

Abstract
Obesity has reached epidemic proportions in the Western society and is increasing in the developing world. It is considered as one of the major contributors to the global burden of disability and chronic diseases, including autoimmune, inflammatory and degenerative diseases. Research conducted on obesity and its complications over the last two decades has transformed the outdated concept of white adipose tissue (WAT) merely serving as an energy depot. WAT is now recognized as an active and inflammatory organ capable of producing a wide variety of factors known as adipokines. These molecules participate through endocrine, paracrine, autocrine or juxtacrine crosstalk mechanisms in a great variety of physiological or pathophysiological processes, regulating food intake, insulin sensitivity, immunity and inflammation. Although initially restricted to metabolic activities (regulation of glucose and lipid metabolism), adipokines currently represent a new family of proteins that can be considered key players in the complex network of soluble mediators involved in the pathophysiology of immune/inflammatory diseases. However, the complexity of the adipokine network in the pathogenesis and progression of inflammatory diseases has posed, since the beginning, the important question of whether it may be possible to target the mechanism(s) by which adipokines contribute to disease selectively without suppressing their physiological functions. Here, we explore in depth the most recent findings concerning the involvement of adipokines in inflammation and immune responses, in particular in rheumatic, inflammatory and degenerative diseases. We also highlight several possible strategies for therapeutic development and propose that adipokines and their signalling pathways may represent innovative therapeutic strategies for inflammatory disorders.
Abbreviations
- ADAMTS
a disintegrin and metalloproteinase with thrombospondin motifs
- AMPK
AMP‐activated protein kinase
- CPCs
chondrogenic progenitor cells
- DCs
dendritic cells
- LCN2
lipocalin‐2
- OA
osteoarthritis
- PGRN
progranulin
- RA
rheumatoid arthritis
- SOCS‐3
suppressor of cytokine signalling 3
- STAT
signal transducer and activator of transcription
- Th
T helper cells
- TIMP
tissue inhibitors of metalloproteinases
- Treg
T regulatory cells
- VCAM
vascular cell adhesion protein
- WAT
white adipose tissue
Introduction
Obesity, the major public health problem in the Western world, has reached epidemic proportions and continues to rise in developing countries. Being itself one of the major contributors to disability, obesity is associated with several chronic autoimmune and inflammatory diseases, such as Type 2 diabetes mellitus, cardiovascular disease, osteoarthritis (OA) and rheumatoid arthritis (RA), and thus has a high socio‐economic impact (Zhang et al., 2014). For decades, researchers have focused on the identification of risk factors, preventive measures and treatments for obesity. However, public health policies centred on diet and physical activity have been largely ineffective. Furthermore, pharmacological approaches have not provided any safe and long‐term therapies (Zhang et al., 2014). Consequently, it is urgent to gain a deeper understanding of the development of obesity‐associated pathologies and to focus on adipose tissue biology in this context. White adipose tissue (WAT) is now recognized as an active endocrine organ, apart from serving an energy storage tissue, and a source of adipose tissue‐derived factors (adipokines). These adipokines have been described as pleiotropic molecules, contributing importantly to low‐grade systemic inflammation in obese subjects (Tilg and Moschen, 2006). http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5015, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3726, lipocalin‐2 (LCN2) and progranulin (PGRN) are adipokines thought to be crucial links between obesity and immune system, and are thus attractive therapeutic targets in obesity‐associated diseases, such as OA and RA.
White adipose tissue as a pro‐inflammatory tissue in obesity
WAT is an active endocrine organ consisting of mature and developing adipocytes, as well as fibroblasts, endothelial cells and a broad array of immune cells, namely, adipose tissue macrophages, neutrophils, eosinophils, mast cells, T and B cells (Huh et al., 2014). Thus, WAT is now considered as a bona fide immunometabolic endocrine organ (Vieira‐Potter, 2014).
In WAT of lean individuals, the crosstalk between adipocytes and immune cells maintains tissue homeostasis. In particular, eosinophils and T regulatory cells (Treg), the main resident T cell population, secreted anti‐inflammatory cytokines (IL‐10 and IL‐4) that polarize adipose tissue macrophages towards an anti‐inflammatory phenotype (i.e. M2 or alternatively activated macrophages), thus maintaining a tolerogenic environment (Exley et al., 2014; Huh et al., 2014). Moreover, ‘lean’ WAT secretes more adiponectin that enhances the sensitivity to insulin. A positive energy balance results in adipocyte expansion, which leads to increased leptin secretion and infiltration of inflammatory cells. Adipocyte hypoxia, apoptosis and cell stress were able to induce the expression of chemoattractant molecules with the consequent recruitment of macrophages, T and B cells (Exley et al., 2014; Huh et al., 2014). T cells become activated, number of Treg cells were reduced, and there is a macrophage phenotypic switch from M2 to M1, which accumulate around necrotic adipocytes forming ‘crown‐like structures’ and producing large amounts of pro‐inflammatory cytokines, such as http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4998 and TNF‐α (Vieira‐Potter, 2014). Additionally, obesity is characterized by a dysregulated secretion of WAT adipokines, such as leptin, adiponectin, LCN2 and PGRN, which have emerged as crucial regulators of the innate and adaptive immune system (Tilg and Moschen, 2006; Scotece et al., 2014; Abella et al., 2017b). Altogether, these data have shown WAT to be an important contributor to local and systemic inflammation in obesity (Figure 1).
Figure 1.
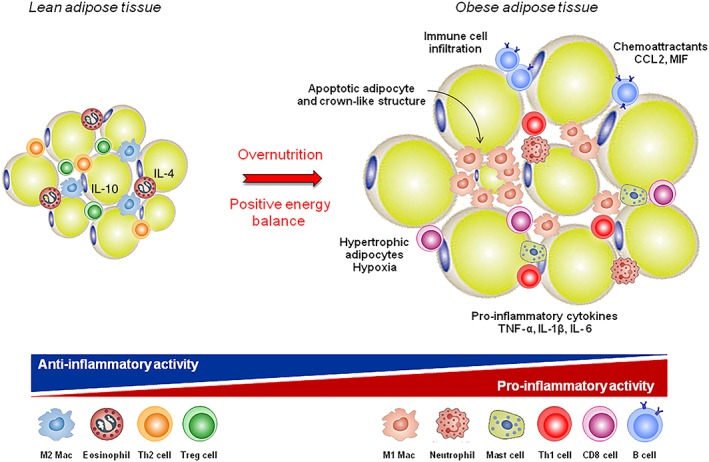
White adipose tissue (WAT) as a pro‐inflammatory tissue. In lean adipose tissue, the crosstalk between adipocytes and immune resident cells maintains tissue homeostasis. In particular, anti‐inflammatory cytokines (IL‐10 and IL‐4) that promote M2 macrophage phenotype, are secreted by Treg cells. Overnutrition results in WAT expansion and adipocyte hypoxia, with consequent production of chemoattractants and infiltration of immune cells. B and T cells become activated, and there is a phenotypic switch from M2 to M1 macrophages, which accumulate around necrotic adipocytes forming ‘crown‐like structures’.
The adipokine superfamily
Adipokines are low MW, pharmacologically active proteins that possess pleiotropic activity. Acting in the hypothalamic region as orexigenic and anorexigenic hormones, adipokines play a crucial role in energy metabolism by communicating the nutrient status of the organism (Al‐Suhaimi and Shehzad, 2013). Furthermore, adipokines are currently considered as key players in inflammation and immunity, as most of them are increased in obesity and contribute to the ‘low‐grade inflammatory state’ associated with obesity (Tilg and Moschen, 2006).
Leptin, the first of the adipokine family, centrally regulates body weight by linking nutritional status and neuroendocrine function. Obese individuals exhibit enhanced circulating leptin levels but, due to leptin resistance (unresponsive state to leptin) in the hypothalamus, leptin fails to increase energy expenditure and to reduce food intake, with consequent body weight gain (Conde et al., 2013a, 2013b). Leptin can also stimulate the production of pro‐inflammatory cytokines and enhance T helper cells (Th)1 immune response, thus linking nutrition, metabolism and immune homeostasis (Abella et al., 2017b). Adiponectin is an intriguing adipokine that is related to insulin sensitivity, anti‐atherogenic actions, regulation of metabolic homeostasis and modulation of the immune system (Liu and Liu, 2014). In recent years, two novel adipokines, LCN2 and PGRN, have emerged as regulators of metabolism and immune function, bridging obesity and inflammatory pathologies that affect bones and joints (Villalvilla et al., 2016; Abella et al., 2017a).
Leptin and leptin receptors
Leptin is a 16 kDa non‐glycosylated cytokine‐like hormone encoded by the LEP gene (the human homologue of the murine ob gene) located on chromosome 7q31.3 (Green et al., 1995) and the best‐characterized member of the adipokine family (Scotece et al., 2014). It is mainly produced by adipocytes, and at low levels by skeletal muscle, intestine, gastric epithelium, placenta, mammary glands, brain, joint tissues and bone (Scotece et al., 2014). In physiological conditions, leptin circulating levels are positively correlated with the WAT mass and body mass index, but its synthesis is also modulated by inflammatory factors (Conde et al., 2011). This hormone has a central role in body weight homeostasis by inducing anorexigenic factors (such as cocaine‐amphetamine‐related transcript) and suppressing orexigenic neuropeptides (such as neuropeptide Y) in the hypothalamus (Zhou and Rui, 2014). Therefore, central leptin resistance, caused by impairment of leptin transportation, leptin signalling and leptin target neural circuits, is considered the main risk factor in the pathogenesis of obesity (Zhou and Rui, 2014). Leptin also affects other physiological functions, like bone metabolism, inflammation, infection and immune responses. Accordingly, http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=307#1712 are expressed throughout the cells of both innate (NK cells, granulocytes, monocytes, macrophages and dendritic cells [DCs]) and adaptive (B and T cells) immune systems (Abella et al., 2017b).
Leptin receptors and signalling
Leptin receptors (or Ob receptors) belong to the class I cytokine receptor family and are products of the diabetes (db) gene in mice (Münzberg and Morrison, 2015). Alternative splicing of the db gene produces at least six isoforms, which possess identical extracellular binding domains but differ by the length of the cytoplasmic domain: a long isoform (Ob‐Rb), four short isoforms (Ob‐Ra, Ob‐Rc, Ob‐Rd and Ob‐Rf) and a soluble isoform (Ob‐Re) (Zhou and Rui, 2014; Münzberg and Morrison, 2015). Leptin exerts its biological actions through activation of the long‐form receptor (OB‐Rb), which has the full intracellular domain with the typical signalling elements of cytokine receptors (Zhou and Rui, 2014; Münzberg and Morrison, 2015). This receptor does not have intrinsic tyrosine kinase (TK) activity but, after leptin binding, Ob‐Rb‐associated http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2048 becomes activated by auto‐phosphorylation or cross‐phosphorylation, which are facilitated by the formation of leptin receptor homodimers. The cytoplasmic domain of the receptor is then phosphorylated in tyrosine residues (Tyr974, 985, 1077, 1138), each one functioning as docking sites for cytoplasmic adaptors, like signal transducer and activator of transcription (STAT), particularly STAT3 (Zhou and Rui, 2014). Besides the canonical JAK/STAT pathway, leptin receptors also signal via alternative pathways including http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=514, http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=519, http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=518 http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=286&familyType=ENZYME, SHP2/GRB2 and http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=781/http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=285 pathways (Scotece et al., 2014; Zhou and Rui, 2014).
In mouse models, mutations in either ob or db gene result in leptin or leptin receptor ablation and severe obesity. However, in humans, common obesity is often characterized by hyperleptinemia, and administration of exogenous leptin does not result in weight loss, indicating an unresponsive state to leptin. Possible mechanisms of leptin resistance include decreased levels of cell surface Ob‐Rb, up‐regulation of negative regulators and down‐regulation of positive regulators. The best‐characterized mechanism of leptin resistance in the CNS is the feedback loop inhibition of leptin signalling by binding of suppressor of cytokine signalling 3 (SOCS‐3) to phosphorylated tyrosines of Ob‐Rb. http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2976 is also involved and acts as a negative regulator of leptin signalling through JAK2 dephosphorylation (Zhou and Rui, 2014; Münzberg and Morrison, 2015). In the periphery, leptin is increased in chondrocytes from obese OA patients (Pallu et al., 2010), and SOCS‐3 expression is lower in cartilage from obese than non‐obese individuals (Vuolteenaho et al., 2012). Thus, leptin responsiveness should be considered when interpreting the leptin effects in peripheral tissues and in the development of leptin‐directed therapeutic approaches.
Leptin in innate and adaptive immune system
Leptin has been described as a potent enhancer of the immune system (Abella et al., 2017b). In innate immunity, leptin augments the cytotoxicity of NK cells, and the activation of granulocytes (neutrophils, basophils and eosinophils), macrophages and DCs, thus exacerbating inflammatory responses (Abella et al., 2017b). Obese hyperleptinemic individuals have lower NK function compared with lean subjects, probably due to leptin resistance (Laue et al., 2015), neutrophils with augmented superoxide release and chemotactic activity (Brotfain et al., 2015), and eosinophils with greater adhesion and chemotaxis towards eotaxin and CCL5 (Grotta et al., 2013). Leptin‐stimulated human macrophages have increased M2‐phenotype surface markers but were able to secrete M1‐typical cytokines (TNF‐α, IL‐6 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4974β), indicating that leptin influences the phenotype of adipose tissue macrophages (Acedo et al., 2013). In DCs, leptin modulated their activation, chemoattraction and survival, with possible implications for DCs maturation and migration (Moraes‐Vieira et al., 2014). Moreover, http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=316), which play a critical role in innate immune system, have been described as important players in adipose tissue, obesity‐associated inflammation and leptin biology (Kim et al., 2012). In particular, leptin‐deficient obese mice showed increased expression of TLR1–9 and TLR11–13 as well as downstream signalling molecules and target cytokines (Kim et al., 2012).
In the adaptive immune system, leptin augments the proliferation of naïve T and B cells whereas it decreases Treg cells (Abella et al., 2017b). Accordingly, morbidly obese children (congenitally leptin‐deficient) had reduced number of circulating CD4+ T cells, and impaired T cell proliferation and cytokine release, which were rescued by administration of recombinant human leptin (Farooqi et al., 2002), and obese individuals presented reduced number of Treg cells (Wagner et al., 2013). Leptin also polarizes Th cells towards a proinflammatory (Th1, which secretes IFNγ) rather than anti‐inflammatory phenotype (Th2, which secretes IL‐4) (Martín‐Romero et al., 2000). In a preclinical model of collagen‐induced arthritis in mice, articular injection of leptin enhanced Th17 cells in joint tissues, with consequent exacerbation of inflammation and early onset of arthritis (Deng et al., 2012). Therefore, leptin decreases Treg cell proliferation, whereas it increases Th17 cell proliferation and responsiveness, indicating the therapeutic potential of the leptin system in inflammation and autoimmunity.
Leptin and osteoarthritis
Osteoarthritis, the most common joint disease, is a degenerative and multifactorial pathology triggered by inflammatory and metabolic imbalances affecting the entire joint structure (articular cartilage, meniscus, ligaments, bone and synovium) (Loeser et al., 2012). Leptin has been associated with OA and cartilage metabolism, as its levels are increased in serum, infrapatellar fat pad (IPFP), synovial tissues and cartilage of OA patients compared with healthy individuals (Conde et al., 2013a, 2013b). Additionally, Ob‐Rb is expressed in chondrocytes and is functional (Figenschau et al., 2001). Recently, a microarray analysis associated the leptin‐induced OA phenotype with the up‐regulation of inflammatory factors, MMPs, growth factors and osteogenic genes (Fan et al., 2018). Our group has demonstrated that leptin, in synergy with IL‐1β, induces the expression of pro‐inflammatory factors, including http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1250, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1376, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1883, IL‐6 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=821 in chondrocytes (Gomez et al., 2011). Chondrocyte‐synovial fibroblast crosstalk mediates leptin‐induced IL‐6 production in OA patients (Pearson et al., 2017). Moreover, leptin modulates the production of inflammatory mediators (IL‐6, IL‐8 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=756) by CD4+ T cells in OA patients, but not in healthy subjects (Scotece et al., 2017) hence demonstrating new insights into the action of leptin in the immune system and OA pathophysiology.
Leptin can also promote OA‐related joint destruction by directly inducing the expression of several http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=738 (MMP‐1, ‐2, ‐3, ‐9 and ‐13, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1677 and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1678), whereas http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4924 and proteoglycan were down‐regulated (Scotece et al., 2014). Moreover, leptin can perpetuate the cartilage‐degradation processes via induction of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6758 in human primary chondrocytes, which attracts leukocytes and monocytes to inflamed joints through the action of chemoreceptors (Conde et al., 2012).
MicroRNAs (miRNAs), small single‐stranded non‐coding segments of RNA, are increasingly recognized as regulatory molecules involved in disease processes, including OA, inflammation and obesity. The levels of miR‐27, which directly targets the 3′‐untranslated region of leptin, are low in OA chondrocytes and injection with miR‐27 lentiviral overexpression vector in a preclinical model of OA in rats resulted in decreased levels of IL‐6 and IL‐8, as well as MMP‐9 and ‐13, thus indicating the protective action of miR‐27 in OA, possibly by targeting leptin (Zhou et al., 2017).
Chondrogenic progenitor cells (CPCs) are cartilage seed cells crucial to maintain cartilage homeostasis and replace damaged tissue. Leptin reduces CPC migratory ability and their chondrogenic potential and induces CPC senescence and osteogenic transformation, thus changing the pattern of CPC differentiation (Zhao et al., 2016). Leptin also regulates bone metabolism via induction of abnormal osteoblast function, which is associated with joint destruction in OA patients (Conde et al., 2015).
Taken together, these results indicated a key role of leptin in OA pathophysiology by influencing pro‐inflammatory status, cartilage catabolic activity, as well as cartilage and bone remodelling.
Leptin and rheumatoid arthritis
Rheumatoid arthritis (RA) is a chronic inflammatory joint disease defined by synovial membrane inflammation and hyperplasia (‘swelling’), production of autoantibodies – autoimmune disease and destruction of cartilage and bone (‘deformity’) (Smolen et al., 2016). Leptin levels were augmented in RA patients, and its serum and synovial fluid (SF) levels were associated with disease duration, parameters of RA activity and radiographic joint damage (Rho et al., 2009; Olama et al., 2012), although there are controversial results and large cohort studies are necessary. Leptin‐deficient mice demonstrate a less severe antigen‐induced arthritis, decreased levels of TNF‐α and IL‐1β in synovium from the knee, a defective cell‐mediated immunity and a shift towards Th2 cell response (Busso et al., 2002). Moreover, reducing leptin levels in RA patients by fasting improves the clinical symptoms of the disease (Fraser et al., 1999). Leptin protein mutants with antagonist activity and monoclonal antibodies against human leptin receptors or leptin itself are promising therapeutic approaches to RA (Tian et al., 2014). Of note, clinical studies evaluating the effect of insulin sensitivity modulators (affected by leptin levels), such as the PPARγ agonists, are ongoing as a new potential treatment to improve the inflammatory status and cardiovascular outcome in RA patients (Chimenti et al., 2015). Further knowledge of the mechanisms of leptin's actions would be important for RA treatment.
Therefore, leptin can be identified as a link between immune tolerance, metabolic function and autoimmunity, and approaches directed to leptin signalling could provide future innovative therapies for autoimmune disorders like RA.
Adiponectin and adiponectin receptors
Adiponectin (also known as GBP28, apM1, Acrp30 or AdipoQ) is a 244‐residue protein with structural homology to collagen type VIII and X, and complement factor C1q. It is mainly synthesized in adipose tissue and is found in several configurations: the http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3727, the full‐length adiponectin (fAPN), the low MW (LMW) adiponectin, the medium MW (MMW) adiponectin, the high MW (HMW) adiponectin, and the serum albumin bonded LMW form (Alb‐LMW) (Sun et al., 2009; Liu and Liu, 2014). In morbidly obese patients, circulating adiponectin levels tend to be low, increasing with weight loss and with use of thiazolidinediones (PPAR agonists), which enhance insulin sensitivity (Liu and Liu, 2014). Adiponectin acts as an endogenous insulin sensitizer by stimulating glucose uptake through its ability to increase fatty acid oxidation and to reduce the synthesis of glucose in the liver. Human adiponectin is encoded by the ADIPOQ gene, which is located on chromosome 3q27 a locus linked with susceptibility to diabetes and cardiovascular disease (Liu and Liu, 2014).
Adiponectin receptors and signalling
Adiponectin acts specifically via two receptors, the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=649 predominantly found in skeletal muscle and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=650&familyId=106&familyType=OTHER, mainly present in the liver. Signalling pathways from the Adipo receptors lead to activation of http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1540&familyId=474&familyType=ENZYME, PPAR‐α, and PPAR‐γ (Liu and Liu, 2014). AMPK activation by adiponectin has been implicated in its insulin‐sensitizing activity in liver and muscles, while AMPK, Ca2+ and PPAR‐α are involved in the regulation of glucose and fatty acid metabolism by adiponectin. Ceramide and MAPK signalling pathways also mediate the actions of adiponectin (Liu and Liu, 2014). Increasing evidence reveals the importance of adiponectin in inflammation‐related pathologies, including cardiovascular disease, endothelial dysfunction, Type 2 diabetes, metabolic syndrome, OA and RA (Liu and Liu, 2014), probably due to its modulation of the innate immune response, as well as B and T cells (Luo and Liu, 2016).
Adiponectin in innate and adaptive immune system
Adiponectin has been recognized as a key regulator of the immune system, playing a major role in the progression of inflammatory and metabolic disorders (Luo and Liu, 2016). Nevertheless, whether adiponectin behaves as an anti‐inflammatory or pro‐inflammatory factor is still a matter of intense debate. This adipokine suppresses the differentiation and the classical activation of M1 macrophages by down‐regulating pro‐inflammatory cytokines (TNF‐α, CCL2 and IL‐6), while it promotes M2 macrophage proliferation and expression of anti‐inflammatory M2 markers (http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1244, Mgl‐1, and IL‐10) (Luo and Liu, 2016). Adiponectin also modulated the activity of eosinophils, neutrophils, NK cells and DCs (Tilg and Moschen, 2006; Luo and Liu, 2016), but it remains unclear whether adiponectin positively or negatively regulates their function. These paradoxical, dual effects might result from different functions of different adiponectin configurations (Sun et al., 2009). For example, HMW and gAPN but not MMW and LMW adiponectin increased NF‐κB activity in monocytic cells while gAPN but not fAPN decreased LPS‐stimulated ERK1/2 pathway in Kupffer cells (Sun et al., 2009).
In the adaptive immune system, adiponectin activates plasma B cells and stimulates the secretion of the B cell‐derived peptide PEPITEM, which inhibits the migration of memory T cells (Chimen et al., 2015). In T cells, Adipo receptors are up‐regulated after its activation and adiponectin decreased antigen‐specific T cell proliferation and cytokine production, via enhancement of T cell apoptosis (Procaccini et al., 2013). Adiponectin also enhances Th1 differentiation and adiponectin‐treated DCs significantly induced both Th1 and Th17 responses in allogenic T cells, contributing to enhanced pro‐inflammatory responses (Procaccini et al., 2013). A greater knowledge of adiponectin's effects on B and T cells and its mechanism of action will be important for developing new therapeutic strategies aimed at the adiponectin system.
Adiponectin and osteoarthritis
There is considerable evidence for the involvement of adiponectin in OA pathophysiology. Serum and plasma adiponectin levels are increased in OA patients, compared with healthy individuals, and its levels in OA synovial fluid were correlated with aggrecan degradation. Moreover, adiponectin was expressed by synovial fibroblasts, infrapatellar fat pad, osteophytes, cartilage and bone tissues within the joint (Scotece et al., 2014). Adiponectin has been associated with erosive OA and with OA severity assessed radiologically, leading to a proposed use as a biomarker for OA (Poonpet, 2014). However, a recent study reported no association between serum adiponectin levels and erosive or non‐erosive hand OA, while resistin and visfatin were suggested as possible OA biomarkers (Fioravanti et al., 2017).
Spontaneous animal models of OA (STR/Ort mice) present with lower serum adiponectin levels than controls. Adiponectin has been reported to have a protective action by inhibiting IL‐1β‐induced MMP‐13 expression and to up‐regulate tissue inhibitors of metalloproteinases (TIMP)‐2 production in human chondrocytes (Scotece et al., 2014). However, most of the data indicated a pro‐inflammatory and catabolic role for adiponectin in OA cartilage by increasing the production of NO, IL‐6, IL‐8, VCAM‐1, TIMP‐1, MMP‐1, ‐3 and ‐13 (Kang et al., 2010; Scotece et al., 2014), which lead to cartilage degradation and OA pathogenesis. Furthermore, adiponectin modulates bone metabolism by stimulation of human osteoblast proliferation and mineralization, via p38 MAPK signalling pathway and bone morphogenetic protein‐2 (http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4881: Scotece et al., 2014). Nevertheless, there are contradictory results published, and further studies are of the utmost importance to clarify the exact role of adiponectin in the joint cartilage and bone and in the pathogenesis of OA.
Adiponectin and rheumatoid arthritis
Clinical evidence suggests that adiponectin levels are increased in serum and synovial fluid from RA patients, compared with healthy subjects, and that baseline serum adiponectin levels are predictive of RA radiographic progression (Giles et al., 2009; Rho et al., 2009; Chen et al., 2013). Adiponectin, alone or in combination with IL‐1β, induces IL‐6, IL‐8 and PGE2 production in RA synovial fibroblasts, with IL‐6 production being dependent on a Adipo1 receptor/AMPK/p38 MAPK/NF‐κB signalling pathway (Chen et al., 2013; Scotece et al., 2014). Of note, anti‐IL‐6 receptor monoclonal antibody has been approved in several countries for the treatment of RA. Adiponectin also increased the production of MMP‐1, ‐13 and VEGF in synovial cells and promoted joint inflammation via attraction of immune cells into the synovium and induction of cytokine production (Scotece et al., 2014), thus indicating a crucial role of adiponectin in synovitis and joint destruction in RA.
Although in vitro data indicate adiponectin as a potential mediator of RA, in vivo this adipokine exhibits quite different effects. In a preclinical model of collagen‐induced arthritis in mice, adiponectin treatment mitigated the severity of arthritis along with a decrease in the expression of TNF‐α, IL‐1β and MMP‐3 in joint tissues (Lee et al., 2008). More data from basic research and from clinical observations in large‐scale cohort studies are important to a further elucidation of the roles and mechanisms of adiponectin in inflammation‐related pathologies, such as RA.
Other adipokines
Lipocalin‐2
Lipocalin‐2 (LCN2; also known as neutrophil gelatinase‐associated lipocalin, 24p3, p25, migration‐stimulating factor inhibitor, human neutrophil lipocalin, α‐1‐microglobulin‐related protein, siderocalin or uterocalin) is a glycoprotein encoded by a gene located at the chromosome locus 9q34.11 (Abella et al., 2015). Originally identified in mouse kidney cells and human neutrophil granules, Lipocalin‐2 is also expressed in immune cells, liver, spleen and chondrocytes, although WAT is its major source (Abella et al., 2015). Two receptors for LCN2 have been proposed: the http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=200#1035 (24p3R) that binds to mouse LCN2 and the megalin/glycoprotein GP330, an LDL receptor that binds human LCN2 protein. LCN2 circulates as a 25 kDa monomer, a 46 kDa homodimer and in a covalent complex with MMP‐9, which blocks MMP‐9 auto‐degradation (Villalvilla et al., 2016) The members of the lipocalin family contain a hydrophobic ligand binding pocket, which confers the ability to bind and transport steroids, LPS, fatty acids, iron, and in the case of LCN2, siderophores. LCN2 has also been involved in the induction of apoptosis in haematopoietic cells, modulation of inflammation and metabolic homeostasis (Abella et al., 2015). Of note, thiazolidinedione treatment reverses obesity‐induced LCN2 expression (Abella et al., 2015), and there is increasing evidence suggesting that LCN2 contributes to obesity‐related disorders, such as Type 2 diabetes and non‐alcoholic fatty liver disease (Moschen et al., 2017).
LCN2 binds to enterobactin, a siderophore present in Gram‐negative bacteria, which transports iron into the bacteria. By depleting bacterial iron stores necessary for their growth, LCN2 exhibits bacteriostatic effects (Abella et al., 2015), which have been implicated in the protection of gastrointestinal tract against various pathogens (Moschen et al., 2017). The promoter region of LCN2 contains binding sites for key inflammatory transcription factors, including NF‐κB, STAT1, STAT3, and C/EBP. Accordingly, LCN2 acts as an anti‐inflammatory regulator of M1/M2 macrophage polarization via NF‐κB/STAT3 loop activation (Guo et al., 2014). In adaptive immunity, LCN2 induced human leukocyte antigen G, a well‐known tolerogenic mediator, on CD4+ T cells, and up‐regulated the expansion of Treg cells in healthy subjects (Abella et al., 2015). Taking into account the role of LCN2 in the modulation of inflammatory and immune response, future studies should investigate the therapeutic potential of LCN2 in immunosuppressive therapy efficacy, tolerance induction in transplanted patients, and to other inflammatory/immune system disorders, such as OA and RA.
In joint tissues, LCN2 is produced as a mechano‐responsive adipokine whose expression can be induced by inflammatory mediators. In osteoblasts, the absence of mechanical loading stimulates LCN2 expression, probably contributing to bone metabolism via stimulation of pro‐osteoclastogenic factors, receptor activator of NF‐κB ligand and IL‐6, and inhibition of the anti‐osteoclastogenic factor, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1882 (Abella et al., 2015). The expression of LCN2 is also augmented by inflammatory cytokines (TNF‐α and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4982) in osteoblasts, while in chondrocytes, it is induced by stimulated osteoblast conditioned medium, IL‐1β, adipokines (leptin and adiponectin), LPS and dexamethasone (Villalvilla et al., 2016). Interestingly, NO is able to exert a control on LCN2 expression in chondrocytes, suggesting the existence of a feedback loop regulating its expression.
In OA patients, LCN2 levels are increased in synovial fluid and cartilage, where it is involved in cartilage degradation via blocking MMP‐9 auto‐degradation and reduction of chondrocyte proliferation (Gupta et al., 2007; Abella et al., 2015). Recently, glucocorticoids (commonly used to treat OA and RA), alone or in combination with IL‐1, have been reported to induce LCN2 expression through the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=625&familyId=98&familyType=NHR and PI3K, ERK1/2 and JAK2 pathways in a mouse chondrogenic cell line (Conde et al., 2017). The transcription factors E74‐like factor 3 and NF‐κB were also reported as modulators of LCN2 expression in chondrocytes (Conde et al., 2016). Together, these data reveal that LCN2 acts as a sensor of mechanical load and inflammatory status of the joint, leading to alterations in subchondral bone, cartilage and bone‐cartilage crosstalk underlying OA pathophysiology. However, one study has shown that LCN2 overexpression in mouse cartilage did not induce OA pathogenesis and that knockout of LCN2 in mice, did not affect the OA cartilage destruction, induced by destabilization of the medial meniscus (Choi and Chun, 2017). Thus, although LCN2 can contribute to OA pathophysiology, it is not sufficient by itself to induce OA cartilage destruction in mice. Further studies are necessary to fully elucidate the role of LCN2 in OA development in humans.
Patients with RA exhibit higher levels of LCN2 in synovial fluid than OA patients (Katano et al., 2009). Using proteome analysis, it has been demonstrated that GM‐CSF contributed to RA pathophysiology through up‐regulation of LCN2 in neutrophils, followed by induction of transitional endoplasmic reticulum ATPase, cathepsin D, and transglutaminase 2 in synoviocytes, with potential implications for the proliferation of synovial cells and infiltration of inflammatory cells into the synovium (Katano et al., 2009). Nevertheless, the actions of LCN2, relevant to RA pathophysiology remain largely unknown.
Progranulin
Progranulin (PGRN; also known as granulin‐epithelin precursor (GEP), proepithelin, GP88, PC‐cell‐derived growth factor or acrogranin) is a cysteine‐rich, secreted protein encoded by the GRN gene, located on chromosome 17q21.32 (Abella et al., 2017a). It is a 68–88 kDa secreted glycoprotein that can undergo enzymic proteolysis into small homologous subunits – granulins or epithelins (Wei et al., 2016). PGRN is produced by a wide range of cells, including epithelial cells, macrophages, chondrocytes and also adipocytes. Recently identified as an adipokine, PGRN has been implicated in inflammation, wound healing, obesity and rheumatic diseases (OA and RA), thus having a potential role as therapeutic target and biomarker in inflammatory diseases (Abella et al., 2017a).
PGRN is a key regulator of inflammation, at least in part, due to direct interaction with TNF receptors (possessing higher affinity than TNF‐α, especially for http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1871), and consequently acting as an antagonist of TNF/TNFR pro‐inflammatory signalling pathway (Jian et al., 2016; Wei et al., 2016). PGRN also binds to http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1883, which is involved in various inflammatory disorders (Jian et al., 2016). Furthermore, PGRN suppressed the production of two chemokines, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=837 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=835, through the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1870&familyId=334&familyType=CATALYTICRECEPTOR pathway, and induced Treg populations and IL‐10 production (Jian et al., 2016; Wei et al., 2016). PGRN can be degraded by several proteinases, like MMP‐9, ‐12 and ‐14, http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=739#1680, elastase and proteinase‐3, to generate granulins, which have pro‐inflammatory activity and may counteract the anti‐inflammatory action of intact PGRNs (Jian et al., 2016). Atsttrin, an engineered PGRN‐protein, effectively prevents the onset and progression of inflammatory arthritis in several preclinical animal models (Liu and Bosch, 2012). Hence, identification of PGRN and the discovery of atsttrin as antagonists of the TNF‐α/TNFR pathway may lead to new therapeutic interventions for TNF‐α‐mediated pathologies, including rheumatic diseases.
Expression of PGRN is augmented during chondrocyte differentiation in vitro, as well as in cartilage, synovial and infrapatellar fat pad samples from OA patients (Abella et al., 2016). Deficiency of PGRN results in an OA‐like phenotype in aged mice, and both recombinant PGRN and atsttrin protect against OA development (Jian et al., 2016). PGRN exhibits anti‐inflammatory properties in OA by promoting anabolic metabolism via TNFR2, and by inhibiting IL‐1β‐mediated catabolic metabolism (suppression of NOS2, COX‐2, MMP‐13 and VCAM‐1) through TNFR1 binding and blocking of TNF‐α mediated activation of NF‐κB, thus inhibiting MMPs and ADAMTS expression, and cartilage degradation (Abella et al., 2016; Jian et al., 2016). PGRN also plays a crucial role in the differentiation and proliferation of chondrocytes and in endochondral ossification of growth plate during development (Feng et al., 2010). Intra‐articular injection of mesenchymal stem cells that express recombinant atsttrin prevents the progression of degenerative changes in a surgically induced preclinical OA mouse model (Xia et al., 2015). Moreover, intra‐articular injection of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6789 (a fusion‐soluble TNFR2 protein that inhibits TNF‐α and has therapeutic activity in RA patients) induces more severe joint destruction in a preclinical OA mouse model, because it blocks PGRN binding to TNFR2 (Jian et al., 2016). Therefore, PGRN directed therapeutic approaches seem to be promising for OA treatment.
In RA patients, circulating and synovial fluid levels of PGRN are elevated and are associated with disease activity (Cerezo et al., 2015). Furthermore, TNF‐α‐driven inflammation and cartilage destruction are critical events in the development of RA pathology, and TNF biological therapies are considered as the most effective treatments for this autoimmune disease (Jian et al., 2016). As a TNF/TNFR inhibitor, PGRN‐derived atsttrin has been extensively studied on RA pathophysiology. In particular, atsttrin administration reduced the disease severity in preclinical collagen‐induced arthritis and collagen‐antibody‐induced arthritis mouse models, being even more effective than PGRN, possibly due to the absence of a complete granulin domain with pro‐inflammatory activity (Jian et al., 2016; Abella et al., 2017a). Interestingly, atsttrin was more effective in reducing inflammation than etanercept and demonstrated high stability and was well absorbed when administered intraperitoneally in mice (Jian et al., 2016). Although the experimental data of atsttrin for RA are promising, so far no clinical trials have been performed.
Future prospects for therapy
Great strides have been made in recent years to elucidate the role of adipokines as mechanistic drivers of obesity, inflammation and immunity. This review has summarized the increasing evidence of the role of the two classical adipokines, leptin and adiponectin, as well as LCN2 and PGRN in obesity‐associated inflammatory and autoimmune diseases, namely, OA and RA (Figure 2). Current OA drug therapies used in clinical practice are mainly based on the use of nonsteroidal anti‐inflammatory drugs and intra‐articular administration of corticosteroids, and there are no disease‐modifying drug therapies for OA, with the ability to inhibit structural damage of articular cartilage (Glyn‐Jones et al., 2015). RA treatment options include anti‐TNF‐α and anti‐IL‐1/IL‐1R biological agents, but their high cost, and short half‐life has prompted the development of alternative strategies against new therapeutic targets, such as adipokines, multiligand receptor for advanced glycation end products as well as inflammatory mediators and signalling pathway components (Alghasham and Rasheed, 2014). In fact, the development of promising therapeutic approaches targeting the adipokine network is already underway.
Figure 2.
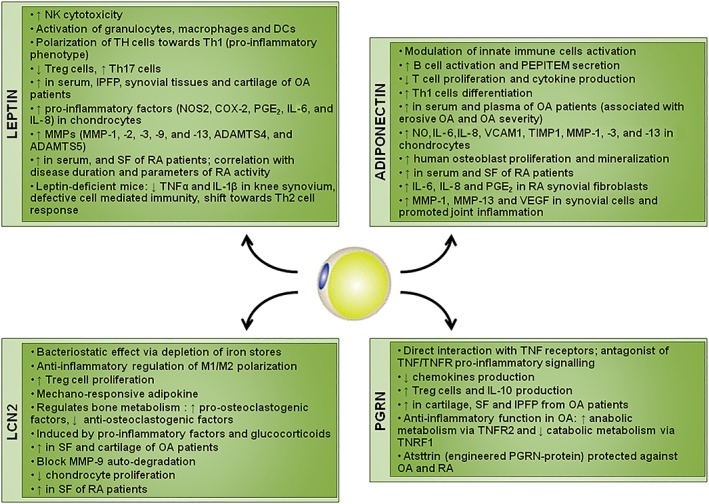
Schematic representation of the effects of the adipokines on inflammatory diseases. IPFP, infrapatellar fat pad; SF, synovial fluid
There is now good evidence that elevated leptin levels are linked to immune system derangements and obesity‐associated disorders (Abella et al., 2017b). Therefore, control of bioactive levels of leptin, by high‐affinity leptin‐binding molecules, monoclonal humanized antibodies blocking leptin receptors, administration of leptin receptor antagonists, or miRNAs targeting leptin, are likely to be feasible therapeutic options (Otvos et al., 2011). In the case of adiponectin, only a few protein‐based biological modulators have been developed due to the extreme insolubility of its C‐terminal domain and larger peptide fragments thereof (Otvos et al., 2014). LCN2 has been implicated in articular cartilage degradation (Villalvilla et al., 2016). Silencing RNAs or miRNAs, promotion of protein sumoylation and nanoparticle delivery systems for drug administration may be useful approaches to regulate LCN2 levels and activity (Meszaros and Malemud, 2012). Moreover, the PGRN‐derived engineered protein atsttrin prevents inflammation in arthritis models (Liu and Bosch, 2012). However, given the pleiotropic action of adipokines, a systematic approach to modulate their levels and thus prevent obesity‐associated disorders, such as OA and RA, might be, for the moment, unavailable. Instead, the local inhibition of adipokines at sites of joint injury or targeting of specific receptor isoforms could be viable options.
Given the role of adipokines in OA and RA pathophysiology, these molecules have been singled‐out as possible biomarkers for monitoring disease onset and progression, as well as the effectiveness of therapeutic interventions (Poonpet, 2014; Abella et al., 2015). However, further evaluations will be necessary to establish adipokines as biomarkers for use in clinical OA and RA.
Altogether, the data presented and reviewed in this paper propose adipokines as emerging biomarkers and therapeutic targets for immune disorders. However, the adipokine network is complex and further insights into the pathophysiological role of adipokines in the immune system as well as in the development of obesity‐associated disorders will be crucial for the development of novel therapeutic approaches.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a, b, c, d, e)
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
O.G. and F.L. are staff personnel of Xunta de Galicia (Servizo Galego de Saude, SERGAS) through a research‐staff stabilization contract (ISCIII/SERGAS). V.F. is a ‘Sara Borrell’ researcher funded by ISCIII and FEDER. R.G. is a ‘Miguel Servet’ researcher funded by Instituto de Salud Carlos III (ISCIII) and FEDER. O.G., M.A.G.G. and R.G. are members of RETICS Programme, RD16/0012/0014 (RIER: Red de Investigación en Inflamación y Enfermedades Reumáticas) via Instituto de Salud Carlos III (ISCIII) and FEDER. F.L. is a member of CIBERCV (Centro de Investigación Biomédica en Red de Enfermedades Cardiovasculares). The work of O.G. (PIE13/00024, PI14/00016 and PI17/00409), F.L. (PI15/00681 and CB16/11/00226) and R.G. (PI16/01870 and CP15/00007) was funded by Instituto de Salud Carlos III and FEDER. O.G. is a beneficiary of a project funded by Research Executive Agency of the European Union in the framework of MSCA‐RISE Action of the H2020 Programme (project number 734899). A.M. has received funding from the European Commission Framework 7 programme (EU FP7; HEALTH.2012.2.4.5‐2, project number 305815; Novel Diagnostics and Biomarkers for Early Identification of Chronic Inflammatory Joint Diseases) plus generous support from the Innovative Medicines Initiative Joint Undertaking under grant agreement no. 115770, resources of which are composed of financial contribution from the European Union's Seventh Framework programme (FP7/2007‐2013) and EFPIA companies' in kind contribution. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Francisco, V. , Pino, J. , Gonzalez‐Gay, M. A. , Mera, A. , Lago, F. , Gómez, R. , Mobasheri, A. , and Gualillo, O. (2018) Adipokines and inflammation: is it a question of weight?. British Journal of Pharmacology, 175: 1569–1579. doi: 10.1111/bph.14181.
References
- Abella V, Pino J, Scotece M, Conde J, Lago F, Gonzalez‐Gay MA et al (2017a). Progranulin as a biomarker and potential therapeutic agent. Drug Discov Today 22: 1557–1564. [DOI] [PubMed] [Google Scholar]
- Abella V, Scotece M, Conde J, Gómez R, Lois A, Pino J et al (2015). The potential of lipocalin‐2/NGAL as biomarker for inflammatory and metabolic diseases. Biomarkers 20: 565–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abella V, Scotece M, Conde J, López V, Pirozzi C, Pino J et al (2016). The novel adipokine progranulin counteracts IL‐1 and TLR4‐driven inflammatory response in human and murine chondrocytes via TNFR1. Sci Rep 6: 20356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abella V, Scotece M, Conde J, Pino J, Gonzalez‐Gay MA, Gómez‐Reino JJ et al (2017b). Leptin in the interplay of inflammation, metabolism and immune system disorders. Nat Rev Rheumatol 13: 100–109. [DOI] [PubMed] [Google Scholar]
- Acedo SC, Gambero S, Cunha FGP, Lorand‐Metze I, Gambero A (2013). Participation of leptin in the determination of the macrophage phenotype: an additional role in adipocyte and macrophage crosstalk. Vitr Cell Dev Biol ‐ Anim 49: 473–478. [DOI] [PubMed] [Google Scholar]
- Al‐Suhaimi EA, Shehzad A (2013). Leptin, resistin and visfatin: the missing link between endocrine metabolic disorders and immunity. Eur J Med Res 18: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Other proteins. Br J Pharmacol 174: S1–S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Catalytic receptors. Br J Pharmacol 174: S225–S271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017c). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Cidlowski JA, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017d). The Concise Guide to PHARMACOLOGY 2017/18: Nuclear hormone receptors. Br J Pharmacol 174: S208–S224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD et al (2017e). The Concise Guide to PHARMACOLOGY 2017/18: Transporters. Br J Pharmacol 174: S360–S446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alghasham A, Rasheed Z (2014). Therapeutic targets for rheumatoid arthritis: progress and promises. Autoimmunity 47: 77–94. [DOI] [PubMed] [Google Scholar]
- Brotfain E, Hadad N, Shapira Y, Avinoah E, Zlotnik A, Raichel L et al (2015). Neutrophil functions in morbidly obese subjects. Clin Exp Immunol 181: 156–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busso N, So A, Chobaz‐Peclat V, Morard C, Martinez‐Soria E, Talabot‐Ayer et al (2002). Leptin signaling deficiency impairs humoral and cellular immune responses and attenuates experimental arthritis. J Immunol 168: 875–882. [DOI] [PubMed] [Google Scholar]
- Cerezo LA, Kuklová M, Hulejová H, Vernerová Z, Kaspříková N, Veigl D et al (2015). Progranulin is associated with disease activity in patients with rheumatoid arthritis. Mediators Inflamm 2015: 740357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Lu J, Bao J, Guo J, Shi J, Wang Y (2013). Adiponectin: a biomarker for rheumatoid arthritis? Cytokine Growth Factor Rev 24: 83–89. [DOI] [PubMed] [Google Scholar]
- Chimen M, Mcgettrick HM, Apta B, Kuravi SJ, Yates CM, Kennedy A et al (2015). Homeostatic regulation of T cell trafficking by a B cell derived peptide is impaired in autoimmune and chronic inflammatory disease Europe PMC Funders Group. Nat Med 21: 467–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chimenti MS, Triggianese P, Conigliaro P, Candi E, Melino G, Perricone R (2015). The interplay between inflammation and metabolism in rheumatoid arthritis. Cell Death Dis 6: e1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi WS, Chun JS (2017). Upregulation of lipocalin‐2 (LCN2) in osteoarthritic cartilage is not necessary for cartilage destruction in mice. Osteoarthritis Cartilage 25: 401–405. [DOI] [PubMed] [Google Scholar]
- Conde J, Gomez R, Bianco G, Scotece M, Lear P, Dieguez C et al (2011). Expanding the adipokine network in cartilage: identification and regulation of novel factors in human and murine chondrocytes. Ann Rheum Dis 70: 551–559. [DOI] [PubMed] [Google Scholar]
- Conde J, Lazzaro V, Scotece M, Abella V, Villar R, López V et al (2017). Corticoids synergize with IL‐1 in the induction of LCN2. Osteoarthritis Cartilage 25: 1172–1178. [DOI] [PubMed] [Google Scholar]
- Conde J, Otero M, Scotece M, Abella V, López V, Pino J et al (2016). E74‐like factor 3 and nuclear factor‐κB regulate lipocalin‐2 expression in chondrocytes. J Physiol 594:6133–6146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conde J, Scotece M, Abella V, López V, Pino J, Gómez‐Reino JJ et al (2015). Basic aspects of adipokines in bone metabolism. Clin Rev Bone Miner Metab 13: 11–19. [Google Scholar]
- Conde J, Scotece M, López V, Abella V, Hermida M, Pino J et al (2013a). Differential expression of adipokines in infrapatellar fat pad (IPFP) and synovium of osteoarthritis patients and healthy individuals. Ann Rheum Dis 73:631. [DOI] [PubMed] [Google Scholar]
- Conde J, Scotece M, López V, Gómez R, Lago F, Pino J et al (2013b). Adipokines: novel players in rheumatic diseases. Discov Med 15: 73–83. [PubMed] [Google Scholar]
- Conde J, Scotece M, López V, Gómez R, Lago F, Pino J et al (2012). Adiponectin and leptin induce VCAM‐1 expression in human and murine chondrocytes. PLoS One 7: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng J, Liu Y, Yang M, Wang S, Zhang M, Wang X et al (2012). Leptin exacerbates collagen‐induced arthritis via enhancement of Th17 cell response. Arthritis Rheum 64: 3564–3573. [DOI] [PubMed] [Google Scholar]
- Exley MA, Hand L, O'Shea D, Lynch L (2014). Interplay between the immune system and adipose tissue in obesity. J Endocrinol 223: R41–R48. [DOI] [PubMed] [Google Scholar]
- Fan Q, Liu Z, Shen C, Li H, Ding J, Jin F et al (2018). Microarray study of gene expression profile to identify new candidate genes involved in the molecular mechanism of leptin‐induced knee joint osteoarthritis in rat. Hereditas 155: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farooqi IS, Matarese G, Lord GM, Keogh JM, Lawrence E, Agwu C et al (2002). Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. J Clin Invest 110: 1093–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng JQ, Guo F‐J, Jiang B‐C, Zhang Y, Frenkel S, Wang D‐W et al (2010). Granulin epithelin precursor: a bone morphogenic protein 2‐inducible growth factor that activates Erk1/2 signaling and JunB transcription factor in chondrogenesis. FASEB J 24: 1879–1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figenschau Y, Knutsen G, Shahazeydi S, Johansen O, Sveinbjörnsson B (2001). Human articular chondrocytes express functional leptin receptors. Biochem Biophys Res Commun 287: 190–197. [DOI] [PubMed] [Google Scholar]
- Fioravanti A, Cheleschi S, De Palma A, Addimanda O, Mancarella L, Pignotti E et al (2017). Can adipokines serum levels be used as biomarkers of hand osteoarthritis? Biomarkers : 1–6 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- Fraser DA, Thoen J, Reseland JE, Førre, Kjeldsen‐Kragh J (1999). Decreased CD4+ lymphocyte activation and increased interleukin‐4 production in peripheral blood of rheumatoid arthritis patients after acute starvation. Clin. Rheumatology 18: 394–401. [DOI] [PubMed] [Google Scholar]
- Giles JT, Allison M, Bingham CO, Scott WM, Bathon JM (2009). Adiponectin is a mediator of the inverse association of adiposity with radiographic damage in rheumatoid arthritis. Arthritis Rheum 61: 1248–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glyn‐Jones S, Palmer AJR, Agricola R, Price AJ, Vincent TL, Weinans H et al (2015). Osteoarthritis. Lancet 386: 376–387. [DOI] [PubMed] [Google Scholar]
- Gomez R, Scotece M, Conde J, Gomez‐Reino JJ, Lago F, Gualillo O (2011). Adiponectin and leptin increase IL‐8 production in human chondrocytes. Ann Rheum Dis 70: 2052–2054. [DOI] [PubMed] [Google Scholar]
- Green ED, Maffei M, Braden VV, Proenca R, DeSilva U, Zhang Y et al (1995). The human obese (OB) gene: RNA expression pattern and mapping on the physical, cytogenetic, and genetic maps of chromosome 7. Genome Res 5: 5–12. [DOI] [PubMed] [Google Scholar]
- Grotta MB, Squebola‐Cola DM, Toro AA, Ribeiro MA, Mazon SB, Ribeiro JD et al (2013). Obesity increases eosinophil activity in asthmatic children and adolescents. BMC Pulm Med 13: 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Jin D, Chen X (2014). Lipocalin 2 is a regulator of macrophage polarization and NF‐κB/STAT3 pathway activation. Mol Endocrinol 28: 1616–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta K, Shukla M, Cowland JB, Malemud CJ, Haqqi TM (2007). Neutrophil gelatinase‐associated lipocalin is expressed in osteoarthritis and forms a complex with matrix metalloproteinase 9. Arthritis Rheum 56: 3326–3335. [DOI] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh JY, Park YJ, Ham M, Kim JB (2014). Crosstalk between adipocytes and immune cells in adipose tissue inflammation and metabolic dysregulation in obesity. Mol Cells 37: 365–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jian J, Li G, Hettinghouse A, Liu C (2016). Progranulin: a key player in autoimmune diseases. Cytokine 101: 48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang EH, Lee YJ, Kim TK, Chang CB, Chung JH, Shin K et al (2010). Adiponectin is a potential catabolic mediator in osteoarthritis cartilage. Arthritis Res Ther 12: R231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katano M, Okamoto K, Arito M, Kawakami Y, Kurokawa MS, Suematsu N et al (2009). Implication of granulocyte‐macrophage colony‐stimulating factor induced neutrophil gelatinase‐associated lipocalin in pathogenesis of rheumatoid arthritis revealed by proteome analysis. Arthritis Res Ther 11: R3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S‐J, Choi Y, Choi Y‐H, Park T (2012). Obesity activates toll‐like receptor‐mediated proinflammatory signaling cascades in the adipose tissue of mice. J Nutr Biochem 23: 113–122. [DOI] [PubMed] [Google Scholar]
- Laue T, Wrann CD, Hoffmann‐Castendiek B, Pietsch D, Hübner L, Kielstein H (2015). Altered NK cell function in obese healthy humans. BMC Obes 2 (1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SW, Kim JH, Park MC, Park YB, Lee SK (2008). Adiponectin mitigates the severity of arthritis in mice with collagen‐induced arthritis. Scand J Rheumatol 37: 260–268. [DOI] [PubMed] [Google Scholar]
- Liu CJ, Bosch X (2012). Progranulin: a growth factor, a novel TNFR ligand and a drug target. Pharmacol Ther 133: 124–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Liu F (2014). Regulation of adiponectin multimerization, signaling and function. Best Pract Res Clin Endocrinol Metab 28: 25–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeser RF, Goldring SR, Scanzello CR, Goldring MB (2012). Osteoarthritis: a disease of the joint as an organ. Arthritis Rheum 64 (6): 1697–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, Liu M (2016). Adiponectin: a versatile player of innate immunity. J Mol Cell Biol 8: 120–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martín‐Romero C, Santos‐Alvarez J, Goberna R, Sánchez‐Margalet V (2000). Human leptin enhances activation and proliferation of human circulating monocytes. Cell Immunol 199: 15–24. [DOI] [PubMed] [Google Scholar]
- Meszaros E, Malemud CJ (2012). Prospects for treating osteoarthritis: enzyme–protein interactions regulating matrix metalloproteinase activity. Ther Adv Chronic Dis 3: 219–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moraes‐Vieira PMM, Larocca RA, Bassi EJ, Peron JPS, Andrade‐Oliveira V, Wasinski F et al (2014). Leptin deficiency impairs maturation of dendritic cells and enhances induction of regulatory T and Th17 cells. Eur J Immunol 44: 794–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moschen AR, Adolph TE, Gerner RR, Wieser V, Tilg H (2017). Lipocalin‐2: a master mediator of intestinal and metabolic inflammation. Trends Endocrinol Metab 28: 388–397. [DOI] [PubMed] [Google Scholar]
- Münzberg H, Morrison CD (2015). Structure, production and signaling of leptin. Metabolism 64: 13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olama SM, Senna MK, Elarman M (2012). Synovial/serum leptin ratio in rheumatoid arthritis: the association with activity and erosion. Rheumatol Int 32: 683–690. [DOI] [PubMed] [Google Scholar]
- Otvos L, Knappe D, Hoffmann R, Kovalszky I, Olah J, Hewitson TD et al (2014). Development of second generation peptides modulating cellular adiponectin receptor responses. Front Chem 2: 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otvos L, Shao W‐H, Vanniasinghe AS, Amon MA, Csilla Holub M, Kovalszky I et al (2011). Toward understanding the role of leptin and leptin receptor antagonism in preclinical models of rheumatoid arthritis. Peptides 32: 1567–1574. [DOI] [PubMed] [Google Scholar]
- Pallu S, Francin P‐J, Guillaume C, Gegout‐Pottie P, Netter P, Mainard D et al (2010). Obesity affects the chondrocyte responsiveness to leptin in patients with osteoarthritis. Arthritis Res Ther 12: R112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson MJ, Herndler‐Brandstetter D, Tariq MA, Nicholson TA, Philp AM, Smith HL et al (2017). IL‐6 secretion in osteoarthritis patients is mediated by chondrocyte‐synovial fibroblast cross‐talk and is enhanced by obesity. Sci Rep 7: 3451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poonpet T (2014). Adipokines: biomarkers for osteoarthritis? World J Orthop 5: 319–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Procaccini C, De Rosa V, Galgani M, Carbone F, La Rocca C, Formisano L et al (2013). Role of adipokines signaling in the modulation of T cells function. Front Immunol 4: 332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rho YH, Solus J, Sokka T, Oeser A, Chung CP, Gebretsadik T et al (2009). Adipocytokines are associated with radiographic joint damage in rheumatoid arthritis. Arthritis Rheum 60: 1906–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scotece M, Conde J, Vuolteenaho K, Koskinen A, López V, Gómez‐Reino J et al (2014). Adipokines as drug targets in joint and bone disease. Drug Discov Today 19 (3): 241–258. [DOI] [PubMed] [Google Scholar]
- Scotece M, Pérez T, Conde J, Abella V, López V, Pino J et al (2017). Adipokines induce pro‐inflammatory factors in activated Cd4+ T cells from osteoarthritis patient. J Orthop Res 35: 1299–1303. [DOI] [PubMed] [Google Scholar]
- Smolen JS, Aletaha D, McInnes IB (2016). Rheumatoid arthritis. Lancet 388: 2023–2038. [DOI] [PubMed] [Google Scholar]
- Sun Y, Xun K, Wang C, Zhao H, Bi H, Chen X et al (2009). Adiponectin, an unlocking adipocytokine. Cardiovasc Ther 27: 59–75. [DOI] [PubMed] [Google Scholar]
- Tian G, Liang J‐N, Wang Z‐Y, Zhou D (2014). Emerging role of leptin in rheumatoid arthritis. Clin Exp Immunol 177: 557–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilg H, Moschen AR (2006). Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nat Rev Immunol 6: 772–783. [DOI] [PubMed] [Google Scholar]
- Vieira‐Potter VJ (2014). Inflammation and macrophage modulation in adipose tissues. Cell Microbiol 16: 1484–1492. [DOI] [PubMed] [Google Scholar]
- Villalvilla A, García‐Martín A, Largo R, Gualillo O, Herrero‐Beaumont G, Gómez R (2016). The adipokine lipocalin‐2 in the context of the osteoarthritic osteochondral junction. Sci Rep 6: 29243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuolteenaho K, Koskinen A, Moilanen T, Moilanen E (2012). Leptin levels are increased and its negative regulators, SOCS‐3 and sOb‐R are decreased in obese patients with osteoarthritis: a link between obesity and osteoarthritis. Ann Rheum Dis 71: 1912–1913. [DOI] [PubMed] [Google Scholar]
- Wagner NM, Brandhorst G, Czepluch F, Lankeit M, Eberle C, Herzberg S et al (2013). Circulating regulatory T cells are reduced in obesity and may identify subjects at increased metabolic and cardiovascular risk. Obesity 21: 461–468. [DOI] [PubMed] [Google Scholar]
- Wei J, Hettinghouse A, Liu C (2016). The role of progranulin in arthritis. Ann N Y Acad Sci 1383: 5–20. [DOI] [PubMed] [Google Scholar]
- Xia P, Wang X, Lin Q, Li X (2015). Efficacy of mesenchymal stem cells injection for the management of knee osteoarthritis: a systematic review and meta‐analysis. Int Orthop 39: 2363–2372. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu J, Yao J, Ji G, Qian L, Wang J et al (2014). Obesity: pathophysiology and intervention. Forum Nutr 6: 5153–5183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Dong Y, Zhang J, Li D, Hu G, Yao J et al (2016). Leptin changes differentiation fate and induces senescence in chondrogenic progenitor cells. Cell Death Dis 7: e2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B, Li H, Shi J (2017). miR‐27 inhibits the NF‐κB signaling pathway by targeting leptin in osteoarthritic chondrocytes. Int J Mol Med 40: 523–530. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Rui L (2014). Leptin signaling and leptin resistance. Front Med 7: 207. [DOI] [PMC free article] [PubMed] [Google Scholar]