

Summary
Misfolded proteins in the endoplasmic reticulum (ER) are destroyed by ER-associated degradation (ERAD). Although the retrotranslocation of misfolded proteins from the ER has been reconstituted, how a polypeptide is initially selected for ERAD remains poorly defined. To address this question while controling for the diverse nature of ERAD substrates, we constructed a series of truncations in a single ER-tethered domain. We observed that the truncated proteins exhibited variable degradation rates and discovered a positive correlation between ERAD substrate instability and detergent insolubility, which demonstrates that aggregation-prone species can be selected for ERAD. Further, Hsp104 facilitated degradation of an insoluble species, consistent with the chaperone’s disaggregase activity. We also show that retrotranslocation of the ubiquitinated substrate from the ER was inhibited in the absence of Hsp104. Therefore, chaperone-mediated selection frees the ER membrane of potentially toxic, aggregation-prone species.
Keywords: molecular chaperone, ERAD, retrotranslocation, ubiquitin, proteasome, yeast
eTOC
Misfolded proteins are destroyed in the endoplasmic reticulum by the ERAD pathway. Preston et al demonstrate that the propensity for misfolded membrane proteins to aggregate dictates their rate of clearance. The Hsp104 chaperone is required to retrotranslocate these substrates and is recruited to the ER membrane when they accumulate.
Introduction
In eukaryotic cells, approximately one-third of all proteins enter the secretory pathway at the endoplasmic reticulum (ER). After passing quality control checkpoints, properly folded proteins proceed through the secretory pathway and are delivered to their ultimate location. However, environmental stressors, genetic mutations, or attenuated folding efficiency leads to the destruction of these nascent proteins via the ER-associated degradation (ERAD) pathway (Christianson and Ye, 2014; Ruggiano et al., 2014). Numerous factors have been identified that play key roles in the recognition, ubiquitination, retrotranslocation, and proteasome-dependent degradation of ERAD substrates, and a growing number of ERAD substrates are linked to human diseases (Guerriero and Brodsky, 2012).
In contrast, significantly less is known about the structural characteristics of substrates that result in their identification. Evidence suggests that quality control factors recognize exposed amphipathic helices or hydrophobic motifs (Arteaga et al., 2006; Furth et al., 2011; Gilon et al., 1998; Johnson et al., 1998; Stein et al., 2014). These studies utilized a limited number of diverse “degrons”, which function as transferrable degradation signals (Dohmen et al., 1994; Levy et al., 1999); therefore, it has proved difficult to generalize rules underlying substrate selection.
Once an ER-resident protein has been recognized, it must be ubiquitinated and retrotranslocated from the ER. In yeast, ERAD substrates are primarily ubiquitinated by one of the two ER resident E3 ligases, Hrd1 (Bordallo et al., 1998) or Doa10 (Ravid et al., 2006). Distinct E3 dependencies arise from the location of the misfolded lesion. If the lesion resides in the ER membrane (ERAD-M) or ER lumen (ERAD-L), the substrate requires Hrd1. However, if the lesion resides in the cytoplasmic portion of the protein (ERAD-C), Doa10 is required (Carvalho et al., 2006; Denic et al., 2006; Huyer et al., 2004; Vashist and Ng, 2004). Doa10 also selects certain ERAD-M substrates for degradation (Habeck et al., 2015; Ruggiano et al., 2016). Once ubiquitinated, the Cdc48/p97 complex is recruited, which retrotranslocates the substrate from the ER for degradation by the 26S proteasome (Bays et al., 2001; Rabinovich et al., 2002; Ye et al., 2001). Soluble ERAD substrates in the ER must be retained in an aggregation-free state prior to retrotranslocation (Nishikawa et al., 2001), and cytosolic chaperones, most notably Hsp70, Ssa1, and Hsp40s, Ydj1 and Hlj1, facilitate the degradation of ERAD-C substrates (Han et al., 2007; Nakatsukasa et al., 2008). Given the diverse structures of ERAD substrates, an understanding of the rules that govern which chaperones mediate substrate selection is in its infancy.
The handling of integral membrane proteins after retrotranslocation is particularly problematic since these aggregation-prone substrates may reside in the cytoplasm (Garza et al., 2009; Leichner et al., 2009; Nakatsukasa et al., 2008; Wiertz et al., 1996). The cytoplasmic Bag6 complex in mammalian cells was identified as a “holdase” (Claessen and Ploegh, 2011; Wang et al., 2011) and associates with Ubl4A, TRC35, and SGTA, which interact with an ER-resident E3, gp78 (Xu et al., 2013b). In the absence of SGTA, the unfolded protein response (UPR) is induced, suggesting that the Bag6 complex plays a general role in ER homeostasis. However, Bag6-dependence has only been investigated for select substrates. In addition, yeast lack Bag6, indicating that other holdases exist and that alternate pathways to retain aggregation-prone proteins in solution might be at-play.
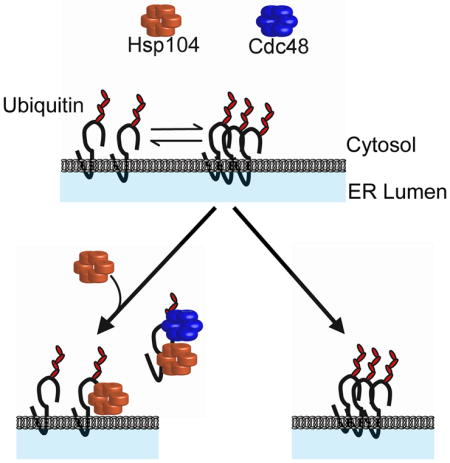
In this study, we investigated the features that dictate substrate recognition by focusing on a single domain, which, when truncated, targets an ER membrane protein (Huyer et al., 2004; Loayza et al., 1998) and a cytoplasmic derivative (Guerriero et al., 2013) for degradation. By characterizing a series of ER-tethered truncations in the domain, we correlated ERAD targeting with detergent insolubility. These data link ERAD substrate selection with aggregation propensity. As anticipated, degradation required select cytosolic chaperones, but unlike any other ERAD substrate characterized to date, we discovered a strong dependence on the Hsp104 disaggregase. Through the use of an in vitro assay, we then established that Hsp104 is required for ubiquitinated substrate retrotranslocation. In addition, Hsp104 was recruited to the ER when an aggregation-prone substrate was expressed. These data indicate that Hsp104, perhaps in conjunction with Cdc48, may help retrotranslocate and maintain the solubility of substrates in the cytosol prior to degradation and that other AAA-ATPases or disaggregases may function similarly in mammals.
Results
Select Truncations in a Nucleotide Binding Domain Decrease Protein Stability
A C-terminal 42 amino acid truncation in the yeast ATP-binding cassette (ABC) transporter, Ste6, leads to the selection of this plasma membrane protein for ERAD, whereas wild type Ste6 is degraded in the vacuole (Kolling and Hollenberg, 1994; Loayza et al., 1998). The destabilizing truncation is in the second nucleotide binding domain (NBD2), which resides in the cytoplasm (black arrowhead, Figure 1A). This ERAD substrate is known as Ste6* (referred to here as “Q1249X”) and many requirements for its degradation have been established (Huyer et al., 2004; Loayza et al., 1998; Nakatsukasa et al., 2008). We wished to determine why this mutation destabilizes Ste6 and introduced truncations into NBD2 both N-and C-terminal of Q1249. Most of the truncations with mutations immediately N-terminal and C-terminal to Q1249 were similarly unstable. In contrast, a more modest C-terminal truncation (R1268X) and, surprisingly, a more severe N-terminal truncation (L1240X) exhibited similar stabilites as the wild type protein (Figure 1B–C). Impressively, truncation mutants differing by as few as two amino acids exhibited distinct metabolic stabilities (for example L1240X and M1242X). These data emphasize the fidelity with which ERAD substrates are selected.
Figure 1. The metabolic stabilities of Ste6 truncations are variable.

(A) Schematic of the yeast 12-transmembrane a-factor transporter, Ste6, with the relative site of the Q1249 truncation (“Ste6*”) marked by an arrowhead. (B) Metabolic stabilities were determined by cycloheximide chase analysis and proteins were visualized by immunoblotting with an anti-HA antibody; t1/2 indicates calculated half-life of each protein. (C) A bar graph depicting the relative half-lives of the Ste6 truncations. Data represent the means of N = 3 independent experiments, +/− SEM.
To investigate the mechanisms underlying recognition of these species in a simplified system—one in which the potential contribution of Ste6’s 12 transmembrane domains might be lessened—NBD2 was tethered to the ER at the C-terminus of a transmembrane hairpin (Figure 2A). Into the resulting chimera we introduced the analogous Ste6 truncations. The truncation analogous to Q1249X, termed Q394X (also known as “Chimera A*” (Guerriero et al., 2017)), was unstable, as were substrates with truncations near this site. Similar to results in Figure 1, the L385X and R413X truncations in the chimera were again as stable as the wild type protein (Figure 2B–C). We also determined the half-life of the wild type chimera and Q394X by pulse chase, and the turnover rates were similar to those observed in the cycloheximide chase assays (Figure S1A). Thus, the truncation series provides a unique, simplified system to investigate how the ERAD machinery distinguishes stable and unstable substrates.
Figure 2. The metabolic stabilities of truncation mutants in a chimera reflect the half-lives of the analogous Ste6 truncation mutants.

(A) Schematic of the yeast dual pass transmembrane protein with the relative site of the Q394 truncation marked by an arrowhead. The chimera consists of the first transmembrane domain of Ste6 followed by a second poly Ala/Leu transmembrane domain annealed to NBD2 from Ste6. The full length species (Wild Type) and the Q394X protein were also recently referred to as “Chimera A” and “Chimera A*” (Guerriero et al., 2017). (B) Metabolic stabilities were determined by cycloheximide chase analyses and proteins were visualized by immunoblotting with an anti-HA antibody. Differences in stabilities between the Ste6 (Figure 1) and chimera (this figure) truncation series are likely due to expression level differences: Ste6 is expressed under the control of an endogenous promotor but the proteins depicted in this figure are under the control of a higher expression (PGK) promoter. (C) A bar graph depicts the relative half-lives of the chimeras. Data represent the means of at least 3 independent experiments, +/− SEM.
To offer a glimpse of the putative structures adopted by the truncation mutants, we created a homology model for NBD2 from Ste6 based on murine P-glycoprotein (Figure S1B; Movie 1). Subsequent homology models from Phyre2 (Kelley et al., 2015) and Uniprot (www.uniprot.org) produced similar models. Each destabilizing truncation occurs within a βαβ turn, whereas the stable truncations are located at either the beginning (L1240X/L385X) or end (R1268X/R413X) of this motif. Destabilizing mutations may also give rise to unaccommodated β strands, which might be aggregation-prone.
The Degradation of an ER Membrane Protein Correlates with Substrate Insolubility
We next confirmed that more stable (wild type chimera and L385X) and less stable (Q394X) substrates reside in the ER (Figure 3A). Moreover, their degradation was proteasome-dependent since MG132 slowed turnover (Figure 3B). Degradation of Q394X is vacuole independent and requires Doa10 and Cdc48 (Guerriero et al., 2017), and L385X requires Doa10 (data not shown).
Figure 3. Protein half-life correlates with detergent solubility.

(A) The cellular localization of the wild type chimera, Q394X, and L385X was determined by indirect immunofluorescence microscopy. Proteins were detected with anti-HA antibody, anti-Kar2 antiserum was used to mark the ER, and the nucleus was visualized with DAPI. (B) Yeast strains lacking PDR5 and expressing either the wild type chimera, Q394X, or L385X were treated with DMSO (closed circles) or 100 μM MG132 (open circles) for 30 min prior to a cycloheximide chase analysis performed at 26°C. Data represent the means of N = 9 (L385X) or N = 12 (wild type chimera and Q394X) independent experiments, +/− SEM; Wild type chimera, P < 0.001 for all time points, Q394X, P < 0.01 for all time points, L385X, P < 0.001 at the 30 and 60 min time points, for data ±MG132. (C) The metabolic stabilities of the truncation mutants were determined by cycloheximide chase analyses and protein solubilities were determined after treatment with the non-ionic detergent, dodecyl-maltoside (DDM). Data represent the means of N = 4–6 independent experiments for stability measurements and N = 4 independent determinations for protein solubility. (D) Wild type microsomes containing Q394X were treated with DDM or DDM in the presence of 6 M urea, and protein residence in the supernatant (S) and pellet (P) fractions was analyzed after centrifugation and immunoblotting. Sec61 was used as a control for membrane solubilization.
We then asked why the truncation mutants bin into two groups based on their stabilities. To assess whether the chimeras may exhibit different solubilities, we prepared ER-enriched microsomes and measured solubility after treatment with detergent and centrifugation. Dodecyl maltoside (DDM) was chosen because the hydrophilic-lipophilic balance (HLB) is in the optimal range for membrane protein solubilization (Helenius and Simons, 1975), and the detergent differentiates between soluble and insoluble populations of integral membrane proteins (Geertsma et al., 2008). Strikingly, a positive correlation between substrate stability and detergent solubility was evident (Figure 3C, Figure S2). In contrast, the solubility of the ER-resident protein, Sec61, was unchanged. Further analysis of the oligomeric state of Q394X after solubilization demonstrated that Q394X resides in an ~1.2 MDa complex (Figure S1C). To determine whether the decreased solubility of Q394X arises from precipitation after membrane extraction, we measured solubility in the presence of DDM and urea. However, the solubility of both the wild type chimera and Q394X were rescued to a similar relative degree by urea (Figure 3D). These results indicate that ERAD substrate selection can depend on a measureable biochemical parameter, one that takes into account the protein’s aggregation-propensity. Moreover, the ability of urea to augment substrate solubility suggests that chaperones prevent aggregation.
Degradation of an Aggregation-Prone ERAD Substrate is Hsp104-Dependent
The cytosolic Hsp70, Ssa1, and Hsp40, Ydj1, chaperones play vital roles during the degradation of ERAD-C substrates (Han et al., 2007; Zhang et al., 2001), including Ste6*(Huyer et al., 2004). We asked whether these same chaperones facilitate Q394X degradation. When expressed at high levels, a strong dependence on Ssa1 and Ydj1 (Figures S3A and S3B) was evident, consistent with preliminary studies with this substrate expressed from a low copy plasmid (Guerriero et al., 2017). Ssa1 and Ydj1 function with Sse1 as a disaggregase (Kaimal et al., 2017; Nillegoda et al., 2015). Based on the relative insolubility of Q394X, we reasoned it may require such activity for degradation and indeed found Q394X was stabilized in sse1Δ yeast (Figure 4A). Sse1 also functions as a nucleotide exchange factor (NEF) for Ssa1 (Raviol et al., 2006), and Q394X was stabilized in yeast lacking another NEF, Fes1 (Figure S3C) (Kabani et al., 2002), suggesting that the Ssa1-dependent degradation of the substrate requires an Ssa1-asociated NEF.
Figure 4. The ERAD of an unstable, aggregation-prone substrate is Hsp104-dependent.

The stabilities of Q394X in the indicated wild type and mutant strains were determined by cycloheximide chase analyses at 37°C. (A) The yeast cytosolic Hsp110, Sse1; N = 3 independent experiments, +/− SEM. (B) The yeast cytosolic disaggregase, Hsp104; N = 3–6 independent experiments, +/− SEM; black asterisks apply to the HSP104 vs hsp104Δ comparison, and the red asterisk applies to the HSP104 vs HSP104 OE comparison. In all panels, *P < 0.05, **P < 0.01, ***P < 0.001.
Based on its disaggregase activity and association with the Hsp70-Hsp40 machinery (Cashikar et al., 2005; Glover and Lindquist, 1998; Kummer et al., 2016), we next investigated whether the Hsp104 chaperone contributed to Q394X degradation. Yeast lacking Hsp104 exhibit a modest decrease in the turnover of a GFP-tagged ER-membrane protein, but no effect was evident at some time points in a cycloheximide chase analysis (Taxis et al., 2003). In contrast, we observed strong and consistent stabilization of Q394X in the hsp104Δ strain (Figure 4B, open circles). Moreover, in line with Hsp104 playing a direct role in the ERAD of this aggregation-prone protein, Q394X degradation was accelerated when HSP104 was over-expressed (Figure 4B, solid inverted triangles).
Other cytosolic chaperones also prevent protein aggregation, some acting in concert with Hsp104. However, as shown in Figure S4, Q394X degradation was unaffected by: 1) loss of the small heat shock proteins, which facilitate Hsp104-dependent substrate disaggregation (Cashikar et al., 2005; Haslbeck et al., 2005); 2) a mutation in the Ssa1-interacting CCT complex (Melville et al., 2003); 3) deletion of SGT2, the yeast homolog of a holdase that functions during ERAD substrate retrotranslocation in mammals (Melville et al., 2003; Xu et al., 2013b); or 4) loss of the yeast UBQLN2 homolog, Dsk2, (examined in a dsk2Δ rad23Δ strain due to redundancy between the two proteins), as UBQLN2 was implicated in the degradation of select ERAD substrates (Lim et al., 2009; Rao and Sastry, 2002).
We next characterized the action of Hsp104 during ERAD. First, Q394X degradation was not restored when yeast expressed an ATPase deficient Hsp104 mutant (Figure S5A), consistent with its role as an energy-dependent disaggregase. Second, Hsp104 primarily acted on the more aggregation-prone ERAD substrates: As noted above, Hsp104 over-expression accelerated Q394X degradation (Figure 4), but the turnover of a more soluble/stable truncation, L385X, was not accelerated upon Hsp104 over-expression (Figure S5B). Third, we asked whether Hsp104 acts up- or down-stream of Q394X ubiquitination. Q394X was expressed in HSP104 and hsp104Δ strains along with myc-tagged ubiquitin. After the substrate was immunoprecipitated, Q394X ubiquitination was measured but was unaffected by the loss of Hsp104 (Figure S5C). Q394X ubiquitination was also measured in an ssa1-45 strain. As observed when Ste6* ubiquitination was assessed (Nakatsukasa et al., 2008), Q394X ubiquitin conjugation was compromised when the cells were inactivated at the non-permissive temperature. Interestingly, only a miminal effect on Q394X ubiquitination was apparent in a strain lacking Sse1 (see Discussion).
Does the expression of Q394X recruit Hsp104 to the ER membrane? Control or Q394X-expressing plasmids were introduced into a MG132-sensitive strain containing genome-integrated Hsp104-GFP (HSP104-GFP pdr5Δ). The number of cells containing Hsp104-positive puncta rose when Q394X was expressed, and the number rose further when cells were pre-treated with MG132 (Figure 5A, Movies 2 and 3). To determine if the puncta resided at the ER, a modified HSP104-GFP pdr5Δ strain was employed that expressed the mCherry-Scs2-tm construct (see Methods). When the proteasome was inhibited, we again saw an increase in the number of cells containing Hsp104 puncta (Figure 5B, Movies 4 and 5, and legend). Of those cells with Hsp104 puncta, ~33% contained puncta that colocalized with the ER in the control but ~78% of the puncta colocalized with the ER when Q394X was expressed.
Figure 5. Hsp104-GFP forms ER puncta in yeast expressing an aggregation-prone membrane protein.

HSP104-GFP pdr5Δ and HSP104-GFP pdr5Δ mCherry-Scs2-tm yeast containing either an empty vector (EV) or a Q394X expression vector were incubated at 37°C for 1 hr. (A) HSP104-GFP pdr5Δ cells containing either an EV or the Q394X expression vector were treated with DMSO or MG132. Data to the right represent the averages of at least 500 cells from at least 3 independent transformations. (B) HSP104-GFP pdr5Δ cells expressing the integrated mCherry-Scs2-tm ER marker and containing either an EV or the Q394X expression vector were treated with MG132. Based on analysis of >590 cells obtained from at least 2 independent transformations, the percentage of cells with punctae in the presence of the vector control was 3% (18/590), and when containing the expression vector, the percentage was 31% (380/1244).
To generalize our findings, we constructed an aggregation-prone ERAD substrate derived from a temperature-sensitive cytosolic enzyme, Ubc9ts (Betting and Seufert, 1996; Kaganovich et al., 2008). Frydman and colleagues utilized Ubc9ts to establish the presence of a cellular compartment, the Q-body, which is dynamic and houses cytoplasmic protein aggregates (Escusa-Toret et al., 2013). Several chaperones from the cytosolic Hsp70 network, including Ssa1, Sse1, and Ydj1, were required for Q-body biogenesis, and Hsp104 localized to Q-bodies. Therefore, Ubc9ts was tethered to the ER via the same dual pass membrane spans used to construct the truncation series above (Figure S6A). We hypothesized that our substrate, termed TM-Ubc9ts, would similarly exhibit temperature-sensitive aggregation and Hsp104-dependent degradation.
We first demonstrated that TM-Ubc9ts was resistant to carbonate extraction, and was thus membrane-integrated (Figure S6B). We also demonstrated that TM-Ubc9ts is an ERAD-C substrate, as degradation was slowed in a doa10Δ strain (Figure S6C). As hypothesized, TM-Ubc9ts degradation was also significantly slowed when Hsp104 was absent (Figure S6D). Moroever, in yeast lacking Hsp104, high molecular weight species were evident, especially in a 6% polyacrylamide gel, when membranes were treated with DDM and lysates were resolved by SDS-PAGE (Figure S6E). To confirm that TM-Ubc9ts also aggregated in vivo, indirect immunofluorescence microscopy indicated that TM-Ubc9ts puncta amassed in the ER membrane only when Hsp104 was absent (Figure S6F). These combined data indicate that Hsp104 is required for the degradation of another aggregation-prone ERAD substrate. However, we also note that ERAD-C substrates do not generally require Hsp104 function: The degradation of other ERAD-C substrates, KWS (Vashist and Ng, 2004) and CTG* (Taxis et al., 2003) was Hsp104-independent (data not shown).
Q394X aggregates in the ER membrane in the absence of Hsp104
Because Hsp104 augments Q394X degradation, concentrates at the ER when Q394X is expressed, and acts after Q394X has been ubiquitinated, Hsp104 may disaggregate Q394X prior to degradation. To test this hypothesis, we utilized a flotation assay (Nakatsukasa and Kamura, 2016) (Figure 6A) and prepared lysates from wild type and hsp104Δ yeast expressing Q394X. We found that a population of Q394X reproducibly resided in a heavier fraction in strains lacking Hsp104 (Figure 6B, right graph, fraction 7). By immunoprecipitating the substrate from each fraction and subsequently probing for ubiquitin, we also observed an increase in ubiquitinated substrate in fraction 7. This was especially notable when the relative amounts of signal were compared between lysates from hsp104Δ and wild type yeast, respectively (Figure 6C, boxed fractions 6 and 7, HSP104 vs hsp104Δ). In contrast, the migration of Sec61 was unaffected when lysates were prepared from HSP104 and hsp104Δ cells (Figure 6D). These data are consistent with an Hsp104-dependent reduction in Q394X oligomers.
Figure 6. Hsp104 disaggregates ubiquitinated Q394X.

(A) A schematic of the flotation assay used to measure substrate oligomerization. Cell lysates from HSP104 or hsp104Δ strains expressing the Q394X mutant incubated at 37°C for 1.5 hrs were prepared and analyzed by equilibrium sucrose density centrifugation. Proteins in each fraction were TCA precipitated and immunoblotted with anti-HA antibody to detect (B) Q394X, or (C) were denatured and immunoprecipitated to detect ubiquitinated Q394X. For (C), compare the boxed regions in lanes 6 and 7 from wild type and hsp104Δ cells, as well as the quantification to the right. (D) The migration of Sec61 was used to mark the ER. Data represent the means of N = 4–5 independent experiments, +/− SEM; * P < 0.05.
Since Hsp104 acts downstream of Q394X ubiquitination (Figure S5C), it was possible that Hsp104 facilitates Q394X retrotranslocation. We therefore used a previously established in vitro assay in which ubiquitination and retrotranslocation can be visualized after membranes containing an ERAD substrate are incubated with radiolabeled ubiquitin, an ATP regenerating system, and yeast cytosol [Figure 7A; (Nakatsukasa et al., 2008)]. After centrifugation, the retrotranslocation of ubiquitinated Q394X was diminished in the presence of hsp104Δ (compare ”S” and “P” for HSP104 vs. hsp104Δ; Figure 7B); a high molecular weight species in the pellet fraction was also apparent only when hsp104Δ-derived components were used (asterisk). Although the supernatant was defined as the soluble fraction after an 18,000 × g centrifugation step in this experiment, the same Hsp104-dependence was observed after a 100,000 × g centrifugation step (data not shown). As a control, retrotranslocation was unaltered when HSP26 HSP42 or SSE1 were deleted (Figure S7).
Figure 7. Hsp104 acts prior to substrate retrotranslocation.

(A) A schematic of the in vitro ubiquitination/retrotranslocation assay. (B) Ubiquitinated Q394X was immunoprecipitated from the retrotranslocated supernatant (S) fractions and the membrane bound pellet (P) fractions after immunoprecipitation. Reactions were run in the presence of either wild type (HSP104) or hsp104Δ cytosol, and in the presence or absence of ATP, as indicated. The native molecular weight (arrow) and high molecular weight species (asterisk) of Q394X are marked. (C) The in vitro assay was scaled up 8-fold with cytosol purified from BY4742 wild type yeast and ER-enriched microsomes purified from BY4742 yeast containing either an empty vector (E) or the Q394X expression vector (Q). The substrate was immunoprecipitated from the pellet (membrane) or supernatant (cytosol, retrotranslocated) fractions and the indicated components were subsequently examined by immunoblot. (D) The membrane bound (P) fraction in a ubiquitination/retrotranslocation assay was collected and washed in 6 M urea to differentiate between membrane-integrated (P) and retrotranslocated, membrane-associated Q394X (US). (E) ER-enriched microsomes were washed with 6 M urea and collected by centrifugation. Samples were also either diluted in buffer or incubated in the presence of SDS. The solubilized fractions (S) and the re-suspended pelleted fractions (P) were TCA precipitated and resolved by SDS-PAGE. Blots were incubated with antibodies against the HA tag in Q394X, the ER resident integral membrane protein, Sec61, and the ER-associated cytosolic proteins, Cdc48 and Hsp104. (F) The stability of Q394X was determined in mutant cdc48-2 yeast containing either an empty vector (EV) or a vector to over-express Hsp104 (OE104). Cultures were shifted to 39°C for 2.5 hrs and chased for 1 hr at 39°C. N = 3 independent experiments. +/− SEM. **P < 0.01, ***P<0.001.
We next scaled-up the in vitro assay and immunoprecipitated Q394X from both the membrane (unretrotranslocated) and soluble (retrotranslocated) fractions (Figure 7C). After immunoblot analysis, a significantly greater fraction of Hsp104 interacted with the retrotranslocated Q394X supernatant pool than the unretrotanslocated pellet. Some Hsp104 also interacted with the membrane pool, but this population was only ~2-fold above background (see Hsp104, “high”, pellet fractions). We believe that the Hsp104-Q394X complex at the ER membrane is transient because DSP crosslinking led to a significant increase in the complex (data not shown). The immunoprecipitation analysis also indicated that Cdc48 preferentially interacted at steady-state with the pool of retrotranslocated Q394X. Based on these data and the results shown in Figure S1C, Q394X resides in a large complex that includes Hsp104 and Cdc48. An identification of the spectrum of associated Q394X partners represents an important future goal.
The shift of Q394X to a more dense, ER-resident fraction when hsp104Δ lysates were examined (Figure 6B), prompted us to ask whether Hsp104 acts on aggregated Q394X in the ER membrane prior to retrotranslocation, or on aggregated Q394X that had first been retrotranslocated. The Q394X-containing pellets were washed with urea and recollected after centrifugation at 18,000 × g. Negligible amounts of ubiquitinated Q394X were solubilized by urea in reactions in the presence or absence of Hsp104 (Figure 7D, “US” lanes), suggesting that the pelleted Q394X had not been retrotranslocated. As a control, Cdc48 and Hsp104 were efficiently released from membranes after urea treatment, while Sec61 remained membrane-embedded (Figure 7E). Overall, Hsp104 appears to act transiently on the aggregation-prone pool of Q394X prior to Cdc48-mediated retrotranslocation, but associates more avidly with the solubilized substrate. More generally, these data define a role for the Hsp104 chaperone and suggest that related disaggregases support the ERAD of aggregation-prone substrates in mammals.
Discussion
The biochemical features that lead to the selection of an ERAD substrate have been difficult to establish. The timing of ERAD substrate degradation is clearly fine-tuned by the modification of appended N-linked and O-linked glycans (Hebert et al., 2005; Xu et al., 2013a). Moreover, many substrates, especially those that lack glycans, can be recognized by ER lumenal molecular chaperones, such as BiP and Herp (Nishikawa et al., 2001; Okuda-Shimizu and Hendershot, 2007; Ushioda et al., 2013). Yet, it has remained mysterious whether the presentation of a chaperone-binding hydrophobic “patch” is sufficient for substrate selection or whether unique structural motifs serve as selection modules. This question has been difficult to answer since myriad substrates, with radically different biochemical and biophysical properties, have been examined, thereby limiting our ability to generalize the nature of substrate selection.
In this study, we focused on a single truncated domain that acts as a degron (Guerriero et al., 2013). Previous work indicated that removing the C-terminal 42 amino acids from NBD2 destabilizes the resulting Ste6* protein (Loayza et al., 1998). Truncations ranging from 23–51 amino acids confer unexpected changes in protein stability. In contrast to what might be expected—and what was demonstrated for another plasma membrane transporter (Pma1), in which extended C-terminal truncations further destabilized the protein (Mason et al., 2014)—truncations on either side of the Ste6* truncation site attenuate ERAD. To define the nature of this phenomenon, we introduced analogous Ste6 truncations into a model transmembrane substrate and demonstrated that the analogous truncations in the chimera behaved similarly to their Ste6 counterparts. The chimera provided us a simplified system in which ERAD substrate selection could be defined, obviating confounding effects that may arise from comparisons between diverse substrates.
Because the destabilizing truncations clustered within a predicted β-sheet, and given the preponderance of aggregation-prone proteins linked to unaccommodated β-sheet-rich regions (Pechmann and Frydman, 2013; Ross and Poirier, 2004; von Bergen et al., 2005), we hypothesized that the instability of an ERAD substrate is associated with its tendency to form insoluble oligomers. A correlation between detergent solubility and ERAD substrate stability has not been established, but the degradation of insoluble species might temper the toxic effects of protein aggregates in the ER. In contrast, insoluble inclusions in the cytosol are protective, and formally the formation of ER-membrane aggregates might also be protective (Haass and Selkoe, 2007; Ogen-Shtern et al., 2016). It is important to note that the correlation between solubility and stability could be influenced by the nature of our substrates, which deposit most of their mass in the cytosol where critical components of the ERAD machinery reside. In fact, an ERAD substrate that lacks a membrane spanning domain, such as the Z-variant of alpha-1-antitrypsin, is more aggregation-prone than another variant, Null Hong Kong alpha 1 antitrypsin, yet both are targeted for ERAD. Nevertheless, the Z variant is also selected for ER-phagy (Hidvegi et al., 2010; Kruse et al., 2006), perhaps because ER lumenal aggregates cannot be accommodated by the retrotranslocation channel.
Other hints in the literature have suggested that ERAD substrate selection might be linked to protein insolubility. Gilon et al. (1998) found that select hydrophobic peptides, when fused to the C-terminus of two model substrates led to ERAD. In addition, a more aggregation-prone version of CPY* has a higher affinity for the E3 ubiquitin ligase, Hrd1, than CPY* (Stein et al., 2014). Yet another ERAD substrate is selected by virtue of an amphipathic helix with hydrophobic character (Arteaga et al., 2006). Similarly, an amphipathic helix in NBD2 may facilitate the ERAD of Ste6* (Ravid et al., 2006), but this motif was present in both the stable and unstable constructs. Finally, during nuclear protein quality control, aggregation propensity plays a role during selection by the E3, San1, as well as Cdc48 (Fredrickson et al., 2013; Gallagher et al., 2014).
Another surprising discovery is our finding that Hsp104 facilitates substrate degradation and acts after substrate ubiquitination. In contrast, Ssa1/Ydj1 act prior to substrate ubiquitination, but have also been shown to act after the ubiquitination of another ERAD substrate (Needham et al., 2011). Based on previous data (Nakatsukasa et al., 2008), Ssa1 and Ydj1 deliver substrates to the ubiquitination machinery, and we suggest that the chaperone ensemble—which includes Hsp104—then plays a key role during and/or after retrotranslocation. We also suggest that Hsp104 activity is required for the turnover of Q394X and TM-Ubc9ts based on their relative insolubilities. Because Hsp104 forms puncta at the ER in the presence of Q394X and a subpopulation of ubiquitinated Q394X oligomerizes in the absence of Hsp104 function, Hsp104: (1) directly maintains the retrotranslocation-competence of substrates; (2) acts in conjunction with Cdc48 to directly retrotranslocate substrates, and/or (3) maintains substrate solubility after retrotranslocation. Based on the in vitro ubiquitination/retrotranslocation assay (Figure 7), we favor the first or second scenario. However, retrotranslocated Q394X bound Hsp104 to a greater degree than the membrane pool of Q394X. These data favor the third scenario, but it is possible that this phenomenon arises due to the fact that substrate degradation is absent in the in vitro retrotranslocation assay (Nakatsukasa et al., 2008). Consequently, Q394X may act as a sink for the chaperone. We also note that Hampton and colleagues showed that Cdc48 is the only chaperone required to maintain the solubility of retrotranslocated integral membrane substrates (Neal et al., 2017). Consistent with this view, yeast lack the Bag6 complex (Wang et al., 2011), which binds retrotranslocated substrates in the cytosol and augments solubility prior to degradation. Therefore, Bag6 might function akin to Hsp104 in yeast. Moreover, because the dense population of Q394X in the gradient assay co-localized with Sec61, the aggregating substrate appears to oligomerize in the ER membrane when Hsp104 is absent. Therefore, we suggest that Hsp104 appears to act upstream of Cdc48 during the liberation of aggregation-prone species from the ER membrane. In support of this hypothesis, over-expression of Hsp104 is unable to rescue a cdc48-2 degradation defect (Figure 7F). Similarly, Hsp104 disaggregates proteins bound to the mitochondrial membrane and helped transport substrates into the mitochondria for degradation (Ruan et al., 2017). Because Q394X contains a cytoplasmic degron, Hsp104 might act similarly at the ER membrane, facilitating retrotranslocation prior to proteasomal degradation.
Sse1, the yeast Hsp110 which is a NEF for Ssa1 as well as a protein holdase, facilitated Q394X degradation but acted after substrate ubiquitination. Work from Bukau and colleagues showed partial disaggregation of luciferase by the Ssa1/Ydj1 complex upon Sse1 addition, supplementing the reaction with Hsp104 led to rapid, optimal disaggregation (Rampelt et al., 2012). Furthermore, Andreasson and colleagues demonstrated that Sse1 is required for both proper targeting to Hsp104 and disaggregation after heat shock (Kaimal et al., 2017). In the future, the specific role of Sse1 during Q394X degradation will need to be better defined.
In sum, the generation of a series of model ERAD substrates indicates how ERAD substrates can potentially be selected and that Hsp104 disaggregase activity allows for Cdc48-dependent degradation of aggregation-prone substrates. It will be exciting to test whether other ERAD substrates are similarly selected for destruction based on their relative insolubilities and whether related disaggregases in mammalian cells are required for ERAD.
STAR Methods
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| 12CA5 anti-HA (mouse) | Roche Applied Science | Cat# 11583816001 |
| Anti-HA-Peroxidase High Affinity (3F10, Rat) | Roche Diagnostics | Cat# 12013819001 |
| Anti-glucose-6-phosphate-dehydrogenase (Rabbit) | Sigma Aldrich | Cat# A9521 |
| Anti-rabbit, HRP-linked antibody (Donkey) | Cell Signaling Technology | Cat# 7074S |
| Anti-mouse, HRP-linked antibody (Horse) | Cell Signaling Technology | Cat# 7076S |
| Anti-Kar2 primary antibody (Rabbit) | Brodsky Lab | N/A |
| Anti-rabbit, Alexa 568 (Goat) | Thermo Fisher Scientific | Cat# A-11011 |
| Anti-mouse, Alexa 488 (Goat) | Thermo Fisher Scientific | Cat# A-11001 |
| Anti-Sec61 (Rabbit) | Brodsky Lab | N/A |
| Anti-ubiquitin (P4D1, Mouse) | Santa Cruz Biotechnology | Cat# sc-8017 |
| Anti-Hsp104 (Rabbit) | Glover Lab | N/A |
| Biological Samples | ||
| Ubiquitin from bovine erythrocytes | Sigma Aldrich | Cat# U6253 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Cycloheximide | Sigma Aldrich | Cat# C7698 |
| SuperSignal Chemiluminescence | Thermo Fisher Scientific | Cat# 34077 |
| Z-Leu-Leu-Leu-al (MG132) | Selleckchem | Cat# S2619 |
| Zymolyase 20T | MP Biomedicals | Cat# 320921 |
| Dodecyl-beta-D-Maltoside (DDM) | Calbiochem | Cat# D310 |
| cOmplete™, EDTA-free Protease Inhibitor Cocktail | Roche | Cat# 11836170001 |
| Apyrase | Sigma Aldrich | Cat# A7646 |
| Leupeptin | Sigma Aldrich | Cat# L9783 |
| Pepstatin A | Sigma Aldrich | Cat# P5318 |
| N-ethylmaleimide (NEM) | Sigma Aldrich | Cat# E1271 |
| Poly-L-lysine, hydrobromide | Sigma Aldrich | Cat# P2636 |
| 4′,6-Diamidino-2-phenylinadole (DAPI) | Sigma Aldrich | Cat# D8417 |
| Critical Commercial Assays | ||
| EasyTag™ EXPRESS 35S Protein Labeling Mix | PerkinElmer Life Sciences | Cat# NEG772007MC |
| BioRad Protein Assay Reagent | Bio-Rad | Cat# 500-0006 |
| Gibson Assembly Master Mix | New England Biolabs | Cat# E2611S |
| Deposited Data | ||
| http://dx.doi.org/10.17632/zw2ff8yyg7.2 | ||
| Experimental Models: Organisms/Strains | ||
| See Table S1 | ||
| Oligonucleotides | ||
| See Table S2 | ||
| Recombinant DNA | ||
| See Table S3 | ||
| Software and Algorithms | ||
| Quantity One software | Bio-Rad Laboratories | CAT# 1709600 |
| ImageJ 1.48V | Wayne Rasband, NIH | www://imagej.nih.gov/ij |
| Pymol version 1.7.4.5 Edu | The PyMOL Molecular Graphics System, Schrodinger, LLC. | http://www.pymol.org |
| Excel 2013 | Microsoft | https://www.microsoftstore.com |
| Sigma Plot 11.0 | Systat | https://systatsoftware.com/products/sigmaplot/ |
| Other | ||
| Prolong Gold Antifade Reagent | Molecular probes | Cat# P10144 |
| Anti-HA conjugated resin | Roche | Cat# 11815016001 |
| Iodine-125 Radionuclide | PerkinElmer | Cat# NEZ033005MC |
| Sephacryl S-300HR | GE Healthcare Life Sciences | Cat# 17059910 |
Contact for Reagent and Resource Sharing
All requests for materials can be directed to the corresponding author, Dr. Jeffrey L. Brodsky, University of Pittsburgh (jbrodsky@pitt.edu)
Experimental Model and Subject Details
Yeast strains were propagated through the use of solid and liquid drop-out media, which were prepared as described previously (Michaelis and Herskowitz, 1988). Yeast strains and cultures utilized for the Ste6 data were grown at 30°C, except for temperature-sensitive strains, which were grown at room temperature (25°C) or then shifted to 37°C, as indicated. Yeast utilized to study the solubility, stability, and localization of the chimera truncations were grown at room temperature (23.5°C). Mutant strains utilized to investigate Q394X degradation, retrotranslocation, and localization were grown at 23.5°C and shifted to 37°C before each experiment and remained at 37°C during the experiment.
Method Details
Plasmid Construction
Plasmids expressing the Ste6 truncation mutants were derived from pSM694 (2μ LEU2 STE6::HA) and contain the triply iterated hemagglutinin (HA) epitope tag between amino acids 68 and 69 (the ecto position) in Ste6 (Berkower et al., 1994). All of the STE6 truncation mutations were constructed by homologous recombination in yeast, where a PCR product containing the desired truncation mutation was generated and co-transformed with a linearized gapped version of the wild type STE6 plasmid (pSM694). To create the chimera truncation series, primer oMP01 was used in combination with oMP02 through oMP09, resulting in the production of pMP01 through pMP08. Because pCG19 expresses the wild type chimera with an internal triple HA tag, the resulting pMP01 through pMP08 plasmids are also engineered to express triply HA-tagged proteins. For vectors designed to overexpress Hsp104, primers oMP17 and oMP18 were used with oMP19 to create two forms of the HSP104 over-expression plasmid: 1) Expression of HSP104 from the endogenous HSP104 promoter in the pRS425 vector (Mumberg et al., 1995) and 2) Expression of HSP104 from the GPD promoter for constitutive expression in the pRS425 vector. All plasmids were sequence verified prior to use. The TM-Ubc9ts construct was created by use of primers oMP13-oMP16 and products from these reactions were utilized to create the construct through Gibson Assembly (NEB).
Cycloheximide Chase Assays
2x azide stop mix
20 mM NaN3, 0.5 mg/ml BSA
Lysis buffer 1
400 mM NaOH, 7% 2-mercaptoethanol, 20 mM Tris pH8, 20 mM EDTA, 1 mM PMSF, 2 mM leupeptin, and 0.7 mM pepstatin A
Trichloroacetic acid sample buffer
3.5% SDS, 0.5M DTT, 80 mM Tris, pH 8, 8 mM EDTA, 15% glycerol, 0.1 mg/ml bromophenol blue, 5% fresh 2-mercaptoethanol
Trichloroacetic acid precipitation (TCAP) Sample Buffer
80 mM Tris, pH 8, 8 mM EDTA, 3.5% SDS, 15% glycerol, 0.08% Tris base, 0.01% bromphenol blue, 5% fresh 2-mercaptoethanol
To determine the effects of C-terminal truncations on Ste6 turnover rates, cycloheximide chase analysis was performed as described previously (Metzger et al., 2008) with modifications. Briefly, cells were grown to logarithmic phase in synthetic media and cycloheximide was added to a final concentration of 100 μg/mL to inhibit further protein synthesis. For the 0 min time point, 500 μL of cells were immediately harvested by addition to an equal volume of 2x azide stop mix on ice. Cells were incubated during the chase at 30°C. Cells were then pelleted and lysed by the addition of a lysis buffer and proteins were precipitated in 5% TCA. Protein pellets were resuspended in TCA sample buffer and proteins were analyzed by SDS-PAGE and immunoblotting. Blots were visualized on an imager with ECL and then quantified using Quantity One software. Half-lives were calculated from exponential curve fits.
To determine the effects of C-terminal truncations on the turnover rates of the chimeras, cycloheximide chases were performed similarly to previously described methods (Zhang et al., 2002). Briefly, cells were grown to logarithmic phase in SC-ura media and incubated at 25°C. Cycloheximide was added to a final concentration of 200 μg/mL to each culture, with cells harvested at each time point and added to sodium azide (final concentration of 17 mM) on ice. Cells were lysed, treated with 8% TCA, and protein pellets were resuspended in TCAP with a mechanical pestle for 20 sec and the samples were incubated at 37°C. Resuspended pellets were analyzed by SDS-PAGE. Blots were visualized on an imager with ECL and then quantified using ImageJ. Half-lives were calculated from exponential curve fits.
To determine the dependence of the designated chimera truncations on the 26S proteasome for degradation, BY4742 pdr5Δ cultures expressing the indicated truncations were grown at 26°C. The 26S proteasome inhibitor, MG132, was added to half of each culture to a final concentration of 100 μM. To the other half of each culture an equal volume of DMSO was added. Cultures were then incubated for 30 min at 25°C. Cycloheximide was next added to a final concentration of 200 μg/ml and the chase was performed as described above.
To analyze the contribution of specific factors on the degradation of Q394X and the contribution of Hsp104 on the degradation of TM-Ubc9ts, the designated strains were temperature shifted to 37°C for 30 min prior to the addition of cycloheximide and remained at 37°C during the chase. Cycloheximide was then added to a final concentration of 200 μg/ml and the samples were processed essentially as described above.
Pulse Chase Assays
Pulse chase UMCC
SC-ura, 13 mM methionine, 40 mM cysteine, and 200 μg/ml cycloheximide
Buffer 88
20 mM HEPES, pH 6.8, 150 mM KOAc, 250 mM sorbitol, and 5 mM MgOAc
Extract buffer
50 mM Tris-HCl pH 7.4, 1 mM EDTA, 1% SDS, 3mM PMSF, 6 mM leupeptin, and 2.1 mM pepstatin A
Immunoprecipitation (IP) wash buffer
50 mM Tris-HCl pH 7.4, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, and 0.2% SDS
TAE buffer
50 mM Tris-HCl pH 7.4, 150 mM NaCl, and 5 mM EDTA
SDS sample buffer
65 mM Tris-HCl pH 6.8, 5 mg/mL bromophenol blue, 2% SDS, 1% β-mercaptoethanol, and 10% glycerol
Pulse chase analyses were performed similarly to previously published methods (Huyer et al., 2004). Briefly, cultures were grown to log phase in SC –ura at 22.5°C. The cells were then labeled with Express 35S for 15 min at 26°C. The cells were harvested, washed in SC-ura, and finally resuspended in Pulse chase UMCC. The cells were collected at 0, 15, 30, and 60 min after the addition of cycloheximide and resuspended in 10 mM sodium azide on ice. The cells were harvested as above and washed in Buffer 88 and resuspended in Extract buffer. Cells were lysed, the debris was removed by centrifugation, and the lysate was mixed with 350 μl of IP wash buffer. The mixture were precleared for 3 hrs at 4°C with 35 μl of a 50/50 slurry of protein A-sepharose beads. Samples were then centrifuged briefly, and the supernatant was incubated overnight at 4°C with an anti-HA antibody and 35 μl of fresh 50/50 slurry protein A-sepharose beads. The beads were collected by centrifugation and then washed in IP wash buffer and IP wash buffer containing 2 M urea. Finally, beads were washed with TAE and resuspended in SDS sample buffer. Samples were resolved by SDS-PAGE, and gels were dried and exposed to a phosphorfilm for 1–2 days, which was then scanned using a GE Typhoon FLA 7000 (Boston, MA). Signals were quantified using ImageJ.
Indirect Immunofluorescence
KM
40 mM KPO4 and 0.5 mM MgCl2
KMS
40 mM KPO4, 0.5 mM MgCl2, and 1.2 M sorbitol
PBS blocking solution
1X PBS, 0.5% BSA, 0.5% ovalbumin, 0.66% fish-skin gelatin, and 0.1% Triton X-100
Samples were prepared similarly to a previously published protocol (Amberg et al., 2005). Briefly, cultures were grown in SC-ura media until reaching early log phase, and then incubated for 10 min at 22.5°C in SC-ura containing 4% formaldehyde. The cells were resuspended in 4% formaldehyde and incubated for 1 h at 30°C. After collecting the cells by centrifugation, they were washed in KM and resuspended in KMS. The cell walls were digested with zymolyase 20T at 37°C for 20, 25, or 30 min, and the yeast were collected by centrifugation, resuspended in KMS, and added to poly-lysine treated slides. The adhered cells were washed in ice cold methanol and acetone and then blocked using PBS blocking solution. The permeabilized, fixed cells were incubated with primary antibodies against HA and Kar2. The samples were washed with PBS blocking solution and incubated with DAPI to visualize DNA, along with secondary antibodies Alexa 568 and Alexa 488. Samples were mounted using Prolong Gold Antifade Reagent and imaged with an Olympus FV1000, x100 UPlanSApo oil immersion objective, numerical aperture 1.40.
Live Cell Imaging
Yeast strains lacking the PDR5 gene and expressing an integrated, C-terminal GFP-tagged copy of Hsp104, and where indicated that also contained an integrated mCherry-Scs2-tm ER marker were transformed with either the Q394X expression vector or a vector control. Overnight cultures were grown in SC –ura media and diluted to early log phase the next day. At that time, MatTek glass bottom microwell dishes were treated with 1 mg/mL concanavalin A (ConA) and allowed to dry at 22°C for 1.5 hrs. Immediately prior to imaging, the cultures were treated with either DMSO or a final concentration of 100 μM of MG132 and placed in wells in the MatTek dishes. Cells were shifted to 37°C and imaged from multiple sections of the plate every 5 min for 1 hr. Images were taken on a point scanning Nikon A1 confocal.
Isolation of ER Microsomes
Lyticase buffer
10 mM Tris-HCl, pH 7.4, 1.5% peptone, 0.75% yeast extract, 0.7 M sorbitol, and 0.5% glucose
Cushion 1
20 mM HEPES, pH 7.4, 0.8 M sucrose, and 1.5% Ficoll 400
Lysis buffer 2
20 mM HEPES, pH 7.4, 50 mM KOAc, 2 mM EDTA, 0.1 M sorbitol, 1 mM of freshly added DTT, 1 mM PMSF, 2 mM leupeptin, and 0.7 mM pepstatin A
Cushion 2
20 mM HEPES, pH 7.4, 50 mM KOAC, 1 M sucrose, and 1 mM of freshly added DTT
Cultures expressing the chimera truncations were incubated at 25°C for 1 h while cultures expressing TM-Ubc9ts were incubated at 37°C for 1.5 hrs. Cells were harvested by centrifugation and yeast ER-enriched microsomes were purified using a modified version of a previously described large-scale technique (Nakatsukasa et al., 2008). Here, the cultures were resuspended in 100 mM Tris-HCl, pH 9.4, plus 10 mM DTT and the cells were collected and resuspended in lyticase buffer. Roughly 10–20 units of lyticase was added to each culture and the solution was incubated at 26°C for 1 hr for the chimera truncations and for 30 min at 37°C for TM-Ubc9ts. The digested cells were then overlaid onto an equal volume of Cushion 1 and the samples were centrifuged. Pellets were resuspended in ice-cold Lysis buffer and lysed using a motor driven Potter-Elvehejm homogenizer. The samples were then overlaid onto an equal volume of Cushion 2 and centrifuged. The ER-enriched fraction was removed and centrifuged. The pellet was then washed in Buffer 88 and re-centrifuged. The final pellet was resuspended in Buffer 88 to a final concentration of 10 mg/ml as determined spectrophotometrically (A280) in a solution of 1% SDS.
Detergent Solubility Assay
Solubilization assays were performed by adding Buffer 88, the indicated concentration of DDM, and ER-enriched microsomes to a final concentration of 0.5 mg/ml of protein. When designated, these samples also contained 6 M urea. The samples were incubated at room temperature (~22°C) for 30 min and then centrifuged at 18,000 × g or 100,000 × g for 10 min. The supernatant was removed and dispensed into an Eppendorf tube containing 5x SDS sample buffer. In turn, the pellet was resuspended to an equal final volume of 1x sample buffer by pipetting. The samples were then incubated at 37°C for 30 min followed by a brief centrifugation and subjected to SDS-PAGE and immunoblotting with anti-HA as described above. Blots were also probed against the ER integral membrane protein Sec61. This served as a control for microsome solubilization and protein extraction. Blots were visualized and then quantified using ImageJ.
Size Exclusion Chromatography
ER-enriched microsomes containing Q394X were solubilized in DDM as described above. After solubilization, samples were loaded onto a 25mL Kontes Flex-Column packed with Sephacryl S-300HR. Samples were run under gravity through the column, and 0.5 ml fractions were collected. A portion of the samples was mixed with 5x SDS sample buffer and resolved by SDS-PAGE and immunoblotting as described above. Known molecular weight standards (RNase A, ovalbumin, aldolase, thyroglobulin, and blue dextran) were loaded and run through the column under the same conditions as Q394X. Elution peaks for the standards were determined by SDS-PAGE and Coomassie staining. Elution peaks for standards were used to calculate the predicted molecular mass of Q394X when solubilized from microsomes with DDM.
Floatation Assays
Lysis buffer 3
20 mM HEPES, pH 7.4, 50 mM KOAc, 2 mM EDTA, 0.1 M sorbitol, 1 mM of freshly added DTT, 20 μM MG132, 10 mM NEM, and 1 x Roche Complete EDTA-Free PI cocktail
Flotation buffer
50 mM HEPES pH 7.4, 150 mM NaCl, 5 mM EDTA, freshly added 1 mM DTT, 1mM PMSF, and 1 x Roche Complete EDTA-Free PI cocktail
Denaturing buffer
2x TBS pH 7.4, 2% SDS, 8M urea, 20 mM NEM, and 2x Roche Complete EDTA-Free PI cocktail
Dilution buffer
1 x TBS pH 7.4, 2% Triton, 10 mM EDTA, 0.5% deoxycholate, 10 mM NEM, and 1x Roche Complete EDTA-Free PI cocktail
To investigate the aggregation state of Q394X in the HSP104 and hsp104Δ strains after heat shock, strains were transformed with pCG12 and pKN31 and grown in selective media. A modified version of a previously published flotation method was utilized (Nakatsukasa and Kamura, 2016). Roughly 30 ODs of cells were temperature-shifted to 37°C for 1.5 hrs, 1 hr of which was in the presence of copper sulfate (to induce expression of myc-ubiquitin), and then centrifuged. Pellets were resuspended in lysis buffer 3. Cultures were lysed and samples were removed from the beads and placed in a fresh 1.5 mL Eppendorf tube. The beads were washed with Buffer 88 and collected into the tubes containing the samples. The combined samples were centrifuged at for 5 minutes at 300 × g and 4°C to pellet unbroken cells. A portion of the supernatant was mixed with a 2.3 M sucrose solution in Flotation buffer. Gradients were layered from bottom to top with 600 μL of 2.3 M sucrose in Flotation buffer, 800 μL sample, 1.2 mL of 1.5 M sucrose in Flotation buffer, and 1 mL of 0.25 M sucrose in Flotation buffer. Gradients were centrifuged at 77,000 × g for 16 hrs at 4°C. A total of 12 fractions were taken and put in fresh tubes. Half of each fraction was added to an equal volume of Denaturing buffer and heated for 30 min at 42°C. Samples were centrifuged and samples were added to dilution buffer. HA-conjugated beads were added to the samples and incubated overnight at 4°C on a rotator. TCA was added to the remaining portion of each fraction to a final volume of 20% TCA and incubated on ice. Samples were centrifuged and pellets were washed with ice old acetone and centrifuged again. Supernatants were aspirated and pellets allowed to dry briefly. The final pellets were resuspended in 1 x SDS sample buffer by mechanical disruption. In parallel, the IPs were washed in IP wash buffer, followed by resuspension and brief agitation of the beads in TCAP. IPs and TCA samples were analyzed by SDS-PAGE. IP blots were boiled in water for 1 hr, and after immunoblotting signals were visualized and then quantified using ImageJ.
In vivo Ubiquitination Assays
RIPA buffer
10 mM Tris-Cl pH 8.0, 140 mM NaCl, 1 mM EDTA, 1 % Triton-X100, 0.1% Sodium Deoxycholate, 0.1% SDS, 1mM PMSF, 2mM leupeptin, and 0.7mM pepstatin A
To investigate the role of select factors on the ubiquitination of Q394X, the designated strains were transformed with pCG12 and pKN31 and grown in selective media. Briefly, 15 ODs of cells were temperature shifted to 37°C for 1.5 hrs, of which 1 hr was in the presence of 100 μM copper sulfate, and collected as described above. Cells were resuspended in 1 mL of RIPA buffer and lysed with glass beads. Supernatants were removed and placed in fresh 1.5 mL Eppendorf tubes. Beads were washed with 500 μL of RIPA buffer, the buffer was removed and pooled with the supernatants. Samples were centrifuged at 18,000 × g and protein levels were determined by measuring the A280nm in 2% SDS. Equal amounts of total protein were added to be immunoprecipitated in a final volume of 1 mL. A total of 40 μL of HA-conjugated beads was added to each IP sample and incubated over-night at 4°C on a rotator. IPs were washed, then IP and protein samples were resolved and quantified as described above.
Yeast Cytosol Preparation
Purified yeast cytosol was prepared using liquid nitrogen via a modified version of a previously published technique (Nakatsukasa et al., 2008). After cells were grown at 37°C for 1 hr, they were harvested, washed in Buffer 88, and flash frozen in liquid nitrogen. The frozen cells were then lysed using a cold mortar and pestle for 1 min in the presence of liquid nitrogen for a total of 6 rounds of grinding. The resulting powder was thawed on ice and DTT was added to a final concentration of 1 mM. Samples were centrifuged to spin out unbroken cells and the resulting supernatants were centrifuged at 80,000 × g for 1 hr at 4°C. Clarified cytosol was harvested and a Bradford assay was performed to determine cytosol concentrations.
In vitro Ubiquitination Assays
ATP Regenerating System
1 mM ATP, 40 mM creatine phosphate, 0.2 mg/mL creatine phosphokinase in Buffer 88
SDS stop solution
50 mM Tris–Cl, pH 7.4, 150 mM NaCl, 5 mM EDTA, 1.25% sodium dodecyl sulfate (SDS), 1 mM PMSF, 2 mM leupeptin, 0.7 mM pepstatin A, and 10 mM N-ethylmaleimide (NEM)
Triton solution
50 mM Tris–Cl, pH 7.4, 150 mM NaCl, 5 mM EDTA, 2% Triton X-100, 1 mM PMSF, 2 mM leupeptin, 0.7 mM pepstatin A, and 10 mM NEM
In vitro ubiquitination conjugation reactions were performed using a variation of a previously described method (Nakatsukasa et al., 2008) and consisted of 1 mg/ml microsomes purified from the designated strains expressing Q394X, 1 mg/ml cytosol, and an ATP-regenerating system in a final volume of 40 μl. Addition of apyrase instead of the ATP-regenerating system served as a negative control. Samples were pre-incubated at room temperature and incubated at the same temperature after 125I-ubiquitin was added to a final concentration of 1.5 mg/ml ubiquitin. The samples were then incubated at 37°C for 45 min before 125 μl of a 1.25% SDS stop solution was added to the reactions, which were incubated at 37°C for 30 min. When retrotranslocation efficiency was investigated, samples were centrifuged for 10 min at 18,000 × g and 4°C prior to the addition of the 1.25% SDS stop solution. Supernatants were collected in separate tubes while pellets were resuspended in an equal volume of Buffer 88. The 1.25% SDS stop solution was added to each and samples were incubated at 37°C for 30 min A total of 400 μl of a Triton solution, 35 μl of a 50/50 protein A-sepharose slurry, and anti-HA antibody were then added to each reaction and samples were immunoprecipitated overnight at 4°C. After centrifugation, the pellets were washed in an IP wash buffer and TCA sample buffer was added to each sample. The samples were briefly agitated and incubated at 37°C for 30 min. The precipitated chimeras were then analyzed by SDS-PAGE. One gel was transferred to nitrocellulose and was visualized to control for IP levels. The second gel was dried and exposed to a phosphorfilm for 2–3 d. Imaging and quantification were performed as described above.
Quantification and Statistical Analysis
All statistical analysis in this work was determined by the use of unpaired t-tests. All statistical details for this work can be found in the figures and figure legends. In this study, N represents an individual transformed colony of yeast. Data presented in graphs consists of the means of all replicates performed and contains error bars that represent the standard error of the mean (SEM). Significance is defined by a P < 0.05. However, significance is presented in three variations. *P < 0.05, **P < 0.01, and ***P < 0.001. This is used to show variations in the level of significance for all data.
Supplementary Material
Movie 1. A homology model for the second nucleotide binding domain (NBD2) of Ste6, focusing on the amino acids at which truncations were examined in the Ste6 and Chimera A proteins, Related to Figures 1 and 2.
Movie 2. Hsp104-GFP is diffuse after proteasome inhibition when cells contain an empty vector control, Related to Figure 5.
Movie 3. Hsp104-GFP localizes to punctae upon proteasome inhibition when cells express Q394X, Related to Figure 5.
Movie 4. Hsp104-GFP does not co-localize with the ER after proteasome inhibition when cells contain an empty vector control, Related to Figure 5.
Movie 5. Hsp104-GFP positive punctae co-localize with the ER after proteasome inhibition when cells express Q394X, Related to Figure 5.
Highlights.
The stability and solubility of an integral membrane ERAD substrate correlate
Hsp104-dependent disaggregation is required to degrade insoluble substrates
Hsp104 facilitates the retrotranslocation of a ubiquitinated insoluble substrate
Acknowledgments
This work was supported by grants GM75061 and DK79307 from the National Institutes of Health to J.L.B, grant R883-CR07 from the Cystic Fibrosis Research Development Program to J.L.B, grant DK101584 to C.J.G., and grant GM051508 to S.M. G.M.P. acknowledges training grants DK061296 from the National Institutes of Health and PRESTO15H0 from the Cystic Fibrosis Foundation. The Center for Biologic Imaging is supported by P30 DK072506 from the National Institutes of Health. We thank Kunio Nakatsukasa, Thomas Nystrom, Patrick Thibodeau, Arohan Subramanya, Yong Wan, Andrew VanDemark, Marijn Ford, Kurt Weiberth, Joel Rosenbaum, Zhihao Sun, Patrick Needham, Judith Frydman, John Glover, and Rong Li for reagents, helpful discussions, and/or sharing preliminary data, and Gaby Nijbroek Longsworth for her initial findings on position-dependent degradation rates in Ste6.
Footnotes
Authors Contributions
G.M.P., C.J.G., M.B.M., S.M., and J.L.B designed, conducted, and interpreted the results of the experiments in the current study. M.B.M and S.M. first identified the variable metabolic stabilities of the Ste6 truncations. C.J.G. contributed conceptually to this work, initially constructed the wild type and Q394X Chimera A constructs, identified the role of Fes1 in the degradation of Q394X, and assisted in the co-IP assays. G.M.P. performed all other experiments.
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amberg DC, Burke DJ, Strathern JN. Methods in Yeast Genetics. Cold Spring Harbor, NY, USA: Cold Spring Harbor Press; 2005. [Google Scholar]
- Arteaga MF, Wang L, Ravid T, Hochstrasser M, Canessa CM. An amphipathic helix targets serum and glucocorticoid-induced kinase 1 to the endoplasmic reticulum-associated ubiquitin-conjugation machinery. Proc Natl Acad Sci U S A. 2006;103:11178–11183. doi: 10.1073/pnas.0604816103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bays NW, Wilhovsky SK, Goradia A, Hodgkiss-Harlow K, Hampton RY. HRD4/NPL4 is required for the proteasomal processing of ubiquitinated ER proteins. Mol Biol Cell. 2001;12:4114–4128. doi: 10.1091/mbc.12.12.4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker J, Walter W, Yan W, Craig EA. Functional interaction of cytosolic hsp70 and a DnaJ-related protein, Ydj1p, in protein translocation in vivo. Mol Cell Biol. 1996;16:4378–4386. doi: 10.1128/mcb.16.8.4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkower C, Loayza D, Michaelis S. Metabolic instability and constitutive endocytosis of STE6, the a-factor transporter of Saccharomyces cerevisiae. Mol Biol Cell. 1994;5:1185–1198. doi: 10.1091/mbc.5.11.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betting J, Seufert W. A yeast Ubc9 mutant protein with temperature-sensitive in vivo function is subject to conditional proteolysis by a ubiquitin- and proteasome-dependent pathway. J Biol Chem. 1996;271:25790–25796. doi: 10.1074/jbc.271.42.25790. [DOI] [PubMed] [Google Scholar]
- Bordallo J, Plemper RK, Finger A, Wolf DH. Der3p/Hrd1p is required for endoplasmic reticulum-associated degradation of misfolded lumenal and integral membrane proteins. Mol Biol Cell. 1998;9:209–222. doi: 10.1091/mbc.9.1.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caplan AJ, Cyr DM, Douglas MG. YDJ1p facilitates polypeptide translocation across different intracellular membranes by a conserved mechanism. Cell. 1992;71:1143–1155. doi: 10.1016/s0092-8674(05)80063-7. [DOI] [PubMed] [Google Scholar]
- Carvalho P, Goder V, Rapoport TA. Distinct ubiquitin-ligase complexes define convergent pathways for the degradation of ER proteins. Cell. 2006;126:361–373. doi: 10.1016/j.cell.2006.05.043. [DOI] [PubMed] [Google Scholar]
- Cashikar AG, Duennwald M, Lindquist SL. A chaperone pathway in protein disaggregation. Hsp26 alters the nature of protein aggregates to facilitate reactivation by Hsp104. J Biol Chem. 2005;280:23869–23875. doi: 10.1074/jbc.M502854200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson JC, Ye Y. Cleaning up in the endoplasmic reticulum: ubiquitin in charge. Nat Struct Mol Biol. 2014;21:325–335. doi: 10.1038/nsmb.2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claessen JH, Ploegh HL. BAT3 guides misfolded glycoproteins out of the endoplasmic reticulum. PLoS One. 2011;6:e28542. doi: 10.1371/journal.pone.0028542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denic V, Quan EM, Weissman JS. A luminal surveillance complex that selects misfolded glycoproteins for ER-associated degradation. Cell. 2006;126:349–359. doi: 10.1016/j.cell.2006.05.045. [DOI] [PubMed] [Google Scholar]
- Dohmen RJ, Wu P, Varshavsky A. Heat-inducible degron: a method for constructing temperature-sensitive mutants. Science. 1994;263:1273–1276. doi: 10.1126/science.8122109. [DOI] [PubMed] [Google Scholar]
- Escusa-Toret S, Vonk WI, Frydman J. Spatial sequestration of misfolded proteins by a dynamic chaperone pathway enhances cellular fitness during stress. Nat Cell Biol. 2013;15:1231–1243. doi: 10.1038/ncb2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredrickson EK, Gallagher PS, Candadai SVC, Gardner RG. Substrate recognition in nuclear protein quality control degradation is governed by exposed hydrophobicity that correlates with aggregation and insolubility. J Biol Chem. 2013;288:6130–6139. doi: 10.1074/jbc.M112.406710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furth N, Gertman O, Shiber A, Alfassy OS, Cohen I, Rosenberg MM, Doron NK, Friedler A, Ravid T. Exposure of bipartite hydrophobic signal triggers nuclear quality control of Ndc10 at the endoplasmic reticulum/nuclear envelope. Mol Biol Cell. 2011;22:4726–4739. doi: 10.1091/mbc.E11-05-0463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher PS, Candadai SVC, Gardner RG. The requirement for Cdc48/p97 in nuclear protein quality control degradation depends on the substrate and correlates with substrate insolubility. J Cell Sci. 2014;127:1980–1991. doi: 10.1242/jcs.141838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garza RM, Sato BK, Hampton RY. In vitro analysis of Hrd1p-mediated retrotranslocation of its multispanning membrane substrate 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase. J Biol Chem. 2009;284:14710–14722. doi: 10.1074/jbc.M809607200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geertsma ER, Groeneveld M, Slotboom DJ, Poolman B. Quality control of overexpressed membrane proteins. Proc Natl Acad Sci U S A. 2008;105:5722–5727. doi: 10.1073/pnas.0802190105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilon T, Chomsky O, Kulka RG. Degradation signals for ubiquitin system proteolysis in Saccharomyces cerevisiae. EMBO J. 1998;17:2759–2766. doi: 10.1093/emboj/17.10.2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover JR, Lindquist S. Hsp104, Hsp70, and Hsp40: a novel chaperone system that rescues previously aggregated proteins. Cell. 1998;94:73–82. doi: 10.1016/s0092-8674(00)81223-4. [DOI] [PubMed] [Google Scholar]
- Guerriero CJ, Brodsky JL. The delicate balance between secreted protein folding and endoplasmic reticulum-associated degradation in human physiology. Physiol Rev. 2012;92:537–576. doi: 10.1152/physrev.00027.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerriero CJ, Reutter KR, Augustine AA, Preston GM, Weiberth KF, Mackie TD, Cleveland-Rubeor HC, Bethel NP, Callenberg KM, Nakatsukasa K, et al. Transmembrane helix hydrophobicity is an energetic barrier during the retrotranslocation of integral membrane ERAD substrates. Mol Biol Cell. 2017;28:2076–2090. doi: 10.1091/mbc.E17-03-0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerriero CJ, Weiberth KF, Brodsky JL. Hsp70 targets a cytoplasmic quality control substrate to the San1p ubiquitin ligase. J Biol Chem. 2013;288:18506–18520. doi: 10.1074/jbc.M113.475905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- Habeck G, Ebner FA, Shimada-Kreft H, Kreft SG. The yeast ERAD-C ubiquitin ligase Doa10 recognizes an intramembrane degron. J Cell Biol. 2015;209:261–273. doi: 10.1083/jcb.201408088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S, Liu Y, Chang A. Cytoplasmic Hsp70 promotes ubiquitination for endoplasmic reticulum-associated degradation of a misfolded mutant of the yeast plasma membrane ATPase, PMA1. J Biol Chem. 2007;282:26140–26149. doi: 10.1074/jbc.M701969200. [DOI] [PubMed] [Google Scholar]
- Haslbeck M, Braun N, Stromer T, Richter B, Model N, Weinkauf S, Buchner J. Hsp42 is the general small heat shock protein in the cytosol of Saccharomyces cerevisiae. EMBO J. 2004;23:638–649. doi: 10.1038/sj.emboj.7600080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haslbeck M, Miess A, Stromer T, Walter S, Buchner J. Disassembling protein aggregates in the yeast cytosol. The cooperation of Hsp26 with Ssa1 and Hsp104. J Biol Chem. 2005;280:23861–23868. doi: 10.1074/jbc.M502697200. [DOI] [PubMed] [Google Scholar]
- Hebert DN, Garman SC, Molinari M. The glycan code of the endoplasmic reticulum: asparagine-linked carbohydrates as protein maturation and quality-control tags. Trends Cell Biol. 2005;15:364–370. doi: 10.1016/j.tcb.2005.05.007. [DOI] [PubMed] [Google Scholar]
- Helenius A, Simons K. Solubilization of membranes by detergents. Biochim Biophys Acta. 1975;415:29–79. doi: 10.1016/0304-4157(75)90016-7. [DOI] [PubMed] [Google Scholar]
- Hidvegi T, Ewing M, Hale P, Dippold C, Beckett C, Kemp C, Maurice N, Mukherjee A, Goldbach C, Watkins S, et al. An autophagy-enhancing drug promotes degradation of mutant alpha1-antitrypsin Z and reduces hepatic fibrosis. Science. 2010;329:229–232. doi: 10.1126/science.1190354. [DOI] [PubMed] [Google Scholar]
- Huyer G, Piluek WF, Fansler Z, Kreft SG, Hochstrasser M, Brodsky JL, Michaelis S. Distinct machinery is required in Saccharomyces cerevisiae for the endoplasmic reticulum-associated degradation of a multispanning membrane protein and a soluble luminal protein. J Biol Chem. 2004;279:38369–38378. doi: 10.1074/jbc.M402468200. [DOI] [PubMed] [Google Scholar]
- Johnson PR, Swanson R, Rakhilina L, Hochstrasser M. Degradation signal masking by heterodimerization of MATα2 and MATa1 blocks their mutual destruction by the ubiquitin-proteasome pathway. Cell. 1998;94:217–227. doi: 10.1016/s0092-8674(00)81421-x. [DOI] [PubMed] [Google Scholar]
- Kabani M, Beckerich JM, Brodsky JL. Nucleotide exchange factor for the yeast Hsp70 molecular chaperone Ssa1p. Mol Cell Biol. 2002;22:4677–4689. doi: 10.1128/MCB.22.13.4677-4689.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaganovich D, Kopito R, Frydman J. Misfolded proteins partition between two distinct quality control compartments. Nature. 2008;454:1088–1095. doi: 10.1038/nature07195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaimal JM, Kandasamy G, Gasser F, Andreasson C. Coordinated Hsp110 and Hsp104 Activities Power Protein Disaggregation in Saccharomyces cerevisiae. Mol Cell Biol. 2017:37. doi: 10.1128/MCB.00027-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJE. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protocols. 2015;10:845–858. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolling R, Hollenberg CP. The ABC-transporter Ste6 accumulates in the plasma membrane in a ubiquitinated form in endocytosis mutants. EMBO J. 1994;13:3261–3271. doi: 10.1002/j.1460-2075.1994.tb06627.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse KB, Brodsky JL, McCracken AA. Characterization of an ERAD gene as VPS30/ATG6 reveals two alternative and functionally distinct protein quality control pathways: one for soluble Z variant of human alpha-1 proteinase inhibitor (A1PiZ) and another for aggregates of A1PiZ. Mol Biol Cell. 2006;17:203–212. doi: 10.1091/mbc.E04-09-0779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kummer E, Szlachcic A, Franke KB, Ungelenk S, Bukau B, Mogk A. Bacterial and Yeast AAA+ Disaggregases ClpB and Hsp104 Operate through Conserved Mechanism Involving Cooperation with Hsp70. J Mol Biol. 2016;428:4378–4391. doi: 10.1016/j.jmb.2016.09.003. [DOI] [PubMed] [Google Scholar]
- Leichner GS, Avner R, Harats D, Roitelman J. Dislocation of HMG-CoA reductase and Insig-1, two polytopic endoplasmic reticulum proteins, en route to proteasomal degradation. Mol Biol Cell. 2009;20:3330–3341. doi: 10.1091/mbc.E08-09-0953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy F, Johnston JA, Varshavsky A. Analysis of a conditional degradation signal in yeast and mammalian cells. Eur J Biochem. 1999;259:244–252. doi: 10.1046/j.1432-1327.1999.00024.x. [DOI] [PubMed] [Google Scholar]
- Lim PJ, Danner R, Liang J, Doong H, Harman C, Srinivasan D, Rothenberg C, Wang H, Ye Y, Fang S. Ubiquilin and p97/VCP bind erasin, forming a complex involved in ERAD. J Cell Biol. 2009;187:201–217. doi: 10.1083/jcb.200903024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loayza D, Tam A, Schmidt WK, Michaelis S. Ste6p mutants defective in exit from the endoplasmic reticulum (ER) reveal aspects of an ER quality control pathway inSaccharomyces cerevisiae. Mol Biol Cell. 1998;9:2767–2784. doi: 10.1091/mbc.9.10.2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason AB, Allen KE, Slayman CW. C-terminal truncations of the Saccharomyces cerevisiae PMA1 H+-ATPase have major impacts on protein conformation, trafficking, quality control, and function. Eukaryot Cell. 2014;13:43–52. doi: 10.1128/EC.00201-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melville MW, McClellan AJ, Meyer AS, Darveau A, Frydman J. The Hsp70 and TRiC/CCT chaperone systems cooperate in vivo to assemble the von Hippel-Lindau tumor suppressor complex. Mol Cell Biol. 2003;23:3141–3151. doi: 10.1128/MCB.23.9.3141-3151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger MB, Maurer MJ, Dancy BM, Michaelis S. Degradation of a cytosolic protein requires endoplasmic reticulum-associated degradation machinery. J Biol Chem. 2008;283:32302–32316. doi: 10.1074/jbc.M806424200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelis S, Herskowitz I. The a-factor pheromone of Saccharomyces cerevisiae is essential for mating. Mol Cell Biol. 1988;8:1309–1318. doi: 10.1128/mcb.8.3.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumberg D, Muller R, Funk M. Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene. 1995;156:119–122. doi: 10.1016/0378-1119(95)00037-7. [DOI] [PubMed] [Google Scholar]
- Nakatsukasa K, Huyer G, Michaelis S, Brodsky JL. Dissecting the ER-associated degradation of a misfolded polytopic membrane protein. Cell. 2008;132:101–112. doi: 10.1016/j.cell.2007.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsukasa K, Kamura T. Subcellular Fractionation Analysis of the Extraction of Ubiquitinated Polytopic Membrane Substrate during ER-Associated Degradation. PLoS One. 2016;11:e0148327. doi: 10.1371/journal.pone.0148327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal SE, Mak R, Bennett EJ, Hampton R. A “Retrochaperone” Function for Cdc48: the Cdc48 Complex is Required for Retrotranslocated ERAD-M Substrate Solubility. J Biol Chem. 2017 doi: 10.1074/jbc.M116.770610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Needham PG, Mikoluk K, Dhakarwal P, Khadem S, Snyder AC, Subramanya AR, Brodsky JL. The thiazide-sensitive NaCl cotransporter is targeted for chaperone-dependent endoplasmic reticulum-associated degradation. J Biol Chem. 2011;286:43611–43621. doi: 10.1074/jbc.M111.288928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nillegoda NB, Kirstein J, Szlachcic A, Berynskyy M, Stank A, Stengel F, Arnsburg K, Gao X, Scior A, Aebersold R, et al. Crucial HSP70 co-chaperone complex unlocks metazoan protein disaggregation. Nature. 2015;524:247–251. doi: 10.1038/nature14884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa SI, Fewell SW, Kato Y, Brodsky JL, Endo T. Molecular chaperones in the yeast endoplasmic reticulum maintain the solubility of proteins for retrotranslocation and degradation. J Cell Biol. 2001;153:1061–1070. doi: 10.1083/jcb.153.5.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogen-Shtern N, Ben David T, Lederkremer GZ. Protein aggregation and ER stress. Brain Res. 2016;1648:658–666. doi: 10.1016/j.brainres.2016.03.044. [DOI] [PubMed] [Google Scholar]
- Okuda-Shimizu Y, Hendershot LM. Characterization of an ERAD pathway for nonglycosylated BiP substrates, which require Herp. Mol Cell. 2007;28:544–554. doi: 10.1016/j.molcel.2007.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pechmann S, Frydman J. Evolutionary conservation of codon optimality reveals hidden signatures of cotranslational folding. Nat Struct Mol Biol. 2013;20:237–243. doi: 10.1038/nsmb.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovich E, Kerem A, Frohlich KU, Diamant N, Bar-Nun S. AAA-ATPase p97/Cdc48p, a cytosolic chaperone required for endoplasmic reticulum-associated protein degradation. Mol Cell Biol. 2002;22:626–634. doi: 10.1128/MCB.22.2.626-634.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rampelt H, Kirstein-Miles J, Nillegoda NB, Chi K, Scholz SR, Morimoto RI, Bukau B. Metazoan Hsp70 machines use Hsp110 to power protein disaggregation. EMBO J. 2012;31:4221–4235. doi: 10.1038/emboj.2012.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao H, Sastry A. Recognition of specific ubiquitin conjugates is important for the proteolytic functions of the ubiquitin-associated domain proteins Dsk2 and Rad23. J Biol Chem. 2002;277:11691–11695. doi: 10.1074/jbc.M200245200. [DOI] [PubMed] [Google Scholar]
- Ravid T, Kreft SG, Hochstrasser M. Membrane and soluble substrates of the Doa10 ubiquitin ligase are degraded by distinct pathways. EMBO J. 2006;25:533–543. doi: 10.1038/sj.emboj.7600946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raviol H, Sadlish H, Rodriguez F, Mayer MP, Bukau B. Chaperone network in the yeast cytosol: Hsp110 is revealed as an Hsp70 nucleotide exchange factor. EMBO J. 2006;25:2510–2518. doi: 10.1038/sj.emboj.7601139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med. 2004;10(Suppl):S10–17. doi: 10.1038/nm1066. [DOI] [PubMed] [Google Scholar]
- Ruan L, Zhou C, Jin E, Kucharavy A, Zhang Y, Wen Z, Florens L, Li R. Cytosolic proteostasis through importing of misfolded proteins into mitochondria. Nature. 2017;543:443–446. doi: 10.1038/nature21695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggiano A, Foresti O, Carvalho P. ER-associated degradation: Protein quality control and beyond. J Cell Biol. 2014;204:869–879. doi: 10.1083/jcb.201312042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggiano A, Mora G, Buxo L, Carvalho P. Spatial control of lipid droplet proteins by the ERAD ubiquitin ligase Doa10. EMBO J. 2016;35:1644–1655. doi: 10.15252/embj.201593106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner L, Trott A, Goeckeler JL, Brodsky JL, Morano KA. The function of the yeast molecular chaperone Sse1 is mechanistically distinct from the closely related hsp70 family. J Biol Chem. 2004;279:21992–22001. doi: 10.1074/jbc.M313739200. [DOI] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein A, Ruggiano A, Carvalho P, Rapoport TA. Key steps in ERAD of luminal ER proteins reconstituted with purified components. Cell. 2014;158:1375–1388. doi: 10.1016/j.cell.2014.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taxis C, Hitt R, Park SH, Deak PM, Kostova Z, Wolf DH. Use of modular substrates demonstrates mechanistic diversity and reveals differences in chaperone requirement of ERAD. J Biol Chem. 2003;278:35903–35913. doi: 10.1074/jbc.M301080200. [DOI] [PubMed] [Google Scholar]
- Ursic D, Culbertson M. The yeast homolog to mouse Tcp-1 affects microtubule-mediated processes. Mol Cell Biol. 1991;11:2629–2640. doi: 10.1128/mcb.11.5.2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ushioda R, Hoseki J, Nagata K. Glycosylation-independent ERAD pathway serves as a backup system under ER stress. Mol Biol Cell. 2013;24:3155–3163. doi: 10.1091/mbc.E13-03-0138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vashist S, Ng DT. Misfolded proteins are sorted by a sequential checkpoint mechanism of ER quality control. J Cell Biol. 2004;165:41–52. doi: 10.1083/jcb.200309132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Bergen M, Barghorn S, Biernat J, Mandelkow EM, Mandelkow E. Tau aggregation is driven by a transition from random coil to beta sheet structure. Biochim Biophys Acta. 2005;1739:158–166. doi: 10.1016/j.bbadis.2004.09.010. [DOI] [PubMed] [Google Scholar]
- Wang Q, Liu Y, Soetandyo N, Baek K, Hegde R, Ye Y. A ubiquitin ligase-associated chaperone holdase maintains polypeptides in soluble states for proteasome degradation. Mol Cell. 2011;42:758–770. doi: 10.1016/j.molcel.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiertz EJ, Jones TR, Sun L, Bogyo M, Geuze HJ, Ploegh HL. The human cytomegalovirus US11 gene product dislocates MHC class I heavy chains from the endoplasmic reticulum to the cytosol. Cell. 1996;84:769–779. doi: 10.1016/s0092-8674(00)81054-5. [DOI] [PubMed] [Google Scholar]
- Xu C, Wang S, Thibault G, Ng DT. Futile protein folding cycles in the ER are terminated by the unfolded protein O-mannosylation pathway. Science. 2013a;340:978–981. doi: 10.1126/science.1234055. [DOI] [PubMed] [Google Scholar]
- Xu Y, Liu Y, Lee J-g, Ye Y. A ubiquitin-like domain recruits an oligomeric chaperone to a retrotranslocation complex in endoplasmic reticulum-associated degradation. J Biol Chem. 2013b;288:18068–18076. doi: 10.1074/jbc.M112.449199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y, Meyer HH, Rapoport TA. The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature. 2001;414:652–656. doi: 10.1038/414652a. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Michaelis S, Brodsky JL. CFTR expression and ER-associated degradation in yeast. Cystic Fibrosis Methods and Protocols. 2002:257–265. doi: 10.1385/1-59259-187-6:257. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Nijbroek G, Sullivan ML, McCracken AA, Watkins SC, Michaelis S, Brodsky JL. Hsp70 molecular chaperone facilitates endoplasmic reticulum-associated protein degradation of cystic fibrosis transmembrane conductance regulator in yeast. Mol Biol Cell. 2001;12:1303–1314. doi: 10.1091/mbc.12.5.1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C, Slaughter BD, Unruh JR, Guo F, Yu Z, Mickey K, Narkar A, Ross RT, McClain M, Li R. Organelle-based aggregation and retention of damaged proteins in asymmetrically dividing cells. Cell. 2014;159:530–542. doi: 10.1016/j.cell.2014.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie 1. A homology model for the second nucleotide binding domain (NBD2) of Ste6, focusing on the amino acids at which truncations were examined in the Ste6 and Chimera A proteins, Related to Figures 1 and 2.
Movie 2. Hsp104-GFP is diffuse after proteasome inhibition when cells contain an empty vector control, Related to Figure 5.
Movie 3. Hsp104-GFP localizes to punctae upon proteasome inhibition when cells express Q394X, Related to Figure 5.
Movie 4. Hsp104-GFP does not co-localize with the ER after proteasome inhibition when cells contain an empty vector control, Related to Figure 5.
Movie 5. Hsp104-GFP positive punctae co-localize with the ER after proteasome inhibition when cells express Q394X, Related to Figure 5.