dr. Xiaobing Zhang, Molecular Science and Biomedicine Laboratory, State Key Laboratory for Chemo/Bio-Sensing and Chemometrics, College of Chemistry and Chemical Engineering, College of Biology and Collaborative Research Center of Molecular Engineering for Theranostics, Hunan University, Changsha 410082 (China)
Prof. Dr. Weihong Tan, Molecular Science and Biomedicine Laboratory, State Key Laboratory for Chemo/Bio-Sensing and Chemometrics, College of Chemistry and Chemical Engineering, College of Biology and Collaborative Research Center of Molecular Engineering for Theranostics, Hunan University, Changsha 410082 (China) Department of Chemistry, Department of Physiology and Functional Genomics, Center for Research at Bio/Nano Interface, Shands Cancer Center, University of Florida Genetics Institute and McKnight Brain Institute, University of Florida, Gainesville, FL 32611-7200 (USA).
References
- 1.a) Zhang XW, Yan XJ, Zhou ZR, Yang FF, Wu ZY, Sun HB, Liang WX, Song AX, Lalle-mand-Breitenbach V, Jeanne M, Zhang QY, Yang HY, Huang QH, Zhou GB, Tong JH, Zhang Y, Wu JH, Hu HY, de Thé H, Chen SJ, Chen Z. Science. 2010;328:240–243. doi: 10.1126/science.1183424. [DOI] [PubMed] [Google Scholar]; b) Mi JQ, Chen SJ, Zhou GB, Yan XJ, Chen Z. J. Intern. Med. 2015;278:627–642. doi: 10.1111/joim.12376. [DOI] [PubMed] [Google Scholar]
- 2.Miller WH, Jr, Schipper HM, Lee JS, Singer J, Waxman S. Cancer Res. 2002;62:3893–3903. [PubMed] [Google Scholar]
- 3.a) Sahu GR, Jena RK. Am. J. Hematol. 2005;78:113–116. doi: 10.1002/ajh.20235. [DOI] [PubMed] [Google Scholar]; b) Murgo AJ. Oncologist. 2001;6:22–28. doi: 10.1634/theoncologist.6-suppl_2-22. [DOI] [PubMed] [Google Scholar]
- 4.Chen GQ, Zhu J, Shi XG, Ni JH, Zhong HJ, Si GY, Jin XL, Tang W, Li XS, Xong SM, Shen ZX, Sun GL, Ma J, Zhang P, Zhang TD, Gazin C, Naoe T, Chen SJ, Wang ZY, Chen Z. Blood. 1996;88:1052–1061. [PubMed] [Google Scholar]
- 5.a) Chen H, MacDonald RC, Li S, Krett NL, Rosen ST, O'Halloran TV. J. Am. Chem. Soc. 2006;128:13348–13349. doi: 10.1021/ja064864h. [DOI] [PubMed] [Google Scholar]; b) Chen H, Pazicni S, Krett NL, Ahn RW, Penner-Hahn JE, Rosen ST, O'Halloran TV. Angew. Chem. Int. Ed. Engl. 2009;48:9295–9299. doi: 10.1002/anie.200903655. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Chen H, Ahn R, Van den Bossche J, Thompson DH, O'Halloran TV. Mol. Cancer Ther. 2009;8:1955–1963. doi: 10.1158/1535-7163.MCT-09-0045. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Zhao Z, Wang X, Zhang Z, Zhang H, Liu H, Zhu X, Li H, Chi X, Yin Z, Gao J. ACS Nano. 2015;9:2749–2759. doi: 10.1021/nn506640h. [DOI] [PubMed] [Google Scholar]; e) Zhang Q, Vakili MR, Li XF, Lavasanifar A, Le XC. Biomaterials. 2014;35:7088–70100. doi: 10.1016/j.biomaterials.2014.04.072. [DOI] [PubMed] [Google Scholar]; f) Yang Z, Yang M, Peng J. Drug Dev. Ind. Pharm. 2008;34:834–839. doi: 10.1080/03639040801926733. [DOI] [PubMed] [Google Scholar]
- 6.a) Sugio S, Kashima A, Mochizuki S, Noda M, Kobayashi K. Protein Eng. 1999;12:439–446. doi: 10.1093/protein/12.6.439. [DOI] [PubMed] [Google Scholar]; b) Li R, Zheng K, Hu P, Chen Z, Zhou S, Chen J, Yuan C, Chen S, Zheng W, Ma E, Zhang F, Xue J, Chen X, Huang M. Theranostics. 2014;4:642–659. doi: 10.7150/thno.8527. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Petitpas I, Petersen CE, Ha CE, Bhattacharya AA, Zunszain PA, Ghuman J, Bhagavan NV, Curry S. Proc. Natl. Acad. Sci. USA. 2003;100:6440–6445. doi: 10.1073/pnas.1137188100. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Jurkowski W, Porebski G, Obtułowicz K, Roterman I. Curr. Drug Metab. 2009;10:448–458. doi: 10.2174/138920009788897984. [DOI] [PubMed] [Google Scholar]
- 7.a) Fanali G, di Masi A, Trezza V, Marino M, Fasano M, Ascenzi P. Mol. Aspects Med. 2012;33:209–290. doi: 10.1016/j.mam.2011.12.002. [DOI] [PubMed] [Google Scholar]; b) Liu Z, Chen X. Chem. Soc. Rev. 2016;45:1432–1456. doi: 10.1039/c5cs00158g. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zheng YR, Suntharalingam K, Johnstone TC, Yoo H, Lin W, Brooks JG, Lippard SJ. J. Am. Chem. Soc. 2014;136:8790–8798. doi: 10.1021/ja5038269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kratz F. J. Control Release. 2008;132:171–183. doi: 10.1016/j.jconrel.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 9.Wang W, Huang Y, Zhao S, Shao T, Cheng Y. Chem. Commun (Camb) 2013;49:2234–2236. doi: 10.1039/c3cc38397k. [DOI] [PubMed] [Google Scholar]
- 10.Duncan R. Nat. Rev. Drug Discov. 2003;2:347–360. doi: 10.1038/nrd1088. [DOI] [PubMed] [Google Scholar]
- 11.Chen Q, Liang C, Wang C, Liu Z. Adv. Mater. 2015;27:903–910. doi: 10.1002/adma.201404308. [DOI] [PubMed] [Google Scholar]
- 12.a) Shen S, Li XF, Cullen WR, Weinfeld M, Le XC. Chem. Rev. 2013;113:7769–7792. doi: 10.1021/cr300015c. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Cline DJ, Thorpe C, Schneider JP. J. Am. Chem. Soc. 2003;125:2923–2929. doi: 10.1021/ja0282644. [DOI] [PubMed] [Google Scholar]; c) Touw DS, Nordman CE, Stuckey JA, Pecoraro VL. Proc. Natl. Acad. Sci. USA. 2007;104:11969–11974. doi: 10.1073/pnas.0701979104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Riener CK, Kada G, Gruber HJ. Anal. Bioanal. Chem. 2002;373:266–276. doi: 10.1007/s00216-002-1347-2. [DOI] [PubMed] [Google Scholar]
- 14.a) Zhang Y, Wang S, Chen C, Wu X, Zhang Q, Jiang F. Bioinorg. Chem. Appl. 2015;2015:751013. doi: 10.1155/2015/751013. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wang W, Lv FF, Du Y, Li N, Chen Y, Chen L. Cancer Cell Int. 2015;15:10. doi: 10.1186/s12935-015-0158-4. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Lyu FF, Du Y, Li NN, Wang W. J. Exp. Hematol. 2015;23:106–110. doi: 10.7534/j.issn.1009-2137.2015.01.020. [DOI] [PubMed] [Google Scholar]
- 15.a) Ross JF, Wang H, Behm FG, Mathew P, Wu M, Booth R, Ratnam M. Cancer. 1999;85:348–357. doi: 10.1002/(sici)1097-0142(19990115)85:2<348::aid-cncr12>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]; b) Wang H, Zheng X, Behm FG, Ratnam M. Blood. 2000;96:3529–3536. [PubMed] [Google Scholar]
- 16.Pan XQ, Zheng X, Shi G, Wang H, Ratnam M, Lee RJ. Blood. 2002;100:594–602. doi: 10.1182/blood.v100.2.594. [DOI] [PubMed] [Google Scholar]
- 17.Shen J, Putt KS, Visscher DW, Murphy L, Cohen C, Singhal S, Sandusky G, Feng Y, Dimitrov DS, Low PS. Oncotarget. 2015;6:14700–14709. doi: 10.18632/oncotarget.3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.