

Summary
Objective
Pathogenic SLC6A1 variants were recently described in patients with myoclonic atonic epilepsy (MAE) and intellectual disability (ID). We set out to define the phenotypic spectrum in a larger cohort of SCL6A1-mutated patients.
Methods
We collected 24 SLC6A1 probands and 6 affected family members. Four previously published cases were included for further electroclinical description. In total, we reviewed the electroclinical data of 34 subjects.
Results
Cognitive development was impaired in 33/34 (97%) subjects; 28/34 had mild to moderate ID, with language impairment being the most common feature. Epilepsy was diagnosed in 31/34 cases with mean onset at 3.7 years. Cognitive assessment before epilepsy onset was available in 24/31 subjects and was normal in 25% (6/24), and consistent with mild ID in 46% (11/24) or moderate ID in 17% (4/24). Two patients had speech delay only, and 1 had severe ID. After epilepsy onset, cognition deteriorated in 46% (11/24) of cases. The most common seizure types were absence, myoclonic, and atonic seizures. Sixteen cases fulfilled the diagnostic criteria for MAE. Seven further patients had different forms of generalized epilepsy and 2 had focal epilepsy. Twenty of 31 patients became seizure-free, with valproic acid being the most effective drug. There was no clear-cut correlation between seizure control and cognitive outcome. Electroencephalography (EEG) findings were available in 27/31 patients showing irregular bursts of diffuse 2.5–3.5 Hz spikes/polyspikes-and-slow waves in 25/31. Two patients developed an EEG pattern resembling electrical status epilepticus during sleep. Ataxia was observed in 7/34 cases. We describe 7 truncating and 18 missense variants, including 4 recurrent variants (Gly232Val, Ala288Val, Val342Met, and Gly362Arg).
Significance
Most patients carrying pathogenic SLC6A1 variants have an MAE phenotype with language delay and mild/moderate ID before epilepsy onset. However, ID alone or associated with focal epilepsy can also be observed.
Keywords: epilepsy, epilepsy genetics, MAE, SLC6A1
1 | INTRODUCTION
The voltage-dependent γ-aminobutyric acid (GABA) transporter 1 (GAT-1), encoded by SLC6A1, is one of the major GABA transporters of the human central nervous system.1 GAT-1 is expressed primarily in the nerve terminals of GABAergic interneurons but can also be found in astrocytes, and has been shown to be crucial for the reuptake of GABA from the synapses and for its clearing from the extracellular space.2 In addition to the neocortex, GAT-1 is found in abundance in the cerebellum.3 GAT-1 knockout mice exhibit spontaneous spike-wave discharges (SWDs) and absence seizures.4
In 2014, Dikow et al.,5 described 3 patients with intellectual disability (ID), epilepsy/electroencephalography (EEG) abnormalities, ataxia and stereotypic hand movements, and a microdeletion at 3p25.3, including the 2 GABA transporter genes, SLC6A1 and SLC6A11. Recently, de novo SLC6A1 variants discovered by large exome sequencing studies were reported in 3 patients with ID, myoclonic atonic (MA) seizures, delayed speech and delayed walking, autism spectrum disorders, and absence seizures, as well as in a patient with epileptic encephalopathy.6–8 In 2015, we reported de novo inactivating variants in SLC6A1 in up to 4% of patients with myoclonic atonic epilepsy (MAE), suggesting that pathogenic SLC6A1 variants might be specific for MAE.9 MAE is a syndrome characterized by the presence of myoclonic–atonic seizures, usually in an otherwise normal child, which may typically develop cognitive impairment after seizure onset (reviewed in Guerrini and Aicardi).10 Extensive searches in other epileptic encephalopathy cohorts, such as epileptic spasms or Lennox-Gastaut syndrome, failed to detect pathogenic variants in this gene.
Here we aim to better characterize the clinical presentation and electroencephalographic features associated with pathogenic SLC6A1 variants.
2 | MATERIAL AND METHODS
Patients with presumed pathogenic variants in SLC6A1 were collected through an international collaboration between epilepsy genetics centers worldwide. The cohorts included for screening consisted primarily of patients with ID or childhood epilepsies including childhood encephalopathies, without a previously identified genetic cause. Probands and families, when available, underwent clinical, neurophysiological, and genetic examinations. All the available EEG data were collected and reviewed by a single epileptologist (EG). Seizures were classified according to the International League Against Epilepsy (ILAE) classification.11 Atypical absences were defined as absence seizures with additional clinical features (eg, myoclonic jerks) and with an EEG pattern of diffuse spike-wave <3 c/s with a slower onset and offset compared to typical absence seizures.12 Cognitive development was based mainly on the assessment of the psychomotor development by the referring physicians; only in a few cases was a formal neuropsychological testing performed.
For the diagnosis of MAE, we adopted the following diagnostic criteria: (1) usually normal development before epilepsy onset and absence of brain structural abnormalities; (2) onset of myoclonic, myoclonic–atonic, or atonic seizures between 7 months and 6 years of age; (3) EEG: 2–3 Hz generalized spike/polyspikes-and-slow waves; (4) diagnoses of benign myoclonic epilepsy of infancy (BMEI), severe myoclonic epilepsy of infancy (SMEI), and Lennox-Gastaut syndrome (LGS) have been excluded. (https://www.epilepsydiagnosis.org/syndrome/epilepsy-myoclonic-atonic-overview.html).
All probands and/or their parents or legal guardians signed an informed consent form. The study was approved by the local ethics committees.
Variants were classified as pathogenic if they were (1) nonsynonymous, splice-site altering, or truncating changes; (2) predicted damaging by one or more prediction software programs (PolyPhen-2 [http://genetics.bwh.harvard.edu/pph2/], SIFT [http://sift.jcvi.org], and MutationTaster [http://www.mutationtaster.org]); (3) not present in >60 000 of nonepileptic controls in the ExAC browser (the exome aggregation consortium browser, see Web Resources); and (4) were either de novo changes, inherited from an unaffected mosaic parent, or inherited from an affected parent. We used Sanger sequencing to confirm all variants and to perform segregation analysis.
3 | RESULTS
We identified 30 unpublished patients with a pathogenic SCL6A1 variant (24 probands and 6 affected family members). Moreover, we included additional clinical information of 4 previously published cases. One (Patient 25) had only briefly been described previously and the 3 remaining cases were included for electroencephalographic characterization (Patients 5, 28, and 34) (Table 1). In total, we studied 34 SLC6A1-positive patients. Gender ratio was 19 female to 15 male patients.
TABLE 1.
Clinical characteristics of SLC6A1 variant carriers
| Patient no. | Gender/ age at inclusion |
Family history |
Cognition before epilepsy onset |
Age at epilepsy onset |
Seizure type | EEG | Epilepsy syn- drome at diagnosis |
Cognition after seizure onset |
Behavioral problems |
Neurological findings |
Effective AED | Mutation |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F/6 y | None | NA | 5 mo | Atypical absences atonic, eyelid myoclonia | BG slowing bilat. occipital +/− generalized epileptic discharges | EOAE (atypical form) | Moderate ID | NA | Hypotonia | Sz free on VPA + CLZ | c.104dupA p.Lys36GluFsTer171 de novo |
| 2 | M/13 y | Father has spastic paraparesis (SP) | Mild ID | 4 y | Absences (up to 100/day) | Generalized atypical SW | Unclassified generalized | Mild ID | None | Mild walking difficulties related to the SP inherited from his father | VPA (partially effective), sz free with VPA + LTG | c.223G>A p.Gly75Arg de novo |
| 3 | F/17 y | None | Speech delay | 2 y | Myoclonic atonic, myoclonic, absences, nonconvulsive status | Generalized epileptiform activity | MAE | Mild to moderate ID | None | Normal | Sz free | c.419A>G p.Tyr140Cys de novo |
| 4 | F/28 mo | None | — | — | None | NA (referred as normal) | — | Mild ID | Autism, irritability | Mild hypotonia, ataxia, chorea | — | c.434C>T p.Ser145Phe de novo |
| 5 Prev.publ. in Carvill et al.9 | F/12 y | None | Mild ID | 3 y, 2 mo | Absences, myoclonic atonic | BG slowing generalized SW (3 y, 6 mo) –> normal (4 y) | MAE- | Mild ID | Mild autistic traits | Normal | ESM, CBZ (partially effective), sz free with VPA | c.578G>A p.Thp193Ter de novo |
| 6 | M/10 y | None | Mild ID | 2 y | Atypical absences (at onset) | Centrotemporal spikes (at evolution) Extreme activation during NREM sleep (7 y) |
MAE evolving to aBECTS with an ESES-like pattem | Mild ID | None | Normal | NA | c.695G>T, p.Gly232Val de novo |
| 7 Family 1, proband | M/17 y | Yes | NA | 1 y | Absences, atonic | BG slowing Generalized SW (3 Hz) |
MAE | Moderate ID | None | Mild ataxia | VPA, ESM, LTG (partially effective) | c.695G>T p.Gly232Val maternal |
| 8 Family 1, brother | F/10 y | Yes | Normal | 13 mo | Absences, atonic | BG slowing Generalized SW/poly-SW |
MAE | Mild ID | None | Normal | Sz free with ESM | c.695G>T p.Gly232Val maternal |
| 9 Family 1, mother | F/NA | NA | — | — | None | NA | — | Learning disabilities | None | NA | — | c.695G>T p.Gly232Val |
| 10 | M/7 y | None | Speech delay | 18 mo | Atypical absences | Occipital notched rhythmic delta SW (2–2.5 Hz) |
Unclassified generalized | Mild ID | ADHD, irritability | Normal | TPM, VPA (partially effective) | c.809T>C p.Phe270Ser de novo |
| 11 | M/8 y | None | Mild ID | 22 mo | Absences, atonic, myoclonic | generalized SW (3 Hz) | MAE | Mild ID | None | Normal | Sz free with VPA, LTG | C.863C>T p.Ala288Val de novo |
| 12 | F/7 y | Psychiatric disease | NA | 5 y | Focal (at onset), absences (at evolution) | Initially centrotemporal spikes → generalized SW | Initially BECTs-like → unclassified generalized | Moderate to severe ID | Autistic features, aggressive behaviors, stereotypies | Normal | LTG (partially effective) | C.863C>T p.Ala288Val unknown |
| 13 | M/12 y | None | Mild ID | 18 mo | Atypical absences atonic, Myoclonic | BG slowing Generalized SW |
MAE | Moderate ID | Attention deficit | Mild ataxia | Sz free with LTG + ESM | c.881_883del p.Phe294del de novo |
| 14 | F/11 y | None | Mild ID | 5.5 y | Falls (seizure type not specified) | BG slowing Generalized slow waves and SW |
MAE | Mild ID | Hyperkinetic, aggressive behaviors, excess, smiling | Normal | Sz free with VPA | С.987C>А p.Cys329Ter de novo |
| 15 Family 2, proband | M/19 y | Yes | Normal | 2 y | Atypical absences Atonic, GTCS | BG slowing Generalized SW Multifocal spikes |
MAE | Severe ID | Attention deficit | Tremor | VPA + ESM (partially effective) | c.1024G>A p.Val342Met paternal |
| 16 Family 2, brother | M/12 y | Yes | Mild ID | 15 mo | Atypical absences, atonic, myoclonic atonic | BG slowing Generalized SW Multifocal spikes |
MAE | Moderate ID | Autistic features, aggressive behaviors | Mild hypotonia | Sz free with ESM+ZSM | c.1024G>A p.Val342Met paternal |
| 17 Family 2, brother | M/28 y | Yes- | Normal | childhood | Absences, GTCS | Generalized SW (3 Hz) | CAE | Normal | NA | Normal | VPA (partially effective) | c,1024G>A p.Val342Met paternal |
| 18 Family 2, sister | F/NA | Yes | — | — | None | NA | — | Mild ID | None | Normal | — | c.1024G>A p.Val342Met paternal |
| 19 Family 2, father | M/NA | None | NA | 3 y | Absences | NA | Unclassified generalized | Learning disability | NA | NA | Treated with ESM till age of 17 y, now sz free | c,1024G>A p.Val342Met de novo |
| 20 | M/8 y | None | Moderate ID | 5 y | Atypical absences | Generalized SW (3 Hz) | Unclassified generalized | Moderate ID | Rigidness, autism, stereotypies | NA | Sz free with VPA | c.1024G>A p.Val342Met de novo |
| 21 | F/5 y | None | Moderate ID | 2 y | Absences, atonic, myoclonic | Generalized SW and poly-SW (2.5–3.5 Hz) | MAE | Moderate ID | Mood swings, ADHD | Verbal dyspraxia, weak fine motor skills | NA | c,1024G>A p.Val342Met de novo |
| 22 | F/10 | Autism, mother’s side | Mild ID | 13 mo | Eyelid myoclonia, absences | GSW and 3 Hz runs with occipital spikes; high voltage notched delta esp occipital. | Eyelid myoclonia with absences | Moderate ID | Autism spectrum | Ataxia | Sz free with VPA | c.l024G>A p.Val342Met de novo |
| 23 | F/9 y | None | Moderate ID | 11 mo | Atypical absences, atonic | Generalized delta activity and SW SW left occipital Spikes/SW left centrotemporal |
MAE | Mild ID | Hand stereotypies | Microcephaly Unsteady gait | Sz free with LEV + LTG | C.1070C>T p.Ala357Val de novo |
| 24 | F/21 y | NA | Mild ID | 7 y | Focal, GTCS | Spikes left frontotemporal Generalized SW and generalized 3–4 Hz activity on HV |
TLE | Moderate ID | None | NA | NA | c.1084g>a p.Gly362Arg unknown |
| 25 Prev.publ. in Hal vors en et al.8 | M/NA | None | NA | 1 y | Atypical absences, Atonic, GTCS | Mild BG slowing 2 Hz generalized poly-SW | Lennox-Gastaut syndrome | Moderate ID | None | NA | None effective, VNS not effective | c.1084G>A p.Gly362Arg from mosaic mother |
| 26 | M/10 y | None | Normal | 15 mo | Myoclonic atonic, myoclonic | 3–4 second spike and wave complexes over posterior third of head provoked by eye closure and complete darkness | MAE | Mild ID | Autistic spectrum disorder | Broad based gait | Sz free with LEV | C.1155C>G p.Phe385Leu de novo |
| 27 | F/10 y | None | Moderate ID | 3 y | Atonic, myoclonic, tonic/myoclonic | Generalized and lateralized (L or R) delta activity and SW | MAE | Moderate ID (nonverbal) | Autism, self-stim behavior, bruxism, rep. night waking | Brisk reflexes, unsteady gait (walked at 3.75 y) | Sz free with CLZ | c.1342A>T p.Lys448Ter de novo |
| 28 Prev.publ. in Carvill et al.9 | M/12 y | NA | Mild ID | 3 y | Absences, atonic, myoclonic atonic → perioral myoclonia mainly during sleep, rare GTCS | Generalized poly-SW (at onset) Centrotemporal spikes (at evolution) Extreme activation during NREM sleep Now normal |
MAE → aBECTs + ESES-like | Mild ID | None | Unsteady gait/balance problems | Sz free with LEV | c.1369_1370 delGG Gly457HisFsTer10 de novo |
| 29 | F/13 y | None | Severe ID | 5 y | Absences | Generalized spikes/poly spikes | Unclassified generalized | Severe ID (almost nonverbal) | Aggressive behaviors, stereotypies | Normal (walked at 2 y) | Sz free with VPA | C.1377C>G p.Ser459Arg de novo |
| 30 | M/5 y | None | Normal | 1 y | Atypical absences | BG slowing Bursts of irregular, diffuse spike and wave activity followed by diffuse delta Stereotyped focal spikes |
Unclassified generalized | Mild ID (verbal) | ADHD | Normal | Sz improved with VPA/LTG | C.1531G>A p.Val511Met de novo |
| 31 | F/5 y | None | Mild ID | 2 y | Atypical absences, atonic, myoclonic, eyelid myoclonia | BG slowing Bi occipital delta activity Generalized SW |
MAE | Mild ID | ADD | Mild ataxia, dyskinesia | Sz free with ESM, LTG | C.1600C>T p.Gln534Ter de novo |
| 32 | F/4 y | NA | Normal | 16 mo | Absences, atonic, myoclonic atonic | Generalized delta activity and SW (posterior prev.) | MAE | Mild ID | NA | Mild fine motor delay | Sz free with LEV + VPA | c.850-2A>G de novo |
| 33 | F/12 y | Sister with a 22ql3 deletion | NA | 6 y | Absences | SW (3 Hz) Rare bifrontal spikes |
CAE | Mild ID | NA | Action tremor | Sz free with VPA | c.6,1528-1G>C de novo |
| 34 Prev.publ. in Carvill et al.9 | F/7 y | None | NA | 3 y | Absences, myoclonic atonic | BG slowing Generalized delta activity and SW |
MAE | Mild/moderate ID | None | Normal | Sz free with VPA, LEV | 3p25.3 del. including SLC6A11 and SLC6A1 (exon 1) de novo |
Prev.publ., previously published; aBECTs, atypical benign childhood epilepsy with centrotemporal spikes; Ab, absence seizures; A, atonic seizures; AA, atypical absence seizures; ADD, attention deficit disorder; ADHD, attention-deficit/hyperactivity disorder; BG, background; CAE, childhood absence epilepsy; CLZ, clonazepam; ESES, electrical status epilepticus during sleep; rep., repeated; ESM, ethosuximide; EOAE, early onset absence epilepsy; F, female; GTC, generalized tonic–clonic; ID, intellectual disability; LEV, levetiracetam; LTG, lamotrigine; MAE, myoclonic atonic epilepsy; M, Male; m, months; NA, not available; NREM, non–rapid eye movement; Sz, seizure; SW, spike wave; TPM, topiramate; VPA, valproic acid; VNS, vagus nerve stimulation; y, years; ZSM, zonisamide; del., deletion.
3.1 | Genetics
We report on a cohort of 24 previously unpublished probands with pathogenic SLC6A1 variants. Twenty variants occurred de novo in the affected proband, and 2 variants segregated in a dominant manner with the phenotype in the affected family (Figure 1). In 2/24 probands (Patients 12 and 24), segregation analysis could not be performed, and the inheritance mode remained unclear. However, both probands had missense variants identical to those found in other affected probands in this study. We found 18 missense variants, 2 splice-site variants, 1 frameshift variant, 3 nonsense variants, and 1 in-frame deletion. The 18 missense variants all affect highly conserved nucleotides and are absent from control databases (Figure 2). Overall, the variants suggest that loss of function of GAT-1 is the likely disease mechanism, as reported previously.
FIGURE 1.
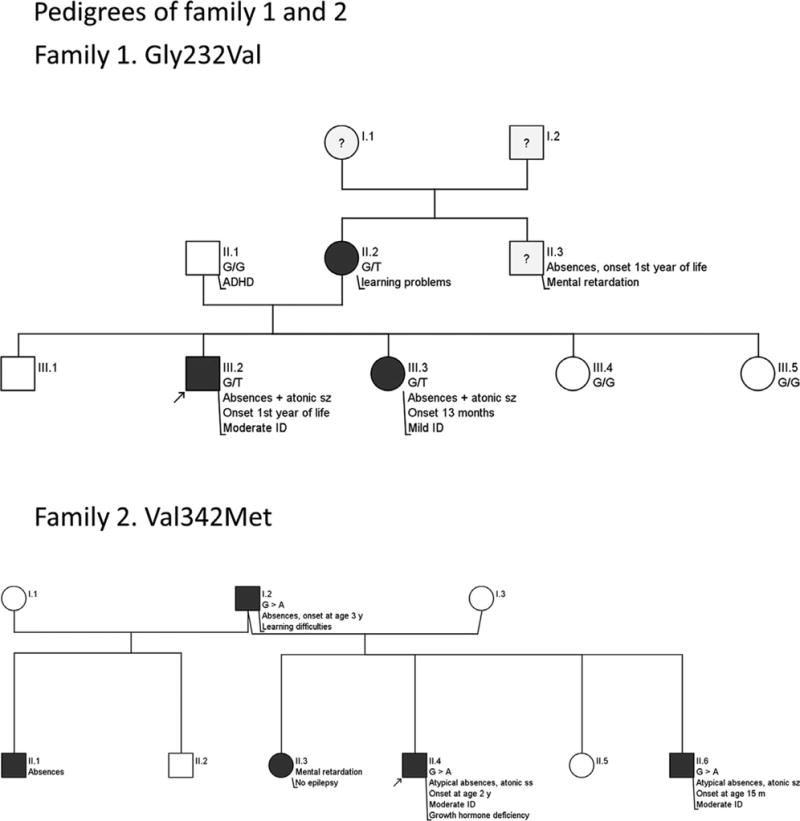
Pedigrees of families 1 and 2
FIGURE 2.

The SLC6A1 gene with the missense variants found in this study. Recurrent variants are highlighted
3.2 | Cognitive development
Data on cognitive performance before epilepsy onset were available in 24/31 patients with epilepsy. Cognition before epilepsy onset was normal in 5/24 (Patients 8, 15, 17, 26, and 30), mildly impaired in 12/24 (Patients 2, 5, 6, 11, 13, 14, 16, 22, 24, 28, and 21), and moderately impaired in 4/24 (Patients 20, 21, 23, and27); 2 Patients had speech delay (3 and 10) and 1 had severe ID (Patient 29),. After epilepsy onset, cognition deteriorated in 11 of them (Patients 3, 8, 10, 13, 15, 16, 22, 24, 26, 30, and 32).
All subjects but 1 (Patient 17) had cognitive impairment at the time of the inclusion in the study.
Sixteen of 34 patients had mild ID, 12 had moderate ID, 3 had severe ID, and 2 had learning disabilities. The most common feature was language delay, described in 16/34, whereas 4 probands presented mainly nonverbal deficits (Patients 1, 25, 27, and 29). There was no apparent correlation between seizure control and cognitive profile.
A large proportion of the 34 SLC6A1 mutation carriers had behavioral problems, such as aggressive behavior/irritability (Patients 4, 10, 12, 14, 16, and 29), attention deficit disorders (Patients 10, 13, 15, 21, 30, and 31), stereotypies/automatisms (Patients 12, 20, 23, 27, and 29), and autistic features (Patients 4, 5, 12, 16, 20, 22, 26, and 27).
3.3 | Other clinical features
Neurological examination revealed mild ataxia (Patients 4, 7, 13, 22, and 31) or unsteady gait (Patients 23, 26, 27, and 28) in 9/34 patients, hypotonia in 3 cases (Patients 1, 4, and 16), and impairment of fine motor skills (Patients 21 and 32) and tremor (Patients 15 and 33) in 2 probands each. One patient each had chorea (4), verbal dyspraxia (21) and dyskinesia (31). No consistent dysmorphisms or malformations were present.
3.4 | Epilepsy phenotype
Thirty-one of 34 individuals had epilepsy, namely 27 of 28 probands (27 unpublished and 4 previously published cases) and 4 of 6 family members. Mean age at seizure onset was 3.7 years (range 0.5–7 years); 2 probands had epilepsy onset before the age of 1 year. Mean age at follow-up was 11.3 years (range 2–21 years).
Affected individuals presented with a variety of seizure types: absences not further specified (14/31), atypical absences (10/31), atonic seizures (14/31), and myoclonic or myoclonic–atonic seizures (12/31) were the most common. A few subjects (5/31) also had generalized tonic–clonic seizures (GTCS) (Table 1).
At the time of diagnosis, epilepsy syndromes were described as MAE in 16 patients and LGS in 1 patient (Patient 25), early onset absence epilepsy (EOAE) in its atypical form (Patient 1), eyelid myoclonia with absences (Patient 22), and childhood absence epilepsy (CAE) (Patients 17 and 33). Seven further patients presented with a generalized epilepsy difficult to classify because of inconsistencies of the electroclinical picture such as absence seizures (up to 100/day), atypical EEG pattern, or because further clinical details and EEG data were not available. Finally, 3 patients were diagnosed with focal epilepsy: 1 had temporal lobe epilepsy (Patient 24) and the other 2 had atypical childhood epilepsy with centrotemporal spikes (aBECTS) (Patients 6 and 28). The diagnosis of aBECTS was based on the description of focal motor seizures during sleep and on the presence of centrotemporal spikes on EEG, together with several atypical features (ID with autistic features or mild neurological deficits, the epilepsy history, and the topography of the EEG abnormalities).
Twenty of 31 patients with epilepsy became seizure-free. Ten of them were treated with valproic acid (VPA), either as monotherapy or in combination with clonazepam (CLZ), lamotrigine (LTG), or levetiracetam (LEV). The remaining 10 subjects became seizure-free on LTG, ethosuximide (ESM), LEV, and LTG or CLZ, alone or in combination. Five further patients were treated with VPA, all achieving partial seizure control. Vagus nerve stimulation (VNS) was attempted in 1 patient with LGS (Patient 25) without benefit. None of the patients reported herein has tried a ketogenic diet, recently described as potentially effective for MAE.
3.5 | Neurophysiological and imaging characteristics
EEG data were available in 27/34 SLC6A1-positive patients, all with epilepsy (23 novel cases and 4 previously published), and were abnormal in all cases. A slowing of the background activity was observed in 11/28 subjects and bursts of irregular generalized 2.5–3.5 Hz spikes/polyspikes and slow waves with a gradual onset in the occipital regions were present in 25/28 patients (Figure 3). In some instances, these epileptic discharges correlated with staring and loss of consciousness with or without a myoclonic or atonic component, in different combinations, resulting in atypical absences, myoclonic–atonic seizures, atonic seizures/drop attacks, or myoclonic seizures. Two further probands had focal epileptiform abnormalities in the centrotemporal (Patient 12) or frontotemporal (Patient 23) regions (the latter occasionally presenting generalized spike and slow waves during the hyperpnea). Of interest, in Patient 12, epilepsy evolved presenting generalized spike and slow waves and atypical absences at the latest observations.
FIGURE 3.
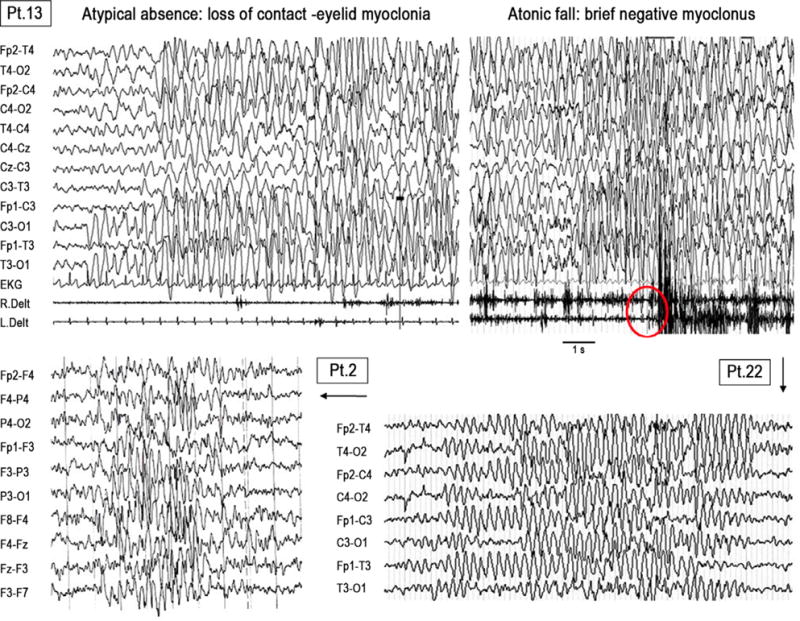
Similar EEG pattern in 3 different patients diagnosed as MAE-like (Patient 20), unclassified generalized (Patient 2), and typical MAE (Patient 31). The EEG abnormalities consisted of prolonged bursts of irregular generalized 2.5–3.5 Hz slow waves with and without prominent spikes/polyspikes component, typically starting gradually in the occipital regions. In Patient 20, roughly the same EEG pattern can give rise to clinical manifestations consisting of atypical absence (to the left), or to a seizure with a brief atonic and myoclonic component (to the right). EEG parameters are the following. Patient 20: band pass filter 1–35 Hz, time constant 0.1 s, sensitivity 20 μV/mm; Patient 2: band pass filter 1–70 Hz time constant 0.3 s, sensitivity 10 μV/mm; Patient 31: band pass filter 1–30, time constant 0.16 s, sensitivity 10 μV/mm
Two patients (6 and 28) initially presenting with an MAE-like phenotype and irregular generalized spikes and slow waves, transitioned to showing only focal epilepti-form abnormalities in the centro-parietotemporal regions at the time of the inclusion in this study (school age). In both cases, the epileptiform abnormalities were markedly enhanced by sleep, as it occurs in encephalopathy with status epilepticus during sleep (ESES). In 1 patient, the spike-wave index during non–rapid eye movement (NREM) sleep was 82% (Figure 4) and in the other, the EEG was reported as “almost continuous epileptic activity” during NREM sleep. In neither patient was this EEG pattern associated with cognitive/behavioral deterioration as seen in ESES.
FIGURE 4.

Nocturnal sleep EEG in Patient 26 at the age of 4 y and 5 mo. During NREM sleep (to the left) the EEG is characterized by almost continuous spike and slow waves in the right hemisphere, with occasional spreading diffuse (spike–wave index 82%). During REM sleep (to the right) the epileptiform abnormalities are less frequent and more focal in the temporocentral region. This EEG picture strongly resembles hemispheric ESES, although the clinical correlate is vague. In the right upper corner, the amplitude map corresponding to the pick of the spike shows a radial orientation of the dipole. EEG parameters: band pass filter 1–70 Hz, time constant 0.1 s
Brain magnetic resonance imaging (MRI) was unremarkable in the majority of SLC6A1 mutation carriers for whom data were available (14/19). Five cases had nonspecific MRI abnormalities, such as mild vermian hypoplasia or enlarged frontal spaces.
4 | DISCUSSION
In the present study, we describe the clinical features of 34 patients with pathogenic variants in SLC6A1 (24 probands, 6 family members, as well as 4 cases previously published8,9) and the EEG features of 28 of them.
The identified variants include several truncating variants supporting loss of function of GAT-1 as a disease mechanism, as reported previously.6 Within this study, we found 4 recurring missense variants (Gly232Val, Ala288-Met, Val342Met, and Gly362Arg), suggesting possible mutational “hot spots” in SLC6A1.
In contrast to the previously published cases with SLC6A1 variants, this cohort displays a broader phenotypic spectrum sharing some common key features.
4.1 | Cognitive and behavioral deficits
The clinical hallmark of the SCL6A1 variant carriers is cognitive impairment. Almost all subjects (33/34) presented certain degrees of cognitive deficits at the time of observation, consisting of mild-to-moderate ID in 82% of the cases (28/34). Speech difficulties were a common feature, as reported previously, present in 17/34 of patients, whereas pure nonverbal deficits were uncommon. In half of the cases (17/34), we also observed behavioral problems, namely, attention deficit, hyperactivity, aggressive behavior, hand stereotypies, and autistic features in different combinations. This observation is supported by the data in the Scl6a1 mouse model.13 Recently, a study found that selected SLC6A1 variants may correlate with a higher risk of Attention-deficit/hyperactivity disorder (ADHD) in humans.14
Of interest, 3/34 SLC6A1 variant carriers never had epileptic seizures, but manifested only learnings disabilities or mild ID with or without autistic features, which represent a previously undescribed phenotype associated with SLC6A1. Two of these individuals had relatives, carrying the same variant, with an MAE phenotype, suggesting variable expressivity possibly due to either genetic or environmental modifying factors, and indicating that genotype–phenotype correlations are not straightforward.
4.2 | Epilepsy
Thirty-one of 34 patients had epilepsy, with a mean age at onset of 3.7 years. The majority (24/30) of the patients displayed absences as the main seizure type, in agreement with studies in mice, that have shown an electroclinical phenotype consistent with atypical absence seizures.4 We identified 16 subjects classified as MAE. Doose et al. stated that MAE usually begins in previously normally developing children.15 However, he did not propose a “rigid” syndrome, but rather a condition with unifying features plus a recognized phenotypic variability attributed to a multifactorial background. In our cohort, the most common phenotype associated with SLC6A1 could indeed be classified as MAE, associated in most of the patients with various degrees of ID before epilepsy onset, as also observed previously.9
Five patients were diagnosed as having a variety of other generalized epilepsy syndromes, including phenotypes resembling CAE (Patient 25) and EOAE (Patient 1), in both cases associated with mild-to-moderate ID. The phenotypes of CAE and EOAE have previously been observed in SLC2A1, GABRB3, and CACNA1H,16–18 but we now extend the genetic etiology of these 2 epilepsy phenotypes to include SLC6A1. Finally, in 4 patients, diagnosed as “unclassified generalized,” the inconsistencies between the clinical features and the EEG findings did not allow a syndromic classification.
Two patients presented with a phenotype resembling aBECTS, although with atypical electroclinical features (Patients 6 and 28), and 1 had temporal lobe epilepsy (Patient 24). This has not been described in any of the Slc6a1 animal models or human SLC6A1 variant carriers.19 The subjects with aBECTS presented an extreme activation of the epileptiform abnormalities during NREM sleep resembling an ESES pattern. Surprisingly, both subjects developed aBECTS as an evolution of an MAE-like phenotype. A third subject presenting with aBECTS at epilepsy onset (Patient 12), evolved into a generalized epilepsy difficult to classify later, suggesting that the electroclinical features of SLC6A1-related epilepsy are variable and can change over time. Finally, 1 patient (25) was diagnosed with an epileptic encephalopathy of the LGS type.
Data about cognition before epilepsy onset were available in 24/31 patients. Only 5 had normal cognitive development, whereas 64% of the subjects (16/24) presented mild-to-moderate ID. After epilepsy onset, cognition deteriorated in 44% of cases (11/24) and did not improve after seizure control was achieved. Therefore, the contribution of epilepsy to the development of the cognitive impairment in patients with a pathogenic SLC6A1 variant is uncertain.
4.3 | EEG data
Most patients exhibited a similar EEG pattern consisting of irregular, generalized 2.5–3.5 Hz spike/polyspikes-and-waves, providing a neurophysiological link to the animal studies. The clinical correlate of these EEG discharges could be an absence seizure, in most cases with atypical features, and with atonic and/or myoclonic components (16/28 cases). In our cohort, differences in the EEG pattern and in the associated clinical manifestations led to different phenotypical classification such as MAE, LGS, EOAE, or CAE. In 3 patients, the epileptiform abnormalities transitioned from generalized to strictly focal in the centro-parietotemporal regions (Patients 6 and 28) and vice versa (Patient 12) during the disease.
4.4 | Epilepsy treatment
The limited data thus far available suggest a good response to VPA, either as monotherapy or in combination with other antiepileptic drugs (AEDs). Ten of 15 patients treated with VPA became seizure-free, and the remaining 5 had a partial benefit. VPA is thought to have a positive effect on the GABA system (possibly increasing the GABA concentration in the human brain.20), which could be part of the explanation for the favorable response to this drug.
4.5 | Neurological signs
Electroclinical studies in mice have suggested that the tonic inhibition in the brain also leads to motor disturbances, such as ataxia, which correlates well with the observation that the GABA transporters are highly present in the cerebellum as well as in cortical neurons.1 We found that 29% (9/31) of the patients had mild ataxia or unsteady gait. A few patients displayed other cerebellar signs such as mild hypotonia, tremor, and fine-motor impairment, in accordance with the hypothesis of a slight perturbation of motor control in animal models.4 Overall, we can conclude that humans carrying pathogenic SLC6A1 variants do not seem to display severe neurological symptoms.
These data combined with those of Carvill et al.9 suggest that typically SLC6A1-positive patients have mild-to-moderate ID, with language delay being one of the major features, preceding the onset of an epilepsy with an MAE phenotype. Therefore, in children with ID with or without behavioral disturbances, developing polymorphic seizure types, including absences, myoclonic and atonic seizures, and rare or no GTCS, screening for SLC6A1 should be undertaken. In addition, we describe cases never developing epilepsy. Because most patients described to date are children or young adults, longitudinal follow-up will be important to understand how seizures and cognition may change with age.
With this study, we define the phenotypic spectrum of clinical manifestations associated with SLC6A1 variants to include different forms of generalized epilepsies, but also a few cases of focal epilepsies as well as cases with ID without epilepsy. We confirm that the predominant epilepsy phenotype is MAE with preexisting mild-to-moderate ID with or without behavioral symptoms. Further studies are needed to confirm the phenotypic spectrum and to investigate the functional consequences of pathogenic variants in SLC6A1.
Key Points.
SLC6A1 mutations cause a wider phenotype than solely MAE
The SLC6A1 phenotype consists of absence seizures and mild-to-moderate intellectual disability
The common EEG pattern comprises irregular, high ample, and generalized spike-and-waves
Acknowledgments
We thank the patients and their families for the participation in this study.
Funding information
Deutsche Forschungsgemeinschaft, Grant/Award Number: We4896/3-1, He5415/6-1; Ambry Genetics; SNSF Early Postdoc fellowship
Footnotes
DISCLOSURE OF CONFLICT OF INTEREST
ST and KLH are employed by Ambry Genetics. CC was funded by the SNSF Early Postdoc fellowship. YW and IH were funded by DFG We4896/3-1; He5415/6-1. The remaining authors have no conflicts of interest to disclose. added. We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
WEB RESOURCES
The ExAC browser: http://exac.broadinstitute.org; SIFT: http://sift.jcvi.org; Poly-Phen2: http://genetics.bwh.harvard.edu/pph2/; MutationTaster: http://www.mutationtaster.org
ORCID
Pasquale Striano http://orcid.org/0000-0002-6065-1476
Guido Rubboli http://orcid.org/0000-0002-5309-2514
References
- 1.Madsen KK, Hansen GH, Danielsen EM, et al. The subcellular localization of GABA transporters and its implication for seizure management. Neurochem Res. 2015;40:410–9. doi: 10.1007/s11064-014-1494-9. [DOI] [PubMed] [Google Scholar]
- 2.Zhou Y, Danbolt NC. GABA and glutamate transporters in brain. Front Endocrinol (Lausanne) 2013;4:165. doi: 10.3389/fendo.2013.00165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scimemi A. Structure, function, and plasticity of GABA transporters. Front Cell Neurosci. 2014;8:161. doi: 10.3389/fncel.2014.00161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cope DW, Di Giovanni G, Fyson SJ, et al. Enhanced tonic GABAA inhibition in typical absence epilepsy. Nat Med. 2009;15:1392–8. doi: 10.1038/nm.2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dikow N, Maas B, Karch S, et al. 3p25.3 microdeletion of GABA transporters SLC6A1 and SLC6A11 results in intellectual disability, epilepsy and stereotypic behavior. Am J Med Genet A. 2014;164a:3061–8. doi: 10.1002/ajmg.a.36761. [DOI] [PubMed] [Google Scholar]
- 6.Rauch A, Wieczorek D, Graf E, et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet. 2012;380:1674–82. doi: 10.1016/S0140-6736(12)61480-9. [DOI] [PubMed] [Google Scholar]
- 7.Sanders SJ, Murtha MT, Gupta AR, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–41. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Halvorsen M, Petrovski S, Shellhaas R, et al. Mosaic mutations in early-onset genetic diseases. Genet Med. 2015;18:746–9. doi: 10.1038/gim.2015.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carvill GL, McMahon JM, Schneider A, et al. Mutations in the GABA transporter SLC6A1 cause epilepsy with myoclonic-atonic seizures. Am J Hum Genet. 2015;96:808–15. doi: 10.1016/j.ajhg.2015.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guerrini R, Aicardi J. Epileptic encephalopathies with myoclonic seizures in infants and children (severe myoclonic epilepsy and myoclonic-astatic epilepsy) J Clin Neurophysiol. 2003;20:449–61. doi: 10.1097/00004691-200311000-00007. [DOI] [PubMed] [Google Scholar]
- 11.Scheffer IE, Berkovic S, Capovilla G, et al. ILAE classification of the epilepsies: position paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017;58:512–21. doi: 10.1111/epi.13709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fisher RS, Cross JH, French JA, et al. Operational classification of seizure types by the International League Against Epilepsy: position paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017;58:522–30. doi: 10.1111/epi.13670. [DOI] [PubMed] [Google Scholar]
- 13.Chen L, Yang X, Zhou X, et al. Hyperactivity and impaired attention in Gamma aminobutyric acid transporter subtype 1 gene knockout mice. Acta Neuropsychiatr. 2015;27:368–74. doi: 10.1017/neu.2015.37. [DOI] [PubMed] [Google Scholar]
- 14.Yuan FF, Gu X, Huang X, et al. SLC6A1 gene involvement in susceptibility to attention-deficit/hyperactivity disorder: A case-control study and gene-environment interaction. Prog Neuropsy-chopharmacol Biol Psychiatry. 2017;77:202–8. doi: 10.1016/j.pnpbp.2017.04.015. [DOI] [PubMed] [Google Scholar]
- 15.Doose H. Myoclonic-astatic epilepsy. Epilepsy Res Suppl. 1992;6:163–8. [PubMed] [Google Scholar]
- 16.Larsen J, Johannesen KM, Ek J, et al. The role of SLC2A1 mutations in myoclonic astatic epilepsy and absence epilepsy, and the estimated frequency of GLUT1 deficiency syndrome. Epilepsia. 2015;56:e203–8. doi: 10.1111/epi.13222. [DOI] [PubMed] [Google Scholar]
- 17.Tanaka M, Olsen RW, Medina MT, et al. Hyperglycosylation and reduced GABA currents of mutated GABRB3 polypeptide in remitting childhood absence epilepsy. Am J Hum Genet. 2008;82:1249–61. doi: 10.1016/j.ajhg.2008.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liang J, Zhang Y, Chen Y, et al. Common polymorphisms in the CACNA1H gene associated with childhood absence epilepsy in Chinese Han population. Ann Hum Genet. 2007;71:325–35. doi: 10.1111/j.1469-1809.2006.00332.x. [DOI] [PubMed] [Google Scholar]
- 19.Oguni H, Fukuyama Y, Tanaka T, et al. Myoclonic-astatic epilepsy of early childhood–clinical and EEG analysis of myoclonic-astatic seizures, and discussions on the nosology of the syndrome. Brain Dev. 2001;23:757–64. doi: 10.1016/s0387-7604(01)00281-9. [DOI] [PubMed] [Google Scholar]
- 20.Chateauvieux S, Morceau F, Dicato M, Diederich M. Molecular and Therapeutic Potential and Toxicity of Valproic Acid. J Biomed Biotechnol. 2010;2010 doi: 10.1155/2010/479364. pii: 479364. [DOI] [PMC free article] [PubMed] [Google Scholar]