

Abstract
We hypothesize that epileptiform abnormalities (EA) in the electroencephalopgram (EEG) during the acute period following traumatic brain injury (TBI) independently predict first-year post-traumatic epilepsy (PTE1). We analyzed PTE1 risk factors in two cohorts matched for TBI severity and age (n=50). EA independently predict risk for PTE1 (OR 3.16[0.99 11.68]); subdural hematoma is another independent risk factor (OR 4.13 [1.18 39.33]). Differences in EA rates are apparent within 5 days following TBI. Our results suggest increased EA prevalence identifies patients at increased risk for PTE1, and that EA acutely post-TBI can identify patients most likely to benefit from anti-epileptogenesis drug trials.
Keywords: epilepsy, traumatic brain injury, EEG
Abbreviations: PTE, EEG, EA, SDH, TBI
Introduction
Severe brain trauma is a leading cause of death and disability in adults and children worldwide1. Post-traumatic epilepsy (PTE) is one of the most disabling complications in survivors and can be difficult to treat2. PTE rates are reported in up to 20% of patients, with increased risk based on brain injury severity, surgical intervention, time since traumatic brain injury (TBI) and younger age3–6.
While some risk factors are known, we need to better stratify patients at highest risk for PTE to better understand anti-epileptogenesis and develop therapeutic agents While there is great interest in interventions to prevent post-TBI epileptogenesis, clinical trials have been plagued by financial and logistical barriers, with estimates upwards of $20M4,7. Efforts to prevent epileptogenesis would be greatly aided by identification of acute biomarkers that identify patients at high risk for developing PTE, thus enriching the population eligible for clinical trials in a cost effective manner8.
Epileptiform abnormalities (EA), which include sporadic epileptiform discharges (spikes and sharp waves), periodic epileptiform discharges, and rhythmic patterns, are common following all types of acute brain injury, including TBI9. Recent work from our group suggests that EA predict risk for secondary brain injury (Kim et al. 2017) and acute seizures10,11. TBI serves as an excellent acute brain injury model in which to investigate the role of EA as a marker of, and possible contributor to, secondary morbidity in the form of PTE. We aimed to determine whether EA could be used as an early biomarker of elevated risk for PTE1. Such information could be used to specify subpopulations of TBI patients that would benefit from targeted trials of anti-epileptogenic interventions with reduced cost and adverse risk exposures.
Methods
We evaluated EEG reports and medical records from 50 patients with TBI at a tertiary care center (Massachusetts General Hospital Neurosciences and Surgical ICUs) who met study inclusion criteria between 2011 and 2015. Inclusion criteria were: age ≥18 years, TBI on presentation and EEG monitoring during the initial hospital admission for TBI. Retrospective collection and analysis of clinical data were performed under a protocol approved by the local institutional review board. Among patients meeting the inclusion criteria we first evaluated consecutive (based on hospital admission) cases to identify 25 who developed PTE1 (defined below), and subsequently evaluated consecutive cases to identify 25 controls without PTE1, matched by age and admission Glasgow Coma Scores (GCS).
EEG recordings and report review
EEG data was recorded using conventional 10–20 scalp electrode placement. EA were classified according to standardized nomenclature12 as: seizures, sporadic epileptiform discharges (EDs), lateralized or generalized periodic discharges (LPDs and GPDs) and lateralized rhythmic delta activity (LRDA)13. We also analyzed generalized rhythmic delta activity (GRDA), polymorphic generalized and focal slowing but consider these separate from EA. The presence (dark bars) or absence (light bars) of these abnormalities, as documented in daily clinical EEG reports, was tallied for each patient with “day of traumatic brain injury” marked as day 0 (Figure 1A). A histogram representing the EEG distribution is shown in Figure 1B.
Figure 1.
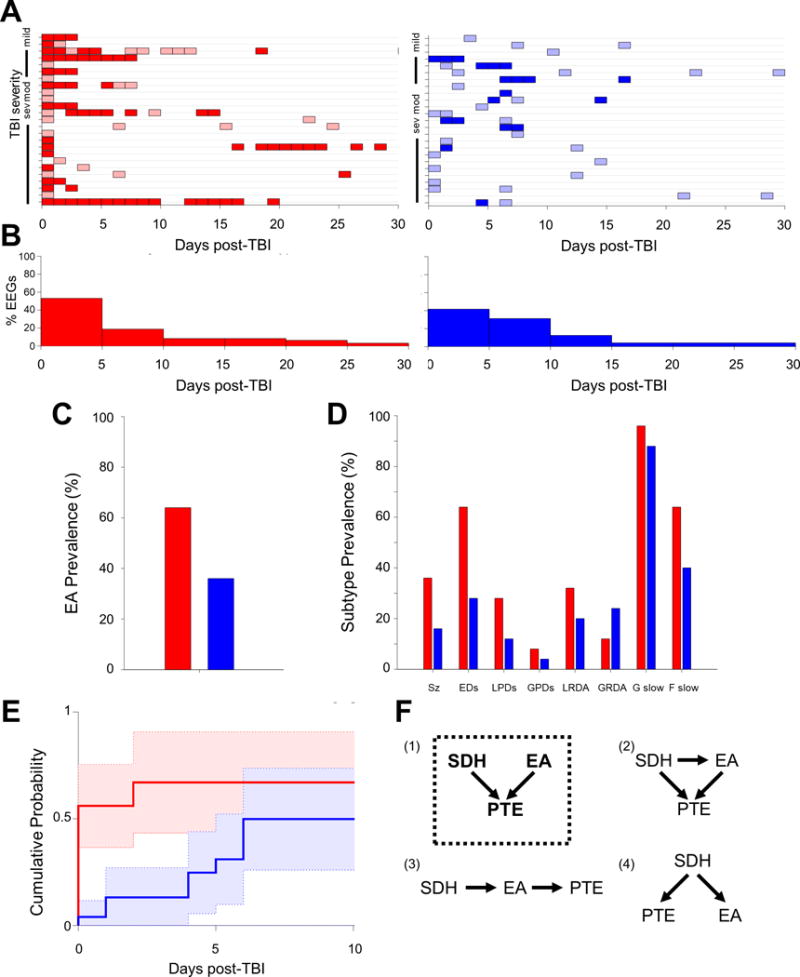
EEG recording distribution and prevalence in PTE1 (red) and non-PTE1 (blue) patients. A) EEG recording days (colored boxes) plotted for individual patients plotted along y-axis based on TBI severity. Shading based upon presence (dark) or absence (light) of EA during that day’s recording. B) Histogram summarizing the proportion of EEGs during each 5 day time-period. C) Prevalence of EA in PTE1 and non-PTE1 groups. D) Prevalence of EA subtypes in PTE1 and non-PTE1 groups. E) Cumulative probability of the first appearance EDs in recordings up to the first 10 days post-TBI. F) Models of possible causal relationship between SDH, EA and PTE1. Model (1) with dotted box outline is the most likely model based upon logistic regression.
PTE1 definition
Patients with at least one seizure 2–12 months post-TBI, based on medical record review. Control subjects were patients meeting the inclusion criteria who had TBI without any documented seizures in the same period, matched for age and admission GCS (Table 1). Patients were excluded if there were insufficient follow up visits in the electronic health record to determine PTE1 status. For practicality, we analyzed up to 12 months, the highest risk period14, while acknowledging this does not fully capture eventual PTE development.
Table 1.
Demographic predictors of PTE1 development.
| Univariate Analysis | PTE (n=25) | No PTE (n=25) | OR ([95% CI]) | P-value |
|---|---|---|---|---|
| Age | 52.5±20.4 | 49.6±25.8 | 1.01 [0.98 1.03] | 0.36 |
| Gender: F/M | 10/15 | 8/17 | 1.42 [0.41 4.85] | 0.25 |
| IPH | 8 (32%) | 3(12%) | 3.45 [0.77 >100] | 0.06 |
| SDH | 21 (84%) | 15(60%) | 4.13 [1.18 39.33] | 0.02* |
| SAH | 14 (56%) | 12(48%) | 1.38 [0.40 4.66] | 0.27 |
| EDH | 5 (20%) | 2(8%) | 2.88 [0.41 >100] | 0.10 |
| Skull Fracture | 14 (56%) | 10(40%) | 1.91 [0.62 6.73] | 0.12 |
| Admission GCS | 6±4.66 | 8±4.53 | 0.94 [0.81 1.07] | 0.25 |
Key: IPH-intraparenchymal hemorrhage, SDH- subdural hemorrhage, SAH-subarachnoid hemorrhage, EDH-epidural hemorrhage, GCS-Glasgow coma scale
Data analysis
For data analysis we used Matlab, including the Matlab Statistics Toolbox (MathWorks; Natick, MA). We employed univariate and multivariate logistic regression to calculate odds ratios of the reported demographic or EEG features (candidate predictor variables for PTE1). In addition to evaluating EA as a group, we analyzed individual EA subtypes (seizures, EDs, LPDs, GPDs and LRDA). Bootstrapping was used to determine 95% confidence intervals and p-values, with a significance threshold of p≤0.05.
Results
Demographic predictors
We calculated associations between demographic variables and PTE1, including age, gender, admission Glasgow coma scale (GCS), presence of intraparenchymal hemorrhage (IPH), subdural hemorrhage (SDH), subarachnoid hemorrhage (SAH), or epidural hemorrhage (EDH) (Table 1). The only demographic variable significantly associated with PTE1 development is subdural hemorrhage (p=0.02, Table 1).
EEG Distribution
EEG acquisition days are shown for each individual and summarized for each cohort (Figure 1A,B). The PTE1 group has more days of EEG monitoring overall, possibly attributable to the continuation of EEG monitoring when epileptiform abnormalities were found.
EEG predictors
EA are more common in patients with PTE1 compared to patients without PTE1 (64% vs 36%; p = 0.04) (Figure 1C). The prevalence of each EA subtype is shown in Figure 1D. EA are significant predictors of PTE1 with an odds ratio of 3.16 [0.99 11.68] (p=0.04) (Table 2).
Table 2.
EEG predictors of PTE1 development.
| Univariate Analysis | OR ([95% CI]) | P-value |
|---|---|---|
| Epileptiform Abnormalities | 3.16 [0.99 11.68] | 0.042* |
| Early Seizures | 2.95 [0.80 24.42] | 0.06 |
| EDs | 4.57 [1.60 21] | 0.007* |
| LPDs | 2.85 [0.71 >100] | 0.10 |
| GPDs | 2.09 [<0.1 >100] | 0.25 |
| LRDA | 1.88 [0.48 8.80] | 0.19 |
| GRDA | 0.43 [<0.1 2.30] | 0.87 |
| Generalized slowing | 3.27 [<0.1 >100] | 0.15 |
| Focal slowing | 2.67 [0.97 10.1] | 0.04* |
| Multivariate Analysis | ||
| EAs adjusted for SDH | 2.97 [0.91 14.18] | 0.03* |
| EDs adjusted for SDH | 3.8 [1.18 18.96] | 0.016* |
Key: EDs-epileptiform discharges, LPDs-lateralized periodic discharges, GPDs-generalized periodic discharges, LRDA-lateralized rhythmic delta activity, GRDA- generalized rhythmic delta activity, EAs-epileptiform abnormalities
When evaluating individual EA subtypes, only EDs (p=0.01) are significantly associated with PTE1 (Table 2). While not classified as an EA, focal slowing (p=0.04) is also significantly associated with PTE1 (Table2). Early seizures and LPDs show positive associations with PTE1 but did not reach significance (p=0.06 and p=0.10, respectively), probably due to small sample size.
The difference in EDs is observed early, ≤5 days after TBI, with 50% occurring on day 0 after TBI (OR 3.67 [1.02 18.76], p=0.04; Figure 1E).
Multivariate analysis
Controlling for SDH, acute EA remains significantly associated with subsequent PTE1 (OR 2.97 [0.91 14.18], p=0.03; Table 2). EDs alone, after adjusting for SDH, have an even stronger effect with an adjusted odds ratio of 3.8 [1.18 18.96] (p=0.016, Table 2).
By comparing multivariate logistic regression models of SDH + EA with the univariate regressions of EA and SDH independently, we find that SDH and EA independently contribute to increased PTE1 risk (p=0.05 and p=0.03, respectively) without any direct relationship to each other (p=0.17), suggesting that model (1) is the most likely relationship between the variables: SDH and ED are independent causal factors for PTE1 (Figure 1F)
Discussion
Our results provide novel evidence that EA may be a useful marker in identifying patients at high risk for PTE1.
SDH and PTE1
SDH is significantly associated with PTE1 in our study, in concordance with multiple other studies15,16. Prior studies also find associations with other variables, such as post-TBI amnesia, alcohol and midline shift, which we did not assess16. Intraparenchymal hemorrhage and skull fractures are also associated with PTE1 in other studies15, and while neither odds ratio in our cohort is significant (p=0.06 and 0.12 respectively), we are underpowered to detect such associations.
EA and PTE1
While the presence and prevalence of EA after TBI has been described17, the association with PTE1 has not been reported. Our results demonstrate that the presence of EA following TBI signals increases risk for the development of PTE1. More specifically, EDs are associated with PTE1 development. Other subtypes of EA in our data, including early seizures and LPDs, show weak associations with PTE1 but do not reach statistical significance, potentially due to small sample size. Interestingly, focal polymorphic slowing is also significantly associated with PTE1. While often considered a non-specific EEG pattern, focal slowing has been observed in PTE previously18 and a recent study showed focal slowing in areas corresponding to blood-brain barrier (BBB) disruption after TBI, which correlated with PTE119.
Our results also show that EA occur early (<5 days) after TBI, suggesting early EEG could be a useful diagnostic tool to assess TBI patients for PTE1 risk. TBI is a defined time-point event in which patients are known to be at risk for epileptogenesis, thus making this group prime for anti-epileptogenesis trials. However, the large patient numbers needed to test interventions and unnecessary exposure to potential adverse effects in low-risk patients has been prohibitive. For example, for an anti-epileptogenesis drug trial that enrolled severe TBI patients with an estimated incidence of PTE1 at 7.1%20, the sample size required to detect a 50% treatment effect is 1364 patients (Fisher’s 2-sided exact test, power 0.8, alpha 0.05). By contrast, if we enroll severe TBI patients with early EAs on EEG, according to our data the incidence of PTE1 rises to 12%, and the required sample size is only 778, a decrease of 43%20. While our sample size is small, retrospective in design and needs further confirmation, our results suggest by using EA as a biomarker to identify the subset of TBI patients at highest risk for PTE1 development, anti-epileptogenesis interventions could be feasible in a cost-effective manner.
Acknowledgments
JAK received funding from NIH-NINDS (R25NS065743) and the Bee Foundation. MBW received funding from NIH-NINDS (1K23NS090900), and the Andrew David Heitman Neuroendovascular Research Fund. ACW is supported by the CDC-NIOSH ERC training-grant (T42 OH008416). KJS received funding from NIH-NINDS (R01NS086364).
Footnotes
Author Contributions
JAK, AJC and MBW contributed to the conception and design of the study. JAK, EB, AW and MBW contributed to the acquisition and analysis of data; JAK, AJC, SZ, SSC, KJS and MBW contributed to drafting the text and preparing the figures.
Potential Conflicts of Interest: None
References
- 1.Wang ML, Li WB. Cognitive impairment after traumatic brain injury: The role of MRI and possible pathological basis. J Neurol Sci. 2016;370:244–250. doi: 10.1016/j.jns.2016.09.049. [DOI] [PubMed] [Google Scholar]
- 2.Curia G, Eastman CL, Miller JW, D’Ambrosio R. Modeling Post-Traumatic Epilepsy for Therapy Development. 2016 [PubMed] [Google Scholar]
- 3.Ritter AC, Wagner AK, Fabio A, et al. Incidence and risk factors of posttraumatic seizures following traumatic brain injury: A Traumatic Brain Injury Model Systems Study. Epilepsia. 2016 doi: 10.1111/epi.13582. [DOI] [PubMed] [Google Scholar]
- 4.Klein P, Tyrlikova I. Prevention of epilepsy: Should we be avoiding clinical trials? Epilepsy Behav. 2017;72:188–194. doi: 10.1016/j.yebeh.2017.05.024. [DOI] [PubMed] [Google Scholar]
- 5.Annegers J, Hauser W, Coan S, Rocca WA. Population-Based Study of Seizures after Traumatic Brain Injuries. N Engl J Med. 1998 doi: 10.1056/NEJM199801013380104. [DOI] [PubMed] [Google Scholar]
- 6.Piccenna L, Shears G, O’brien TJ. Management of post-traumatic epilepsy: An evidence review over the last 5 years and future directions. Epilepsia Open. 2017;2(2):123–144. doi: 10.1002/epi4.12049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schmidt D, Friedman D, Dichter MA. Anti-epileptogenic clinical trial designs in epilepsy: issues and options. Neurotherapeutics. 2014;11(2):401–11. doi: 10.1007/s13311-013-0252-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Terrone G, Pauletti A, Pascente R, Vezzani A. Preventing epileptogenesis: A realistic goal? Pharmacol Res. 2016;110:96–100. doi: 10.1016/j.phrs.2016.05.009. [DOI] [PubMed] [Google Scholar]
- 9.Vespa P, Tubi M, Claassen J, et al. Metabolic Crisis occurs with Seizures and Periodic Discharges after Brain Trauma. Ann Neurol. 2016;79(4):579–90. doi: 10.1002/ana.24606. [DOI] [PubMed] [Google Scholar]
- 10.Shafi MM, Westover MB, Cole AJ, et al. Absence of early epileptiform abnormalities predicts lack of seizures on continuous EEG. Neurology. 2012;79(17):1796–1801. doi: 10.1212/WNL.0b013e3182703fbc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Westover MB, Shafi MM, Bianchi MT, et al. The probability of seizures during EEG monitoring in critically ill adults. Clin Neurophysiol. 2015;126(3):463–471. doi: 10.1016/j.clinph.2014.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hirsch LJ, LaRoche SM, Gaspard N, et al. American Clinical Neurophysiology Society’s Standardized Critical Care EEG Terminology: 2012 version. J Clin Neurophysiol. 2013;30(1):1–27. doi: 10.1097/WNP.0b013e3182784729. [DOI] [PubMed] [Google Scholar]
- 13.Ruiz AR, Vlachy J, Lee JW, et al. Association of periodic and rhythmic electroencephalographic patterns with seizures in critically ill patients. JAMA Neurol. 2017;74(2):181–188. doi: 10.1001/jamaneurol.2016.4990. [DOI] [PubMed] [Google Scholar]
- 14.Yeh C-C, Chen T-L, Hu C-J, et al. Risk of epilepsy after traumatic brain injury: a retrospective population-based cohort study. J Neurol Neurosurg Psychiatry. 2013;84(4):441–5. doi: 10.1136/jnnp-2012-302547. [DOI] [PubMed] [Google Scholar]
- 15.Liesemer K, Bratton SL, Zebrack CM, et al. Early Post-Traumatic Seizures in Moderate to Severe Pediatric Traumatic Brain Injury: Rates, Risk Factors, and Clinical Features. J Neurotrauma. 2011;28(5):755–762. doi: 10.1089/neu.2010.1518. [DOI] [PubMed] [Google Scholar]
- 16.Xu T, Yu X, Ou S, et al. Risk factors for posttraumatic epilepsy: A systematic review and meta-analysis. Epilepsy Behav. 2017;67:1–6. doi: 10.1016/j.yebeh.2016.10.026. [DOI] [PubMed] [Google Scholar]
- 17.Vespa P, Tubi M, Claassen J, et al. Metabolic crisis occurs with seizures and periodic discharges after brain trauma. Ann Neurol. 2016;79(4):579–590. doi: 10.1002/ana.24606. [DOI] [PubMed] [Google Scholar]
- 18.Koufen H, Dichgans J. Haufigkeit und Ablauf von traumatischen EEGVeranderungen und ihre klinischen Korrelationen: systematische verlaufsuntersuchungen bei 344 Erwachsenen. Fortschr Neurol Psychiatr. 1978;46:165–177. [PubMed] [Google Scholar]
- 19.Tomkins O, Shelef I, Kaizerman I, et al. Blood-brain barrier disruption in post-traumatic epilepsy. J Neurol Neurosurg Psychiatry. 2008;79(7):774–777. doi: 10.1136/jnnp.2007.126425. [DOI] [PubMed] [Google Scholar]
- 20.Annegers JF, Grabow JD, Groover RV, et al. Seizures after head trauma: a population study. Neurology. 1980;30(7 Pt 1):683–9. doi: 10.1212/wnl.30.7.683. [DOI] [PubMed] [Google Scholar]