

Abstract
Objective
Voltage-gated sodium (Na+) channels underlie action potential generation and propagation and hence are central to the regulation of excitability in the nervous system. Mutations in the genes SCN1A, SCN2A, and SCN8A, encoding the Na+ channel pore-forming (α) subunits Nav1.1, 1.2, and 1.6, respectively, and SCN1B, encoding the accessory subunit β1, are established causes of genetic epilepsies. SCN3A, encoding Nav1.3, is known to be highly expressed in brain, but has not previously been linked to early infantile epileptic encephalopathy. Here, we describe a cohort of four patients with epileptic encephalopathy and heterozygous de novo missense variants in SCN3A (p.Ile875Thr in two cases, p.Pro1333Leu, and p.Val1769Ala).
Methods
All patients presented with treatment-resistant epilepsy in the first year of life, severe to profound intellectual disability, and, in two cases (both with the variant p.Ile875Thr), diffuse polymicrogyria.
Results
Electrophysiological recordings of mutant channels revealed prominent gain of channel function, with a markedly increased amplitude of the slowly inactivating current component, and, for two of three mutants (p.Ile875Thr and p.Pro1333Leu), a left-shift in the voltage dependence of activation to more hyperpolarized potentials. Gain of function was not observed for Nav1.3 variants known or presumed to be inherited (p.Arg1642Cys and p.Lys1799Gln). The anti-seizure medications phenytoin and lacosamide selectively blocked slowly inactivating over transient current in wild-type and mutant Nav1.3 channels.
Interpretation
These findings establish SCN3A as a new gene for infantile epileptic encephalopathy and suggest a potential pharmacologic intervention. These findings also reinforce the role of Nav1.3 as an important regulator of neuronal excitability in the developing brain while providing additional insight into mechanisms of slow inactivation of Nav1.3.
Introduction
Voltage-gated sodium (Na+) channels mediate the generation and propagation of electrical signals in excitable tissues including brain, spinal cord and peripheral nerve, and muscle.1–3 Sodium channels are composed of a pore-forming α and auxiliary β subunits. In mammals, nine α subunits (Nav1.1-1.9) have been identified (encoded by the genes SCN1A-5A and SCN8A-11A).4 These α subunits consist of four domains (I-IV), each with six transmembrane segments (S1-S6), with S1-S4 forming the voltage sensor and S5-S6 of each domain contributing to the central ion-conducting pore.1 Mutation of sodium channels is a known cause of abnormal excitability underlying human disease including epilepsy, cardiac arrhythmia, periodic paralysis, and pain syndromes.2 Interestingly, all voltage-gated sodium channels, except Nav1.3, have known, well-established human disease associations.
The predominant Na+ channels expressed in brain are Nav1.1, 1.2, 1.3, and 1.6.1 Mutations in the genes encoding three of these four channels are highly associated with epilepsy: heterozygous loss of function mutation of SCN1A (encoding Nav1.1) is the major cause of Dravet syndrome spectrum disorders.5,6 Mutations of SCN2A7–9 (encoding Nav1.2) and SCN8A10,11 (encoding Nav1.6) are also well-established causes of genetic epilepsy, associated with a wider phenotypic spectrum that includes early infantile epileptic encephalopathy. However, a link between Nav1.3 and epilepsy has been less clear.
SCN3A encodes the type 3 voltage gated Na+ channel α subunit Nav1.3, which is known to be highly expressed in embryonic brain.12,13 Postnatal expression levels in rodent is low or undetectable,14,15 although Nav1.3 is upregulated in response to various insults to the nervous system including nerve injury16,17 and in some rodent models of epilepsy.18,19 However, Nav1.3 knockout mice appear healthy.16 A specific function of Nav1.3 in brain is not clear. However, it has been consistently noted that Nav1.3 current includes a relatively prominent slowly inactivating/”persistent” current component (INaP) of between 0.5-10.3% of peak transient sodium current (INaT).20–22
Previous studies have reported heterozygous variants in SCN3A in association with less severe forms of epilepsy; 21–25 however, de novo mutation in SCN3A as a cause of early infantile epileptic encephalopathy has yet to be described. Prior reports include cases of focal epilepsy, with onset after infancy, and not consistently associated with developmental delay/intellectual disability; variants were in most instances not shown definitively to be de novo mutations. Here, we describe a cohort of four patients with infantile onset epileptic encephalopathy due to verified de novo mutation of SCN3A, and characterize the functional consequences of these mutations electrophysiologically. We also assess the electrophysiological properties of two SCN3A variants that were either demonstrated or suspected to be inherited. These latter variants did not show the gain of function seen in patients with a proven de novo mutation and early infantile epileptic encephalopathy. Our results support the status of SCN3A as an early infantile epileptic encephalopathy gene and further suggest that the mechanism of severe epilepsy is gain of function; our results show a prominent increase in INaP and, for two of the three mutants, an apparent left-shift in the voltage dependence of channel activation.
Subjects and Methods
Study subjects
Patients included in the study (Table 1) were either seen and evaluated at The Children's Hospital of Philadelphia, Philadelphia, PA, U.S.A, the University Hospitals Cleveland Medical Center, Cleveland, OH, U.S.A., or the Clinical Institute of Medical Genetics, University Medical Centre Ljubljana, Ljubljana, Slovenia, or were ascertained through a collaboration with Ambry Genetics, Aliso Viejo, CA. Informed consent was obtained from all parent(s) where applicable. This study was approved by the local institutional review boards at each of the participating centers. All patients were unrelated and pregnancy and birth history were unremarkable for all patients.
Table 1.
Epilepsy-associated Nav1.3 mutant channels – present report and review of the literature
| Patient | Amino acid change | Domain | Segment | Inheritance pattern | Epilepsyage of onset | EEG classification | Seizure semiology | DD | MRI |
|---|---|---|---|---|---|---|---|---|---|
| 1 | p.I875T | II | 4-5 linker | de novo | 2 w | Multifocal | Tonic or myoclonic motor onset | Profound | PMG |
| 2 | p.I875T | II | 4-5 linker | de novo | 2 w | Multifocal | Tonic or generalized tonic-clonic motor onset | Profound | PMG |
| 3 | p.P1333L | III | 4-5 linker | de novo | 1-3 d | Hypsarrhythmia;Multifocal | Tonic, focal motor | Severe | Normal |
| 4 | p.V1769A | IV | 6 | de novo | < 1 y | Multifocal | NA | Yes | NA |
| 5 | p.R1642C | IV | 5 | Unknown* | 18 mo | NA | Focal onset w/ impaired awareness | Yes | Normal |
PMG, polymicrogyria. NA, not available for review.
Non-maternal; father not available for testing.
Patient 1 was a male with epilepsy onset at age 2 weeks, with continued intractable epilepsy at last follow up (at age 13 years), accompanied by microcephaly, profound global developmental delay and central hypotonia. There were multiple seizure types including tonic and myoclonic seizures. At last follow up, the patient remained non-verbal, non-ambulatory, with dysphagia and failure to thrive requiring exclusive feeding via gastrostomy tube. MRI of the brain showed extensive frontoparietal polymicrogyria with thickening of the cerebral cortex (FIG 2A). Trio whole exome sequencing analysis (WES) revealed a de novo heterozygous c.2624T>C missense mutation in SCN3A leading to an p.Ile875Thr amino acid substitution (based on NCBI Reference Sequence NP_008853.3 for human Nav1.3 isoform 1; see below).
Figure 2. Magnetic resonance imaging scans of patients with SCN3A-p.Ile875Thr mutation.
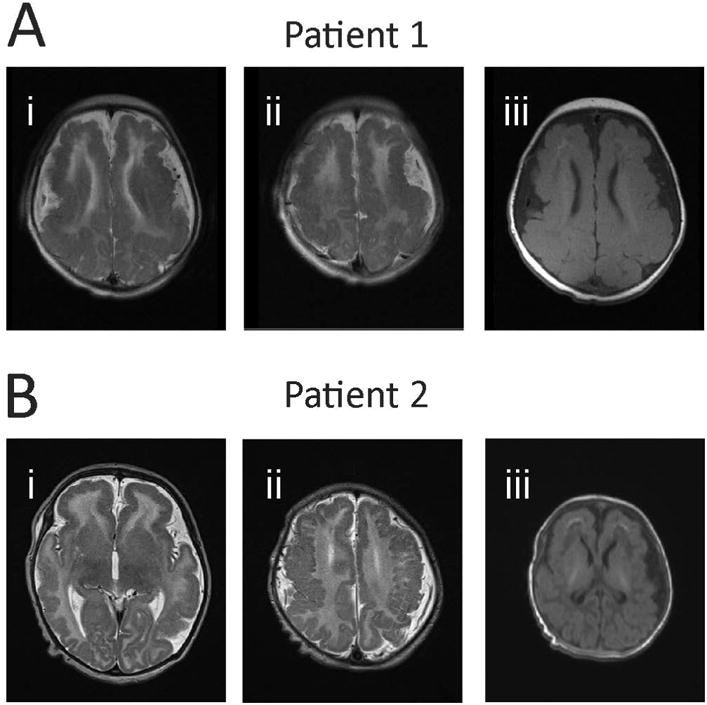
(A) MRI for Patient 1. Axial T2 MRI images (Ai-ii) at two rostrocaudal levels demonstrating extensive, bilaterally symmetric, predominantly frontoparietal polymicrogyria. There is incomplete opercularization of the insula with slightly dysplastic sylvian fissures. There is extensive foci of frontal subcortical and periventricular T1 shortening representing calcifications (Aiii). (B) MRI for Patient 2. Axial T2 MRI images (Bi-ii) show diffuse polymicrogyria. T1 images show apparent subcortical calcifications in the frontal lobes (Biii).
Patient 2 was a female born at term after an uncomplicated pregnancy and delivery, with onset of seizures at age 2 weeks that were described as generalized tonic. MRI of the brain showed diffuse, bilateral polymicrogyria, abnormal white matter, and thin corpus callosum (FIG 2B). The patient was also found to have a de novo c.2624T>C missense mutation leading to an p.Ile875Thr amino acid substitution. As of last follow up, at age 3 years, the patient exhibited severe global developmental delay, central hypotonia, spastic tetraparesis, and intractable epilepsy.
Patient 3, a male born at full term, had onset of seizures on day of life 1 (36 hours), characterized by bradycardia, oxygen desaturation, and tonic unilateral upper extremity movements. An initial EEG performed at age 2 weeks showed hypsarrhythmia, with high-amplitude multifocal epileptiform discharges along with eye deviation or tonic unilateral limb movements associated with electrodecrement; classical flexor or extensor spasms were not noted. Repeat EEG at age 6 months showed multifocal sharp waves and spikes and multifocal areas of intermittent slowing. Epilepsy remained refractory to treatment with last follow up at approximately 2 years of age, at which time the patient exhibited severe global developmental delay, central hypotonia, and cortical blindness. Seizure types included unilateral clonic or tonic-clonic movements of an extremity, ipsilateral facial twitching, and/or upward or lateral eye deviation. MRI of the brain showed thinning of the corpus callosum and low-normal myelination of the cerebral white matter; head circumference was normal. WES revealed a de novo heterozygous c.3998C>T missense mutation in SCN3A leading to a p.Pro1333Leu amino acid substitution.
Patient 4, a female, had epilepsy onset within the first year of life, although exact age of unset is unknown. There was continued developmental delay and intractable epilepsy at age 4 years. EEG showed multifocal epileptiform discharges. Whole exome sequencing demonstrated a de novo c.5306T>C missense mutation in SCN3A leading to a Val1769Ala amino acid substitution. MRI of the brain was not performed.
Patient 5, a male, had normal early childhood development, with epilepsy onset at 18 months. Epilepsy became intractable by age 3 years, with developmental arrest and regression of speech/language and motor development. At last follow up at age 32 years, the patient continued to suffer from intractable epilepsy with minimal independent ambulation and severe intellectual impairment. Seizures have been of apparent focal onset, with arrest of activity and staring, sometimes progressing to limb stiffening and occasional fall. MRI of the brain was normal, showing only a likely posterior fossa arachnoid cyst. Neurological examination was significant for low axial tone and peripheral spasticity. Whole exome sequencing revealed a c.4924C>T missense variant leading to a p.Arg1642Cys substitution in SCN3A that was shown to be not inherited from the patient's mother; however, the inheritance pattern could not be strictly established as the patient's biological father was not available for testing.
Patient 6 had onset of infantile spasms at age 4 months accompanied by developmental arrest. By age 17 years, the patient exhibited severe intellectual impairment and was nonverbal, with ataxic gait, continued seizures, and a hyperkinetic movement disorder. Whole exome sequencing analysis revealed a c.5395A>C variant in SCN3A leading to a p.Lys1799Gln amino acid substitution in Nav1.3 that was subsequently shown to be maternally-inherited and was hence considered a variant of uncertain significance.
Exome analysis in all cases included more detailed evaluation of variants in genes associated with developmental delay, epilepsy, and brain malformation. All variants in SCN3A were confirmed via Sanger sequencing.
Cell culture, plasmid preparation, and transfection
Transfections and electrophysiological experiments were performed using tsA-201 cells. Cells were grown at 37°C with 5% CO2 in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 2 mM L-glutamine, and penicillin (50 U/mL)-streptomycin (50 μg/mL).
A plasmid encoding the major splice isoform of human SCN3A (isoform 2, which is 49 amino acids longer than isoform 1) was used (Reference Sequence NP_001075145.1), and variants were introduced by site-directed mutagenesis. Constructs were propagated in STBL2 cells at 30 °C (Invitrogen). All plasmid preparations were resequenced prior to transfection. Plasmids encoding human sodium channel auxiliary subunits β1 (hβ1-V5-2A-dsRed) or β2 (pGFP-IRES- hβ2) were co-transfected in vectors containing marker genes facilitating the identification of cells expressing all three constructs (Nav1.3, Navβ1, and Navβ2). We performed transient transfection of 2 μg total cDNA at a ratio of Nav1.3, &beta1, and β2 of 10:1:1 using Lipofectamine-2000 (Invitrogen) transfection reagent. Cells were incubated for 48 hours after transfection prior to electrophysiological recording. Transfected cells were dissociated by brief exposure to trypsin/EDTA, re-suspended in supplemented DMEM medium, plated on 15 mm glass coverslips, and allowed to recover for at least 4 hours at 37 °C in 5% CO2 prior to recording.
Electrophysiology
Glass coverslips were placed in a chamber housed on the stage of an upright epifluorescence microscope for electrophysiological recording. Extracellular solution contained, in mM: NaCl, 145; KCl, 4.0; CaCl2, 1.8; MgCl2, 1.0; HEPES, 10. pH was adjusted to 7.30 with NaOH. Intracellular pipette-filling solution contained, in mM: CsF, 110; NaF, 10; CsCl, 20; EGTA, 2.0; HEPES, 10. pH was adjusted to 7.35 with CsOH and osmolarity to 305 mOsm/L with sucrose. All chemicals were produced by Sigma Aldrich (St. Louis, MO). Recordings were performed using an agar bridge (2% in extracellular solution) reference electrode.
Recording pipettes were fashioned from thin-walled borosilicate glass (Sutter Instruments, Novato, CA) using a two-stage upright puller (PC-10, Narishige, Tokyo, Japan), fire-polished using a microforge (MF-830; Narishige), and coated with Sylgard 182 (Dow Corning, Midland, MI). The resistance of pipettes when placed in extracellular solution was 2.18 ± 0.04 MΩ. Cells that were positive for both RFP (β1 expression) and GFP (β2 expression) and exhibited fast transient inward current consistent with a voltage-gated sodium current were selected for subsequent analysis.
Whole-cell patch clamp recordings were performed at room temperature (22-24° C) using a MultiClamp 700B amplifier (Molecular Devices, Sunnyvale, CA). Pipette capacitance was zeroed after formation of a gigaseal. Access resistance was 4.75 ± 0.16 MΩ and recording was initiated after 10 minutes of equilibration after which recorded currents were found to be stable, and voltage errors were reduced via partial series resistance compensation. Voltage clamp pulses were generated using Clampex 10.6, acquired at 10 kHz, and filtered at 5 kHz. Leak was subtracted either using a standard P/4 protocol or post-hoc using a similar method involving a small prepulse. Cells were rejected from analysis if access resistance changed more than 15% during the recording.
The activation protocol was performed using 20 millisecond steps from a holding potential of -120 mV to potentials ranging from -80 mV to a maximum of 50 mV, in 5 mV increments, with a 10 second intersweep interval. Current was converted to current density (pA/pF) by normalizing to cell capacitance. Conductance was derived using G = I / (V - ENa) where G is conductance, I is current, V is voltage, and ENa the calculated equilibrium potential for sodium (+68.0 mV). Conductance was normalized to maximum conductance, which was fit with a Boltzmann function to determine the voltage at half-maximal channel activation (V1/2 of activation) and slope factor k. Persistent current was measured as the average value of the current response in the last 10 ms of a 200 ms test pulse to -10 mV. The prepulse voltage dependence was determined using a 100 millisecond prepulse to various potentials from a holding potential of -120 mV, followed by a 20 millisecond test pulse to -10 mV. Normalized conductance was plotted against voltage and fit with a Boltzmann function to determine the voltage at half-maximal inactivation (V1/2 of inactivation) and slope factor k. Kinetics of recovery from channel inactivation was determined using a 1 second prepulse to -10 mV from a holding potential of -120 mV followed by a variable time delay to a 10 ms test pulse to -10 mV. Data was fit with a double exponential function to determine the first (τ1) and second (τ2) time constants of recovery and their relative weights.
Ramp currents were obtained using a voltage ramp from -120 to +40 mV at 0.8 mV/ms. Peak current and total charge (area under the curve; in Coulombs) were calculated.
Pharmacologic experiments were performed via bath perfusion. 5,5-Diphenylhydantoin (phenytoin) was from Sigma Aldrich. Lacosamide (L098500) was purchased from Toronto Research Chemicals (Toronto, Ontario, Canada), dissolved in methanol, and stored as 100 mM stock solution prior to 1:1000 dilution in ACSF. For such experiments, control extracellular solution contained the same final concentration of methanol (0.0998%).
All recordings and data analysis were performed blind to experimental group.
Data analysis
Data for standard electrophysiological parameters was obtained from at least n = 12 cells (1 from each coverslip) from at least 6 separate transfections for each experiment. Data were analyzed using Clampfit 10.6 (Molecular Devices) or using custom scripts written in Matlab (Mathworks, Natick, MA) and statistics were generated and plotted using Statistical Package for the Social Sciences version 14 (SPSS), Microsoft Excel (Microsoft, Seattle, WA), GraphPad Prism (Graphpad Software, Inc, La Jolla, CA) and Sigma Plot 11 (Systat Software, Inc., San Jose, CA, USA). Results are presented as the mean ± standard error of the mean (SEM) and statistical significance was established using the p value calculated from a paired or unpaired Student's t-test, or via one-way ANOVA with Bonferroni correction for multiple comparisons, as appropriate, with the p value specified at the p < 0.05, 0.01, or 0.001 level, or reported exactly.
Results
We identified a cohort of four patients with severe infantile onset epileptic encephalopathy and de novo heterozygous variants in SCN3A encoding the type 3 voltage gated sodium (Na+) channel α subunit Nav1.3 (FIG 1; Table 1). Two of the patients (Patients 1 and 2) carried the same de novo p.Ile875Thr variant, while the other two patients harbored unique de novo variants p.Pro1333Leu and p.Val1769Ala. We also included two patients with SCN3A variants that were presumed (Patient 5) or demonstrated to be inherited (Patient 6). The four patients (Patients 1-4) with confirmed de novo mutation had intractable epilepsy with onset during infancy and severe to profound developmental delay. MRI of the brain was essentially normal in all cases except for the two patients with p.Ile875Thr mutation (Patients 1 and 2; see FIG 2). Previously reported cases of Nav1.3 variants associated with epilepsy are summarized for comparison (Table 2).
Figure 1. Locations of epilepsy-associated Nav1.3 variants.
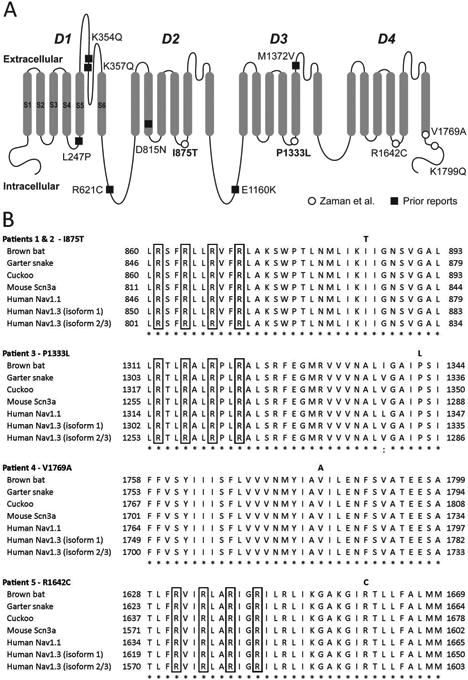
(A) Shown is a schematic of the Nav1.3 voltage-gated Na+ channel α subunit, with domains (D) 1-4, and transmembrane segments (S) 1-6 (shown for D1). Epilepsy-associated variants previously reported in the literature are shown as closed squares, and those described in the present report are shown as open circles. Variants in bold are recurrent. Amino acid location is based on Reference Sequence NP_008853.3 (hNav1.3 isoform 1). (B) Nav1.3 mutations associated with epileptic encephalopathy occur at highly conserved amino acid residues, as shown using ClustalX multiple sequence alignment of human Nav1.3 isoforms 1, 2, and 3, human Nav1.1 homolog, and various species orthologs. The Nav1.3 variants described in this report are indicated in bold above the corresponding amino acid residue. The highly conserved arginine residues of the S4 helix of the voltage sensor of Nav1.3 are indicated by the boxed regions. Notation indicates conservation across human Na+ channel genes and between species: period (.) indicates weakly similar; colon (:), strongly similar; asterisk (*), fully conserved.
Table 2.
Epilepsy-associated Nav1.3 variants in the published literature.
| Reference | Amino acid change | Inheritance pattern | Epilepsyage of onset | Epilepsy type | DD | MRI |
|---|---|---|---|---|---|---|
| Holland et al, 2008 | p.Lys354Gln | Paternal | 2 y | Focal | No | Normal |
| Vanoye et al, 2014 | p.Arg357Gln | Unknown | 4 y | Focal | Yes | Normal |
| Vanoye et al, 2014 | p.Asp815Asn | Unknown | 22 mo | Focal | Mild | Normal |
| Vanoye et al, 2014 | p.Glu1160Lys | Unknown | Neonate | Focal | No | Normal |
| Vanoye et al, 2014 | p.Met1372Val | Unknown | 16 mo | FS; Focal | No | Normal |
| Wang et al, 2017 | p.Arg621Cys | Maternal | Unkown | FS; BECTS | No | Normal |
| Lamar et al, 2017 | p.Leu247Pro | de novo | 18 mo | Focal | Yes | Normal |
| Trujillano et al, 2017 | p.Pro1333Leu | de novo | 1-3 d | Unspecified | Severe | Normal |
Note that all variants are presented in relation to Nav1.3 isoform 1 (Reference Sequence NP_008853.3) FS, febrile seizures; BECTS, centrotemporal spikes on EEG.
Na+ channels are formed by a single polypeptide composed of four repeated domains (D1-4), with each domain comprising six transmembrane segments (S1-S6); S1-S4 represent the voltage-sensing region, while S5-6 forms the conducting pore. Identified variants in Patients 1-5 were in the S4-S6 domains (Table 1). Figure 1B shows the sequence alignment between isoform 1 and isoforms 2 and 3 of hNav1.3 (see Subjects and Methods), other human voltage-gated Na+ channel α subunits, as well as across phylogeny. p.Ile875Thr identified in Patients 1 and 2 produces substitution of a hydrophobic isoleucine to polar uncharged threonine at a highly evolutionarily conserved residue located in the S4-5 linker of Domain II. The p.Pro1333Leu mutation found in Patient 3 represents the substitution of a small, aliphatic amino acid proline to hydrophobic leucine at a residue located in the S4-5 linker of Domain III that is highly conserved between human Na+ channels Nav1.1-1.9 as well as across phylogeny. The p.Val1769Ala mutation in Patient 4 results in substitution of an alanine for valine at a highly conserved residue in the S6 segment of Domain IV. And the p.Arg1642Cys variant of unknown inheritance detected in the more mildly affected Patient 5 produces a substitution of a positively charged arginine for cysteine at a conserved residue in the S4-5 linker of Domain IV.
Reference to in silico prediction software PolyPhen-2 and SIFT supports the pathogenic nature of the variants identified in the patients in the cohort with early infantile epileptic encephalopathy (Table 4). Variants were absent from control databases including the genome Aggregation Database (gnomAD) and the Exome Variant Server (EVS). We also queried ClinVar for mutations in the other epilepsy-associated sodium channel genes SCN1A, 2A, and 8A that were homologous to the SCN3A mutations reported here (Table 5) and determined that, for example, Nav1.1-p.Val1784Ala (an identical substitution at the position homologous to amino acid 1769 in Nav1.3) has been reported in a patient with Severe Myoclonic Epilepsy of Infancy (Dravet syndrome). A recent report of 9 patients with an early profound form of SMEI26 reported one patient with an Nav1.1-p.Pro1345Ser mutation, an identical substitution at the amino acid residue homologous to position 1333 in SCN3A that is mutated in Patient 3 in our cohort. It should be noted that prior work has shown that homologous mutations in SCN1A and 3A can have divergent effects on channel physiology.27 Search of the online SCN1A database (http://www.molgen.ua.ac.be/SCN1AMutations/Home) revealed an additional case of a patient with Genetic Epilepsy with Febrile Seizures Plus (GEFS+) due to familial p.Arg1657Cys missense mutation,28,29 an identical amino acid substitution at the position homologous to residue 1642 in Nav1.3 that was identified in Patient 5.
Table 4.
ClinVar* entries for homologous variants in other epilepsy-associated sodium channel genes.
| SCN1A | SCN2A | SCN8A | |||||||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| a.a. | Variants | Phenotype | a.a. | Variants | Phenotype | a.a. | Variants | Phenotype | |
| p.Ile875Thr | 871 | N | NA | 873 | N | NA | 867 | N | NA |
| p.Pro1333Leu | 1345 | P1345S | SMEI | 1335 | N | NA | 1325 | N | NA |
| p.Val1769Ala | 1784 | V1784A | SMEI | 1774 | N | NA | 1764 | N | NA |
| p.Arg1642Cys | 1657 | N | NA | 1647 | R1647H | Nonspecific | 1638 | R1638H | Not provided |
| p.Lys1799Gln | 1814 | N | NA | 1804 | N | NA | 1794 | N | NA |
a.a., homologous amino acid residue; SMEI, severe myoclonic epilepsy of infancy (Dravet syndrome).
Table 5.
Epilepsy-associated Nav1.3 mutant channels: present report and review of the literature.
| Patient | Amino acid change | ExAc* | gnomAD** | PolyPhen-2 | |||
|---|---|---|---|---|---|---|---|
|
|
|
|
|||||
| Allele count | Allele frequency | Allele count | Allele frequency | Score | Prediction | ||
|
|
|
|
|
|
|||
| 1-2 | p.Pro1333Leu | 0 | -- | 0 | -- | 1.000 | Probably damaging |
| 3 | p.Ile875Thr | 0 | -- | 0 | -- | 1.000 | Probably damaging |
| 4 | p.Val1769Ala | 0 | -- | 0 | -- | 0.999 | Probably damaging |
| 5 | p.Arg1642Cys | 0 | -- | 2/277,136 | 0.000007 | 1.000 | Probably damaging |
| Holland et al, 2008 | p.Lys354Gln | 0 | -- | -- | 0.999 | Probably damaging | |
| Vanoye et al, 2014 | p.Arg357Gln | 8/121,404 | 0.000066 | 14/277,216 | 0.000051 | 0.003 | Benign |
| Vanoye et al, 2014 | p.Asp815Asn | 2/121,172 | 0.000017 | 4/274,730 | 0.000015 | 0.984 | Probably damaging |
| Vanoye et al, 2014 | p.Glu1160Lys | 2/121,302 | 0.000016 | 6/276,996 | 0.000022 | 0.996 | Probably damaging |
| Vanoye et al, 2014 | p.Met1372Val | 1/121,392 | 0.000008 | 7/277,124 | 0.000025 | 0.003 | Benign |
| Wang et al, 2017 | p.Arg621Cys | 0 | -- | 0 | -- | 1.00 | Probably damaging |
| Lamar et al, 2017 | p.Leu247Pro | 0 | -- | 0 | -- | 1.00 | Probably damaging |
Exome Aggregation Consortium (http://exac.broadinstitute.org/)
genome Aggregation Database (http://gnomad.broadinstitute.org/)
To assess the functional effects of these variants, we performed whole-cell voltage-clamp electrophysiological recordings of wild-type hNav1.3 and mutant hNav1.3 expressed in tSA-201 cells co-expressed with wild-type human β1 and β2 auxiliary subunits. Representative rapidly activating and rapidly inactivating inward currents are shown (FIG 3A) along with plots of average current density vs. voltage (FIG 3B and Table 4). Peak current density was 374.1 ± 18.5 pA/pF for wild-type (n = 27); 391.1 ± 28.6 pA/pF for p.Ile875Thr (n = 14), 411.5 ± 55.5 pA/pF for p.Pro1333Leu (n = 10), which was not different between groups (p = 0.27 and 0.20 for wild-type vs. p.Ile875Thr and p.Pro1333Leu, respectively). Rise time was also not statistically different between groups: time to peak current (in ms) at -10 mV was 0.82 ± 0.05 for wild-type (n = 27), 0.75 ± 0.08 for Nav1.3-p.Ile875Thr (n = 14; p = 0.43 vs. wild-type via unpaired two-tailed t-test), 0.69 ± 0.06 for Nav1.3-p.Pro1333Leu (n = 10; p = 0.18), 0.74 ± 0.04 for Nav1.3-p.Val1769Ala (n = 18; p = 0.32), and 0.72 ± 0.05 for Nav1.3-p.Arg1642Cys (n = 5; p = 0.43).
Figure 3. Epilepsy-associated mutations alter function of the Nav1.3 channel.
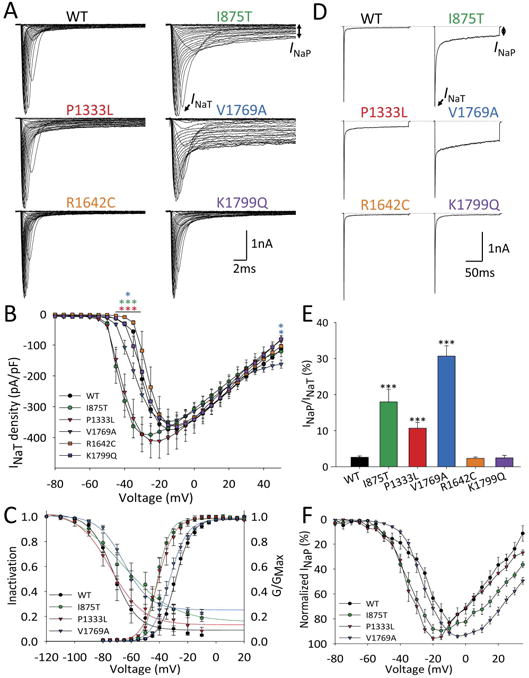
(A) Representative single leak-subtracted traces showing families of Na+ currents elicited by 20 ms depolarizing voltage steps from -80 to +50 mV in 5 mV increments from a holding potential of -120 mV with a 10 second intersweep interval for wild-type and each Nav1.3 variant co-expressed with β1 and β2. (B) Normalized I-V curves showing peak INaT current density (in pA/pF) vs. voltage for wild-type (n = 27), p.Ile875Thr (n = 14), p.Pro1333Leu (n = 10), p.Arg1642Cys (n = 8), p.Val1769Ala (n = 27), and p.Lys1799Gln (n = 13), presented as mean ± SEM. (C) Prepulse voltage-dependence of channel inactivation and conductance-voltage relationships for wild-type (n = 27), p.Ile875Thr (n = 14), p.Pro1333Leu (n = 10) and p.Val1769Ala (n = 19 respectively) were fit by a Boltzmann function.
(D) Representative individual traces showing INaT and INaP in response to a 200 ms voltage step from -120 to -10 mV for wild-type Nav1.3 and p.Ile875Thr, p.Pro1333Leu, p.Arg1642Cys, p.Val1769Ala, and p.Lys1799Gln. (E) Bar graph showing ratio of INaP/INaT for wild-type (n = 27), p.Ile875Thr (n =12), p.Pro1333Leu (n = 14), p.Arg1642Cys (n = 12), p.Val1769Ala (n = 25), and p.Lys1799Gln (n = 13), presented as mean ± SEM. (F) I-V curves of INaP in wild-type (n = 27) and Nav1.3 variants p.Ile875Thr (n = 14), p.Pro1333Leu (n = 10), and p.Val1769Ala (n = 13). Note that INaP at each voltage is normalized to the maximum value. *, p < 0.05; **, p < 0.01; ***, p < 0.001 vs. wild-type via one-way ANOVA with Bonferroni correction for multiple comparisons.
Slightly higher peak current density values for p.Ile875Thr and p.Pro1333Leu are likely a consequence of a hyperpolarizing leftward-shift in the voltage dependence of activation (FIG 3B-C). In these two mutant channels, the voltage at which maximal inward current was observed was shifted in the hyperpolarized direction, being -15.9 ± 1.6 mV for wild-type hNav1.3 (n = 27), -28.9 ± 2.4 mV for p.Ile875Thr (n = 14; p < 0.01 vs. wild-type via one-way ANOVA with Bonferroni correction for multiple comparisons), and -26.0 ± 2.8 for p.Pro1333Leu (n = 10; p < 0.01 vs. wild-type). In contrast, we did not observe a change in the voltage dependence of activation for Nav1.3-p.Val1769Ala (-29.4 ± 1.1 mV; n = 27; p = 0.079 vs. wild-type via one-way ANOVA).
We noted striking differences in the slowly inactivating/”persistent” Na+ current (INaP) between groups, defined here as residual non-inactivating current at 200 ms following a voltage step from -120 mV (FIG 3D-F). INaP was 2.6 ± 0.4 % (n = 27) of peak transient current (INaT) for wild-type channels, but was 17.9 ± 3.4 % for p.Ile875Thr (n = 12; p < 0.001 vs. wild-type), 10.7 ± 1.6 % for p.Pro1333Leu (n = 14; p < 0.0001 vs. wild-type), and 30.7 ± 2.8 % for p.Val1769Ala (n = 25; p < 0.0001 vs. wild-type). We noted that this current was not persistent per se,30–32 but rather inactivated with slow kinetics over the course of seconds for both wild-type or mutant Nav1.3 channels: for example, remaining current at 1 second following a voltage step to -10 mV was 0.7 ± 0.2% of peak INaT for wild-type Nav1.3 channels (n = 7), and 5.3 ± 1.4% of peak for Nav1.3-p.Val1769Ala. We also found that Nav1.3-p.Val1769Ala mutant channels displayed markedly larger ramp currents (wild-type: 197.1± 26.0 pA, n = 7; p.Val1769Ala: 1370.0 ± 305.0 pA, n = 12; p = 0.013) and larger total charge (wild-type: 0.96± 0.13 nanoCoulomb (nC), n = 7; p.Val1769Ala: 9.0± 2.0 nC, n = 12; p = 0.012) in response to 0.8 mV/ms slow depolarizations (FIG 4C).
Figure 4. Anti-seizure medications partially normalize pathological persistent current in Nav1.3 mutant channels.

(A) Traces showing INaT and INaP of wild-type and mutant NaV1.3 channels before (black) and after (red traces) application of 100 μM lacosamide (A1) and phenytoin (A2). (B) Bar graph showing % blockade of INaP by lacosamide (wild-type, n = 10; p.Ile875Thr, n = 6; p.Pro1333Leu, n = 6; p.Val1769Ala, n = 10) and phenytoin (wild-type, n = 3; p.Val1769Ala, n = 7). (C) Representative examples of Nav1.3 currents evoked by a voltage ramp from -120 to +40 mV at 0.8 mV/ms in wild-type Nav1.3 (top left) and p.Val1769Ala mutant channels (top right), with block by phenytoin (bottom). (D) Quantification of charge (in nanocoulombs; nC) between wild-type Nav1.3 (n = 5) and p.Val1769Ala (n = 5) before and after drug application. All data are presented as means ± SEM. * indicates p < 0.05 and ** indicates p < 0.01 via paired two-tailed t-test.
Targeting persistent current is a proposed therapeutic strategy in epilepsy.33,34 As Nav1.3 mutants p.Ile875Thr, p.Pro1333Leu, and p.Val1769Ala showed increased persistent current relative to wild-type Nav1.3, we hypothesized that pharmacological agents that target INaP might normalize pathological epilepsy-associated Na+ channel current, and could represent potential treatments for SCN3A encephalopathy. We tested agents known to exert activity on this current component. The INaP component of Nav1.3 has been shown to be blocked by various anti-seizure medications including phenytoin, lacosamide, topiramate, and carbamazepine.20,35 We first tested lacosamide, shown previously to act as a channel blocker primarily of Nav1.7,35–37 but also of Nav1.3 and Nav1.8.35
Consistent with previous results from wild-type Nav1.3,35 we found that 100 μM lacosamide had no effect on peak transient Na+ current (before, 435.2 ± 24.4 pA/pF; after, 447.4 ± 21.2 pA/pF; n =10, p = 0.41). We did find that lacosamide produced a small but statistically significant block of persistent current, as measured at 200 ms during a depolarizing pulse from -120 to -10 mV (from 2.2 ± 1.0 to 0.8 ± 0.2% or 52.5 ± 6.9% block; n = 10; p = 0.019 via paired two-tailed t-test). This effect did not wash out; however, the effect was seen after an incubation time of as little as 1 minute. We confirmed that this purported block by lacosamide was not due to rundown of INaP as persistent current was stable for at least 15 minutes in control experiments performed without addition of lacosamide to the extracellular solution.
We then tested lacosamide on heterologously-expressed Nav1.3 mutants and found that it produced a similar proportional decrease in INaP. In Nav1.3-p.Val1769Ala mutants, which exhibit the largest persistent current, 100 μM lacosamide produced a 48.1 ± 5.1% block in INaP, from 33.0 ± 4.1% to 17.7 ± 2.8% of peak transient current (n = 10; p < 0.001). Similar effects (63.7 ± 1.2% block and 49.9 ± 6.7% block) were seen in p.Ile875Thr and p.Pro1333Leu mutants (FIG 4A-B).
We next tested phenytoin, also known to block persistent sodium current.30,38 In rat neocortical pyramidal neurons, phenytoin produces a hyperpolarizing shift in the voltage dependence of INaP inactivation.30 100 μM phenytoin had a small effect on peak transient sodium current of wild-type Nav1.3 channels (control, 403.9 ± 34.7 pA/pF; phenytoin, 318.5 ± 58.3 pA/pF; n =5, p = 0.14), but produced a decrease in INaP from 1.3 ± 0.3% to 0.8 ± 0.2% of peak transient current (n = 5; p = 0.20). In Nav1.3-p.Val1769Ala mutant channels, phenytoin also had minimal effect on INaT (control, 389.9.7 ± 36.7 pA/pF; phenytoin, 365.7 ± 29.7 pA/pF; n =7, p = 0.27), but produced a striking reduction of INaP (from 24.0 ± 3.6 to 6.5 ± 0.9% of the transient current, a 68.4 ± 7.9% block; n = 7; p = < 0.01 via paired t-test) (FIG 4C-D). Phenytoin also reduced peak currents (control, 3,002.6 ± 696.0 pA; phenytoin, 1,149.0 ± 244.0 pA; n = 5; p = 0.026) and total charge (control, 19.8± 4.7 nC; phenytoin, 8.4± 2.0 nC; n = 5; p = 0.029) in response to 0.8 mV/ms slow ramp depolarizations from -120 mV (FIG 4C-DB).
Discussion
Here we demonstrate that heterozygous missense mutations in the gene SCN3A encoding the voltage-gated Na+ channel α subunit Nav1.3 are a cause of early infantile epileptic encephalopathy, identifying three causative variants in four patients. This conclusion is rigorously supported by multiple lines of evidence: the mutations are de novo and are absent from various control databases; the mutations occur at residues that exhibit a high level of evolutionary conservation; and functional studies demonstrate marked channel dysfunction. Mutations in other sodium channel genes SCN1A, 2A, and 8A, encoding Nav1.1, 1.2, and 1.6, respectively, have been previously determined to be important causes of severe epilepsy with onset in early childhood. With this report, the four major neuronally-expressed Na+ channel genes have all been implicated as causes of early infantile epileptic encephalopathy.
By identifying patients with SCN3A-related early infantile epileptic encephalopathy (SCN3A encephalopathy), we extend prior reports that have linked SCN3A variants with milder forms of epilepsy. Holland et al. (2008) reported a developmentally normal child with mild partial epilepsy onset at age 2 years with a heterozygous p.Lys354Gln variant in Nav1.3, which was inherited from the patient's unaffected father.23 Estacion et al. (2010) characterized rat Scn3a harboring the corresponding variant p.Lys343Gln (rScn3aK343Q) in heterologous systems and found increased persistent current (from 4.4 ± 0.4 to 8.1 ± 1.4% of INaT).21 Vanoye et al. (2014) performed specific targeted sequencing of SCN3A in a cohort of epilepsy patients and reported four Nav1.3 variants associated with childhood-onset epilepsy,22 although none of the patients had early-onset epileptic encephalopathy and inheritance pattern was not available. One variant, p.Glu1160Lys (p.Glu1111Lys in hNav1.3 isoform 2), showed increased persistent current (1.2 ± 0.2% for p.Glu1111Lys versus 0.5 ± 0.1% for wild-type). Lamar et al. (2017) reported a patient with a de novo p.Leu247Pro mutation in Nav1.3 with developmental delay and focal epilepsy onset at 18 months;24 functional studies showed decreased current density as well as cell surface expression suggesting loss of function. Trujillano et al (2017) reported a single patient with febrile seizures and epilepsy with a de novo SCN3A p.Pro1333Leu mutation, which is the same mutation seen in Patient 3 in our study, although additional details regarding the patient phenotype were not provided.39
Taken together, the results of our study combined with previous investigations suggest a spectrum of SCN3A-related phenotypes, ranging from mild epilepsy with normal neurocognitive development to early infantile epileptic encephalopathy. Our study suggests that SCN3A mutations causative of epileptic encephalopathy exhibit prominent gain of function with markedly increased non-inactivating Na+ current, while variants identified in patients with milder phenotypes may result in loss of function or more subtle gain of function. For many of the previously reported cases of epilepsy associated with variants in SCN3A, inheritance could not be demonstrated, and such variants may represent rare population variants that could act as modifiers of disease or risk alleles rather than being causative of highly penetrant monogenic epilepsy per se.
Patients 1 and 2 in this study exhibited an abnormal MRI showing polymicrogyria (see FIG 2). This was not a consistent feature of the cohort of patients with SCN3A encephalopathy, but was instead only seen in the two patients, who were unrelated, with the recurrent p.Ile875Thr variant, suggesting a potential mutation-specific effect on structural brain development. The basis of any association between SCN3A mutation and malformation of cerebral cortical development is unclear. Such an association could reflect a non-canonical ion channel function of Nav1.3, such as in mediating cell-cell interactions during cerebral cortical development, perhaps via interaction (or abnormal interaction) with or by auxiliary β subunits, which are known to have roles in cell adhesion. Alternatively, this could be due to an early deleterious effects of abnormal electrogenesis on neuronal migration and cerebral cortical development. It should be noted that we cannot absolutely rule out the unlikely possibility of a contribution of a second, perhaps somatic, mutation,40 or mutation in an unknown gene, or of an early life insult such as infection or hypoxic ischemic brain injury, as contributory. Ascertainment of additional cases will be required to begin to clarify the issue of genotype:phenotype correlation and potential role of Nav1.3 in structural brain development. Of note, developmental brain malformations have also been found in patients with epilepsy with identified causative de novo mutations in other ion channels.41,42
Our findings offer the suggestion that SCN3A encephalopathy with onset in the first two months of life can be clearly differentiated from Dravet syndrome, which is mainly due to heterozygous loss of function of SCN1A.5,6 This presumably reflects the differential developmental expression pattern of these two genes, with SCN3A being expressed at high levels during embryogenesis and during early postnatal life, falling to near-undetectable levels by adulthood in rodent brain (although low-level expression persists in adult brain tissue13). In contrast, SCN1A expression is at low levels at birth, increasing in infancy and young childhood.12
The p.Ile875Thr and p.Pro1333Leu de novo mutations produce a marked left-shift in the voltage dependence of activation of approximately 10 mV. This finding is consistent with the location of these mutations in the S4-5 linker, close to the positively charged arginine residues of the S4 helix domain of the voltage sensor. In addition, these mutations are located in DII and III, respectively: the first three domains are thought to be responsible for fast activation, which may account for the left-shift in the voltage dependence of activation observed for these variants. The mutation identified in Patient 4 (p.Val1769Ala), which was not associated with a hyperpolarizing shift in the G-V curve (albeit smaller in magnitude), is located in DIV.43,44 While the exact mechanism of voltage-dependent activation of Na+ channels is not fully determined, it is thought that the S4-S5 linker translates voltage-dependent shifts of the S4 segment into opening of the pore,45 with motion of S4-S5 coupled to that of neighboring S5 and S6 segments.46
We also found increased slowly inactivating/”persistent” current in p.Ile875Thr, p.Pro1333Leu, and p.Val1769Ala, corresponding to the three de novo mutations identified in the patients with early-onset epileptic encephalopathy in this cohort. The persistent sodium current INaP remains a somewhat enigmatic feature of Na+ channels and continues to be a subject of intense investigation. INaP is thought to be important for various cellular functions including regulation of repetitive firing, boosting of synaptic inputs, and in the generation of subthreshold membrane potential oscillations.31,32,47,48 Nav1.3 is known to exhibit INaP in heterologous systems as well as in neurons under normal and pathological conditions.21,49,50 The value for persistent current recorded here for wild-type Nav1.3 (4.4%) is higher that reported by Vanoye et al. (2014), similar to that previously by Estacion et al. (2010), and lower than reported by Sun et al. (2007). This current may not be truly “persistent,” but rather exhibits slow inactivation on the order of seconds or tens of seconds,30,32,47,51–53 as we have also shown here. We further found that the anti-seizure medications lacosamide and phenytoin selectively blocked INaP over INaT and that block was proportional to the amplitude of the persistent current.
Previous data indicate that mutations in various sodium channels across all four domains including intracellular loops could affect the amplitude of INaP.54 The finding that Nav1.3-p.Ile875Thr (located in DII) and p.Val1769Ala (located in DIV) were associated with the largest increase in INaP is consistent with a recent report using the tarantula toxin RTX-VII. This toxin increases Nav1.3-mediated INaP via interaction with DII and DIV. 55 Interestingly, focal application of RTX-VII to the surface of the cerebral cortex of adult rats induced seizures, consistent with a potentially ictogenic effect of increased Nav1.3 INaP and supporting the pathogenic mechanism proposed here. Why the p.Pro1333Leu mutation, located in DIII, also increased INaP, remains unclear. While additional work is required to further elucidate the structural basis of Nav1.3-mediated INaP, our results may provide some additional insight into this phenomenon.
We noted that the I-V relation of Na+ channels containing Nav1.3-p.Val1769Ala exhibited a somewhat flat appearance at depolarized potentials. The basis of this observation is unclear and would require additional investigation, and may be attributable to allosteric modification of voltage-dependent gating by the Nav1.3-p.Val1769Ala mutation.
The inheritance pattern of the p.Arg1642Cys variant could not be definitively established. We did note that the p.Arg1642Cys variant is homologous to an p.Arg1657Cys missense mutation described in a family with GEFS+, and this difference between Dravet syndrome and the more mild GEFS parallels the difference in disease severity between Patient 5 and the more severe Patients 1-4 with proven de novo mutations. We did find that Nav1.3-p.Arg1642Cys channels appeared to recover from inactivation more rapidly, which might be predicted to increase neuronal excitability by supporting the ability to discharge action potentials repetitively. Further work will be required to better delineate the relationship of this variant to epilepsy and the mechanism whereby faster recovery from Na+ inactivation might produce cellular hyperexcitability.
The establishment of a gene-specific diagnosis is important as it may have current or future treatment implications. For example, clinical data indicates that epilepsy in patients with Dravet syndrome responds best to specific anti-seizure medications, while anti-seizure medications with a predominant mechanism of action of Na+ channel blockade may exacerbate seizures and are considered to be contraindicated.56,57 In contrast, recent reports suggest that epilepsy in some patients with SCN8A encephalopathy may respond well to sodium channel antagonists such as phenytoin.7,58 Our data raises the possibility that early introduction of agents specifically targeting Nav1.3-mediated persistent Na+ current, during the transient developmental time period within which this channel is expressed, represents a potential strategy for treatment of SCN3A encephalopathy.
In conclusion, we report a cohort of four patients with infantile onset epileptic encephalopathy due to de novo heterozygous gain-of-function missense mutations in the gene SCN3A encoding the type 3 voltage gated sodium channel Nav1.3. Mutant channels exhibited various mechanisms of gain-of-function, including activation at hyperpolarized potentials, and enhanced persistent current. We hypothesize that SCN3A encephalopathy presents in the early infantile period due to the developmental expression pattern of SCN3A, combined with prominent gain-of-function effects predicted to increase neuronal excitability. We further show that the enhanced persistent current seen in mutant channels may be selectively inhibited with the anti-seizure medications lacosamide and phenytoin. Given the transient developmental trajectory of SCN3A, it is hopeful that partial normalization of pathological epilepsy-associated ion channel activity in the neonatal period in patients with SCN3A encephalopathy could protect the newborn brain until Nav1.3 is replaced by Nav1.1, 1.2, and 1.6, later in infancy.
Table 3.
Biophysical properties of wild-type Nav1.3 channels and Nav1.3 variants.
| Current density (at -10 mV) | Voltage dependence of activation | Voltage dependence of inactivation | Recovery from inactivation | Persistent current | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| pA/pF | n | V1/2 (mV) | k | n | V1/2 (mV) | k | n | τ1 (ms) | τ2 (ms) | n | % of peak | n | |
| WT | 373.1 ± 16.8 | 27 | -26.2 ± 1.3 | 5.1 ± 0.3 | 27 | -69.7 ± 2.2 | 10.2 ± 1.2 | 9 | 3.9 ± 0.6 | 63.6 ± 20.9 | 4 | 2.6 ± 0.4 | 27 |
| p.Ile875Thr | 391.1 ± 28.6 | 15 | -37.1 ± 1.4* | 4.9 ± 0.5 | 14 | -49.2 ± 6.4† | 19.9 ± 3.2† | 5 | 4.5 ± 1.5 | 47.6 ± 20.7 | 4 | 18.0 ± 3.5* | 12 |
| p.Pro1333Leu | 384.4 ± 53.1 | 12 | -37.7 ± 2.2* | 4.7 ± 0.6 | 10 | -58.7 ± 10.6 | 9.5 ± 1.3 | 4 | 2.6 ± 0.6 | 25.7 ± 4.4 | 6 | 10.7 ± 1.6* | 14 |
| p.Val1769Ala | 364.1 ± 10.7 | 19 | -29.4 ± 1.1 | 5.5 ± 0.3 | 27 | -49.6 ± 3.9** | 16.9 ± 2.1† | 9 | 2.1 ± 0.1 | 71.5 ± 8.6 | 7 | 30.7 ± 2.8* | 25 |
| p.Arg1642Cys | 360.9 ± 43.3 | 8 | -21.6 ± 1.7 | 4.7 ± 0.5 | 6 | -66.9 ± 1.7 | 4.9 ± 1.2* | 5 | 1.2 ± 0.2† | 26.4 ± 7.9 | 7 | 2.3 ± 0.4 | 12 |
| p.V1799Q | 344.9 ± 24.1 | 13 | -24.7 ± 1.2 | 5.3 ± 0.4 | 13 | -73.7 ± 2.1 | 7.8 ± 0.6 | 12 | 3.3 ± 0.4 | 49.5 ± 6.2 | 7 | 2.4 ± 0.7 | 13 |
p < 0.001 vs. wild-type (one-way ANOVA with Bonferroni correction for multiple comparisons).
p < 0.01 vs. wild-type
p < 0.05 vs. wild-type
Acknowledgments
We thank Toshinori Hoshi, Dejian Ren, and Eric D. Marsh for discussions and a critical reading of the manuscript, Manuel Covarrubias for a tSA-201 cell line, Lori L. Isom for the gift of a β-1 cDNA clone, and Alfred L. George for the gift of a β-2 cDNA clone. This work was supported by NIH NINDS K08 NS097633 and the Burroughs Wellcome Fund Career Award for Medical Scientists to EMG.
Footnotes
Author Contributions: TZ, IH, and EMG, contributed to the conception and design of the study; TZ, IH, IBB, SD, ACB, KW, LM, AM, BP, KLH, XZ, and EMG contributed to the acquisition and analysis of data; TZ and EMG contributed to drafting the text and preparing the figures.
Potential Conflicts of Interest: Nothing to report.
References
- 1.Catterall WA. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron. 2000;26(1):13–25. doi: 10.1016/s0896-6273(00)81133-2. [DOI] [PubMed] [Google Scholar]
- 2.Catterall WA. Voltage-gated sodium channels at 60: structure, function and pathophysiology. [Internet] J Physiol. 2012;590(11):2577–2589. doi: 10.1113/jphysiol.2011.224204. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3424717&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O'Malley HA, Isom LL. Sodium Channel β Subunits: Emerging Targets in Channelopathies. Annu Rev Physiol. 2015;77(1):481–504. doi: 10.1146/annurev-physiol-021014-071846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Catterall WA, Goldin AL, Waxman SG International Union of Pharmacology. International Union of Pharmacology. XXXIX. Compendium of Voltage-Gated Ion Channels: Sodium Channels [Internet] Pharmacol Rev. 2003;55(4):575–578. doi: 10.1124/pr.55.4.7. Available from: http://www.ncbi.nlm.nih.gov/pubmed/14657413. [DOI] [PubMed] [Google Scholar]
- 5.Claes L, Del-Favero J, Ceulemans B, et al. De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. [Internet] Am J Hum Genet. 2001;68(6):1327–1332. doi: 10.1086/320609. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3424717&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Claes L, Ceulemans B, Audenaert D, et al. De novo SCN1A mutations are a major cause of severe myoclonic epilepsy of infancy. [Internet] Hum Mutat. 2003;21(6):615–621. doi: 10.1002/humu.10217. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12754708. [DOI] [PubMed] [Google Scholar]
- 7.Wolff M, Johannesen KM, Hedrich UBS, et al. Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders [Internet] Brain. 2017;140(5):1316–1336. doi: 10.1093/brain/awx054. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28379373. [DOI] [PubMed] [Google Scholar]
- 8.Kearney JA, Plummer NW, Smith MR, et al. A gain-of-function mutation in the sodium channel gene Scn2a results in seizures and behavioral abnormalities [Internet] Neuroscience. 2001;102(2):307–317. doi: 10.1016/s0306-4522(00)00479-6. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11166117. [DOI] [PubMed] [Google Scholar]
- 9.Sugawara T, Tsurubuchi Y, Agarwala KL, et al. A missense mutation of the Na+ channel alpha II subunit gene Na(v)1.2 in a patient with febrile and afebrile seizures causes channel dysfunction. [Internet] Proc Natl Acad Sci USA. 2001;98(11):6384–6389. doi: 10.1073/pnas.111065098. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3424717&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Larsen J, Carvill GL, Gardella E, et al. The phenotypic spectrum of SCN8A encephalopathy [Internet] Neurology. 2015;84(5):480–489. doi: 10.1212/WNL.0000000000001211. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25568300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Veeramah KR, O'Brien JE, Meisler MH, et al. De Novo Pathogenic SCN8A Mutation Identified by Whole-Genome Sequencing of a Family Quartet Affected by Infantile Epileptic Encephalopathy and SUDEP. Am J Hum Genet. 2012;90(3):502–510. doi: 10.1016/j.ajhg.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheah CS, Westenbroek RE, Roden WH, et al. Correlations in timing of sodium channel expression, epilepsy, and sudden death in Dravet syndrome. Channels. 2013;7(6):468–472. doi: 10.4161/chan.26023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Whitaker WR, Clare JJ, Powell AJ, et al. Distribution of voltage-gated sodium channel alpha-subunit and beta-subunit mRNAs in human hippocampal formation, cortex, and cerebellum. [Internet] J Comp Neurol. 2000;422(1):123–139. doi: 10.1002/(sici)1096-9861(20000619)422:1<123::aid-cne8>3.0.co;2-x. Available from: http://www.ncbi.nlm.nih.gov/pubmed/10842222. [DOI] [PubMed] [Google Scholar]
- 14.Beckh S, Noda M, Lübbert H, Numa S. Differential regulation of three sodium channel messenger RNAs in the rat central nervous system during development. [Internet] EMBO J. 1989;8(12):3611–3616. doi: 10.1002/j.1460-2075.1989.tb08534.x. Available from: http://www.ncbi.nlm.nih.gov/pubmed/2555170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Felts PA, Yokoyama S, Dib-Hajj S, et al. Sodium channel alpha-subunit mRNAs I, II, III, NaG, Na6 and hNE (PN1): different expression patterns in developing rat nervous system. Brain Res Mol Brain Res. 1997;45(1):71–82. doi: 10.1016/s0169-328x(96)00241-0. [DOI] [PubMed] [Google Scholar]
- 16.Nassar MA, Baker MD, Levato A, et al. Nerve Injury Induces Robust Allodynia and Ectopic Discharges in Na v1.3 Null Mutant Mice [Internet] Mol Pain. 2006;2:1733–1744. doi: 10.1186/1744-8069-2-33. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17052333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hains BC, Klein JP, Saab CY, et al. Upregulation of sodium channel Nav1.3 and functional involvement in neuronal hyperexcitability associated with central neuropathic pain after spinal cord injury. [Internet] J Neurosci. 2003;23(26):8881–8892. doi: 10.1523/JNEUROSCI.23-26-08881.2003. Available from: http://www.ncbi.nlm.nih.gov/pubmed/14523090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo F, Yu N, Cai JQ, et al. Voltage-gated sodium channel Nav1.1, Nav1.3 and $β$1 subunit were up-regulated in the hippocampus of spontaneously epileptic rat [Internet] Brain Res Bull. 2008;75(1):179–187. doi: 10.1016/j.brainresbull.2007.10.005. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18158113. [DOI] [PubMed] [Google Scholar]
- 19.Li HJ, Wan RP, Tang LJ, et al. Alteration of Scn3a expression is mediated via CpG methylation and MBD2 in mouse hippocampus during postnatal development and seizure condition [Internet] Biochim Biophys Acta - Gene Regul Mech. 2015;1849(1):1–9. doi: 10.1016/j.bbagrm.2014.11.004. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25459751. [DOI] [PubMed] [Google Scholar]
- 20.Sun G, Werkman TR, Battefeld A, et al. Carbamazepine and topiramate modulation of transient and persistent sodium currents studied in HEK293 cells expressing the Na(v)1.3 alpha-subunit. [Internet] Epilepsia. 2007;48(4):774–782. doi: 10.1111/j.1528-1167.2007.01001.x. Available from: http://doi.wiley.com/10.1111/j.1528-1167.2007.01001.x. [DOI] [PubMed] [Google Scholar]
- 21.Estacion M, Gasser A, Dib-hajj SD, Waxman SG. A sodium channel mutation linked to epilepsy increases ramp and persistent current of Nav1.3 and induces hyperexcitability in hippocampal neurons [Internet] Exp Neurol. 2010;224(2):362–368. doi: 10.1016/j.expneurol.2010.04.012. Available from: http://dx.doi.org/10.1016/j.expneurol.2010.04.012. [DOI] [PubMed] [Google Scholar]
- 22.Vanoye CG, Gurnett CA, Holland KD, et al. Neurobiology of Disease Novel SCN3A variants associated with focal epilepsy in children [Internet] Neurobiol Dis. 2014;62:313–322. doi: 10.1016/j.nbd.2013.10.015. Available from: http://dx.doi.org/10.1016/j.nbd.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holland KD, Kearney JA, Glauser TA, et al. Mutation of sodium channel SCN3A in a patient with cryptogenic pediatric partial epilepsy. 2008;433:65–70. doi: 10.1016/j.neulet.2007.12.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lamar T, Vanoye CG, Calhoun J, et al. SCN3A deficiency associated with increased seizure susceptibility [Internet] Neurobiol Dis. 2017;102:38–48. doi: 10.1016/j.nbd.2017.02.006. Available from: http://dx.doi.org/10.1016/j.nbd.2017.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Y, Du X, Bin R, et al. Genetic Variants Identified from Epilepsy of Unknown Etiology in Chinese Children by Targeted Exome Sequencing. [Internet] Sci Rep. 2017;7:40319. doi: 10.1038/srep40319. Available from: http://www.nature.com/articles/srep40319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sadleir LG, Mountier EI, Gill D, et al. Not all SCN1A epileptic encephalopathies are Dravet syndrome. Neurology. 2017;89(10):1035–1042. doi: 10.1212/WNL.0000000000004331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen YJ, Shi YW, Xu HQ, et al. Electrophysiological Differences between the Same Pore Region Mutation in SCN1A and SCN3A [Internet] Mol Neurobiol. 2015;51(3):1263–1270. doi: 10.1007/s12035-014-8802-x. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24990319. [DOI] [PubMed] [Google Scholar]
- 28.Grant AC, Vazquez B. A case of extended spectrum GEFS+. [Internet] Epilepsia. 2005;46(Suppl 1s10):39–40. doi: 10.1111/j.1528-1167.2005.00357.x. Available from: http://doi.wiley.com/10.1111/j.1528-1167.2005.00357.x. [DOI] [PubMed] [Google Scholar]
- 29.Vanoye CG, Lossin C, Rhodes TH, George AL. Single-channel Properties of Human Na V 1.1 and Mechanism of Channel Dysfunction in SCN1A -associated Epilepsy. J Gen Physiol. 2006;127(1):1–14. doi: 10.1085/jgp.200509373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Colombo E, Franceschetti S, Avanzini G, Mantegazza M. Phenytoin Inhibits the Persistent Sodium Current in Neocortical Neurons by Modifying Its Inactivation Properties. PLoS One. 2013;8(1) doi: 10.1371/journal.pone.0055329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crill WE. Persistent Sodium Current in Mammalian. 1996;(31) doi: 10.1146/annurev.ph.58.030196.002025. [DOI] [PubMed] [Google Scholar]
- 32.Fleidervish IA, Friedman A, Gutnick MJ. Slow inactivation of Na+ current and slow cumulative spike adaptation in mouse and guinea-pig neocortical neurones in slices. [Internet] J Physiol. 1996;493:83–97. doi: 10.1113/jphysiol.1996.sp021366. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3424717&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stafstrom CE. Persistent Sodium Current and Its Role in Epilepsy [Internet] Epilepsy Curr. 2007;7(1):15–22. doi: 10.1111/j.1535-7511.2007.00156.x. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17304346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mantegazza M, Curia G, Biagini G, et al. Voltage-gated sodium channels as therapeutic targets in epilepsy and other neurological disorders [Internet] Lancet Neurol. 2010;9(4):413–424. doi: 10.1016/S1474-4422(10)70059-4. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20298965. [DOI] [PubMed] [Google Scholar]
- 35.Sheets PL, Heers C, Stoehr T, Cummins TR. Differential Block of Sensory Neuronal Voltage-Gated Sodium Channels by Lacosamide [(2 R) -2- (Acetylamino) - N -benzyl-3. J Pharmacol Exp Ther. 2008;326(1):89–99. doi: 10.1124/jpet.107.133413. [DOI] [PubMed] [Google Scholar]
- 36.de Greef BTA, Merkies ISJ, Geerts M, et al. Efficacy, safety, and tolerability of lacosamide in patients with gain-of-function Nav1.7 mutation-related small fiber neuropathy: study protocol of a randomized controlled trial–the LENSS study [Internet] Trials. 2016;17(1):306. doi: 10.1186/s13063-016-1430-1. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27363506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jo S, Bean BP. Lacosamide Inhibition of Nav1.7 Voltage-Gated Sodium Channels: Slow Binding to Fast-Inactivated States [Internet] Mol Pharmacol. 2017;91(4):277–286. doi: 10.1124/mol.116.106401. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28119481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lampl I, Schwindt P, Crill W. Reduction of cortical pyramidal neuron excitability by the action of phenytoin on persistent Na+ current. [Internet] J Pharmacol Exp Ther. 1998;284(1):228–237. Available from: http://www.ncbi.nlm.nih.gov/pubmed/9435183. [PubMed] [Google Scholar]
- 39.Trujillano D, Bertoli-Avella AM, Kumar Kandaswamy K, et al. Clinical exome sequencing: results from 2819 samples reflecting 1000 families [Internet] Eur J Hum Genet. 2017;25(2):176–182. doi: 10.1038/ejhg.2016.146. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27848944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jamuar SS, Lam ATN, Kircher M, et al. Somatic Mutations in Cerebral Cortical Malformations [Internet] [cited 2018 Jan 25];N Engl J Med. 2014 371(8):733–743. doi: 10.1056/NEJMoa1314432. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25140959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Platzer K, Yuan H, Schütz H, et al. GRIN2B encephalopathy: novel findings on phenotype, variant clustering, functional consequences and treatment aspects. J Med Genet. 2017:2016–104509. doi: 10.1136/jmedgenet-2016-104509. jmedgenet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barba C, Parrini E, Coras R, et al. Co-occurring malformations of cortical development and SCN1A gene mutations. Epilepsia. 2014;55(7):1009–1019. doi: 10.1111/epi.12658. [DOI] [PubMed] [Google Scholar]
- 43.Capes DL, Goldschen-Ohm MP, Arcisio-Miranda M, et al. Domain IV voltage-sensor movement is both sufficient and rate limiting for fast inactivation in sodium channels [Internet] J Gen Physiol. 2013;142(2):101–112. doi: 10.1085/jgp.201310998. Available from: http://www.jgp.org/lookup/doi/10.1085/jgp.201310998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goldin AL. Mechanisms of sodium channel inactivation. Curr Opin Neurobiol. 2003;13(3):284–290. doi: 10.1016/s0959-4388(03)00065-5. [DOI] [PubMed] [Google Scholar]
- 45.Long SB, Campbell EB, Mackinnon R. Voltage Sensor of Kv1.2: Structural Basis of Electromechanical Coupling [Internet] Science (80) 2005;309(5736):903–908. doi: 10.1126/science.1116270. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16002579. [DOI] [PubMed] [Google Scholar]
- 46.Shen H, Zhou Q, Pan X, et al. Structure of a eukaryotic voltage-gated sodium channel at near-atomic resolution [Internet] Science (80) 2017;355(6328):eaal4326. doi: 10.1126/science.aal4326. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28183995. [DOI] [PubMed] [Google Scholar]
- 47.Do MTH, Bean BP. Subthreshold sodium currents and pacemaking of subthalamic neurons: Modulation by slow inactivation. Neuron. 2003;39(1):109–120. doi: 10.1016/s0896-6273(03)00360-x. [DOI] [PubMed] [Google Scholar]
- 48.Stuart G. Voltage-activated sodium channels amplify inhibition in neocortical pyramidal neurons. Nat Neurosci. 1999;2(2):144–150. doi: 10.1038/5698. [DOI] [PubMed] [Google Scholar]
- 49.Lampert A, Hains BC, Waxman SG. Upregulation of persistent and ramp sodium current in dorsal horn neurons after spinal cord injury [Internet] Exp Brain Res. 2006;174(4):660–666. doi: 10.1007/s00221-006-0511-x. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16718433. [DOI] [PubMed] [Google Scholar]
- 50.Cusdin FS, Nietlispach D, Maman J, et al. The Sodium Channel β3-Subunit Induces Multiphasic Gating in Na V 1.3 and Affects Fast Inactivation via Distinct Intracellular Regions. J Biol Chem. 2010;285(43):33404–33412. doi: 10.1074/jbc.M110.114058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Holtkamp D, Opitz T, Niespodziany I, et al. Activity of the anticonvulsant lacosamide in experimental and human epilepsy via selective effects on slow Na+ channel inactivation. Epilepsia. 2017;58(1):27–41. doi: 10.1111/epi.13602. [DOI] [PubMed] [Google Scholar]
- 52.Aracri P, Colombo E, Mantegazza M, et al. Layer-specific properties of the persistent sodium current in sensorimotor cortex. J Neurophysiol. 2006;95(6):3460–3468. doi: 10.1152/jn.00588.2005. [DOI] [PubMed] [Google Scholar]
- 53.Magistretti J, Alonso A. Biophysical Properties and Slow Voltage-Dependent Inactivation of a Sustained Sodium Current in Entorhinal Cortex Layer-II Principal Neurons. J Gen Physiol. 1999;114(4):491–509. doi: 10.1085/jgp.114.4.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jarecki BW, Piekarz AD, Jackson JO, Cummins TR. Human voltage-gated sodium channel mutations that cause inherited neuronal and muscle channelopathies increase resurgent sodium currents [Internet] J Clin Invest. 2010;120(1):369–378. doi: 10.1172/JCI40801. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20038812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tang C, Zhou X, Zhang Y, et al. Synergetic Action of Domain II and IV Underlies Persistent Current Generation in Nav1.3 as revealed by a tarantula toxin [Internet] Sci Rep. 2015;5(1):9241. doi: 10.1038/srep09241. Available from: http://www.nature.com/articles/srep09241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wirrell EC, Laux L, Donner E, et al. Optimizing the Diagnosis and Management of Dravet Syndrome: Recommendations from A North American Consensus Panel [Internet] Pediatr Neurol. 2017;0(0):136–2336. doi: 10.1016/j.pediatrneurol.2017.01.025. Available from: http://linkinghub.elsevier.com/retrieve/pii/S0887899416310372. [DOI] [PubMed] [Google Scholar]
- 57.Wirrell EC. Treatment of Dravet Syndrome [Internet] Can J Neurol Sci / J Can des Sci Neurol. 2016;43(S3):S13–S18. doi: 10.1017/cjn.2016.249. Available from: http://www.journals.cambridge.org/abstract_S0317167116002493. [DOI] [PubMed] [Google Scholar]
- 58.Boerma RS, Braun KP, van de Broek MPH, et al. Remarkable Phenytoin Sensitivity in 4 Children with SCN8A-related Epilepsy: A Molecular Neuropharmacological Approach [Internet] Neurotherapeutics. 2016;13(1):192–197. doi: 10.1007/s13311-015-0372-8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26252990. [DOI] [PMC free article] [PubMed] [Google Scholar]