

Abstract
Bifidobacteria colonize the human gastrointestinal tract, vagina, oral cavity and breast milk. They influence human physiology and nutrition through health-promoting effects, play an important role as primary colonizers of the newborn gut, and contribute to vaginal microbiome homeostasis by producing lactic acid. Nevertheless, the mechanisms by which bifidobacteria are transmitted from mother to infant remains in discussion. Moreover, studies have suggested that Bifidobacterium spp. have specializations for gut colonization, but comparisons of strains of the same bifidobacteria species from different body sites are lacking. Here, our objective was to compare the genomes of Bifidobacterium breve (n = 17) and Bifidobacterium longum (n = 26) to assess whether gut and vaginal isolates of either species were distinguishable based on genome content. Comparison of the general genome features showed that vaginal and gut isolates did not differ in size, GC content, number of genes and CRISPR, either for B. breve or B. longum. Average nucleotide identity and whole genome phylogeny analysis revealed that vaginal and gut isolates did not cluster separately. Vaginal and gut isolates also had a similar COG (Cluster of Orthologous Group) category distribution. Differences in the accessory genomes between vaginal and gut strains were observed, but were not sufficient to distinguish isolates based on their origin. The results of this study support the hypothesis that the vaginal and gut microbiomes are colonized by a shared community of Bifidobacterium, and further emphasize the potential importance of the maternal vaginal microbiome as a source of infant gut microbiota.
Introduction
Bifidobacterium are Gram-positive, non-motile, anaerobic, non-spore forming rod-shaped bacteria. They belong to the Bifidobacteriaceae family and are characterized by high genomic G+C content (55–67 mol%) [1]. Bifidobacteria are common members of the gastrointestinal tract (GIT), representing ~10% of the adult gut microbiota [2, 3]. Bifidobacteria also colonize the human vagina, oral cavity, and breast milk [1, 4]. Beyond the human microbiome, they can be found in sewage, fermented milk products, and the gastrointestinal tracts of animals including insects [5]. Although members of the genus inhabit a wide range of habitats, most Bifidobacterium spp. are host-specific [1].
Bifidobacteria have been the subject of numerous studies due their probiotic potential and health promoting characteristics, such as immune modulation [6, 7], production of bacteriocins [8] and inhibition of pathogens [9–11]. The precise mechanisms by which bifidobacteria provide these benefits, however, are not fully understood. Bifidobacteria also play an important role as one of the primary colonizers of the neonatal gut, representing 60–91% of fecal bacteria in breast-fed infants [12, 13]. This early microbial colonization is an essential step in the modulation of the neonatal immune system [14, 15] and may be influenced by mode of delivery (vaginal or C-section) and feeding type (breast milk or formula) [16–18].
Studies of the vaginal microbiota using deep sequencing methods have shown that bifidobacteria are the dominant bacteria in the vaginal microbiomes of some reproductive aged women [19]. Culture-based studies have subsequently confirmed that some vaginal Bifidobacterium spp. (Actinobacteria phylum) are able to perform a protective role similar to the beneficial lactobacilli (Firmicutes phylum), which includes the production of lactic acid and hydrogen peroxide [19, 20]. These features prevent the overgrowth of unwanted bacteria and help to maintain the homeostasis of the vaginal microbiome. Common species detected in the vaginal microbiome, based on sequencing [19, 21, 22] and culture methods [19, 23], include Bifidobacterium breve, B. longum, B. bifidum, and B. dentium. Based on the recognition that both the gut and vagina harbour bifidobacteria, a large number of studies have been conducted to investigate and demonstrate the influence of maternal microbiota on the neonatal gut microbiome [4, 17, 18, 24–30], but the specific contribution of each microbial community (vaginal, gut and to a smaller extent, milk) in the mother-to-infant bifidobacteria transmission remains in discussion.
Comparison of Bifidobacterium spp. genome sequences has revealed a high degree of conservation and synteny across their genomes [31]. Nevertheless, phenotypic differences have been described among bifidobacteria species indicating some degree of species adaptation [32]. One indication of these adaptations is the greater percentage of the bifidobacteria genome involved in carbohydrate metabolism in comparison with the genomes of other members of the gut microbiota. Specifically, genome analysis has demonstrated that B. longum has the ability to metabolize a variety of complex sugars, which gives an ecological advantage in the GIT and evidently reflects its gut adaptation [33]. It has also been shown that B. longum subsp. infantis has adaptations for milk utilization [34], and that B. dentium is adapted for the colonization of the human oral cavity [35]. These observations, however, mostly reflect the differences among different bifidobacteria species rather than among strains of the same species from different ecological niches.
The importance of bifidobacteria in adult and neonatal health is evident, although evidence supporting the importance of vertical transmission of maternal microbiota in establishing these populations remains inconclusive. The overlapping occurrence of bacterial species in different body sites is one of the challenges in studying vertical transmission. While several bifidobacteria adaptations for survival in the GIT and oral cavity have been proposed, no study has addressed possible adaptations to the genital tract, in particular, by comparing strains of the same species from different body sites. Here, we compared the genomes of gut and vaginal isolates of B. breve and B. longum to identify evidence of strain specialization that could indicate if vaginal and gut strains represent two distinct, adapted subpopulations. Improved knowledge of bifidobacteria ecology is necessary for a better understanding of mother-to-infant bifidobacteria transmission, a potentially important determinant of infant health.
Material and methods
Bacterial strains
A total of 16 bifidobacteria (7 Bifidobacterium breve and 9 Bifidobacterium longum) were sequenced in this study. Genome sequences from an additional 27 Bifidobacterium spp. were acquired from GenBank for comparative analysis (Table 1). All 16 genomes sequenced in this study were from strains originally isolated from human vaginal microbiota as part of previous studies [19, 20]. Gut and vaginal isolates were from different individuals. Genomic DNA was isolated from cultures grown in Modified Reinforced Clostridial broth using a modified salting out procedure [36]. The integrity of DNA was verified by electrophoresis on 1% agarose gels. Genomic DNA was quantified using Qubit dsDNA BR assay kit (Invitrogen, Burlington, Ontario) and DNA quality was assessed by the A260/A280 ratio using a spectrophotometer.
Table 1. General features of the bifidobacteria genomes included in this study.
| Strain | Ecological origin | Size (Mb) | GC (%) | Total genes | tRNA | CRISPR | N50 / N90 | GenBank | Status (n. scaffolds/ contigs) |
|---|---|---|---|---|---|---|---|---|---|
| B. breve (n = 17) | |||||||||
| B.b.# 30–1 | Vagina | 2.54 | 59.8 | 2346 | 67 | 1 | 125268 / 38442 | * | D (32 / 5) |
| B.b. 91–1 | Vagina | 2.24 | 58.0 | 2215 | 50 | 0 | 188169 / 56499 | * | D (21 / 1) |
| B.b. 322–1 | Vagina | 2.24 | 58.5 | 2026 | 47 | 1 | 29270 / 9437 | * | D (67 / 36) |
| B.b. W20-13 | Vagina | 2.30 | 58.3 | 2113 | 50 | 1 | 43276 / 19775 | * | D (49 / 16) |
| B.b. W56 | Vagina | 2.36 | 57.8 | 2207 | 49 | 2 | 126027 / 46295 | * | D (24 / 13) |
| B.b. N6D12 | Vagina | 2.29 | 58.5 | 2097 | 50 | 1 | 119740 / 29243 | * | D (30 / 12) |
| B.b. 12–4 | Vagina | 2.26 | 58.7 | 2148 | 48 | 3 | 148702 / 48178 | * | D (25 / 7) |
| B.b. ACS-071-V-Sch8b | Vagina | 2.33 | 58.7 | 2046 | 53 | 4 | NA | CP002743 | C |
| B.b. JCM 1192T | Infant feces | 2.27 | 58.9 | 2039 | 53 | 0 | NA | AP012324 | C |
| B.b. UCC2003 | Infant feces | 2.42 | 58.7 | 2131 | 54 | 3 | NA | CP000303 | C |
| B.b. JCM 7017 | Infant feces | 2.29 | 58.7 | 1995 | 54 | 2 | NA | CP006712 | C |
| B.b. JCM 7019 | Adult feces | 2.36 | 58.6 | 2133 | 56 | 2 | NA | CP006713 | C |
| B.b. NCFB 2258 | Infant feces | 2.32 | 58.7 | 2036 | 53 | 2 | NA | CP006714 | C |
| B.b. 689b | Infant feces | 2.33 | 58.7 | 2052 | 53 | 0 | NA | CP006715 | C |
| B.b. S27 | Infant feces | 2.29 | 58.7 | 2005 | 53 | 2 | NA | CP006716 | C |
| B.b. CBT BR3 | Infant feces | 2.43 | 59.1 | 2195 | 54 | 2 | NA | CP010413 | C |
| B.b. LMC520 | Infant feces | 2.40 | 59.0 | 2146 | 55 | 1 | NA | CP019596 | C |
| B. longum (n = 26) | |||||||||
| B.l.# 239–2 | Vagina | 2.28 | 59.1 | 2060 | 50 | 8 | 33459 / 10497 | * | D (60 / 56) |
| B.l. W35-1 | Vagina | 2.30 | 60.3 | 2133 | 49 | 1 | 17831 / 5459 | * | D (81 / 132) |
| B.l. N2E12 | Vagina | 2.34 | 59.1 | 2114 | 51 | 5 | 80659 / 26287 | * | D (43 / 12) |
| B.l. N2F05 | Vagina | 2.33 | 59.6 | 2076 | 55 | 2 | 105634 / 41749 | * | D (30 / 13) |
| B.l. N2G10 | Vagina | 2.28 | 59.6 | 2200 | 38 | 0 | 9284 / 3230 | * | D (165 / 186) |
| B.l. N3A01 | Vagina | 2.31 | 59.7 | 2056 | 51 | 5 | 82928 / 40450 | * | D (33 / 13) |
| B.l. N3E01-2 | Vagina | 2.34 | 59.6 | 2167 | 51 | 3 | 202175 / 50971 | * | D (23 / 1) |
| B.l. N5E04 | Vagina | 2.46 | 58.3 | 2219 | 45 | 0 | 154725 / 47782 | * | D (27 / 3) |
| B.l. N6D05 | Vagina | 2.72 | 60.4 | 2641 | 77 | 3 | 25630 / 6992 | * | D (80 / 105) |
| B.l.l. JCM 1217T | Infant feces | 2.39 | 60.3 | 2090 | 73 | 1 | NA | AP010888 | C |
| B.l.l. JDM301 | Gut | 2.48 | 59.8 | 2156 | 55 | 1 | NA | CP002010 | C |
| B.l.l. BBMN68 | Elderly feces | 2.27 | 59.9 | 1959 | 54 | 3 | NA | CP002286 | C |
| B.l.l. KACC 91563 | Infant feces | 2.40 | 59.8 | 2064 | 56 | 1 | NA | CP002794 | C |
| B.l.l. GT15 | Adult feces | 2.34 | 60.0 | 2021 | 56 | 1 | NA | CP006741 | C |
| B.l.l. NCIMB 8809 | Human feces | 2.34 | 60.1 | 2037 | 56 | 1 | NA | CP011964 | C |
| B.l.l. CCUG 30698 | Gut | 2.46 | 60.2 | 2184 | 72 | 1 | NA | CP011965 | C |
| B.l.l. AH1206 | Infant feces | 2.42 | 60.2 | 2179 | 60 | 2 | NA | CP016019 | C |
| B.l. NCC2705 | Infant feces | 2.26 | 60.1 | 1799 | 57 | 1 | NA | AE014295 | C |
| B.l. DJO10A | Adult feces | 2.39 | 60.1 | 2105 | 58 | 2 | NA | CP000605 | C |
| B.l. 105-A | Human feces | 2.29 | 60.1 | 1950 | 56 | 2 | NA | AP014658 | C |
| B.l. BXY01 | Gut | 2.48 | 59.8 | 2158 | 55 | 1 | NA | CP008885 | C |
| B.l. BG7 | Infant feces | 2.46 | 60.0 | 2128 | 57 | 1 | NA | CP010453 | C |
| B.l. 35624 | Gut | 2.26 | 60.0 | 1942 | 57 | 2 | NA | CP013673 | C |
| B.l.i. JCM 1222T | Infant feces | 2.83 | 59.9 | 2673 | 77 | 0 | NA | CP001095 | C |
| B.l.i. 157F | Infant feces | 2.41 | 60.1 | 2147 | 59 | 1 | NA | AP010890 | C |
| B.l.i. BT1 | Infant feces | 2.58 | 59.4 | 2308 | 56 | 1 | NA | CP010411 | C |
# B.b. = B. breve; B.l. = B. longum; B.l.l. = B. longum subsp. longum; B.l.i. = B. longum subsp. infantis.
* Genome sequenced as part of this study. T = type strain. NA = not applicable. C = complete, D = draft.
Genome sequencing and assembly
Libraries were prepared with 1 ng of genomic DNA using the Nextera XT DNA Library Preparation Kit (Illumina Inc., San Diego, CA) according to the manufacturer’s instructions. After PCR amplification and clean up, the fragment size distribution of the tagmented DNA was analyzed using the High Sensitivity DNA Analysis Kit on Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). PhiX DNA (15% (v/v)) was added to the pooled indexed libraries prior to loading onto the flow cell. The libraries were sequenced using Reagent Kit V2 (500 cycles) on Illumina Miseq platform (Illumina Inc., San Diego, CA).
Raw sequence reads were trimmed for quality using Trimmomatic [37] with a minimum read length of 40 and quality cut-off of Phred score of 20. To estimate genome coverage and calculate average insert size, reads were mapped on to the reference genome of B. breve (Genbank Accession AP012324) or B. longum (Genbank Accession NC_015067.1) using Bowtie2 [38] and the results were converted to BAM format for viewing in Qualimap v2.2.1 [39]. High quality reads were assembled with SOAPdenovo2 [40] using the estimated average insert size from Qualimap analysis.
Genome analysis
Genomes sequenced in this study were annotated using the NCBI (National Center for Biotechnology Information) Prokaryotic Genome Automatic Annotation Pipeline (PGAAP). For all other genomes, the published annotation from the NCBI Genbank or Refseq database was used. CRISPRFinder (http://crispr.i2bc.paris-saclay.fr/) was used to identify CRISPR (clustered regularly interspaced short palindromic repeats) within the genome sequences [41]. Annotated genomes were also submitted to the Joint Genome Institute (JGI - http://jgi.doe.gov/) for COG (Clusters of Orthologous Groups) category assignment.
Overall genome similarities were assessed by calculating the Average Nucleotide Identity by Mummer (ANIm) and tetranucleotide scores (tetra) within JSpecies [42]. ANIm values were visualized as heatmap, generated in R. The ‘vegdist’ function was used to calculate the Euclidean distance between the ANI divergence values, and ‘hclust’ function was used to calculate the complete linkage on the distance matrix, in R.
CSI Phylogeny was used to call SNPs (single-nucleotide polymorphisms) and infer phylogeny based on the concatenated alignment of SNPs. The following settings were used: minimum depth at SNP positions of 10; relative depth at SNP positions of 10; minimum distance between SNPs (prune) of 10; minimum SNP quality of 30; minimum read mapping quality of 25; minimum Z-score: 1.96. A maximum likelihood tree indicating the whole genome phylogeny was also computed within CSI Phylogeny [43].
Pangenome calculation based on a pairwise BLASTp comparison of all predicted proteins from all genomes and rarefaction plots were conducted using the R package micropan [44]. Gene clusters (families) were identified using complete linkage and the resulting table of gene cluster prevalence in individual genomes was used to determine the size of the core genome for each species and the distribution of gene clusters among genomes from gut and vaginal isolates. Gene cluster prevalence data was converted to a binary matrix and similarities in presence/absence patterns were calculated using the Jaccard index. Dendrograms were calculated using the unweighted pair group method with arithmetic mean (UPGMA) in DendroUPGMA ([45] http://genomes.urv.cat/UPGMA/index.php).
Results and discussion
General genome features
We performed a comparative genomic analysis between gut and vaginal isolates for two Bifidobacterium species commonly found in the human gut and vagina: B. breve and B. longum. Thus, all analysis comparing vaginal and gut strains was performed in parallel for these two species. The general features of all genomes included in this study are listed in Table 1.
Seventeen B. breve genomes were analyzed, seven of which were sequenced in this study. We also analyzed the genome sequences of twenty-six B. longum strains, nine of which were sequenced as part of this study. Genome sequence data and annotated assemblies have been deposited in GenBank under BioProject PRJNA387952. The B. breve genomes were sequenced to an average of 97-fold coverage ± 47 (range 14–270), and B. longum genomes were sequenced to an average of 57-fold coverage ± 34 (range 11–182). Currently, B. longum encompass three subspecies: -longum, -infantis and -suis, but it has been previously considered as three separate species (B. longum, B. infantis and B. suis) or as a unified species (B. infantis and B. suis were published as synonyms of B. longum) [46, 47]. Considering this controversial taxonomic history of B. longum, we opted to include in our analysis published complete genomes of B. longum of gut origin regardless of subspecies, which included (n = 8 subsp. longum, n = 3 subsp. infantis and n = 6 for which no subspecies affiliation was reported).
The average genome size of vaginal and gut B. breve was 2.32 ± 0.09 Mb and 2.34 ± 0.05 Mb, respectively; where vaginal strains 91–1 and 322–1 were the smallest (2.24 Mb) and the vaginal strain 30–1 was the largest (2.54 Mb). For B. longum, the average genome sizes of vaginal and gut isolates were 2.37 ± 0.14 Mb and 2.41 ± 0.13 Mb, respectively; the smallest genomes were represented by gut isolates NCC2705 and 35624 (2.26 Mb) and the largest genome was from JCM 1222T (2.83 Mb). All genomes analyzed had high GC content, a known characteristic of the genus Bifidobacterium and previously reported as 55–67 mol% [5]. Vaginal and gut B. breve have 58.5% and 58.8% genomic GC content, respectively; and vaginal and gut B. longum have 59.5% and 60% of GC content, respectively. There were no differences in genome size and GC content between gut and vaginal isolates, either for B. breve or B. longum (t-test, all p>0.05) (Fig 1).
Fig 1. Genome features.

Genome size and GC content of B. breve and B. longum of gut and vaginal origin.
The genomes of B. breve and B. longum contained an average of 2113 and 2137 genes, respectively, which is within the range of number of predicted genes previously reported for Bifidobacterium spp. (1369–2564 genes) [48]. Also, B. breve and B. longum contained an average of 1.6 (range 0–4) and 1.8 (range 0–8) CRISPR, respectively. The inclusion of complete and draft genomes in the analysis and the choice of particular analysis tool may have affected the CRISPR analysis; however, there was no difference in the number of genes and CRISPR between gut and vaginal isolates of either B. breve or B. longum (t-test, all p>0.05). Since gut strains are more likely to be often exposed to viral infection, a greater CRISPR activity might be expected within the gut isolates, which was not the case. CRISPR have been implicated in chromosomal rearrangement, modulation of expression of neighbouring genes, target for DNA binding proteins, and DNA repair [49]. More recently, it has been shown to act as the defense mechanism in bacteria against phages and plasmids by providing adaptive immunity [50]. Notably, vaginal B. longum 239–2 and N2E12 had a total of 8 and 5 CRISPR, respectively, which suggest these strains had an active CRISPR immune system against potentially damaging foreign DNA. Previous studies have shown that CRISPR systems are frequent and diverse in the genus Bifidobacterium and differences in the frequency of CRISPR-Cas systems (CRISPR and CRISPR-associated proteins) within species are an indication that CRISPR distribution is strain-dependent [51, 52].
The overall genome sequence similarity between vaginal and gut strains was assessed based on average nucleotide identity (ANI). A hierarchal clustering based on the distance matrix of ANI divergence values was computed and visualized as a heatmap (Fig 2). All ANI values between isolates of the same species were >95%, consistent with their identification as members of the same species [53]. The genomes of vaginal B. breve did not cluster separately from the gut isolates (Fig 2A). Two clusters of B. longum were apparent but these do not correspond to subspecies longum or infantis based on genomes where a subspecies was identified (Fig 2B). For example, in the upper cluster of five genomes, JDM301 is reported to be subsp. longum, JCM 1222T and BT1 are reported to be subsp. infantis and no subspecies information is available for 239–2 or BXY01 (Table 1). This result is not unexpected since genome-wide ANI may not provide sufficient resolution to discern subspecies and identities between the two B. longum clusters were >95%. Similarly, vaginal B. longum did not form a separate cluster from the gut isolates (Fig 2B). Therefore, vaginal and gut isolates could not be distinguished based on their overall nucleotide identity, either for B. breve or B. longum. The relatedness of isolates was also assessed by calculating the tetranucleotide scores (tetra), which led to similar results as ANI (data not shown).
Fig 2. Average nucleotide identity (ANI).

Heatmap of ANI values between genomes of gut and vaginal origin. (A) B. breve; (B) B. longum. T = type strain.
Whole genome phylogeny
Whole genome phylogeny of isolates was inferred based on the concatenated alignment of SNPs, and a maximum likelihood circular tree was created for phylogeny visualization (Fig 3). The comparison of all genomes revealed an average of 9307 SNPs in B. breve strains and an average of 9050 SNPs in B. longum strains. Phylogenetic analysis indicated that vaginal B. breve isolates did not cluster separately from the gut isolates (Fig 3A). Similarly, the genomes of vaginal B. longum did not form a separate cluster from the genomes of gut strains (Fig 3B).
Fig 3. Whole genome phylogenies based on SNPs.
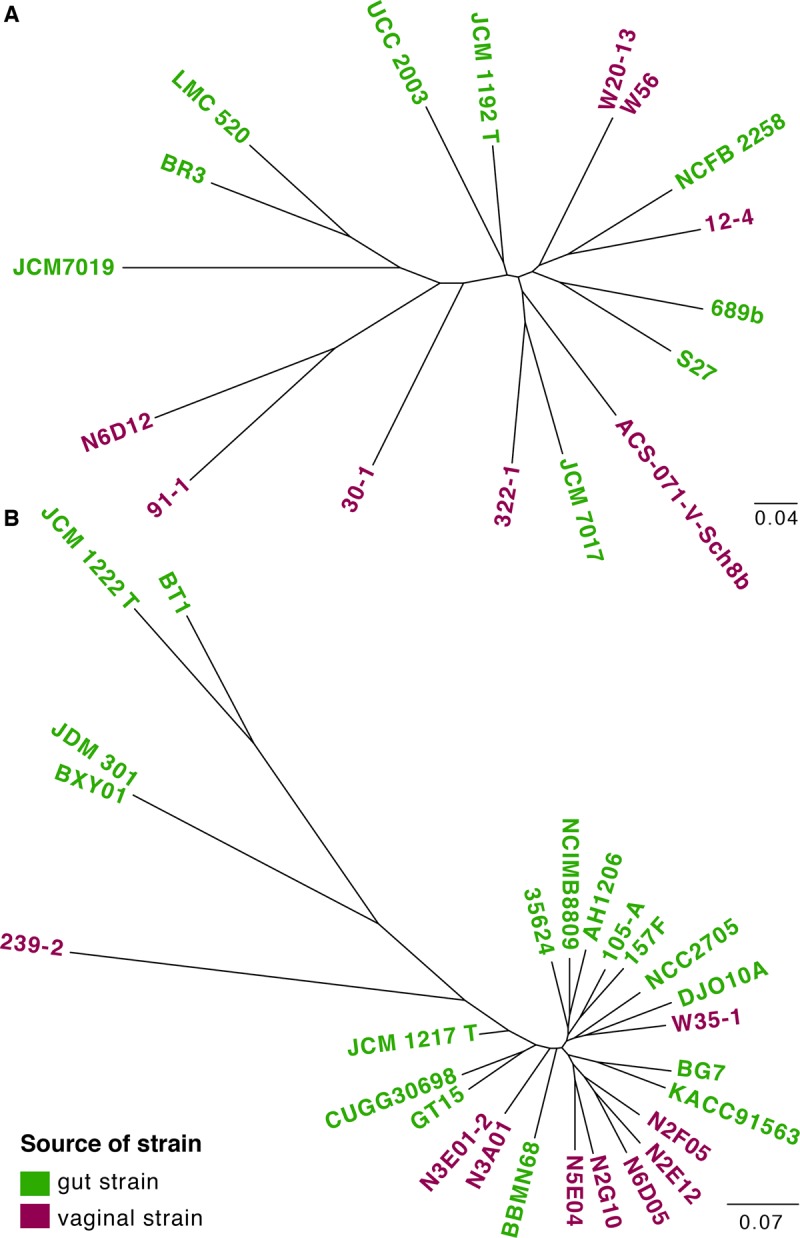
(A) B. breve; (B) B. longum. T = type strain.
COG distribution
To identify significant differences in the predicted functional repertoires of gut or vaginal isolates of B. breve or B. longum, predicted proteins were functionally categorized based on COG (Cluster of Orthologous Group) assignment and the proportions in each category were compared between vaginal and gut isolates (Fig 4). Most sequences in B. breve and B. longum were assigned to the ‘carbohydrate transport and metabolism’ category, followed by ‘amino acid transport and metabolism’ and ‘translation, ribosomal structure and biogenesis’ categories, which is consistent with previous reports [54, 55]. Most importantly, vaginal and gut strains did not differ in terms of COG category distribution. This does not mean that there are not differences in the specific genes within these larger functional categories, but only that the proportion of each genome encoding functions in the broad categories are similar. Considering that carbohydrates are less abundant in quantity and variety in the vagina relative to the gut, we had anticipated that if vaginal isolates have genome adaptations, these would include presence of fewer genes involved in metabolism. Our observation of similar proportions of COGs in category G (‘carbohydrate transport and metabolism’) in vaginal and gut isolates corroborates our previous observation that the carbohydrate utilization profile phenotypes of the vaginal isolates included in this study do not differ from those reported for the type strains of B. breve and B. longum [19].
Fig 4. Clusters of Orthologous Groups (COG).

COG function distribution among genomes of (A) B. breve and (B) B. longum. COG classification: [D] Cell cycle control, cell division, chromosome partitioning; [M] Cell wall/membrane/envelope biogenesis; [N] Cell motility; [O] Post-translational modification, protein turnover, and chaperones; [T] Signal transduction mechanisms; [U] Intracellular trafficking, secretion, and vesicular transport; [V] Defense mechanisms; [W] Extracellular structures; [Y] Nuclear structure; [Z] Cytoskeleton; [A] RNA processing and modification; [B] Chromatin structure and dynamics; [J] Translation, ribosomal structure and biogenesis; [K] Transcription; [L] Replication, recombination and repair; [C] Energy production and conversion; [E] Amino acid transport and metabolism; [F] Nucleotide transport and metabolism; [G] Carbohydrate transport and metabolism; [H] Coenzyme transport and metabolism; [I] Lipid transport and metabolism; [P] Inorganic ion transport and metabolism; [Q] Secondary metabolites biosynthesis, transport, and catabolism; [R] General function prediction only; [S] Function unknown; [X] Mobilome: prophages, transposons.
COGs were also investigated in terms of their presence/absence among isolates. B. breve isolates were represented by a set of 1016 COG, 489 of which were present in all strains (vaginal and gut origin). A total of 30 COG were exclusively present in vaginal isolates, i.e., they were absent in gut isolates and present in at least one vaginal isolate. Notably, the prevalence of these 30 “unique” COG among vaginal isolates was low, ranging from 12.5% (1/8) to 37.5% (3/8), suggesting it is unlikely that these rare COG represent a biologically significant vaginal adaptation. Similarly, a total of 34 COG were exclusively present in gut isolates, but their prevalence was also mostly low, ranging from 11.1% (1/9) to 33.3% (3/9), with only one exception: COG1396 (Transcriptional regulator, contains XRE-family HTH domain) that was present in 55.5% (5/9) of the gut isolates. Two additional COG showed substantial differences in distribution between vaginal and gut B. breve isolates. Glucan phosphorylase (COG0058) was present in only 12.5% (1/8) of vaginal isolates against 88.9% (8/9) of isolates of gut origin. On the other hand, the predicted ABC-type sugar transport system (permease component) (COG4158) was present in 87% (7/8) and 22.2% (2/9) of vaginal and gut isolates, respectively.
For B. longum, a total of 1128 COG were identified, 451 of which were present in all strains (vaginal and gut). Although 26 COG were exclusively present in vaginal isolates, most of them were found in only one or two vaginal isolates, with one exception: COG3695 (Alkylated DNA nucleotide flippase Atl1), which was present in 55.5% (5/9) of vaginal isolates. On the other hand, a total of 113 COG were exclusively associated with isolates of gut origin, although their prevalence was also low, ranging from 5.8% (1/17) to 29.4% (5/17). The only exception was COG1672 (Predicted ATPase), which was found in 76.5% (13/17) of gut strains. Additionally, 5 COG were more frequently found in gut isolates than vaginal isolates: COG0481 (Translation elongation factor EF-4, membrane-bound GTPase), COG0802 (tRNA A37 threonylcarbamoyladenosine biosynthesis protein TsaE), COG1225 (Peroxiredoxin), COG0159 (Tryptophan synthase alpha chain), and COG0732 (Restriction endonuclease S subunit). Four COG were more prevalent in vaginal isolates: COG1327 (Transcriptional regulator NrdR, contains Zn-riB.b.on and ATP-cone domains), COG0328 (Ribonuclease HI), COG1983 (Phage shock protein PspC (stress-responsive transcriptional regulator)), and COG0759 (Membrane-anchored protein YidD, putatitve component of membrane protein insertase Oxa1/YidC/SpoIIIJ). Although there were differences in the presence/absence of COG between vaginal and gut isolates, differences were not systematically concentrated within function categories, and were insufficient to distinguish isolates from the two body sites.
Pangenome analysis
The pangenome is defined as the entire gene set of all isolates, including genes present in all isolates (core genome) and genes present in one or some isolates (accessory genome). B. breve had a pangenome of 3773 gene clusters, consistent with a previous report (3667 in 13 genomes) [54] (Fig 5). The larger pangenome of B. longum (5609 gene clusters) reflects the inclusion of different B. longum subspecies in the analysis and is also consistent with a recent analysis of 20 B. longum genomes (5970 in 37 genomes) [56] (Fig 5). The content of pangenomes of B. breve and B. longum have recently been described in great detail [54–57] and so the focus of this analysis was on our primary objective, to determine if vaginal and gut bifidobacteria could be distinguished based on the presence/absence of components of the pangenome.
Fig 5. Rarefaction plot of B. breve and B. longum pangenome.

Accumulated number of new gene clusters plotted against the number of genomes sequentially added. One hundred permutations were performed at each step.
By strictest definition (gene must be present in all genomes included), the core genomes of B. breve and B. longum contained 916 and 835 gene clusters, respectively. There were 2192 gene clusters shared by at least one vaginal and one gut isolate of B. breve, while vaginal and gut B. longum had a total of 2659 shared gene clusters (Fig 6A). Relatively large numbers of gene clusters were identified exclusively in vaginal or gut bifidobacteria but further investigation showed that the majority of these were present in only one genome and thus could not be used to distinguish vaginal and gut isolates (Fig 6A). In fact, only two gene clusters in the B. breve pangenome, corresponding to a hypothetical protein and a putative fluoride ion transporter (crcB, Fluc family), were found in all (9/9) gut isolate genomes and none (0/8) of the vaginal isolate genomes. A crcB sequence was also found in 16/17 gut B. longum and 0/9 vaginal B. longum isolates. These small integral membrane proteins are important for counteracting toxicity of environmental fluoride anions and are widespread in bacteria [58]. Most of the gene clusters that were present in at least half of the genomes in one group and absent from genomes in the other group were unidentified (hypothetical proteins of unknown function), which is not surprising given that at least 20–40% of genes in sequenced bacterial genomes encode unknown functions [59]. Average numbers of gene clusters that were unique to one genome were 68.1 (range 6–247) for B. longum and 67.8 (range 2–228) for B. breve.
Fig 6. Shared and unique gene clusters.

(A) The numbers of shared (present in at least one genome of each group) and unique (present in at least one genome of one group) gene clusters of B. breve and B. longum isolates from vaginal and gut microbiotas. Histograms below the Venn diagrams show the distribution of group-specific gene clusters among genomes and indicate that most group-specific gene clusters are found in only one genome. (B) Relationships of genomes based on presence/absence of gene clusters identified in the pangenomes for B. breve (3773 gene clusters) and B. longum (5609 gene clusters).
To determine if the vaginal and gut bifidobacteria genomes could be differentiated based on shared and unique gene clusters, hierarchical clustering of the presence/absence patterns of gene clusters in the calculated pangenome was conducted (Fig 6B). Similar to the results of the clustering based on ANI comparisons, the vaginal and gut bifidobacteria did not form separate clusters based on core and accessory genome content, which further emphasizes that although there is strain diversity in genome content, systematic differences that differentiate isolates based on source (vaginal or gut) are not apparent.
Conclusions
In this study, we investigated the genomes of B. breve and B. longum from two different body sites of significant importance in neonatal health: gut and vagina. In all analyses, gut and vaginal strains of B. breve or B. longum were not distinguishable from each other based their genomic content. Consistent with these observations, it has been previously demonstrated that several vaginal and gut Bifidobacterium spp. did not differ based on phenotypic characteristics related to their carbohydrate fermentation patterns, lactic acid production and tolerance to low pH [19]. Our results support the hypothesis that vaginal and gut isolates represent the same bacterial population with similar genetic repertoires that allow them to efficiently colonize both body sites. The genomes included in our study are from isolates recovered from individuals of different ages around the world over many years. While this provides an opportunity to look at these species very broadly, an obvious complimentary study would be a comparison of gut and vaginal isolates from individual women, or paired samples from women and their babies. Both vaginal and gut microbiota are thought to contribute to mother-infant transmission of bifidobacteria, and recognition that vaginal and gut isolates represent the same population is an important step for future studies in maternal-neonatal health.
Acknowledgments
The authors would like to thank Champika Fernando for the help with genome sequencing and Sarah Vancuren for assistance in genome assembly.
Data Availability
All of the sequence data for the vaginal bifidobacteria genome sequencing has been deposited in GenBank under the BioProject Accession PRJNA387952.
Funding Statement
This research is supported by a Discovery Grant from the Natural Sciences and Engineering Research Council of Canada (http://www.nserc-crsng.gc.ca). ACF was supported by scholarships from the University of Saskatchewan and the Saskatchewan Innovation and Opportunity Scholarship program. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Dworkin M. The Prokaryotes Volume 3: Archaea. Bacteria: Firmicutes, Actinomycetes. 3rd ed. Dworkin M, Falkow S, Rosenberg E, Schleifer K-H, Stackebrandt E, editors. New York: Springer; 2006.
- 2.Turroni F, Ribbera A, Foroni E, van Sinderen D, Ventura M. Human gut microbiota and bifidobacteria: from composition to functionality. Antonie Van Leeuwenhoek. 2008;94(1):35–50. doi: 10.1007/s10482-008-9232-4 [DOI] [PubMed] [Google Scholar]
- 3.Arboleya S, Watkins C, Stanton C, Ross RP. Gut Bifidobacteria populations in human health and aging. Frontiers Microbiol. 2016;7:1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gomez-Gallego C, Garcia-Mantrana I, Salminen S, Collado MC. The human milk microbiome and factors influencing its composition and activity. Semin Fetal Neonatal Med. 2016;21(6):400–5. doi: 10.1016/j.siny.2016.05.003 [DOI] [PubMed] [Google Scholar]
- 5.Biavati B, Mattarelli P. Genus Bifidobacterium In: Goodfellow M, Kampfer P, Busse HJ, Trujillo ME, Suzuki K, Ludwig W, editors. Bergey’s Manual of Systematic Bacteriology. Volume 5 New York: Springer; 2012. p. 171–205. [Google Scholar]
- 6.Fanning S, Hall LJ, Cronin M, Zomer A, MacSharry J, Goulding D, et al. Bifidobacterial surface-exopolysaccharide facilitates commensal-host interaction through immune modulation and pathogen protection. Proc Natl Acad Sci U S A. 2012;109(6):2108–13. doi: 10.1073/pnas.1115621109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hart AL, Lammers K, Brigidi P, Vitali B, Rizzello F, Gionchetti P, et al. Modulation of human dendritic cell phenotype and function by probiotic bacteria. Gut. 2004;53(11):1602–9. doi: 10.1136/gut.2003.037325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martinez FA, Balciunas EM, Converti A, Cotter PD, de Souza Oliveira RP. Bacteriocin production by Bifidobacterium spp. A review. Biotchnol Adv. 2013;31(4):482–8. [DOI] [PubMed] [Google Scholar]
- 9.Bernet MF, Brassart D, Neeser JR, Servin AL. Adhesion of human bifidobacterial strains to cultured human intestinal epithelial cells and inhibition of enteropathogen-cell interactions. Appl Environ Microbiol. 1993;59(12):4121–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arboleya S, Ruas-Madiedo P, Margolles A, Solis G, Salminen S, de Los Reyes-Gavilan CG, et al. Characterization and in vitro properties of potentially probiotic Bifidobacterium strains isolated from breast-milk. Int J Food Microbiol. 2011;149(1):28–36. doi: 10.1016/j.ijfoodmicro.2010.10.036 [DOI] [PubMed] [Google Scholar]
- 11.Fukuda S, Toh H, Hase K, Oshima K, Nakanishi Y, Yoshimura K, et al. Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nature. 2011;469(7331):543–7. doi: 10.1038/nature09646 [DOI] [PubMed] [Google Scholar]
- 12.Harmsen HJ, Wildeboer-Veloo AC, Raangs GC, Wagendorp AA, Klijn N, Bindels JG, et al. Analysis of intestinal flora development in breast-fed and formula-fed infants by using molecular identification and detection methods. J Pediatr Gastroenterol Nutr. 2000;30(1):61–7. [DOI] [PubMed] [Google Scholar]
- 13.Turroni F, Peano C, Pass DA, Foroni E, Severgnini M, Claesson MJ, et al. Diversity of bifidobacteria within the infant gut microbiota. PLoS ONE. 2012;7(5):e36957 doi: 10.1371/journal.pone.0036957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rautava S, Luoto R, Salminen S, Isolauri E. Microbial contact during pregnancy, intestinal colonization and human disease. Nat Rev Gastroenterol Hepatol. 2012;9(10):565–76. doi: 10.1038/nrgastro.2012.144 [DOI] [PubMed] [Google Scholar]
- 15.Olszak T, An D, Zeissig S, Vera MP, Richter J, Franke A, et al. Microbial exposure during early life has persistent effects on natural killer T cell function. Science. 2012;336(6080):489–93. doi: 10.1126/science.1219328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Penders J, Thijs C, Vink C, Stelma FF, Snijders B, Kummeling I, et al. Factors influencing the composition of the intestinal microbiota in early infancy. Pediatrics. 2006;118(2):511–21. doi: 10.1542/peds.2005-2824 [DOI] [PubMed] [Google Scholar]
- 17.Dominguez-Bello MG, Costello EK, Contreras M, Magris M, Hidalgo G, Fierer N, et al. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci U S A. 2010;107(26):11971–5. doi: 10.1073/pnas.1002601107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Backhed F, Roswall J, Peng Y, Feng Q, Jia H, Kovatcheva-Datchary P, et al. Dynamics and stabilization of the human gut microbiome during the first year of life. Cell Host Microbe. 2015;17(5):690–703. doi: 10.1016/j.chom.2015.04.004 [DOI] [PubMed] [Google Scholar]
- 19.Freitas AC, Hill JE. Quantification, isolation and characterization of Bifidobacterium from the vaginal microbiomes of reproductive aged women. Anaerobe. 2017;47:145–56. doi: 10.1016/j.anaerobe.2017.05.012 [DOI] [PubMed] [Google Scholar]
- 20.Schellenberg JJ, Dumonceaux TJ, Hill JE, Kimani J, Jaoko W, Wachihi C, et al. Selection, phenotyping and identification of acid and hydrogen peroxide producing bacteria from vaginal samples of Canadian and East African women. PLoS ONE. 2012;7(7):e41217 doi: 10.1371/journal.pone.0041217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chaban B, Links MG, Paramel Jayaprakash T, Wagner EC, Bourque DK, Lohn Z, et al. Characterization of the vaginal microbiota of healthy Canadian women through the menstrual cycle. Microbiome. 2014;2:23 doi: 10.1186/2049-2618-2-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Freitas AC, Chaban B, Bocking A, Rocco M, Yang S, Hill JE, et al. The vaginal microbiome of healthy pregnant women is less rich and diverse with lower prevalence of Mollicutes compared to healthy non-pregnant women. Sci Rep. 2017;7:9212 doi: 10.1038/s41598-017-07790-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Verhelst R, Verstraelen H, Claeys G, Verschraegen G, Van Simaey L, De Ganck C, et al. Comparison between Gram stain and culture for the characterization of vaginal microflora: definition of a distinct grade that resembles grade I microflora and revised categorization of grade I microflora. BMC Microbiol. 2005;5:61 doi: 10.1186/1471-2180-5-61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matamoros S, Gras-Leguen C, Le Vacon F, Potel G, de La Cochetiere MF. Development of intestinal microbiota in infants and its impact on health. Trends Microbiol. 2013;21(4):167–73. doi: 10.1016/j.tim.2012.12.001 [DOI] [PubMed] [Google Scholar]
- 25.Makino H, Kushiro A, Ishikawa E, Kubota H, Gawad A, Sakai T, et al. Mother-to-infant transmission of intestinal bifidobacterial strains has an impact on the early development of vaginally delivered infant's microbiota. PLoS ONE. 2013;8(11):e78331 doi: 10.1371/journal.pone.0078331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gabriel I, Olejek A, Stencel-Gabriel K, Wielgos M. The influence of maternal vaginal flora on the intestinal colonization in newborns and 3-month-old infants. J Matern Fetal Neonatal Med. 2018;31(11):1448–53. doi: 10.1080/14767058.2017.1319352 [DOI] [PubMed] [Google Scholar]
- 27.Hunt KM, Foster JA, Forney LJ, Schutte UM, Beck DL, Abdo Z, et al. Characterization of the diversity and temporal stability of bacterial communities in human milk. PLoS ONE. 2011;6(6):e21313 doi: 10.1371/journal.pone.0021313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Milani C, Mancabelli L, Lugli GA, Duranti S, Turroni F, Ferrario C, et al. Exploring vertical transmission of Bifidobacteria from mother to child. Appl Environ Microbiol. 2015;81(20):7078–87. doi: 10.1128/AEM.02037-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Duranti S, Lugli GA, Mancabelli L, Armanini F, Turroni F, James K, et al. Maternal inheritance of bifidobacterial communities and bifidophages in infants through vertical transmission. Microbiome. 2017;5(1):66 doi: 10.1186/s40168-017-0282-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pannaraj PS, Li F, Cerini C, Bender JM, Yang S, Rollie A, et al. Association between breast milk bacterial communities and establishment and development of the infant gut microbiome. JAMA Pediatr. 2017;171(7):647–54. doi: 10.1001/jamapediatrics.2017.0378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ventura M, Canchaya C, Tauch A, Chandra G, Fitzgerald GF, Chater KF, et al. Genomics of Actinobacteria: tracing the evolutionary history of an ancient phylum. Microbiol Mol Biol Rev. 2007;71(3):495–548. doi: 10.1128/MMBR.00005-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ventura M, O'Flaherty S, Claesson MJ, Turroni F, Klaenhammer TR, van Sinderen D, et al. Genome-scale analyses of health-promoting bacteria: probiogenomics. Nature Rev Microbiol. 2009;7(1):61–71. [DOI] [PubMed] [Google Scholar]
- 33.Schell MA, Karmirantzou M, Snel B, Vilanova D, Berger B, Pessi G, et al. The genome sequence of Bifidobacterium longum reflects its adaptation to the human gastrointestinal tract. Proc Natl Acad Sci U S A. 2002;99(22):14422–7. doi: 10.1073/pnas.212527599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sela DA, Chapman J, Adeuya A, Kim JH, Chen F, Whitehead TR, et al. The genome sequence of Bifidobacterium longum subsp. infantis reveals adaptations for milk utilization within the infant microbiome. Proc Natl Acad Sci U S A. 2008;105(48):18964–9. doi: 10.1073/pnas.0809584105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ventura M, Turroni F, Zomer A, Foroni E, Giubellini V, Bottacini F, et al. The Bifidobacterium dentium Bd1 genome sequence reflects its genetic adaptation to the human oral cavity. PLoS Genet. 2009;5(12):e1000785 doi: 10.1371/journal.pgen.1000785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martin-Platero AM, Valdivia E, Maqueda M, Martinez-Bueno M. Fast, convenient, and economical method for isolating genomic DNA from lactic acid bacteria using a modification of the protein "salting-out" procedure. Anal Biochem. 2007;366(1):102–4. doi: 10.1016/j.ab.2007.03.010 [DOI] [PubMed] [Google Scholar]
- 37.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–20. doi: 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nature Meth. 2012;9(4):357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Okonechnikov K, Conesa A, Garcia-Alcalde F. Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics. 2016;32(2):292–4. doi: 10.1093/bioinformatics/btv566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience. 2012;1(1):18 doi: 10.1186/2047-217X-1-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grissa I, Vergnaud G, Pourcel C. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucl Acids Res. 2007;35:W52–W7. doi: 10.1093/nar/gkm360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Richter M, Rossello-Mora R, Oliver Glockner F, Peplies J. JSpeciesWS: a web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics. 2016;32(6):929–31. doi: 10.1093/bioinformatics/btv681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kaas RS, Leekitcharoenphon P, Aarestrup FM, Lund O. Solving the problem of comparing whole bacterial genomes across different sequencing platforms. PLoS ONE. 2014;9(8):e104984 doi: 10.1371/journal.pone.0104984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Snipen L, Liland KH. micropan: an R-package for microbial pan-genomics. BMC Bioinformatics. 2015;16:79 doi: 10.1186/s12859-015-0517-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Garcia-Vallve S, Palau J, Romeu A. Horizontal gene transfer in glycosyl hydrolases inferred from codon usage in Escherichia coli and Bacillus subtilis. Mol Biol Evol. 1999;16(9):1125–34. doi: 10.1093/oxfordjournals.molbev.a026203 [DOI] [PubMed] [Google Scholar]
- 46.Mattarelli P, Bonaparte C, Pot B, Biavati B. Proposal to reclassify the three biotypes of Bifidobacterium longum as three subspecies: Bifidobacterium longum subsp. longum subsp. nov., Bifidobacterium longum subsp. infantis comb. nov. and Bifidobacterium longum subsp. suis comb. nov. Int J Syst Evol Microbiol. 2008;58(Pt 4):767–72. doi: 10.1099/ijs.0.65319-0 [DOI] [PubMed] [Google Scholar]
- 47.Sakata S, Kitahara M, Sakamoto M, Hayashi H, Fukuyama M, Benno Y. Unification of Bifidobacterium infantis and Bifidobacterium suis as Bifidobacterium longum. Int J Syst Evol Microbiol. 2002;52(Pt 6):1945–51. doi: 10.1099/00207713-52-6-1945 [DOI] [PubMed] [Google Scholar]
- 48.Sun Z, Zhang W, Guo C, Yang X, Liu W, Wu Y, et al. Comparative genomic analysis of 45 type strains of the genus Bifidobacterium: a snapshot of its genetic diversity and evolution. PLoS ONE. 2015;10(2):e0117912 doi: 10.1371/journal.pone.0117912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Horvath P, Barrangou R. CRISPR/Cas, the immune system of bacteria and archaea. Science. 2010;327(5962):167–70. doi: 10.1126/science.1179555 [DOI] [PubMed] [Google Scholar]
- 50.Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315(5819):1709–12. doi: 10.1126/science.1138140 [DOI] [PubMed] [Google Scholar]
- 51.Briner AE, Lugli GA, Milani C, Duranti S, Turroni F, Gueimonde M, et al. Occurrence and diversity of CRISPR-Cas systems in the genus Bifidobacterium. PLoS ONE. 2015;10(7):e0133661 doi: 10.1371/journal.pone.0133661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Milani C, Lugli GA, Duranti S, Turroni F, Bottacini F, Mangifesta M, et al. Genomic encyclopedia of type strains of the genus Bifidobacterium. Appl Environ Microbiol. 2014;80(20):6290–302. doi: 10.1128/AEM.02308-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Richter M, Rossello-Mora R. Shifting the genomic gold standard for the prokaryotic species definition. Proc Nat Acad Sci USA. 2009;106(45):19126–31. doi: 10.1073/pnas.0906412106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bottacini F, O'Connell Motherway M, Kuczynski J, O'Connell KJ, Serafini F, Duranti S, et al. Comparative genomics of the Bifidobacterium breve taxon. BMC Genomics. 2014;15:170 doi: 10.1186/1471-2164-15-170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.O'Callaghan A, Bottacini F, O'Connell Motherway M, van Sinderen D. Pangenome analysis of Bifidobacterium longum and site-directed mutagenesis through by-pass of restriction-modification systems. BMC Genomics. 2015;16:832 doi: 10.1186/s12864-015-1968-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Arboleya S, Bottacini F, O'Connell-Motherway M, Ryan CA, Ross RP, van Sinderen D, et al. Gene-trait matching across the Bifidobacterium longum pan-genome reveals considerable diversity in carbohydrate catabolism among human infant strains. BMC Genomics. 2018;19(1):33 doi: 10.1186/s12864-017-4388-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lugli GA, Milani C, Turroni F, Duranti S, Mancabelli L, Mangifesta M, et al. Comparative genomic and phylogenomic analyses of the Bifidobacteriaceae family. BMC Genomics. 2017;18(1):568 doi: 10.1186/s12864-017-3955-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Baker JL, Sudarsan N, Weinberg Z, Roth A, Stockbridge RB, Breaker RR. Widespread genetic switches and toxicity resistance proteins for fluoride. Science. 2012;335(6065):233–5. doi: 10.1126/science.1215063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xu J. Microbial ecology in the age of genomics and metagenomics: concepts, tools, and recent advances. Mol Ecol. 2006;15(7):1713–31. doi: 10.1111/j.1365-294X.2006.02882.x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All of the sequence data for the vaginal bifidobacteria genome sequencing has been deposited in GenBank under the BioProject Accession PRJNA387952.