

Abstract
Background and Purpose
The anti‐helminthic drug niclosamide regulates multiple cellular signals including STAT3, AMP‐activated protein kinase (AMPK), Akt, Wnt/β‐catenin and mitochondrial uncoupling which are involved in neointimal hyperplasia. Here we have examined the effects of niclosamide on vascular smooth muscle cell proliferation, migration and neointimal hyperplasia and assessed the potential mechanisms.
Experimental Approach
Cell migration was measured by using wound‐induced migration assay and Boyden chamber assay. Protein levels were measured by using Western blot technique. Neointimal hyperplasia in vivo was induced in rats by balloon injury to the carotid artery.
Key Results
Niclosamide treatment inhibited serum‐induced (15% FBS) and PDGF‐BB‐induced proliferation and migration of vascular smooth muscle cells (A10 cells). Niclosamide showed no cytotoxicity at anti‐proliferative concentrations, but induced cell apoptosis at higher concentrations. Niclosamide treatment inhibited serum‐induced (15% FBS) and PDGF‐BB‐induced STAT3 activation (increased protein levels of p‐STAT3 at Tyr705) but activated AMPK, in A10 cells. Niclosamide exerted no significant effects on β‐catenin expression and the activities of ERK1/2 and Akt in A10 cells. Injection (i.p.) of soluble pegylated niclosamide (PEG5000‐niclosamide) (equivalent to niclosamide 25 mg·kg−1) attenuated neointimal hyperplasia following balloon‐injury in rat carotid arteries in vivo.
Conclusions and Implications
Niclosamide inhibited vascular smooth muscle cell proliferation and migration and attenuated neointimal hyperplasia in balloon‐injured rat carotid arteries through a mechanism involving inhibition of STAT3.
Abbreviations
- AMPK
AMP‐activated protein kinase
- UCPs
mitochondrial uncoupling proteins
Introduction
Neointimal hyperplasia is a characteristic common to a variety of vascular disorders, such as restenosis after angioplasty, vein graft stenosis, allograft vasculopathy and atherosclerosis (Kearney et al., 1997; Mitchell and Libby, 2007; Ross, 1993). In response to vascular injury, vascular smooth muscle cells (VSMCs) become highly proliferative and migrate towards the luminal side of the vessel (Dzau et al., 2002; Gomez and Owens, 2012; Owens et al., 2004; Schwartz, 1997), which is a key event in the process of neointimal hyperplasia. Several signal pathways contributing to VSMC proliferation and migration have been identified, including STAT3, AMP‐activated protein kinase (http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=474), http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=285, http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=514 and http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=964 (Kim et al., 2011;Tsaousi et al., 2011) signalling. Inhibition of STAT3 (Daniel et al., 2012; Seki et al., 2000), Akt (Stabile et al., 2003), ERK (Kim et al., 2011) and Wnt/β‐catenin (Tsaousi et al., 2011) or activation of AMPK (Ki et al., 2013; Stone et al., 2013) has been reported to inhibit VSMC proliferation, migration and neointimal hyperplasia.
Recently, mild uncoupling of mitochondria has been considered as a potential therapeutic strategy for various metabolic disorders, such as diabetes, hyperlipidaemia and obesity (Busiello et al., 2015; Cunha et al., 2011). http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=207, members of the mitochondrial anion transporter family, can dissipate the proton gradient generated by the mitochondrial electron transport chain and thus uncouple ATP synthesis from oxidative phosphorylation (Klingenberg, 1999). http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=207#1067 is expressed in VSMCs and endothelial cells, and UCP2 transgenic and knockout models have been used to study the role of mitochondrial uncoupling in vascular dysfunction and remodelling. UCP2 overexpression in human VSMCs inhibits cell proliferation and migration induced by high glucose and angiotensin II (Ang II) (Park et al., 2005). Low MW mitochondrial uncouplers are small molecules that mimic the action of UCPs, enabling protons to enter the mitochondrial matrix and uncoupling the mitochondrial oxidative phosphorylation process. Mitochondrial uncouplers have been reported to reverse hypertriglyceridemia, fatty liver disease, insulin resistance and diabetes (Fu et al., 2013; Perry et al., 2013; Perry et al., 2015; Tao et al., 2014) and protect against heart and renal ischaemic injury (Brennan et al., 2006; Kenwood et al., 2014). We previously found that mitochondrial uncouplers relaxed the constricted arteries through a mechanism involving AMPK activation (Li et al., 2017; Zhang et al., 2017; Zhang et al., 2016). However, the effects of chemical mitochondrial uncouplers on VSMC proliferation and migration have never been identified.
Niclosamide is a clinically used treatment of intestinal infections of tapeworms, due to its mitochondrial uncoupling effects (Frayha et al., 1997). In addition to the mitochondrial uncoupling effect, niclosamide regulates a range of signals including STAT3, AMPK, Akt, ERK and Wnt/β‐catenin signals in cancer cells (Chen et al., 2018; Liu et al., 2017; Liu et al., 2016) and, because of this effect, niclosamide is a novel candidate drug for cancer chemotherapy. On the other hand, these signal pathways regulated by niclosamide are also involved in VSMC proliferation and migration. We therefore tested the possibility that niclosamide might also inhibit VSMC proliferation, migration and the related neointimal hyperplasia.
Methods
Preparation of niclosamide
Niclosamide is insoluble in water and has to be dissolved in DMSO, which limits its use in vivo. Therefore, the soluble pegylated (PEG5000)‐niclosamide (PEG5000‐niclosamide) was synthesized for i.p. injection in vivo by our collaborator from the Institute of Materials Processing and Intelligent Manufacturing and Center for Biomedical Materials and Engineering, Harbin Engineering University. The chemical structure of PEG5000‐niclosamide was characterized by using FTIR spectra and 1H NMR spectra. The acute toxicity of PEG5000‐niclosamide administered by i.p. injection in mice was evaluated. No deaths were observed in the animals with accumulative amount of PEG5000‐ niclosamide equivalent to 1000 mg·kg−1 niclosamide within 24 h.
Niclosamide ethanolamine was also used for i.p injection and was suspended in 0.5% methylcellulose (Tianjin Fuchen Chemical Reagents Factory).
Rats in the control groups were treated with the appropriate volume of solvent solution (0.5% methylcellulose or saline).
Animals
All animal care and experimental protocols complied with the Laboratory Animal Management Regulations in China and were approved by the Institutional Animal Care and Use Committee of Harbin Medical University, PR China. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). We used male Sprague–Dawley rats (270–300 g, 8 weeks old) supplied by Charles River (Charles River Laboratory Animal, Beijing, China). Prior to experiments, the rats were housed for 1 week at 23 ± 1°C, with a relative humidity of 55–60% and a 14 h light and 10 h dark circadian rhythm with free access to water and standard rat chow. They were maintained in groups of six animals per cage (1300 cm2) according to our routine procedure (Zhang et al., 2016).
Rat carotid artery injury model
Rats were anaesthetized by an injection (i.p.) of chloral hydrate (300 mg·kg−1), and the level of anaesthesia was monitored via tail pinch. The surgical site was epilated and sterilized, then a ventral mid‐line incision was made in the neck using micro‐scissors. The left common carotid arteries and its bifurcation into the internal and external branches were exposed. Then, the left common and internal carotid arteries were temporarily occluded by sutures, and the external carotid artery was ligated at the exposed distal end. A Fogarty 2F arterial embolectomy catheter (Edwards Lifesciences, Irvine, CA, USA) was introduced into the common carotid artery through a small window opened in the external carotid artery. The balloon was inflated to a pre‐determined volume (0.2 mL) and then was withdrawn with rotation toward the carotid bifurcation. This procedure was performed three times. When the catheter was removed, the external carotid was ligated at the arteriotomy site, the blood flow to the common and internal carotid arteries was restored by untying the sutures, and the wound was closed. Alcohol (about 70%) was swabbed around the periphery of the wound to reduce likelihood of infection. The rat was returned to the cage with prone position, and sterile gauze pads were placed below the wound during recovery. In addition, saline was used to amend fluid loss and moisten the mouth and tongue of the rat. Breathing rate was monitored during recovery. Sufficient water to drink was provided when the rat was ambulatory. After recovery, the animals were randomly divided into model with solvent groups and model with niclosamide ethanolamine or PEG5000‐niclosamide treatment groups. Weight was monitored during the intervention period. After 4 weeks, the rats were killed by an overdose of anaesthetic (chloral hydrate 500 mg·kg−1), and the left common carotid arteries at the same position were collected for analysis.
Morphometric analysis
For morphological analysis, the carotid arteries were dissected, divided into four specimens and fixed in 4% paraformaldehyde at 4°C. Twenty‐four hours later, the arterial segments were embedded in paraffin and sectioned (5 μm thick). The 5 μm cross sections were mounted on microscope slides and stained with haematoxylin and eosin. The images were obtained using a light microscope (Olympus BX53 with DP80 camera, Japan) at 10× or 20× magnification. The intima, medial and adventitial areas were analysed by ImagePro Plus image analysis software (Media Cybernetics Inc., Silver Spring, MD, USA). The intima area was calculated as the internal elastic lamina area minus the luminal area, and the medial area was calculated as the external elastic lamina area minus the internal elastic lamina area. Intima/media ratio was then calculated.
Cell culture
VSMCs (A10 cells) and HUVECs from ATCC source were purchased from Bioleaf Biotech CO. Ltd (Shanghai, China). The cells were grown in DMEM supplemented with 15% FBS and 1% penicillin‐streptomycin in a humidified incubator at 37°C and 5% CO2.
Cell viability measurement
Cell viability was assessed by using the MTT assay. Briefly, cells were seeded in 96‐well flat‐bottomed plates at 5 × 103 cells per well, treated with or without niclosamide at indicated concentrations for 24 h and then incubated with MTT (5 mg·mL−1) for 4 h at 37°C. The absorbance was then measured at 490 nm using a plate reader (Tecan Infinite m200, Mannedorf, Switzerland).
Live and dead cell staining
The live and dead cells were detected by using the LIVE/DEAD® Viability/Cytotoxicity Assay Kit (Invitrogen, Waltham, MA, USA) as described in our previous study (Xie et al., 2015). Briefly, the cells were seeded into the six‐well plate at 5 × 104 cells per well and incubated with a mixture of 2 μM calcein AM and 4 μM EthD‐1 for 15 min at 37°C. The labelled cells were randomly visualized using a fluorescence microscope (Olympus IX73 with DP73 camera, Japan) at 20× magnification and counted using ImagePro Plus image analysis software (Media Cybernetics Inc.).
DNA synthesis analysis
DNA synthesis was measured by using the 5‐bromo‐deoxyuridine (BrdU) elisa Kit (ab126556, Abcam, Cambridge, MA, USA) according to the manufacturer's instructions. In brief, cells were seeded in a 96‐well plate at 5 × 103 cells per well and incubated with BrdU reagent for 12 h at 37°C. After cells were incubated with fixing solution for 30 min, the anti‐BrdU monoclonal detector antibody was added for 60 min followed by the incubation with peroxidase goat anti‐mouse IgG conjugate for 30 min at room temperature. Cells were then incubated with TMB (Tetraliethylbenzidine) peroxidase substrate for 30 min at room temperature in the dark. The reaction was terminated by the addition of stop solution. The absorbance was then determined at 450 nm using a plate reader (Tecan Infinite m200).
Wound‐induced migration assay
Cells were seeded into a six‐well plate at 5 × 104 cells per well and cultured in DMEM supplemented with 15% FBS and 1% penicillin–streptomycin. Upon reaching 90% confluence, a vertical scratch was made by using a 200 μL pipette tip to create a cell‐free zone, which crossed with the horizontal lines drawn on the back of the plate, prior to cell seeding, to obtain accurate location. The debris was removed by washing the cells gently twice with PBS. The images for wound healing were obtained at 4× magnification under a light microscope (Olympus, CKX41) with the horizontal lines as baseline at indicated time points. Cell migration was quantified by measuring the area of the cell‐free zone using ImagePro Plus image analysis software (Media Cybernetics Inc.).
Boyden chamber assays
Boyden chamber assays were carried out in Transwell chambers (6.5 mm diameter, 8.0 μm pore size, Corning Inc., Corning, NY, USA). The polycarbonate membranes were coated with 0.2% gelatin prior to experiments. Cells were trypsinized and resuspended in DMEM supplemented with 0.2% FBS. The upper chamber was loaded with 1 × 104 cells in 200 μL media. The lower chamber contained 600 μL DMEM supplemented with 15% FBS. After being incubated for 6 h at 37°C and 5% CO2, the cells on the upper side of the filter were removed with a cotton swab. The filter membrane through which the cells migrated were fixed in 4% paraformaldehyde and stained with Crystal Violet. The images were visualized and captured on a light microscope (Olympus IX73 with DP73 camera, Japan) at 4× and 20× magnification. The Crystal Violet stain was extracted from migrated cells with 33% acetic acid. Then, the extracted solution was transferred into a 96‐well plate, and the absorbance was measured at 570 nm using a plate reader (Tecan Infinite m200, Mannedorf, Switzerland).
Western blot analysis
Protein samples from cultured cells were harvested with RIPA buffer containing 1% protease inhibitor and 10% phosphatase inhibitor. After centrifugation at 12 200 × g for 15 min at 4°C, the supernatants were transferred and protein concentrations were assessed using BCA Protein Assay Kit (Bio‐Rad). Equal amounts of protein were analysed by electrophoresis on 8–15% SDS‐PAGE gels and blotted onto nitrocellulose membranes. After blocking with 5% non‐fat milk, the membranes were probed with primary antibodies at 4°C overnight. After washing with TBS‐0.1% Tween 20 (TBST), the membranes were subsequently incubated with fluorescence‐conjugated goat anti‐rabbit IgG or goat anti‐mouse IgG secondary antibody (1, 10 000 dilution, LI‐COR) for 1 h. Blots were quantified using Odyssey infrared imaging system (Li‐Cor Inc., Lincoln, NE, USA) and Odyssey v3.0 software.
TUNEL staining
Cell apoptosis was measured by using the In Situ Cell Death Detection Kit (Roche, Indianapolis, IN, USA) as described in our previous studies (Sun et al., 2013). Briefly, cells were grown in a 24‐well plate at 1 × 104 cells per well. After treatment, cells were fixed in 4% paraformaldehyde for 40 min, blocked with 3% H2O2 for 10 min and permeabilized with 0.1% Triton x‐100 sodium citrate solution for 3 min. Apoptotic cells were labelled with TUNEL reaction mixture, while cell nuclei were counterstained with DAPI. The images were randomly obtained with a fluorescence microscope (Olympus IX73 with DP73 camera, Japan) at 20× magnification, and the counts of labelled cells were performed using ImagePro Plus image analysis software (Media Cybernetics Inc.). The apoptosis rate was calculated as the ratio of TUNEL‐positive cells to total cells.
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). No blinding was undertaken in this study. It is not a usual procedure for this form of study and cannot be applied retrospectively. The exact group size (n) for each experimental group is shown in the legends and ‘n’ refers to independent values, not replicates. Data subjected to statistical analysis have ‘n’ of at least five per group.
In the in vivo experiments, the animals were randomly divided into different groups after operation. In the wound‐induced migration assay, the picture was obtained randomly at 0 h, and the same location was captured again after 12 or 24 h. In addition, the images of live and dead cell staining and TUNEL staining were randomly obtained. Normalization of responses was carried out in some experiments (BrDU incorporation, wound‐induced migration assay, Western blot experiments) to minimize the influence of variable baseline and allow comparison of the data from independent experiments.
All data are presented as mean ± SEM. Statistical analysis of the results was performed with GraphPad Prism version 5.0 (GraphPadSoftware Inc., San Diego, CA, USA). Statistical significance of two groups was determined with Student's t‐test. For two more groups, one‐way ANOVA followed by Holm–Sidak test or Tukey's test was used. Post hoc tests were run only if F achieved P < 0.05 and there was no significant variance in homogeneity. P < 0.05 was considered statistically significant. The group sizes refer to independent values, not replicates; the replicates from a single experiments were averaged.
Materials
Niclosamide was purchased from Jianglai Reagent Company (Shanghai, China). Niclosamide ethanolamine was obtained from Shanghai Rongbai Biological Technology Co. Ltd (Shanghai, China). Recombinant murine PDGF‐BB was obtained from PeproTech (Rocky Hill, NJ, USA). FBS was purchased from Biological Industries (Beit Haemek, Israel). DMEM was purchased from Hyclone (Logan, UT, USA). Anti‐β‐catenin antibody was purchased from Santa Cruz (Eugene, OR, USA). Anti‐p‐Stat3 (Tyr705), anti‐p‐Stat3 (Ser727), anti‐Stat3, anti‐p‐AMPKα(Thr172), anti‐AMPKα, anti‐p‐ERK1/2 (Thr202/Tyr204), anti‐ERK1/2, anti‐p‐Akt(Ser473) and anti‐Akt antibodies were from Cell Signaling Technology (Beverly, MA, USA). Anti‐β‐actin was obtained from Zhong Shan‐Golden Bridge Biological Technology (Beijing, China). MTT was from Amresco (Solon, OH, USA). Haematoxylin and eosin were from Biosharp (Beijing, China).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a,b,c).
Results
Niclosamide inhibits serum‐induced (15% FBS) and PDGF‐BB‐induced proliferation of A10 cells
The A10 cells are nondifferentiated VSMCs and commonly used as the model of neointimal VSMCs (Choe et al., 2015; Munoz et al., 2011; Rao et al., 1997). Hence, A10 cells were used in the present study. Firstly, we used BrdU incorporation to assess the proliferation of A10 cells. As shown in Figure 1A, compared with the culture in 0.2% FBS, DNA synthesis was significantly increased after 24 h treatment in serum‐induced conditions (15% FBS culture), and niclosamide dose‐dependently inhibited the serum‐induced increase of cell proliferation. We further examined the effects of niclosamide on A10 cell proliferation in http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=322‐induced model which is commonly used in VSMC proliferation and migration studies (Park et al., 2017; Fairaq et al., 2017). Results showed that niclosamide also reduced PDGF‐BB‐induced A10 proliferation in a dose‐dependent manner (Figure 1B).
Figure 1.
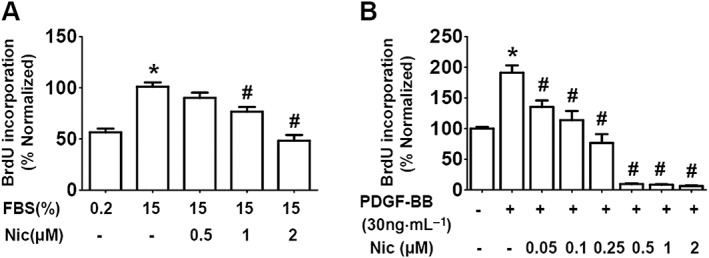
Niclosamide inhibits serum‐induced (15% FBS) and PDGF‐BB‐induced A10 cell proliferation. (A) A10 cells were cultured in DMEM supplemented with 15% FBS in the absence or presence of niclosamide (Nic; 0.5–2 μM) for 24 h, with 0.2% FBS condition as control. n = 8 in each group. *P < 0.05, significantly different from 0.2% FBS (control), # P < 0.05, significantly different from 15% FBS. (B) Quiescent A10 cells were incubated with PDGF‐BB (30 ng·mL−1) in the absence or presence of niclosamide (0.05–2 μM) for 24 h. n = 12 in each group. The control medium contains 0.2% FBS. *P < 0.05, significantly different from control, # P < 0.05, significantly different from PDGF‐BB.
Niclosamide inhibits serum‐induced (15% FBS) and PDGF‐BB‐induced A10 cell migration
Next, we investigated whether niclosamide could suppress serum‐stimulated A10 cell migration. In the wound cell migration assay, the serum‐induced (15% FBS) cell migration was inhibited by niclosamide in a dose‐dependent manner (Figure 2A, B). To confirm the effects of niclosamide on A10 cell migration in response to serum, we also used the Boyden chamber assay. Results showed that the migrated cells were markedly increased at 6 h after incubation with medium containing 15% FBS, while niclosamide (2 μM) significantly reduced the serum‐induced migration of A10 cells (Figure 2C, D).
Figure 2.
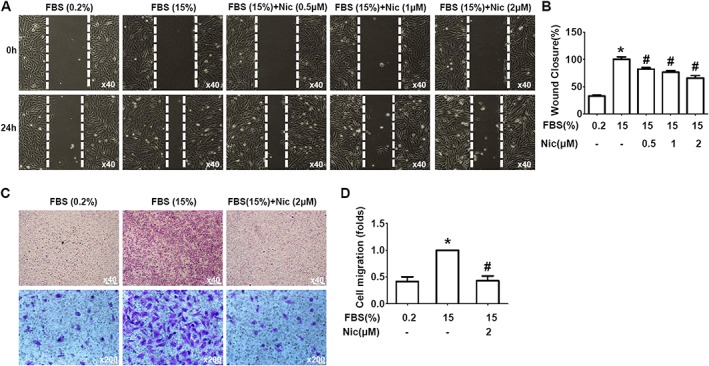
Niclosamide inhibits serum‐induced (15% FBS) A10 cell migration. (A, B) Niclosamide (Nic) inhibited A10 cell migration measured by scratch wound‐healing assay. (A) Representative images showing migration of A10 cells at the time 0 and 24 h. (B) Quantitative data as the percentage of cell migrating into the wound with respect to the cell‐free area at the time 0. n = 12 in each group. *P < 0.05, significantly different from control, # P < 0.05, significantly different from 15% FBS. (C, D) Niclosamide reduced A10 cell migration measured using the modified Boyden chamber assay. (C) Representative images showing transmigrated cells in the absence or presence of niclosamide (2 μM). (D) Quantitative data of A10 cell migration. n = 10 in each group. *P < 0.05, significantly different from control, # P < 0.05, significantly different from 15% FBS.
Additionally, we examined the effects of niclosamide on A10 cell migration in response to PDGF‐BB. Wound‐induced migration assay revealed that PDGF‐BB treatment increased A10 cell migration, which was significantly inhibited by niclosamide treatment for 12 and 24 h (Figure 3A–D).
Figure 3.

Niclosamide inhibits PDGF‐BB‐induced A10 cell migration. A10 cells were incubated with PDGF‐BB (30 ng·mL−1) in the absence or presence of niclosamide (Nic; 0.5–2 μM) for 12 h (A, B) and 24 h (C, D). The control medium contains 0.2% FBS. *P < 0.05, significantly different from control, # P < 0.05, significantly different from PDGF‐BB. n = 10 in each group in (B); n = 12 in (D).
The effects of niclosamide on A10 cell viability
A delicate balance between cell proliferation and apoptosis contributes to tissue growth. Thus, we further investigated whether niclosamide could inhibit A10 cell viability and promote cell apoptosis. Firstly, we examined the effects of niclosamide on A10 cell viability by using the MTT method. Under the normal cell culture condition (15% FBS), the IC50 value of niclosamide on A10 cell viability was 4.430 μM, but in the absence of serum, the IC50 value was 0.68 μM (Figure 4A), indicating that the A10 cells were more susceptible to niclosamide in the serum‐deprived cultures. We further examined the effects of niclosamide on cell viability of HUVECs and found that HUVECs were less sensitive to niclosamide with IC50 more than 40 μM (Figure 4B). These results implied that niclosamide inhibited the proliferation and migration of VSMCs over a particular dose range, without affecting endothelial cells.
Figure 4.
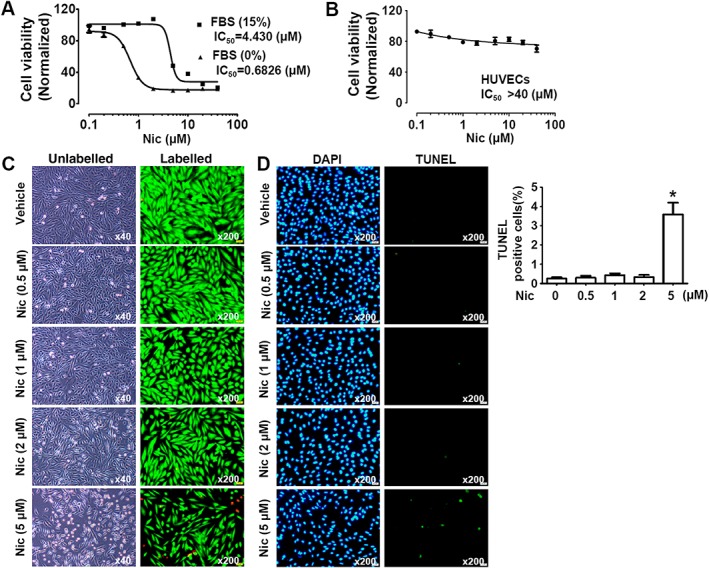
Effects of niclosamide on cell viability. (A) Effects of niclosamide (Nic) on A10 cell viability evaluated by MTT method. The IC50 value was measured under FBS‐free and 15% FBS conditions. n = 6 in each group. (B) Effects of niclosamide on cell viability of HUVECs evaluated by MTT method. The IC50 value was assessed under 15% FBS conditions. n = 6 in each group. (C) The cytotoxicity of niclosamide on A10 cells was evaluated by using the LIVE/DEAD Viability/Cytotoxicity® Assay. The live cells were stained green with calcein AM, and the dead cells were stained red with EthD‐1. The cells were cultured under 15% FBS conditions. (D) High dose of niclosamide (5 μM) induced A10 cell apoptosis. The cells were cultured under 15% FBS conditions. Cell apoptosis was evaluated using TUNEL staining. Quantitative data show the percentage of TUNEL‐positive cells from total number of cells. n = 11 in each group. *P < 0.05, significantly different from untreated cells (control).
We further tested the cytotoxicity of niclosamide by using the LIVE/DEAD Viability/Cytotoxicity® assay. Consistent with the effects on A10 cell proliferation, niclosamide slightly reduced the number of live cells at 2 μM with further cell death when the concentration of niclosamide was increased to 5 μM (Figure 4C). Similarly, TUNEL staining results showed that niclosamide at the higher concentration (5 μM) induced apoptosis of A10 cells (Figure 4D).
Niclosamide suppresses serum‐induced (15% FBS) and PDGF‐BB‐induced STAT3 activation in A10 cells
STAT3 is involved in cell proliferation and migration in vascular injury (Daniel et al., 2012; Dronadula et al., 2005) ad we therefore checked the effects of niclosamide on STAT3 signalling in A10 cells. As shown in Figure 5A–C, niclosamide dose‐dependently reduced the serum‐induced increase of protein level of STAT3 phosphorylation at Tyr705 (p‐Stat3(Tyr705)), without any effects on the protein level of p‐Stat3(Ser727) (Figure 5D–F). Niclosamide (2 μM) significantly reduced the PDGF‐BB‐induced increase of p‐Stat3(Tyr705) protein level in A10 cells (Figure 5G–I).
Figure 5.
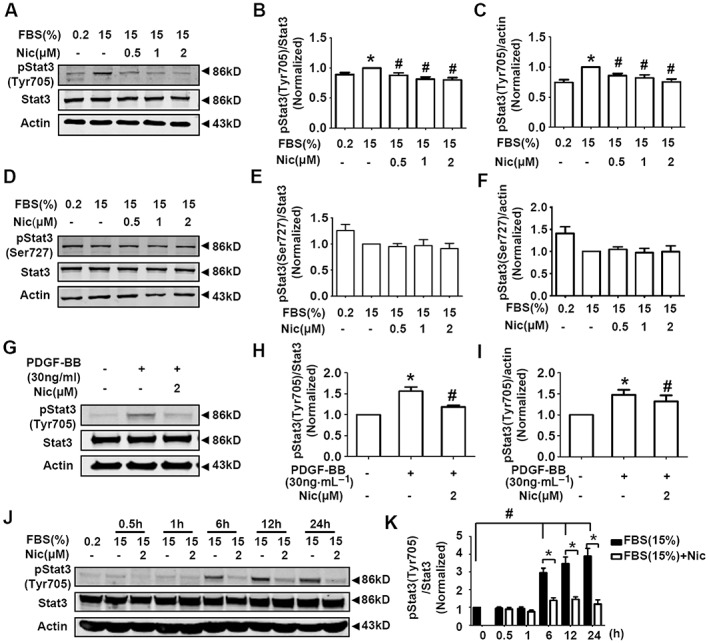
Niclosamide suppresses serum‐induced (15% FBS) and PDGF‐BB‐induced STAT3 activation in A10 cells. (A–C) Serum‐starved A10 cells were grown in DMEM supplemented with 0.2% FBS or 15% FBS in the absence or presence of niclosamide (Nic; 0.5–2 μM) for 24 h. Representative images of the protein levels of STAT3 Tyr705 phosphorylation were shown in (A). STAT3 Tyr705 phosphorylation level was normalized to that of total STAT3 protein level in (B), and STAT3 Tyr705 phosphorylation level was normalized to that of actin protein level in (C). n = 15 in each group. *P < 0.05, significantly different from 0.2% FBS (control), # P < 0.05, significantly different from 15% FBS. (D–F) Serum‐starved A10 cells were grown in DMEM supplemented with 0.2% FBS or 15% FBS in the absence or presence of niclosamide (0.5–2 μM) for 24 h. Representative images of the protein levels of STAT3 Ser727 phosphorylation were shown in (D). STAT3 Ser727 phosphorylation level was normalized to that of total STAT3 protein level in (E), and STAT3 Ser727 phosphorylation level was normalized to that of actin protein level in (F). n = 13 in each group. (G–I) A10 cells were exposed to PDGF‐BB (30 ng·mL−1) in the absence or presence of niclosamide (2 μM) for 30 min. The control medium contains 0.2% FBS. Representative images of the protein levels of STAT3 Tyr705 phosphorylation were shown in (G). STAT3 Tyr705 phosphorylation level was normalized to that of total STAT3 protein level in (H), and STAT3 Tyr705 phosphorylation level was normalized to that of actin protein level in (I). n = 8 in each group. *P < 0.05, significantly different from 0.2% FBS (control), # P < 0.05 versus PDGF‐BB. (J, K) Serum‐starved A10 cells were cultured in DMEM supplemented with 15% FBS in the absence or presence of niclosamide (2 μM). The incubation time was set as 0, 0.5, 1, 6, 12 and 24 h. Representative images of the protein levels of STAT3 Tyr705 phosphorylation were shown in (J). STAT3 Tyr705 phosphorylation level was normalized to that of total STAT3 protein level in (K). n = 10 in each group. *P < 0.05, significantly different from FBS (15%); # P < 0.05, significantly different from 0 h (0.2% FBS).
In addition, the time course of serum‐induced increase of p‐Stat3(Tyr705) protein in A10 cells showed that the levels of p‐Stat3(Tyr705) were increased at 6, 12 and 24 h of 15% FBS treatment. The serum‐induced (15% FBS) STAT3 activation at these time points was reversed by niclosamide (2 μM) (Figure 5J, K).
Niclosamide activates AMPK but shows no effect on the expression of β‐catenin and the activities of ERK1/2 and Akt in A10 cells
AMPK is involved in pathogenesis of neointimal hyperplasia and restenosis (Igata et al., 2005; Nagata et al., 2004; Song et al., 2011). Niclosamide (2 μM) induced AMPK activation (Figure 6A), in line with our previous finding that niclosamide ethanolamine (a salt of niclosamide) induced AMPK activation in A10 cells (Li et al., 2017). We further checked the effects of niclosamide on the expression of β‐catenin and the activities of ERK1/2 and Akt, signalling pathways known to be involved in smooth muscle cell proliferation and migration. As shown in Figure 6B–D, niclosamide (2 μM) had no effect on these signals.
Figure 6.
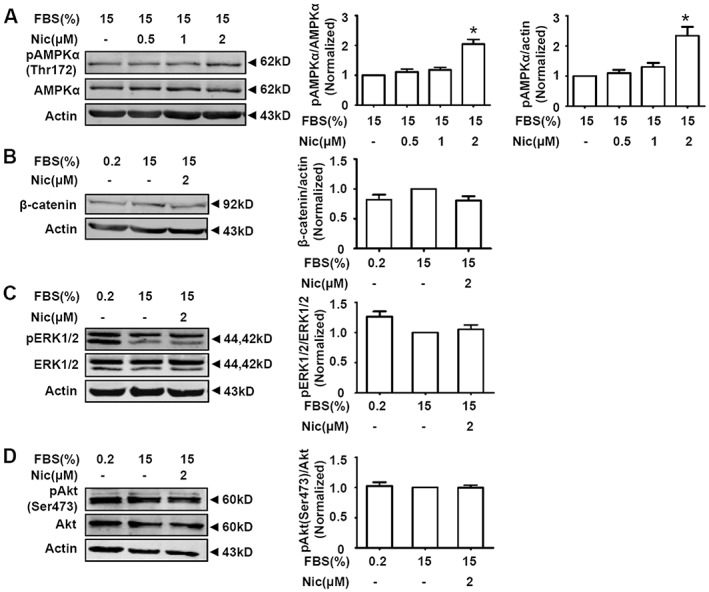
Effects of niclosamide on AMPK, β‐catenin, ERK1/2 and Akt signals in A10 cells. Serum‐starved A10 cells were grown in DMEM supplemented with 15% FBS in the absence or presence of niclosamide (Nic) for 24 h. (A) Niclosamide activated AMPK in the presence of 15% FBS. n = 16 in each group, *P < 0.05, significantly different from 15% FBS. (B) Niclosamide showed no significant effect on β‐catenin expressions in serum‐stimulated A10 cells. n = 6 in each group. (C) Niclosamide had no significant effect on ERK1/2 phosphorylation. n = 12 in each group. (D) Niclosamide had no significant effect on Akt Ser473 phosphorylation. n = 6 in each group.
Niclosamide attenuates neointimal hyperplasia in balloon‐injured rat carotid arteries
Neointimal hyperplasia of rat carotid artery after balloon injury is the most widely used model of vascular remodelling related to VSMC proliferation and migration. We examined the effects of long‐term niclosamide treatment on neointimal hyperplasia in rat carotid artery after balloon injury. Firstly, we studied the effects of intragastric administration of niclosamide ethanolamine on neointimal hyperplasia in our model as this mode of administration of niclosamide ethanolamine improved diabetic symptoms in mice (Tao et al., 2014). However, in our model, intragastric administration of niclosamide ethanolamine (250 mg·kg−1) showed no inhibitory effect on neointimal hyperplasia of rat carotid artery after balloon injury (Figure 7A, B). Because the solubility and bioavailability of both niclosamide and niclosamide ethanolamine are poor; we suspected that the plasma concentration of niclosamide did not reach an effective level. Therefore, the water‐soluble pegylated (PEG5000) niclosamide was synthesized, to be given by i.p. injection in vivo. As shown in Figure 7C, D, the long‐term (4 weeks) treatment with i.p. pegylated‐niclosamide (equivalent to niclosamide 25 mg·kg−1) clearly attenuated neointimal hyperplasia in balloon‐injured rat carotid arteries, indicating that niclosamide with optimal pharmaceutical modification to improve its pharmacokinetic profile would be a potential strategy for treatment of restenosis.
Figure 7.
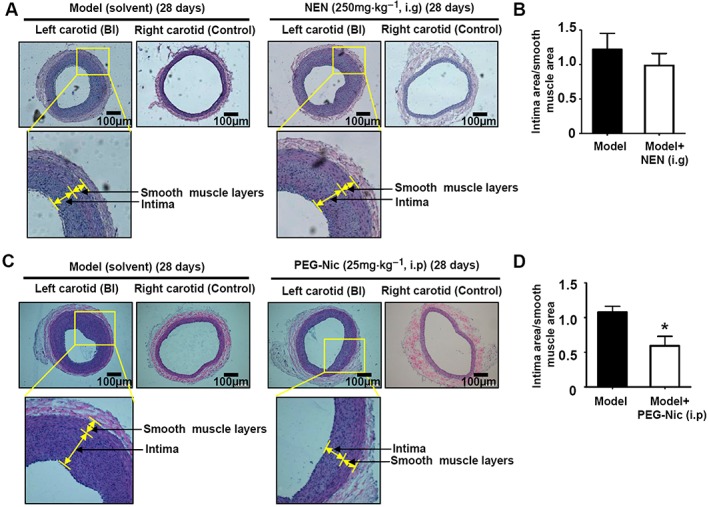
Niclosamide attenuated neointimal hyperplasia in balloon‐injured rat carotid arteries in vivo. (A, B) Intragastric administration of niclosamide ethanolamine (NEN; 250 mg·kg−1) daily for 4 weeks showed no inhibitory effect on neointimal hyperplasia of rat carotid artery after balloon injury. n = 5 in each group. (C, D) Injection (i.p.) of PEG5000‐niclosamide (PEG‐Nic; equivalent to niclosamide 25 mg·kg−1) daily for 4 weeks attenuated neointimal hyperplasia in balloon‐injured rat carotid arteries. n = 6 in each group, *P < 0.05, significant effect of PEG‐Nic.
Discussion
Niclosamide is an oral antihelminthic drug approved by the FDA for treating parasitic infections. Recent work has shown that niclosamide regulates several signalling pathways and has the potential to be applied clinically for systemic diseases including several types of cancers, bacterial and viral infections, metabolic syndrome and neuropathic pain (Chen et al., 2018). Here, we show for the first time that niclosamide inhibited VSMC proliferation and migration and attenuated neointimal hyperplasia in balloon‐injured rat carotid arteries, indicating that niclosamide might be a potential drug for restenosis.
The A10 cells are non‐differentiated VSMCs and commonly used as the model of neointimal VSMCs (Choe et al., 2015; Munoz et al., 2011; Rao et al., 1997). Therefore, in the present study, we used A10 cells and serum‐induced (15% FBS) and PDGF‐BB‐induced models to examine the effects and mechanisms of niclosamide on VSMC proliferation and migration. Niclosamide inhibits both serum‐induced and PDGF‐BB‐induced proliferation and migration of A10 cells. We further examined the effect of niclosamide on viability of A10 cells and found that the IC50 of niclosamide was significantly different in serum‐free and 15% FBS conditions, so that niclosamide was a more potent inhibitor effect on A10 cells in serum‐free cultures. This effect could be due to the buffering of the cytotoxic action of niclosamide by the serum proteins and nutrient factors. We further used the LIVE/DEAD Cell Viability assay and TUNEL staining to study the cytotoxic effect of niclosamide. We found that niclosamide induced cell death and apoptosis only at the higher concentrations, with inhibition of the proliferation and migration of A10 cells at concentrations which showed no significant cytotoxic effect.
Many signalling pathways are involved in VSMC proliferation, migration and intimal thickening, including STAT3 (Daniel et al., 2012; Seki et al., 2000), AMPK (Ki et al., 2013; Stone et al., 2013), Akt (Stabile et al., 2003), ERK (Kim et al., 2011) and Wnt/β‐catenin (Tsaousi et al., 2011) pathways. Several studies have shown that STAT3 is activated in the injured artery (Daniel et al., 2012; Seki et al., 2000), and the activated STAT3 induces trans‐activation of cyclin D1 and http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=889#2795 in SMCs in vitro and in neointimal cells in vivo, thus promoting proliferation and migration of SMCs (Daniel et al., 2012). The STAT3 siRNA can inhibit the proliferation of VSMCs in vivo and in vitro and attenuate neointimal formation (Sun et al., 2012). AMPK is a stress‐activated protein kinase. AMPK activation suppresses VSMC proliferation (Igata et al., 2005; Ki et al., 2013; Nagata et al., 2004), and AMPKα2 deletion exacerbates neointima formation (Song et al., 2011). Although niclosamide inhibits the Wnt/β‐catenin, mTORC1, STAT3, NF‐κB and Notch signalling pathways in cancer cells (Li et al., 2014), we find that niclosamide inhibits STAT3 and activates AMPK but shows no significant effect on the expression of β‐catenin and the activities of Akt and ERK in A10 cells. Furthermore, niclosamide was a more potent inhibitor of STAT3 than an activator of AMPK. For instance, 0.5 μM niclosamide inhibited STAT3 (Figure 5A), but activation of AMPK required 2 μM niclosamide (Figure 6A). Therefore, we suggest that STAT3 inhibition might be the major mechanism by which niclosamide inhibits VSMC proliferation and migration and attenuates neointimal hyperplasia.
Niclosamide is used clinically to treat most tapeworm infections. Although recent studies have demonstrated that niclosamide is a promising agent for systemic diseases, because niclosamide is insoluble, the low bioavailability and the resultant low plasma concentration limit its potential effects in vivo. Therefore, development of novel niclosamide delivery for systemic application is necessary. Covalent attachment of polyethylene glycol (PEG) to proteins or chemicals is a technique known as PEGylation, which is known to increase the solubility of water‐insoluble compounds. In the present study, pegylated (PEG5000) niclosamide was synthesized and i.p. injection of pegylated‐niclosamide attenuated neointimal hyperplasia in balloon‐injured rat carotid arteries. However, administration by mouth is more viable for clinical use, for instance in the treatment of restenosis. Hence, the preparation of a modified niclosamide with better oral absorption will be the next stage is this project.
In summary, we have demonstrated that niclosamide inhibited VSMC proliferation and migration and attenuated neointimal hyperplasia in balloon‐injured rat carotid arteries. We suggest that niclosamide could be a useful drug for restenosis, once the pharmacokinetics of niclosamide are modified with improved oral bioavailability.
Author contributions
X.‐L.X., N.H., X.‐Z.Z. and M.J. performed the cell proliferation, migration and Western blot experiments. C.C. established the rat carotid artery injury model. J.‐L.G. and X.‐C.X. performed the MTT measurement. R.M., Z.‐G.M. and Z.‐J.S. synthesized the pegylated (PEG5000) niclosamide. D.‐L.D. designed the project and wrote the paper.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This http://onlinelibrary.wiley.com/doi/10.1111/bph.13405/abstract acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grants 91739102 and 81773725).
Xiao, X.‐L. , Hu, N. , Zhang, X.‐Z. , Jiang, M. , Chen, C. , Ma, R. , Ma, Z.‐G. , Gao, J.‐L. , Xuan, X.‐C. , Sun, Z.‐J. , and Dong, D.‐L. (2018) Niclosamide inhibits vascular smooth muscle cell proliferation and migration and attenuates neointimal hyperplasia in injured rat carotid arteries. British Journal of Pharmacology, 175: 1707–1718. doi: 10.1111/bph.14182.
References
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Catalytic receptors. Br J Pharmacol 174: S225–S271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD et al (2017c). The Concise Guide to PHARMACOLOGY 2017/18: Other proteins. Br J Pharmacol 174: S1–S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan JP, Southworth R, Medina RA, Davidson SM, Duchen MR, Shattock MJ (2006). Mitochondrial uncoupling, with low concentration FCCP, induces ROS‐dependent cardioprotection independent of KATP channel activation. Cardiovasc Res 72: 313–321. [DOI] [PubMed] [Google Scholar]
- Busiello RA, Savarese S, Lombardi A (2015). Mitochondrial uncoupling proteins and energy metabolism. Front Physiol 6: 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Mook RA Jr, Premont RT, Wang J (2018). Niclosamide: beyond an antihelminthic drug. Cell Signal 41: 89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe N, Kwon JS, Kim YS, Eom GH, Ahn YK, Baik YH et al (2015). The microRNA miR‐34c inhibits vascular smooth muscle cell proliferation and neointimal hyperplasia by targeting stem cell factor. Cell Signal 27: 1056–1065. [DOI] [PubMed] [Google Scholar]
- Cunha FM, Caldeira da Silva CC, Cerqueira FM, Kowaltowski AJ (2011). Mild mitochondrial uncoupling as a therapeutic strategy. Curr Drug Targets 12: 783–789. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel JM, Dutzmann J, Bielenberg W, Widmer‐Teske R, Gunduz D, Hamm CW et al (2012). Inhibition of STAT3 signaling prevents vascular smooth muscle cell proliferation and neointima formation. Basic Res Cardiol 107: 261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dronadula N, Liu Z, Wang C, Cao H, Rao GN (2005). STAT‐3‐dependent cytosolic phospholipase A2 expression is required for thrombin‐induced vascular smooth muscle cell motility. J Biol Chem 280: 3112–3120. [DOI] [PubMed] [Google Scholar]
- Dzau VJ, Braun‐Dullaeus RC, Sedding DG (2002). Vascular proliferation and atherosclerosis: new perspectives and therapeutic strategies. Nat Med 8: 1249–1256. [DOI] [PubMed] [Google Scholar]
- Fairaq A, Shawky NM, Osman I, Pichavaram P, Segar L (2017). AdipoRon, an adiponectin receptor agonist, attenuates PDGF‐induced VSMC proliferation through inhibition of mTOR signaling independent of AMPK: implications toward suppression of neointimal hyperplasia. Pharmacol Res 119: 289–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frayha GJ, Smyth JD, Gobert JG, Savel J (1997). The mechanisms of action of antiprotozoal and anthelmintic drugs in man. Gen Pharmacol 28: 273–299. [DOI] [PubMed] [Google Scholar]
- Fu YY, Zhang M, Turner N, Zhang LN, Dong TC, Gu M et al (2013). A novel chemical uncoupler ameliorates obesity and related phenotypes in mice with diet‐induced obesity by modulating energy expenditure and food intake. Diabetologia 56: 2297–2307. [DOI] [PubMed] [Google Scholar]
- Gomez D, Owens GK (2012). Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc Res 95: 156–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igata M, Motoshima H, Tsuruzoe K, Kojima K, Matsumura T, Kondo T et al (2005). Adenosine monophosphate‐activated protein kinase suppresses vascular smooth muscle cell proliferation through the inhibition of cell cycle progression. Circ Res 97: 837–844. [DOI] [PubMed] [Google Scholar]
- Kearney M, Pieczek A, Haley L, Losordo DW, Andres V, Schainfeld R et al (1997). Histopathology of in‐stent restenosis in patients with peripheral artery disease. Circulation 95: 1998–2002. [DOI] [PubMed] [Google Scholar]
- Kenwood BM, Weaver JL, Bajwa A, Poon IK, Byrne FL, Murrow BA et al (2014). Identification of a novel mitochondrial uncoupler that does not depolarize the plasma membrane. Mol Metab 3: 114–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ki SH, Lee JW, Lim SC, Hien TT, Im JH, Oh WK et al (2013). Protective effect of nectandrin B, a potent AMPK activator on neointima formation: inhibition of Pin1 expression through AMPK activation. Br J Pharmacol 168: 932–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SY, Kwon YW, Jung IL, Sung JH, Park SG (2011). Tauroursodeoxycholate (TUDCA) inhibits neointimal hyperplasia by suppression of ERK via PKCalpha‐mediated MKP‐1 induction. Cardiovasc Res 92: 307–316. [DOI] [PubMed] [Google Scholar]
- Klingenberg M (1999). Uncoupling protein – a useful energy dissipator. J Bioenerg Biomembr 31: 419–430. [DOI] [PubMed] [Google Scholar]
- Li SL, Yan J, Zhang YQ, Zhen CL, Liu MY, Jin J et al (2017). Niclosamide ethanolamine inhibits artery constriction. Pharmacol Res 115: 78–86. [DOI] [PubMed] [Google Scholar]
- Li Y, Li PK, Roberts MJ, Arend RC, Samant RS, Buchsbaum DJ (2014). Multi‐targeted therapy of cancer by niclosamide: a new application for an old drug. Cancer Lett 349: 8–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XL, Zhang XT, Meng J, Zhang HF, Zhao Y, Li C et al (2017). ING5 knockdown enhances migration and invasion of lung cancer cells by inducing EMT via EGFR/PI3K/Akt and IL‐6/STAT3 signaling pathways. Oncotarget 8: 54265–54276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Li Y, Lv C, Wang L, Song H (2016). Anthelmintic drug niclosamide enhances the sensitivity of chronic myeloid leukemia cells to dasatinib through inhibiting Erk/Mnk1/eIF4E pathway. Biochem Biophys Res Commun 478: 893–899. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell RN, Libby P (2007). Vascular remodeling in transplant vasculopathy. Circ Res 100: 967–978. [DOI] [PubMed] [Google Scholar]
- Munoz E, Valero RA, Quintana A, Hoth M, Nunez L, Villalobos C (2011). Nonsteroidal anti‐inflammatory drugs inhibit vascular smooth muscle cell proliferation by enabling the Ca2+‐dependent inactivation of calcium release‐activated calcium/orai channels normally prevented by mitochondria. J Biol Chem 286: 16186–16196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata D, Takeda R, Sata M, Satonaka H, Suzuki E, Nagano T et al (2004). AMP‐activated protein kinase inhibits angiotensin II‐stimulated vascular smooth muscle cell proliferation. Circulation 110: 444–451. [DOI] [PubMed] [Google Scholar]
- Owens GK, Kumar MS, Wamhoff BR (2004). Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev 84: 767–801. [DOI] [PubMed] [Google Scholar]
- Park HS, Quan KT, Han JH, Jung SH, Lee DH, Jo E et al (2017). Rubiarbonone C inhibits platelet‐derived growth factor‐induced proliferation and migration of vascular smooth muscle cells through the focal adhesion kinase, MAPK and STAT3 Tyr705 signalling pathways. Br J Pharmacol 174: 4140–4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JY, Park KG, Kim HJ, Kang HG, Ahn JD, Kim HS et al (2005). The effects of the overexpression of recombinant uncoupling protein 2 on proliferation, migration and plasminogen activator inhibitor 1 expression in human vascular smooth muscle cells. Diabetologia 48: 1022–1028. [DOI] [PubMed] [Google Scholar]
- Perry RJ, Kim T, Zhang XM, Lee HY, Pesta D, Popov VB et al (2013). Reversal of hypertriglyceridemia, fatty liver disease, and insulin resistance by a liver‐targeted mitochondrial uncoupler. Cell Metab 18: 740–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RJ, Zhang D, Zhang XM, Boyer JL, Shulman GI (2015). Controlled‐release mitochondrial protonophore reverses diabetes and steatohepatitis in rats. Science 347: 1253–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao RS, Miano JM, Olson EN, Seidel CL (1997). The A10 cell line: a model for neonatal, neointimal, or differentiated vascular smooth muscle cells? Cardiovasc Res 36: 118–126. [DOI] [PubMed] [Google Scholar]
- Ross R (1993). The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature 362: 801–809. [DOI] [PubMed] [Google Scholar]
- Schwartz SM (1997). Smooth muscle migration in atherosclerosis and restenosis. J Clin Invest 100: S87–S89. [PubMed] [Google Scholar]
- Seki Y, Kai H, Shibata R, Nagata T, Yasukawa H, Yoshimura A et al (2000). Role of the JAK/STAT pathway in rat carotid artery remodeling after vascular injury. Circ Res 87: 12–18. [DOI] [PubMed] [Google Scholar]
- Song P, Wang S, He C, Wang S, Liang B, Viollet B et al (2011). AMPKalpha2 deletion exacerbates neointima formation by upregulating Skp2 in vascular smooth muscle cells. Circ Res 109: 1230–1239. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Stabile E, Zhou YF, Saji M, Castagna M, Shou M, Kinnaird TD et al (2003). Akt controls vascular smooth muscle cell proliferation in vitro and in vivo by delaying G1/S exit. Circ Res 93: 1059–1065. [DOI] [PubMed] [Google Scholar]
- Stone JD, Narine A, Shaver PR, Fox JC, Vuncannon JR, Tulis DA (2013). AMP‐activated protein kinase inhibits vascular smooth muscle cell proliferation and migration and vascular remodeling following injury. Am J Physiol Heart Circ Physiol 304: H369–H381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun B, Huo R, Sheng Y, Li Y, Xie X, Chen C et al (2013). Bone morphogenetic protein‐4 mediates cardiac hypertrophy, apoptosis, and fibrosis in experimentally pathological cardiac hypertrophy. Hypertension 61: 352–360. [DOI] [PubMed] [Google Scholar]
- Sun J, Zheng J, Ling KH, Zhao K, Xie Z, Li B et al (2012). Preventing intimal thickening of vein grafts in vein artery bypass using STAT‐3 siRNA. J Transl Med 10: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao H, Zhang Y, Zeng X, Shulman GI, Jin S (2014). Niclosamide ethanolamine‐induced mild mitochondrial uncoupling improves diabetic symptoms in mice. Nat Med 20: 1263–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsaousi A, Williams H, Lyon CA, Taylor V, Swain A, Johnson JL et al (2011). Wnt4/beta‐catenin signaling induces VSMC proliferation and is associated with intimal thickening. Circ Res 108: 427–436. [DOI] [PubMed] [Google Scholar]
- Xie X, Zhao Y, Ma CY, Xu XM, Zhang YQ, Wang CG et al (2015). Dimethyl fumarate induces necroptosis in colon cancer cells through GSH depletion/ROS increase/MAPKs activation pathway. Br J Pharmacol 172: 3929–3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Zhang X, Zhang Y, Liu M, Jin J, Yan J et al (2017). Mitochondrial uncoupler triclosan induces vasorelaxation of rat arteries. Acta Pharmaceutica Sinica B 7: 623–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YQ, Shen X, Xiao XL, Liu MY, Li SL, Yan J et al (2016). Mitochondrial uncoupler carbonyl cyanide m‐chlorophenylhydrazone induces vasorelaxation without involving KATP channel activation in smooth muscle cells of arteries. Br J Pharmacol 173: 3145–3158. [DOI] [PMC free article] [PubMed] [Google Scholar]