

Abstract
Halogenated pyrrolo[3,2-d]pyrimidine analogues have shown antiproliferative activity in recent studies, with cell accumulation occurring in the G2/M stage without apoptosis. However, the mechanism of action and pharmacokinetic (PK) profile of these compounds has yet to be determined. In order to investigate the PK profile of these compounds, a series of halogenated pyrrolo[3,2-d]pyrimidine compounds was synthesized and first tested for activity in various cancer cell lines followed by a mouse model. EC50 values ranged from 0.014 – 14.5 μM, and maximum tolerated doses (MTD) in mice were between 5–10 mg/kg. This indicates a wide variance in activity and toxicity that necessitates further study. To decrease toxicity, a second series of compounds was synthesized with N5 alkyl substitutions in an effort to slow the rate of metabolism, which was thought to be leading to the toxicity. The N-substituted compounds demonstrated comparable cell line activity (EC50 values between 0.83 – 7.3 μM) with significantly decreased toxicity (MTD = 40 mg/kg). Finally, the PK profile of the active N5-substituted compound shows a plasma half-life of 32.7 minutes, and rapid conversion into the parent unsubstituted analogue. Together, these data indicate that halogenated pyrrolo[3,2-d]pyrimidines present a promising lead into potent antiproliferative agents with tunable activity and toxicity, and rapid metabolism.
Keywords: Pyrrolopyrimidine, triple negative breast cancer, anticancer, prodrug, antiproliferative
Graphical Abstract
N-substituted pyrrolo[3,2-d]pyrimidines are demonstrated to possess low toxicity against a wide range of cancer cell lines while retaining activity, with IC50 values ranging from 0.014 – 14.5 μM. Further, cell studies indicate a DNA-damaging mechanism of action, and pharmacokinetic studies show rapid metabolism (t1/2 = 30.2 min). These data suggest that N-substitution offers a path towards development of anti-proliferative pyrrolo[3,2-d] pyrimidines with ‘tunable’ biological activity.

Introduction
Pyrrolo[3,2-d]pyrimidines, also known as 9-deazapurines, closely resemble natural purine nucleobases and are a medicinally important group of heterocyclic compounds. These interesting heterocycles have shown activity as bactericides[1], protozoicides[2], and as anti-tumor agents through inhibition of a variety of enzymes including tubulin[3], NEDD8-activating enzyme (NAE)[4], and several kinases.[5, 6] However, the properties of these molecules that affect their pharmacokinetic profile is under-investigated. Previous reports have found that the pyrrole moiety in pyrrolo[3,2-d]pyrimidines is partially responsible for their pharmacokinetic profile, with N5 hydroxyalkyl additions leading to increased metabolic stability.[7] A comparison of pyrrolo[3,2-d]pyrimidines and their analogous thieno[3,2-d]pyrimidine equivalents allowed assessment of thieno- and pyrrolopyrimidine compounds that were identical with the exception of the five-membered heterocycle.[8] Results of these previous studies indicated that 2,4-dichloro-5H-pyrrolo[3,2-d]pyrimidine has a cytotoxicity of 6.0 μM against L1210 leukemia cells, whereas 2,4-dichloro-thienopyrimidine has a cytotoxicity (CC50) of 0.32 μM in the same cell line.[9] This supported that the pyrrolopyrimidine structure offers decreased cellular toxicity, making it a more promising lead compound.[8–10]
In an effort to further pursue efficacious pyrrolopyrimidines with antiproliferative properties, we undertook the synthesis of a series of compounds with modifications to the pyrrole ring in the pyrrolo[3,2-d]pyrimidine scaffold. The effect of various pyrrole substituents on compound pharmacokinetics were examined, as well as the compounds’ efficacy against a wide range of cancer cell lines. The target compounds are shown in Figure 1.
Figure 1.

Target compounds with the pyrrolo[3,2-d]pyrimidine structure outlined in red.
Earlier examples of pyrrolopyrimidines such as Immunicillin H and TAK-285 have shown inhibition against various key enzymes, with Immunicillin-H acting as a potent inhibitor of purine nucleoside phosphorylase (PNP), and TAK-285 acting as an epidermal growth factor receptor (EGFR) inhibitor (Figure 2).[11] However, these compounds have extensive modifications to the pyrimidine moiety of the molecule or the addition of an azasugar. In contrast, little work has been done to explore the effects of substituents on the pyrrole ring.
Figure 2.

The pyrrolo[3,2-d]pyrimidine scaffold (outlined in red) in the PNP inhibitor Immunicillin-H (left) and the EGFR inhibitor TAK-285 (right).
In that regard, pyrrolopyrimidines have been previously reported by our laboratory as potential therapeutics against triple negative breast cancer (TNBC), leukemia, non-small cell lung cancer, and pancreatic cancer.[9] A structure-activity relationship (SAR) study found that compound 5 is a potent inhibitor of the TNBC MDA-MB-231 cell line, and further, caused an accumulation of cells in the G2/M stage without causing significant apoptosis.[9] These findings are consistent with earlier reports of other 9-deazapurines finding use as antiproliferative agents.[12]
The introduction of a halogen at C7 resulted in increased activity, with the IC50 against HeLa cells decreasing from 19±3 μM for 5 to 0.92±0.04 μM for 6. The presence of the halogen also altered the apparent mechanism of action, with the majority of cells now undergoing apoptosis, although the mechanism is still unclear.[9] These findings led us to further investigate the effect of substituents on the pyrrole moiety of these compounds with the aim to retain or increase activity while decreasing toxicity. Compounds 5–9 were synthesized and subsequently evaluated for activity against multiple cancer cell lines. Following initial cell line screening, compound 7 was chosen as a candidate for pharmacokinetic studies and further chemical modification.
The initial pharmacokinetic results for compound 7 indicated rapid metabolism, thus derivatives were designed that introduced N5 substituents with the aim to slow metabolic rate compared to the unsubstituted parent compounds. This was based on previous literature that indicates the nature of the N5 substituent can affect varied pharmacokinetic properties: decreased polarity can increase lipophilicity[6], increased polarity can increase affinity for the membrane efflux pump P-glycoprotein[13], and various substituents can alter or increase substrate-ligand hydrogen bonding.[14]
The log P values for compounds 5, 6, and 7 are estimated at 1.66, 2.49, and 3.02, respectively. This increases to 5.11 and 4.95 for 8 and 9, indicating that the N5 substituents are significantly more lipophilic than the parent compounds. Further, it was previously determined that the C7 halogen increases cellular activity and that the C4 chlorine is required for activity[9], thus it was hypothesized that introducing bulky N5 groups with the ability to sterically hinder these positions would decrease the metabolic rate while still the retaining antiproliferative activity.
Results and Discussion
Chemistry
Target compounds 5–9 were synthesized starting with commercially available 6-methyluracil, shown in Scheme 1. Nitration at C5 was accomplished by adding 6-methyluracil to a solution of concentrated sulfuric and nitric acids at 0°C, followed by quenching in ice water to give 2 as a pale yellow solid. This was followed by a modified Batcho-Leimgruber indole synthesis to prepare enamine 3.[9] The Batcho-Leimgruber process relies on the slight acidity of the 6-methyl group, which can be deprotonated to form a carbocation that is resonance stabilized by the nitro group. Nucleophilic attack on the dimethylacetal reagent results in formation of the amino side chain, which is deprotonated to result in the elimination product 3.
Ring cyclization is accomplished by reduction of the nitro moiety to an amino group, with subsequent nucleophilic attack at C8 by the amino lone pair with loss of dimethylamine. The reduction of the nitro group is accomplished by stirring 3 in acetic acid and zinc dust overnight at 80 °C, followed by filtration, washing the solids with water, then stirring the collected solids in 1M NaOH for 30 minutes at 40 °C. Filtration of the undissolved solids, then acidification of the filtrate with acetic acid results in the precipitation of solid 4.
The parent compound 5 was synthesized by first preparing the sodium salt of 4 by stirring compound 4 in 1M NaOH at 40 °C for one hour, followed by crystallization at 0°C and repeated washes with cold water and acetone. Chlorination of the sodium salt of 4 was achieved by refluxing for 5 hours in phenylphosphonic dichloride under a nitrogen atmosphere. Halogenation at C7 is achieved with N-bromosuccimimide (6) or N-iodosuccinimide (7) in anhydrous THF at 0°C. Substitution at N5 was accomplished by N5 deprotonation using sodium hydride at 0°C, followed by addition of toluenesulfonyl chloride (9) or benzylchloromethyl ether (8).
Cancer Cell Line Screening
Initial screening of 5–7 against four cancer cell lines showed IC50 activity as low as 0.014±0.001 μM (Table 1). The cell lines chosen were A549 adenocarcinomic human alveolar basal epithelial cells, MDA-MB-231 triple negative human epithelial breast cells, MIA Pa-Ca-2 human pancreatic carcinoma epithelial cells, and MOLM-14 acute monocytic leukemia cells, representing a diverse set of cancerous cell types. All of the lead compounds demonstrated activity against the four cell lines, suggesting that they have potential as broad spectrum antiproliferative agents.
Table 1.
Cancer cell line activity screening for target compounds 5–7.a
| Compound | A549 | MDA-MB-231 | MIA Pa-Ca 2 | MOLM14 |
|---|---|---|---|---|
| 5 | 14.5±3.5 | 9.7±9.7 | 5.3±1.0 | 9.5±3.1 |
| 6 | 0.05±0.02 | 0.4±0.3 | 0.014±0.001 | 0.023±0.004 |
| 7 | 1.0±0.1 | 1.2±0.4 | 0.37±0.05 | 0.55±0.07 |
| 8 | NDb | 6.1±0.4 | 7.3±0.8 | ND |
| 9 | ND | 0.79±0.04 | 0.5±0.02 | ND |
vales shown in μM.
Not Determined. Assays run in triplicate.
The highest activity was against MIA Pa-Ca-2 pancreatic cancer cells by compound 6. However, compound 7 also displayed submicromolar activity and was subsequently determined to have decreased toxicity compared at 6 (discussed in following section). These combined traits led us to utilize 7 as a lead compound, with prodrug formulation through substitution at N5. The activity of prodrug 8 decreased ten-fold compared to the parent, but it was observed that 9 was comparable (Table 1). Previous work on the pyrrolopyrimidine analogues had indicated that the presence of a halogen at C4 on the pyrimidine was essential for activity and that the presence of a halogen at C7 enhanced the activity[9] – a trend also observed in the current study.
Maximum Tolerated Dose
The maximum tolerated dose (MTD) for compounds 6 and 7 was 5 and 10 mg/kg, respectively (Table 2). The higher toxicity of compound 6 excluded it as a lead compound. In addition, although the toxicity of 6 is only 2-fold higher than 7, significant mortality rate was observed in the mouse model immediately following administration, raising concerns that mortality would interfere with effective dose studies. Comparative values for MTD of pyrrolo[3,2-d]pyrimidine compounds are difficult to find in current literature, but similar compounds have shown MTD of 5 mg/kg (LY231514, a pyrrolo[2,3-d]pyrimidine currently in Phase II)[15] and 150 mg/kg (PKI-116, a pyrrolo[2,3-d]pyrimidine currently in Phase I).[16] The MTD of compound 7 is within this comparative range and still exhibited low micromolar activity, so it was chosen for further modifications in an attempt to decrease toxicity. The addition of a toluenesulfonyl group in 9 raised the MTD from 10 to 40 mg/kg, while maintaining submicromolar levels of activity against all tested cell lines.
Table 2.
Maximum tolerated dose and half-lives of compounds 6, 7, and 9.
vales shown in mg/kg.
Not Determined.
N5 Substitution Cell Line Activity and Toxicity
Addition of a large moiety at N5 was designed to sterically hinder both the C4 chlorine and C7 iodine, as these groups impart the most activity and are likely the sites of protein-molecule interaction. Alkyl and oxyalkyl groups are common nitrogen substituents in pyrrolopyrimidines[17], and sulfonyl groups are not uncommon in FDA approved anticancer drugs, often as salts formations, lending validity to the biological compatibility of these groups.[18] Introduction of a benzyloxymethyl moiety in 8 decreased the efficacy of the compound against all cell lines, and was not pursued further. N5 toluenesulfonyl substitution in compound 9 led to a four-fold increase in maximum tolerated dose compared to parent compound 7, with comparable cell activity. This suggests that substitutions at this position can aide in decreasing cellular toxicity, or that the substitution allows the compound to bypass toxic pathways during delivery to their target site. These and other possibilities will be discussed in later sections.
Aqueous Stability
Aqueous benchtop stability tests were carried out for compounds 6 and 7, by preparing 25 μg/mL solutions in phosphate buffer at pH 5.0, 7.0, and 7.5. Aliquots taken at 0.0, 0.25, 0.5, 1, 4, 8, and 24 hours were initially analyzed by thin-layer chromatography against a verified standard, then extracted with two 250 μl portions of chloroform. Solvent removal and 1H NMR analysis of the residue was performed to verify the TLC results. Similar procedures were carried out with human serum. These experiments indicated that the compounds were stable at each pH tested, and in human serum, with no evidence of hydrolysis or decomposition.
Aqueous benchtop stability of 9 and 7 was also carried out at the CCPC, by spiking 5, 12.5, 25, 50, 125, 250 and 500 ng/mL solutions in 1:1 acetonitrile/Millipore H2O and subsequent analysis by LC/MS for both 9 and 7, quantitating using a previously constructed calibration curve of the same concentrations. Samples spiked with 7 showed expected concentration values within ±5.7% from the aqueous solution. Samples spiked with 9 gave a similar result, with a recovery deviation of ±15.8%. More importantly, there was no observed hydrolysis of 9 to 7 in aqueous solution.
Mouse Model Pharmacokinetic Studies
Mouse studies on 7 were carried out at The Cancer Center Pharmacology Core (CCPC, Colorado State University) by injection of 10 mg/kg of the compound, followed by euthanization at 15, 30, 60, 90, 120, 240, and 480 minutes. The results of the analysis are shown in Figure 3A. The compound is rapidly metabolized in vivo, with a half-life of 30.7 minutes (Table 2 and Figure 3A). Mouse studies on 9 were performed in the same manner and the results are shown in Figure 3B. The half-life of 9 is similar to the parent compound, at 32.7 minutes (Table 2 and Figure 3B).
Figure 3.

Pharmacokinetics of A) compound 7, and B) compound 9. Half-life of the prodrug 9 is similar to the parent 7.
Plasma Stability
Plasma stability was carried out at the CCPC by spiking mouse plasma with 500 ng/mL of 9 and analyzing for both 9 and 7 at 0, 5, 15, 30, 45, 60, and 90 minutes. Significant conversion of 9 to 7 over the time frame of the experiment was observed as shown in Figure 4. This is a strong indication that plasma enzymes are capable of metabolizing the prodrug to its active form. Supporting this hypothesis is the analysis of 7 in plasma, which shows excellent stability with a deviation of ±5.7% over the time frame investigated.
Figure 4.
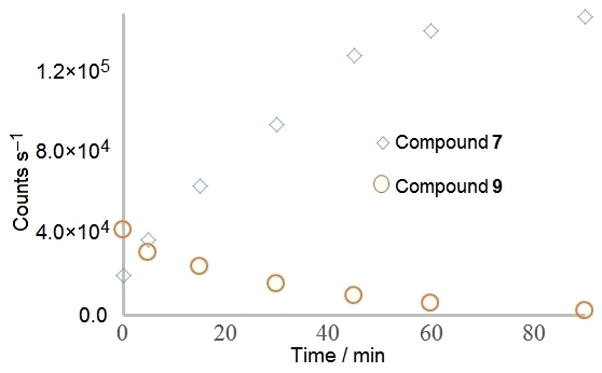
In vitro conversion of compound 9 to compound 7. Data is reported in analyte counts to control for variation between standard curves for the two compounds.
DNA Damage Assay
It was hypothesized that the compounds are acting as DNA alkylators since they are highly electrophilic. This theory was tested by staining MOLM-14 cells for γ-H2Ax (Figure 5). The phosphorylated histone protein γ-H2Ax is formed immediately following double-stranded DNA cleavage, and is known to be an initial step in recruitment of local DNA repair proteins.[19] Fluorescently-tagged antibody staining for γ-H2Ax allows visualization of the extent of DNA damage in cell cultures. The cells were grown in 0.5 and 4 μM 7 for 24 hours and show significant DNA damage (Figure 5B and C). Results indicated that compound 7 appears to be acting as a potent DNA damaging agent.
Figure 5.

DNA damage assay of compound 7 by staining MOLM-14 leukemia cells for phosphorylated γ-H2Ax. A) DMSO control; B) 500 nM 7 (1× IC50 at 24 Hours); C) 4 μM 7 (6× IC50 at 24 Hours); D) Positive control (6 gy radiation).
Conclusions
Initial screening of 5–7 against six cancer cell lines showed IC50 values as low as 0.014±0.0 μM (Table 1). All of the compounds demonstrated activity against all four cell lines, indicating that they have potential as broad spectrum antiproliferative agents. However, the best activity overall was displayed against MIA Pa-Ca-2 pancreatic cancer cells.
Maximum tolerated dose of 6, 7, and 9 was between 5–40 mg/kg, indicative of high toxicity but not outside the range of current epigenetic therapeutics such as paclitaxel (25 mg/kg), docetaxel (20 mg/kg) and vinorelbine (2.5 mg/kg)[20], as well as experimental antiproliferative compound LY231514 (5 mg/kg).[15] However, as these other clinical compounds are all semi-synthetic, complex molecules with different mechanisms of action than DNA damage, it is difficult to compare clinical results between them and the fully synthetic pyrrolo[3,2-d]pyrimidines.
The parent compound 7 displays rapid pharmacokinetics (Figure 3A), with little to no detection of the compound in mouse plasma within 70 minutes. Prodrugs 8 and 9 were designed to decrease the metabolic rate by both sterically hindering access to the reactive C4 and C7 positions as well as by acting as a labile leaving group following metabolism to the active compound 7.
Compound 9 proved stable in aqueous buffer for up to 24 hours, but was undetectable in biological media within 1 hour (Figure 4). A pharmacokinetic profile supported the serum stability studies, indicating a half-life 30.7 minutes for compound 7 and a half-life of 32.7 minutes for 9. This indicates that the N5 substituent does not act as metabolically desired, and suggests that delivery of the compounds to the target cells remains a limiting factor in the use of these compounds as potential antiproliferative agents. The plasma studies confirm that the breakdown product of 9 is the active compound 7.
Halogenated pyrrolo[2,3-d]pyrimidine analogues have been shown to be potent antiproliferative agents against a wide spectrum of cancer cell lines. The effective dose of these compounds is in the low micromolar to nanomolar range, representing a promising avenue of lead compounds. Two disadvantages for these compounds are their toxicity and rapid metabolism. However, by manipulation of the prodrug moieties at the N5 position, both of these have been remediated significantly. Furthermore, this position has not been fully explored in the literature, opening the possibility for further modifications that can potentially decrease the toxicity and improve the pharmacokinetic profile. This would allow the drugs to be more effectively delivered to the target cells in vivo before undergoing metabolism to their active form.
Experimental Section
Cell Lines and Culturing
The human AML cell-line, MOLM-14, was a gift from Dr. Mark Levis from Johns Hopkins University. The human pancreatic MIA Pa-Ca-2, human breast MDA-MB-231 and Non-small cell lung A549 cell lines were purchased from ATCC (ATCC, Manassas, VA). Cell lines were grown in 37 °C with 5% CO2 atmosphere with RPMI 1640 (Life technologies, Carlsbad, CA) or DMEM (Cellgro Mediatech; Manassas, VA) supplemented with heat-inactivated 10% (V/V) fetal bovine serum (Hyclone; Fisher Scientific, Pittsburgh, PA) and 1% glutamine (Cellgro Mediatech). Cell lines were grown and maintained according to ATCC recommendations.
IC50 Proliferation Assay
Cell lines were seeded into 96-well plates the afternoon prior to treatment. Approximately 18 hours later, compounds were semi-serially diluted in dimethyl sulfoxide (DMSO) and then growth medium, and added to cells. Plates were incubated for 72 hours for cell lines and 48 hours for primary cells prior to addition of water-soluble tetrazolium (WST-1) (Clontech, Mountain View, CA). Plates were read after 4 additional hours of incubation at 37 °C using a BioTek Synergy HT plate reader (BioTek, Winooski, VT). Data was analyzed and graphed using GraphPad Prism Software (GraphPad, La Jolla, CA).
Gamma-H2Ax Assay in MOLM-14 Cells
The evening prior to treatment, MOLM-14 cells were seeded at a density of 1×105 cells/mL in RPMI 1640/10%FBS. The following morning, cells were treated with either DMSO, 7 at 1× or 6× IC50, or 6.0 Gy gamma irradiation (followed by a one-hour incubation period). After 24 hours (or 1 hour for gamma-irradiated cells), cells were collected, washed with PBS containing 2% serum, and spun onto cytospin slides at a density of 1.5×105 cells/slide. Slides were fixed for 15 minutes in 4% PFA followed by 3–15 minute washes in PBS. Cells were subsequently permeabilized with permeabilization buffer (50 mM NaCl, 3 mM MgCl2, 10 mM HEPES, 200 mM sucrose and 0.5% Triton X-100 in PBS), and blocked overnight with 10% FBS in PBS containing 0.1% Triton X-100. Slides were subsequently washed, incubated with Gamma-H2Ax primary antibody (Millipore) and goat anti-mouse Dylight secondary antibody (Invitrogen) and mounted with Vectashield containing DAPI (VWR). After drying overnight, slides were sealed with nail polish and observed under a fluorescent scope.
Maximum Tolerated Dose Assay
The tolerability of 6,7, and 9 was tested in female nude mice ages 6 weeks old. Note this strain and sex of mice was used since female nude mice would be used in future efficacy experiments. Mice were dosed via intraperitoneal (IP) administration (10% DMSO/10% Tween 20/80% saline vehicle) 5 days per week for two consecutive weeks and then monitored for two additional weeks. If a single mouse in a group lost greater than 20% body weight, the dose was considered toxic.
Pharmacokinetic Studies
Compounds 7 and 9 were measured using liquid chromatography coupled to tandem mass spectrometry (LC/MS/MS). The analysis was carried out in the Pharmacology Shared Resource (University of Colorado Cancer Center) located in the Flint Animal Cancer Center at the Veterinary Teaching Hospital at Colorado State University. An LC/MS/MS based assay was developed and validated for the analysis of compounds in mouse plasma.
Sample Extraction
Blank mouse plasma was used for the construction of calibration and quality control standards and defined amounts of 7 or 9 added in concentrations ranging from 10–50,000 ng/mL. Samples were prepared for analysis by acetonitrile (ACN) precipitation of plasma proteins. Briefly, 5 (in 50:50 ACN:water) was added as an internal standard to prepared standard or unknown plasma samples at a final concentration of 1000 ng/ml followed by the addition of an equal volume of ACN. Samples were then mixed on a vortex mixer for 10 minutes followed by centrifugation at 14,000 rpm in a microcentrifuge. The supernatants were collected and transferred to HPLC vials for analysis.
LC/MS/MS Analysis
Aliquots (60 μL) were injected for LC/MS/MS analysis. Guidelines for batch acceptance and assay validation were based on accepted practices for quantitative chromatographic assays (Viswanathan et al., 2007), and the assay accuracy and precision were calculated to be 92.6 % ± 5.0 (CV%) for 7 and 90.3% ± 10.2 for 9 (CV%). The reference standard range used was from 1–1,000 ng/mL and quality control samples monitored at 5, 25, and 500 ng/mL for 7 and 10–10,000 ng/mL and quality control samples monitored at 50, 250 and 5,000 ng/mL for 9. Positive ion electrospray ionization mass spectra were obtained with a 3200 Q-TRAP triple quadrupole mass spectrometer with a turbo ionspray source interfaced to an Agilent 1200 Series Binary Pump SL liquid chromatography system and HTC-PAL autosampler. Chromatography was performed on a Waters Sunfire C18, 5 μm, 4.6 × 50 mm column using a gradient of acetonitrile with 0.1% formic acid (solvent A) and 0.1% formic acid (solvent B). The initial flow rate was 40% A:60% B with a linear gradient starting at 0.1 minutes to 98% A:2% B over 1 minute (9) or 2.5 minutes (7) followed by holding at 98% A for 0.75 minutes for 7 and returning to the original 40% A:60% B over 2 minutes. The flow rate was 1 mL/minute, and the total analysis time was 5–6 minutes. The mass spectrometer settings were (7/9): turbo ionspray temperature, 625°C/550°C; ion-spray voltage, 4750/5500 V; with declustering potential, entrance potential, collision energy, collision cell exit potential, and collision cell entrance potential optimized for each ion pair. Analytes were quantified by area of the signal obtained by monitoring the transitions for 7 at m/z 314.0 → 187.2, m/z 314.0 → 287.0, m/z 314.0 → 243.2 and m/z 316.0 → 189.3, for 9 at m/z 467.8 → 313.1 and m/z 467.8 →119.2 and the internal standard (KSR01) at m/z 205.0 → 169.1 and comparison to a standard curve constructed using known standards.
Aqueous Stability Studies
Compounds 6–9 were dissolved in anhydrous DMSO (Sigma-Aldrich, St. Louis, MO) to a final concentration of 1 mg/mL. Phosphate buffer solutions at pH 5.0, 7.0, and 7.5 were prepared and spiked with at 25 μg/mL and stirred at room temperature. 500 μl aliquots taken at 0, 0.25, 0.5, 1, 4, 8, and 24 hours were initially analyzed by thin-layer chromatography against a verified standard, then extracted with two 250 μl portions of chloroform. Solvent removal and 1H NMR analysis of the residue was performed to verify the TLC results. Similar procedures were carried out with pooled human serum (K3 EDTA, Innovative Research, Novi, MI).
Synthesis
6-Methyl-5-nitropyrimidine-2,4(1H,3H)-dione (2)
6-methyluracil (Sigma-Aldrich, St. Louis, MO) (10g, 79.3 mmol) was dissolved in concentrated sulfuric acid at 0 °C, at a rate that maintained the temperature below 20 °C. After complete dissolution, concentrated nitric acid (6 mL) was added dropwise, keeping the temperature below 15 °C. The reaction was brought to room temperature then stirred for 1 h, followed by carefully pouring the reaction mixture into 200 mL of ice-water slurry. The resultant yellow suspension was stirred vigorously for 30 min, then solids collected by vacuum filtration. Solids were washed with ice-cold water until the filtrate was no longer acidic, then dried under vacuum to yield a pale yellow powder (10.8g, 80%). 1H NMR (400 MHz, DMSO-d6): δ 11.76–11.74 (d, 2H), δ 2.28 (s, 3H). 13C NMR (100 MHZ, DMSO-d6): δ156.8, 154.6, 149.5, 127.7, 17.2. APCI-MS m/z for C5H5N3O4 calcd. at 171.03, found 171.91 (M+H)
(E)-6-(2-(Dimethylamino)vinyl)-5-nitropyrimidine-2,4(1H,3H)-dione (3)
6-methyl-5-nitropyrimidine-2,4(1H,3H)-dione (5g, 29.2 mmol) suspended in 25 mL of anhydrous DMF, then warmed to 80 °C under N2. N,N-dimethylformamide-dimethylacetal (8.75 mL, 65.7 mmol) added and the resultant orange solution stirred for 15 min, then heat raised to 140 °C for 30 min. Green/yellow solids formed during this time. Reaction cooled to room temperature, then to 0 °C, followed by vacuum filtration to obtain green-yellow solid that was washed with cold EtOAc and dried under vacuum (3.92 g, 60%). 1H NMR (400 MHz, DMSO-d6): δ10.97 (s, 1H), δ10.67 (s, 1H), δ8.06–8.03 (d, 1H, J=13.28 Hz), δ5.35–5.32 (d, 1H, J=13.13 Hz), δ3.11 (s, 3H), δ2.85 (s, 3H). 13C NMR (100 MHZ, DMSO-d6): δ157.3, 153.7, 151.1, 149.9, 119.1, 81.5, 45.3, 37.3. APCI-MS m/z for C8H10N4O4 calcd. at 226.07, found 226.93 (M+H)
1H-Pyrrolo[3,2-d]pyrimidine-2,4(3H,5H)-dione (4)
(E)-6-(2-(dimethylamino)vinyl)-5-nitropyrimidine-2,4(1H,3H)-dione (5g, 22.1 mmol) was suspended in glacial acetic acid, heated to 80 °C, then −100 mesh zinc dust (7g, 110 mmol) added in five portions over 1h. The reaction was stirred overnight to give a pale yellow to white suspension. Solids were collected by vacuum filtration, dried on filter for 30 minutes, then placed in a beaker and rinsed with cold water. Undissolved solids were collected by vacuum filtration, dried on filter for 45 min, then transferred to a flask with 25 mL of 1M NaOH. The suspension was heated to 80 °C and stirred for 30 minutes, then insoluble material removed by filtration. Glacial acetic acid was added to filtrate until pH=5, then precipitated solids collected by vacuum filtration and washed with ice-cold water and ethanol and dried under vacuum to obtain an off-white powder (2.35g, 70%). %). 1H NMR (400 MHz, DMSO-d6): δ11.81 (s, 1H), δ10. 72 (s, 1H) δ10.54 (s, 1H), δ7.10–7.09 (d, 1H, J=2.7 Hz)), δ5.79 (s, 1H). 13C NMR (100 MHZ, DMSO-d6): δ 156.3, 152.0, 135.1, 127.5, 110.9, 96.5. APCI-MS m/z for C6H5N3O2 calcd. at 151.04, found 151.95 (M+H)
2,4-Dichloro-5H-pyrrolo[3,2-d]pyrimidine (5)
1H-pyrrolo[3,2-d]pyrimidine-2,4(3H,5H)-dione (2g, 13.2 mmol) was dissolved in 15 mL of 1M NaOH at 40 °C and stirred for 30 min, then cooled to room temperature then 0 °C. White crystalline solid collected by vacuum filtration and washed with cold methanol and acetone, then dried under vacuum. Dry solids were suspended in neat phenylphosphonic dichloride (10 mL) and heated to 175 °C to obtain a brown solution. The reaction was stirred for 5 h, then carefully poured into 50 mL of ice-water slurry and stirred for 30 min. The resultant aqueous suspension was extracted with three 25 mL portions of EtOAc, then organics combined and washed with multiple portions of sat. sodium bicarbonate until the pH=7. Organic layer then dried with brine and MgSO4, and loaded onto Celite. The product was purified using silica column chromatography eluting with 4:1 hexanes:EtOAc to obtain a pale yellow powder (1.5 g, 60%). Rf = 0.5 (3:1 hexanes:EtOAc) 1H NMR (400 MHz, CDCl3): δ12.75 (s, 1H), δ8.07–8.06 (d, 1H, J=2.76 Hz), δ6.70–6.69 (d, 1H, J=3.2). 13C NMR (100 MHZ, CDCl3): δ153.2, 150.2, 143.3, 134.9, 123.9, 103.1. APCI-MS m/z for C6H3Cl2N3 calcd. at 186.97, found 187.84 (M+H). Anal. calcd. for C6H3Cl2N3: C, 38.33; H, 1.61; N, 22.35; Cl, 37.71. Found: C, 38.33; H, 1.65; N, 22.27; Cl, 37.73.
7-Bromo-2,4-dichloro-5H-pyrrolo[3,2-d]pyrimidine (6)
2,4-dichloro-5H-pyrrolo[3,2-d]pyrimidine (250 mg, 0.937 mmol) was dissolved in anhydrous THF at room temperature under N2. N-bromosuccinimide (208 mg, 1.17 mmol) was added directly and reaction stirred under N2 for 3 h, upon which TLC indicated reaction completion. The solvent was removed and residue dissolved in EtOAc. This organic solution was washed with aq. Na2S2O3, water, and brine, then dried over MgSO4. Organics were loaded onto Celite and product was purified using silica column chromatography eluting with 4:1 hexanes:EtOAc to obtain a pale orange powder (225 mg, 90%). Rf = 0.65 (3:1 hexanes:EtOAc) 1H NMR (400 MHz, DMSO-d6): δ13.19 (s, 1H), 8.32 (s, 1H). 13C NMR (100 MHZ, DMSO-d6): δ151.9, 150.1, 144.2, 133.0, 123.6, 92.2. APCI-MS m/z for C6H2BrCl2N3 calcd. at 264.88, found 265.77 (M+H). Anal. calcd. for C6H2BrCl2N3: C, 27.00; H, 0.76; N, 15.74. Found: C, 27.02; H, 0.78; N, 15.71.
2,4-Dichloro-7-iodo-5H-pyrrolo[3,2-d]pyrimidine (7)
2,4-dichloro-5H-pyrrolo[3,2-d]pyrimidine (250 mg, 0.937 mmol) was dissolved in anhydrous THF at room temperature under N2. N-iodosuccinimide (263 mg, 1.17 mmol) added directly and reaction stirred under N2 for 2 h, upon which TLC indicated reaction completion. The solvent was removed and residue dissolved in EtOAc. This organic solution was washed with aq. Na2S2O3, water, and brine, then dried over MgSO4. Organics were loaded onto Celite and product was purified using silica column chromatography eluting with 4:1 hexanes:EtOAc to obtain a pale yellow powder (250 mg, 85%). Rf = 0.68 (3:1 hexanes:EtOAc) 1H NMR (400 MHz, DMS0-d6): δ13.17 (s, 1H), 8.26 (s, 1H). 13C NMR (100 MHZ, DMSO-d6): δ152.9, 150.1, 143.3, 139.0, 123.9, 56.3. APCI-MS m/z for C6H2Cl2IN3 calcd. at 312.87, found 313.77 (M+H). ). Anal. calcd. for C6H2ICl2N3: C, 22.96; H, 0.65; N, 13.39. Found: C, 22.97; H, 0.65; N, 13.37.
5-((Benzyloxy)methyl)-2,4-dichloro-7-iodo-pyrrolo[3,2-d]pyrimidine (8)
2,4-dichloro-7-iodo-5H-pyrrolo[3,2-d]pyrimidine (100 mg, 0.318 mmol) was dissolved in 1:1 THF:DMF (anhydrous) at room temperature under N2. NaH (60% in mineral oil, 16 mg, 0.398 mmol) was added directly and reaction stirred for 1.5 h. Benzyl chloromethyl ether (73 μL, 0.398 mmol) was added and reaction stirred for 3 h, upon which TLC indicated reaction completion. The solvent was removed and residue dissolved in EtOAc and washed with water and brine, then dried over MgSO4. Organics were loaded onto Celite and product purified using silica column chromatography eluting with 5:1 hexanes:EtOAc to obtain a white powder (124 mg, 90%). 1H NMR (400 MHz, CDCl3): δ7.63 (s, 1H), δ7.29–7.28 (m, 3H), δ7.25–7.22 (m, 2H), δ5.79 (s, 2H), δ4.54 (s, 2H). 13C NMR (100 MHZ, CDCl3): δ155.4, 150.4, 146.1, 144.8, 143.6, 128.5, 128.1, 127.9, 123.6, 86.1, 77.9, 70.4, 58.8. APCI-MS m/z for C14H10Cl2IN3O calcd. at 432.92, found 434.06 (M+H). ). Anal. calcd. for C14H10Cl2IN3O: C, 38.74; H, 2.32; N, 9.68. Found: C, 38.74; H, 2.34; N, 9.67.
2,4-Dichloro-7-iodo-5-tosyl-pyrrolo[3,2-d]pyrimidine (9)
2,4-dichloro-7-iodo-5H-pyrrolo[3,2-d]pyrimidine (100 mg, 0.318 mmol) was dissolved in 1:1 THF:DMF (anhydrous) at room temperature under N2. NaH (60% in mineral oil, 16 mg, 0.398 mmol) was added directly and reaction stirred for 1.5 h. p-Toluenesulfonyl chloride (76 mg, 0.398 mmol) was added and the reaction stirred overnight. The solvent was removed, and the residue dissolved in EtOAc and washed with water and brine, then dried over MgSO4. Organics were loaded onto Celite and product purified using silica column chromatography eluting with 5:1 hexanes:EtOAc to obtain a white powder (126 mg, 85%). 1H NMR (400 MHz, CDCl3): δ8.82 (s, 1H), δ7.94–7.92 (d, 2H, J=8 Hz), δ7.46–7.44 (d, 2H, J=8 Hz), δ2.43 (s, 3H). 13C NMR (100 MHZ, CDCl3): δ158.1, 154.2, 146.9, 140.9, 137.1, 134.5, 130.5, 127.9, 64.1, 21.9. APCI-MS m/z for C13H8Cl2IN3O2S calcd. at 466.9, found 467.97 (M+H). Anal. calcd. for C14H8Cl2IN3O2S: C, 33.36; H, 1.72; N, 8.98. Found: C, 33.36; H, 1.74; N, 8.96.
Supplementary Material
Scheme 1.

Synthesis of target compounds 5–9. Reagents and Conditions: a) H2SO4, HNO3, rt, 80%; b) DMF·DMA, DMF, 80 °C, 60% ; c) AcOH, Zn, rt, 70%; d) PhPOCl2, reflux, 60% ; e) NBS or NIS, THF, 0°C to rt, 85–90%; f) NaH, Tos-Cl or BOM-Cl, DMF, 0°C to rt, 85–90%.
Acknowledgments
The authors thank the National Institutes of Health for funding - NIH T32 GM066706 (KSR and BMC); NIH R01 GM076345 (KSR) and the Maryland Innovation Initiative (KSR).
Footnotes
Supporting information for this article is given via a link at the end of the document
References
- 1.Trzoss M, Bensen DC, Li X, Chen Z, Lam T, Zhang J, Creighton CJ, Cunningham ML, Kwan B, Stidham M, Nelson K, Brown-Driver V, Castellano A, Shaw KJ, Lightstone FC, Wong SE, Nguyen TB, Finn J, Tari LW. Bioorg Med Chem Lett. 2013;23(5):1537–1543. doi: 10.1016/j.bmcl.2012.11.073. [DOI] [PubMed] [Google Scholar]; Tari LW, Trzoss M, Bensen DC, Li X, Chen Z, Lam T, Zhang J, Creighton CJ, Cunningham ML, Kwan B, Stidham M, Shaw KJ, Lightstone FC, Wong SE, Nguyen TB, Nix J, Finn J. Bioorg Med Chem Lett. 2013;23(5):1529–1536. doi: 10.1016/j.bmcl.2012.11.032. [DOI] [PubMed] [Google Scholar]; Gangjee A, Devraj R, McGuire JJ, Kisliuk RL. J Med Chem. 1995;38(22):4495–4502. doi: 10.1021/jm00022a015. [DOI] [PubMed] [Google Scholar]
- 2.Srinivasan P, Yasgar A, Luci DK, Beatty WL, Hu X, Andersen J, Narum DL, Moch JK, Sun H, Haynes JD, Maloney DJ, Jadhav A, Simeonov A, Miller LH. Nat Commun. 2013;4:2261. doi: 10.1038/ncomms3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scott R, Karki M, Reisenauer MR, Rodrigues R, Dasari R, Smith WR, Pelly SC, van Otterlo WA, Shuster CB, Rogelj S, Magedov IV, Frolova LV, Kornienko A. ChemMedChem. 2014;9(7):1428–1435. doi: 10.1002/cmdc.201300532. [DOI] [PMC free article] [PubMed] [Google Scholar]; Gangjee A, Pavana RK, Li W, Hamel E, Westbrook C, Mooberry SL. Pharm Res. 2012;29(11):3033–3039. doi: 10.1007/s11095-012-0816-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, Brownell JE, Burke KE, Cardin DP, Critchley S, Cullis CA, Doucette A, Garnsey JJ, Gaulin JL, Gershman RE, Lublinsky AR, McDonald A, Mizutani H, Narayanan U, Olhava EJ, Peluso S, Rezaei M, Sintchak MD, Talreja T, Thomas MP, Traore T, Vyskocil S, Weatherhead GS, Yu J, Zhang J, Dick LR, Claiborne CF, Rolfe M, Bolen JB, Langston SP. Nature. 2009;458(7239):732–736. doi: 10.1038/nature07884. [DOI] [PubMed] [Google Scholar]
- 5.Chen Z, Venkatesan AM, Dehnhardt CM, Ayral-Kaloustian S, Brooijmans N, Mallon R, Feldberg L, Hollander I, Lucas J, Yu K, Kong F, Mansour TS. J Med Chem. 2010;53(8):3169–3182. doi: 10.1021/jm901783v. [DOI] [PubMed] [Google Scholar]; Oguro Y, Miyamoto N, Okada K, Takagi T, Iwata H, Awazu Y, Miki H, Hori A, Kamiyama K, Imamura S. Bioorg Med Chem. 2010;18(20):7260–7273. doi: 10.1016/j.bmc.2010.08.017. [DOI] [PubMed] [Google Scholar]; Iwata H, Imamura S, Hori A, Hixon MS, Kimura H, Miki H. Bioorg Med Chem. 2011;19(18):5342–5351. doi: 10.1016/j.bmc.2011.08.002. [DOI] [PubMed] [Google Scholar]
- 6.Kawakita Y, Banno H, Ohashi T, Tamura T, Yusa T, Nakayama A, Miki H, Iwata H, Kamiguchi H, Tanaka T, Habuka N, Sogabe S, Ohta Y, Ishikawa T. J Med Chem. 2012;55(8):3975–3991. doi: 10.1021/jm300185p. [DOI] [PubMed] [Google Scholar]
- 7.Ishikawa T, Seto M, Banno H, Kawakita Y, Oorui M, Taniguchi T, Ohta Y, Tamura T, Nakayama A, Miki H, Kamiguchi H, Tanaka T, Habuka N, Sogabe S, Yano J, Aertgeerts K, Kamiyama K. J Med Chem. 2011;54(23):8030–8050. doi: 10.1021/jm2008634. [DOI] [PubMed] [Google Scholar]
- 8.Temburnikar KW, Zimmermann SC, Kim NT, Ross CR, Gelbmann C, Salomon CE, Wilson GM, Balzarini J, Seley-Radtke KL. Bioorg Med Chem. 2014;22(7):2113–2122. doi: 10.1016/j.bmc.2014.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Temburnikar KW, Ross CR, Wilson GM, Balzarini J, Cawrse BM, Seley-Radtke KL. Bioorg Med Chem. 2015;23(15):4354–4363. doi: 10.1016/j.bmc.2015.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xie H, Zeng L, Zeng S, Lu X, Zhang G, Zhao X, Cheng N, Tu Z, Li Z, Xu H, Yang L, Zhang X, Huang M, Zhao J, Hu W. Eur J Med Chem. 2012;52:205–212. doi: 10.1016/j.ejmech.2012.03.015. [DOI] [PubMed] [Google Scholar]; Bugge S, Buene AF, Jurisch-Yaksi N, Moen IU, Skjønsfjell EM, Sundby E, Hoff BH. Eur J Med Chem. 2016;107:255–274. doi: 10.1016/j.ejmech.2015.11.012. [DOI] [PubMed] [Google Scholar]; Zhang Q, Hu Z, Shen Q, Chen Y, Lu W. Molecules. 2017;22(5) doi: 10.3390/molecules22050788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dowty ME, Lin J, Ryder TF, Wang W, Walker GS, Vaz A, Chan GL, Krishnaswami S, Prakash C. Drug Metab Dispos. 2014;42(4):759–773. doi: 10.1124/dmd.113.054940. [DOI] [PubMed] [Google Scholar]; Sogabe S, Kawakita Y, Igaki S, Iwata H, Miki H, Cary DR, Takagi T, Takagi S, Ohta Y, Ishikawa T. ACS Med Chem Lett. 2013;4(2):201–205. doi: 10.1021/ml300327z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pudziuvelyte E, Ríos-Luci C, León LG, Cikotiene I, Padrón JM. Bioorg Med Chem. 2009;17(14):4955–4960. doi: 10.1016/j.bmc.2009.05.078. [DOI] [PubMed] [Google Scholar]
- 13.Raub TJ. Mol Pharm. 2006;3(1):3–25. doi: 10.1021/mp0500871. [DOI] [PubMed] [Google Scholar]
- 14.Baraldi PG, Romagnoli R, Saponaro G, Aghazadeh Tabrizi M, Baraldi S, Pedretti P, Fusi C, Nassini R, Materazzi S, Geppetti P, Preti D. Bioorg Med Chem. 2012;20(5):1690–1698. doi: 10.1016/j.bmc.2012.01.020. [DOI] [PubMed] [Google Scholar]
- 15.John W, Picus J, Blanke CD, Clark JW, Schulman LN, Rowinsky EK, Thornton DE, Loehrer PJ. Cancer. 2000;88(8):1807–1813. [PubMed] [Google Scholar]
- 16.Mosley RT, Edwards TE, Murakami E, Lam AM, Grice RL, Du J, Sofia MJ, Furman PA, Otto MJ. J Virol. 2012;86(12):6503–6511. doi: 10.1128/JVI.00386-12. [DOI] [PMC free article] [PubMed] [Google Scholar]; Brandt R, Wong AM, Hynes NE. Oncogene. 2001;20(39):5459–5465. doi: 10.1038/sj.onc.1204709. [DOI] [PubMed] [Google Scholar]
- 17.De Coen LM, Heugebaert TS, García D, Stevens CV. Chem Rev. 2016;116(1):80–139. doi: 10.1021/acs.chemrev.5b00483. [DOI] [PubMed] [Google Scholar]
- 18.Spirli C, Morell CM, Locatelli L, Okolicsanyi S, Ferrero C, Kim AK, Fabris L, Fiorotto R, Strazzabosco M. Hepatology. 2012;56(6):2363–2374. doi: 10.1002/hep.25872. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]; Kadivar A, Kamalidehghan B, Javar HA, Davoudi ET, Zaharuddin ND, Sabeti B, Chung LY, Noordin MI. PLoS One. 2015;10(6):e0126874. doi: 10.1371/journal.pone.0126874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuo LJ, Yang LX. In Vivo. 2008;22(3):305–309. [PubMed] [Google Scholar]
- 20.Nervi C, De Marinis E, Codacci-Pisanelli G. Clinical Epigenetics. 2015;7 doi: 10.1186/s13148-015-0157-2. [DOI] [PMC free article] [PubMed] [Google Scholar]; Sirotnak FM, Zakowski MF, Miller VA, Scher HI, Kris MG. Clin Cancer Res. 2000;612:4885–4892. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.