

Abstract
Background and Purpose
Transient receptor potential (TRP) channels are a superfamily of non‐selective cation permeable channels involved in peripheral sensory signalling. Animal studies have shown that several TRPs are important players in pain modulation. Among them, the TRP melastatin 8 (TRPM8) has elicited more interest for its controversial role in nociception. This channel, expressed by a subpopulation of sensory neurons in dorsal root ganglia (DRG) and trigeminal ganglia (TG), is activated by cold temperatures and cooling agents. In experimental neuropathic pain models, an up‐regulation of this receptor in DRG and TG has been observed, suggesting a key role for TRPM8 in the development and maintenance of pain. Consistent with this hypothesis, TRPM8 knockout mice are less responsive to pain stimuli.
Experimental Approach
In this study, the therapeutic potential and efficacy of two novel TRPM8 antagonists, DFL23693 and DFL23448, were tested.
Key Results
Two potent and selective TRPM8 antagonists with distinct pharmacokinetic profiles, DFL23693 and DFL23448, have been fully characterized in vitro. In vivo studies in well‐established models, namely, the wet‐dog shaking test and changes in body temperature, confirmed their ability to block the TRPM8 channel. Finally, TRPM8 blockage resulted in a significant antinociceptive effect in formalin‐induced orofacial pain and in chronic constriction injury‐induced neuropathic pain, confirming an important role for this channel in pain perception.
Conclusion and Implications
Our findings, in agreement with previous literature, encourage further studies for a better comprehension of the therapeutic potential of TRPM8 blockers as novel agents for pain management.
Abbreviations
- CCI
chronic constriction injury
- DPA
Dynamic Plantar Aesthesiometer
- DRG
dorsal root ganglia
- hERG
human ether‐a‐go‐go related gene
- Tb
body temperature
- TRPM8
transient receptor potential melastatin 8
- WDS
wet‐dog shake
Introduction
Transient receptor potential melastatin 8 (http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=500 ) belongs to the http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=78. It is activated by moderate cold and exogenous ‘cooling‐mimetic’ compounds, such as http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2430 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2429 (McKemy et al., 2002; Peier et al., 2002), and is considered the principal sensor for innocuous cold (Takashima et al., 2007; Dhaka et al., 2008). In coherence with its function as a thermal sensory sentinel, several studies have shown that TRPM8 is strongly expressed in a subpopulation of primary sensory neurons within the dorsal root (DRG) and trigeminal ganglia (McKemy et al., 2002). Specifically, it is widely expressed on terminals of thermosensitive neurons that innervate peripheral tissues including the skin, tongue, teeth and cornea (Abe et al., 2005; Parra et al., 2010). Several studies in animals and humans have clearly demonstrated the role of TRPM8 in pain perception (Marwaha et al., 2016); nevertheless, its precise role in pain modulation remains a matter of debate (Namer et al., 2005; Proudfoot et al., 2006). Application of relatively low doses of menthol (TRPM8 agonist) is associated with a cooling sensation and even pain relief, while at higher doses, the same drug causes burning, irritation and pain (Wasner et al., 2004; Hatem et al., 2006). Similarly, both TRPM8 activators and blockers have been demonstrated to attenuate pain sensation in neuropathic pain models (Parks et al., 2011; Sałat and Filipek, 2015). The conflicting results of these studies anticipate a complex and multifaceted role of TRPM8 in the pathophysiology of nociception, but the evidence that TRPM8−/−mice are deficient of innocuous cold and partially of noxious cold sensitivity strongly support the not‐redundant function of this channel (Colburn et al., 2007). Interestingly, preclinical studies have shown that levels of TRPM8 protein and RNA are both increased in sciatic nerve ligated (Frederick et al., 2007; Xing et al., 2007) and spinal nerve ligated rats (Koh et al., 2016). Studies with TRPM8‐deficient mice have also demonstrated the involvement of TRPM8 activation in chemotherapy‐induced hypersensitivity to innocuous and noxious cold stimuli (Descoeur et al., 2011).
The role of TRPM8 in cold hyperalgesia, a frequent symptom of neuropathic pain, has been also confirmed in clinical studies in humans. Topical application of the TRPM8 agonist menthol is a well‐characterized human model of cold pain, and several studies have demonstrated that menthol induces spontaneous burning pain and cold hyperalgesia by activation and sensitization of C‐nociceptors (Wasner et al., 2004; Campero et al., 2009). Thus, whereas converging evidence suggests that TRPM8 inhibition could be a valuable pharmacological strategy for the control of inflammatory and neuropathic pain, the poor selectivity of the agonists/antagonists commonly used in animal models has so far impaired the full assessment of TRPM8 as a potential pharmacological target. To better understand the relevance of TRPM8 modulation, we ran a medicinal chemistry programme that led to the identification of two potent and selective TRPM8 antagonists, with a pharmacological profile suitable for the treatment of pain disorders. The aim of this study was to test the efficacy of these TRMP8 blockers in acute and chronic animal pain models and to evaluate their effects on body temperature and on icilin‐induced wet‐dog shaking.
Methods
In vitro TRPM8 assay
The in vitro human TRPM8 cell‐based assay was performed by Axxam, Italy.
The TRPM8 antagonist activity of the compounds was determined by measuring changes in intracellular calcium levels triggered by the agonists http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2463 and icilin by using a Ca2+ sensitive fluorescent dye. The experiments were performed using HEK‐293 cells stably expressing the human TRPM8. Cells were seeded 10.000 cells per well and grown at 37°C (% CO2) in complete medium in 384‐well plates coated with poly‐D‐lysine (Matrix black/clear bottom; Thermo Scientific, Waltham, MA). Twenty‐four hours after seeding of the cells, cell culture medium was removed; cells were washed with Tyrode's assay buffer and then loaded with the fluorescent Ca2+ indicator Fluo‐4 NW dye (Molecular Probes, Life Technologies, Paisley, UK) supplemented with water‐soluble http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4357 (Molecular Probes). Dye‐loaded cell plates were incubated for 1 h at room temperature. The compounds or vehicle were added, and the kinetic response was monitored by a fluorimetric imaging plate (FLIPRTETRA; Molecular Devices, Sunnyvale, CA) over a period of 5 min. Then an injection of the reference agonist, cooling agent 10, at its EC80 concentration was performed. The signal of the fluorescence emitted was recorded for an additional 3 min. The bioactivity exerted by the compounds or vehicle was expressed as % inhibition, and IC50 values were then calculated. The percentage scale is defined by a 100% inhibition in which the relative fluorescence units (RFUs) of the test were identical to the MIN controls in second injection (http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2461 at IC100, 50 μM) and 0% of inhibition in which the RFUs of the test were identical to the MAX controls in second injection (cooling agent 10 and icilin at EC80, 20–30 μM).
The % activity was calculated according to the following formula:
- <MAX>
median of max signal controls.
- <MIN>
median of min signal controls.
- x
measured value of a well.
Compounds were tested at eight concentrations in quadruplicate to determinate the IC50 towards both cooling agent 10 and icilin activation. Evaluation of compound activity profile was performed with Genedata Screener 12.0.5.
In vitro cold stimulation assay
The in vitro human TRPM8 cell‐based assay with a low temperature stimulation protocol was performed by Axxam, Italy.
The temperature‐activation cell‐based assay described by Aneiros and Dabrowski (2009) was modified and used to assess the ability of DFL23448 and DFL23693 to inhibit cold‐induced TRPM8 stimulation. HEK‐293 cells stably expressing human TRPM8 were seeded (1.5–1.8 × 106 cells) in a T75 flask in complete medium. Three to four days after seeding (approximately 80% confluent cells), the medium was removed, and cells were loaded with Screen QuestTM Fluo‐8 NW dye solution (ABD Bioquest, Sunnyvale, CA, USA). Dye‐loaded cell flasks were incubated for 45 min at room temperature in the dark, and the Fluo‐8 NW solution was then removed, and cells were seeded in 96‐well assay plates (MicroAmpTM Optical 96‐Well Reaction Plates; Applied Biosystems, Life technologies) at 100.000 cells per well (20 μL per well). The compounds were added and incubated at room temperature for 5 min. The signal was recorded for 2 min at 25°C; then the temperature was lowered to sub‐physiological levels, and the signal recorded for 3 min by the ABI Prism® 7900HT Sequence Detection System (Life Technologies). The fluorescence difference (ΔF = fluorescence525nm at 14°C – fluorescence525nm at 25°C) was assessed. The analysis was performed computing ΔF/F0, where F0 was the fluorescent signal at the starting temperature (25°C).
ΔF/F0 was normalized versus Neutral Controls (max signals) and Inhibitor Controls (min signals) in order to obtain % activity according to the following formula:
where
- <MAX>
median of max signal controls.
- <MIN>
median of min signal controls.
- x
measured value of a well.
Compounds were tested at eight concentrations in quadruplicate to determinate the IC50.
IC50 curves were generated by fitting the fluorescence data with a sigmoidal curve equation using GraphPad PRISM® software (version 5, GraphPad Software Inc., La Jolla, CA, USA).
In vitro ion channels selectivity assay
The profiling of the compounds on human http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=485, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=507, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=510 and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=584 ion channels was performed by Axxam, Italy.
TRPA1‐, TRPV1‐, TRPV4‐ and Nav1.7‐expressing HEK‐293 cells were analysed in order to study the response to the compounds using a Ca2+ mobilization‐dependent fluorescence signal in 384 MTP format. Cells were seeded 10 000 cells per well in 384 MTP (Matrix black/clear bottom plates) in complete medium (25 μL per well). Twenty‐four hours after seeding the cells, the medium was removed, and cells were loaded with 20 μL per well of the Fluo‐8 NW dye solution. The dye‐loaded cell plates were incubated for 1 h at room temperature. Test compounds at 3X concentration in 1.5% DMSO Tyrode's buffer were added to the wells of an assay plate, in 10 μL volume (for a final DMSO concentration of 0.5%) and read by the FLIPRTETRA plate. The kinetic response was monitored by the instrument over a period of 3 min (200 s). A second injection of (10 μL per well) of reference agonists (http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2486, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4205, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2420 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2626 for TRPA1, TRPV1, TRPV4 and Nav1.7 respectively) at 4 x concentration in assay buffer (EC80) was added by the FLIPRTETRA. The signal of the emitted fluorescence was recorded for an additional 3 min.
The inhibitory effect (% activity) of the compounds was calculated according to the following formula:
where
- <MAX>
median of max signal controls.
- <MIN>
median of min signal controls.
- x
measured value of a well.
Compounds were tested at eight concentrations in quadruplicate to determinate the IC50 towards the panel of ion channels. The compound curve fitting profile for each dose–response was performed with the Condoseo module of Genedata Screener 13.0.5.
Physicochemical characterization
The main physicochemical properties of the compounds (pKa and solubility) were determined by using the SiriusT3 apparatus (Sirius Analytical Instruments Ltd., East Sussex, UK) equipped with an Ag/AgCl double junction reference pH electrode, a Sirius D‐PAS spectrometer and a turbidity sensing device. The titration experiments were conducted in 0.15 M KCl solution under an argon atmosphere at a temperature of 25 ± 1°C.
All tests were performed using standardized 0.5 M KOH and 0.5 M HCl as titration reagents.
DFL23448 and DFL23693 were analysed in triplicate (n = 3) solutions (Gibco PBS 1X, pH 7.4) at a concentration of 10 μM to test their stability towards the hydrolysis reaction. Incubations were performed at 37°C for 5 days. Samples were then transferred in vials for LC–MS/MS analysis.
Radioligand binding assay for selectivity versus GPCRs
Evaluation of the affinity of DFL23448 and DFL23693 for human http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=4 (http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=22, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=25, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=28 and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=29), http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=33 (http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=262 and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=263), http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=2 (http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=14 and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=15), the neurokinin http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=360, http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=50 (δ, κ and μ), the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=320 (also known as ORL1), the 5‐HT1A http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1, http://www.guidetopharmacology.org/GRAC/FamilyIntroductionForward?familyId=13 (http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=56 and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=57), http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=20 (http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=215 and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=216) and http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=10 (http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=41 and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=42) was performed by Eurofins Cerep SA (France) by radioligand binding assays. Cell membrane homogenates (48 μg protein) were incubated for 60 min at 22°C with the respective reference compound in the absence or presence of the test compound in a buffer containing 50 mM Tris–HCl (pH 7.4), 2 mM MgCl2 and 1 mM EDTA. After incubation, the samples were filtered rapidly under vacuum through glass fibre filters (GF/B, Packard Instruments, Meriden, CT, USA) presoaked with 0.3% polyethylenimine and rinsed several times with ice‐cold 50 mM Tris–HCl using a 96‐sample cell harvester (Unifilter, Packard Instruments). The filters were dried, then counted for radioactivity in a scintillation counter (Topcount, Packard Instruments) using a scintillation cocktail (Microscint 0, Packard Instruments). The results are expressed as % inhibition of the control radioligand specific binding. The compounds were tested at a single concentration of 10 μM in triplicate.
hERG inhibition
The human ether‐a‐go‐go related gene (hERG) inhibition assay was performed by Eurofins Panlabs Inc, USA. Compounds were tested to assess the potential interaction on http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=572 stably transfected in CHO‐K1 cell line. Single cell ionic currents were measured in the automated patch clamp configuration at room temperature by using QPatch 16 instrument. The compounds were tested at five concentrations in triplicate to obtain the IC50 values.
Cytochrome P450 inhibition
The cytochrome P450 inhibition assay was performed by Eurofins Panlabs Inc, USA. The compounds were tested to evaluate the potential direct inhibitory effect on the major human cytochrome P450 enzyme isoforms (http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1337, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1319, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1326, http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=262#1328 and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1329) after incubation with pooled human liver microsomes using http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2858/http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3342, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7402, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2714, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4279 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6953 as probe model substrates for CYP3A4, CYP1A2, CYP2C9, CYP2C19 and CYP2D6 respectively.
Test compounds were incubated for 10 min at 37°C with human liver microsomes (0.1 mg·mL−1) and NADPH (1 mM) in the presence of a cytochrome P450 isoform‐specific probe substrate. The metabolite concentrations were then monitored by LC–MS/MS spectrometry. A decrease in the formation of the metabolite compared with the vehicle control was used to calculate the IC50 value. The effect of DFL23448 and DFL23693 was tested at five concentrations in triplicate.
Plasma protein binding
The evaluation of protein binding of DFL23448 and DFL23693 in rat plasma was performed by Aphad, Italy.
Stock solutions of the compounds and of the reference standards (diclofenac and antipyrine) were prepared in DMSO at a final concentration of 2 mM. Then 1.5 μL of compounds were spiked in 298.5 μL of plasma to obtain a final concentration of 10 μM. Plasma (200 μL) in triplicate for each compound was dialysed in the rapid equilibrium dialysis (RED) inserts (Thermo Scientific) against 350 μL of PBS in the RED plate for 4 h at 37°C, under agitation. At the end of the incubation, 50 μL of plasma was added to 50 μL of blank PBS buffer and 300 μL of acetonitrile (with 2.5 ng·mL−1 of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2406 as internal standard).
Similarly, 50 μL of incubated PBS was added to 50 μL of blank plasma and 300 μL of acetonitrile with verapamil. Samples were centrifuged at 12 000× g for 5 min, and supernatants were transferred in vials for LC–MS/MS analysis.
Drug treatment
DFL23448 and DFL23693 were dissolved in 10% v/v of Solutol HS 15: N‐methyl pyrrolidone in the ratio 2:1 w.v‐1 and mixed at T = 40°C until complete dissolution. Then 90% v.v‐1 of PBS was added to these solutions and mixed (50°C) for 2–3 min; they were adjusted to pH 7.4–7.5 with NaOH (0.1 N) and mixed (40°C) for 2–3 min until complete dissolution. DFL23448 and DFL23693 were prepared daily before use.
For the pharmacokinetic studies, only, DFL23448 was dissolved in 50% PEG400 and 50% PBS, pH 7.4.
Pharmacokinetics studies in the rat
Pharmacokinetic studies were carried out by Aphad, Italy.
PK parameters for DFL23448 were determined following i.v. and p.o. administration at 3 and 10 mg·kg−1 respectively (five male Sprague–Dawley rats, 7–8 weeks of age, were used for each dose). For DFL23693, five male Sprague–Dawley rats were dosed at 5 mg·kg−1 (i.v.) and 10 mg·kg−1 (p.o.). After administration of the compounds, 0.15–0.20 mL of blood were collected via jugular vein catheters with the use of zero‐dead volume disposable syringes (1 mL insulin syringes with attached 25G 5/8 inch needles). Blood samples were taken at 0.12 (i.v. only), 0.25, 0.5, 1, 2, 4, 8, 10 and 24 h post‐dose. After each blood withdrawal, the catheters were washed with 0.15–0.2 mL of sterile physiological saline and filled with 0.03 mL of heparin saline solution (0.25 U·mL−1). The heparin‐treated blood was immediately centrifuged (6000× g for 5 min at approximately 4°C), and the resulting plasma collected and transferred to uniquely labelled eppendorfs and frozen at −20°C until the analysis.
At the end of the study, animals were killed by exsanguination under deep isoflurane anaesthesia.
Calibration curves and QC samples were prepared in rat blank plasma adding 2 μL of each stock solution to 18 μL of plasma. Ten microlitres of spiked plasma was then added to 200 μL of cold acetonitrile with the internal standard at the final concentration of 100 ng·mL−1. Samples were kept under agitation for 15 min and centrifuged for 5 min at 9000× g at 5°C.
DFL23448 and DFL23693 concentrations were quantified by ULPC–MS/MS using a chromatographic method with a lower limit of quantitation of 1 ng·mL−1.
PK parameters were calculated using non‐compartmental analysis in Phoenix WinNonlin, Version 6.3 (Certara, L.P. – USA): AUC (0‐inf) was calculated using a non‐compartmental pharmacokinetic analysis with the linear‐logarithmic trapezoidal rule; half‐life (t 1/2) was calculated by linear regression on semi‐log plot of plasma concentration versus time (at least 3 time points during the terminal elimination phase with R 2 > 0.85).
Animals
The behavioural experiments were performed on male Sprague–Dawley rats (7–9 weeks of age; Charles River, Italy) housed in the animal care facility of the Department of Pharmacy of the University of Naples, Italy. Animals were housed in groups of five, in a room with controlled temperature (22 ± 1°C), humidity (60 ± 10%) and light (12 h per day); food and water were available ad libitum throughout the study. All animals were weighed on the day of each treatment. All behavioural tests were performed between 09:00 and 17:00 h, and the animals were used only once. Animal care and manipulations were conducted in conformity with International and National law and policies (EU Directive 2010/63/EU for animal experiments, ARRIVE guidelines and the Basel declaration including the 3R concept). The procedure reported here was approved by the Institutional Committee on the Ethics of Animal Experiments (CVS) of the University of Naples Federico II and by Ministero della Salute under protocol no. 2014‐00884607. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
For the pharmacokinetic studies, not‐fasted male Sprague–Dawley male rats were used and purchased from Harlan Italy Srl (body weights 250 g at the time of the treatment). The animals were housed, in a group of five, in cages suitable for the species, also during dosing and feeding periods. The animals were acclimatized to local housing conditions for 5 days and housed in a single, exclusive room, air‐conditioned to provide a minimum of 15 air changes per hour. The environmental controls were set to maintain temperature at 22°C and relative humidity within the range 50 to 60% with an approximate 12 h light and 12 h dark cycle that is controlled automatically. Food and water were available ad libitum throughout the study. All animals were weighed on the day of each treatment. Clinical signs were monitored at regular intervals throughout the study in order to assess any reaction to treatment. Each animal was uniquely identified with a coloured spray on the back before the experiment. The experiment was carried on in agreement with the Italian Law D. L.vo 4 Marzo 2014, no. 26.
Experimental groups and procedures
For the behavioural experiments, rats were randomized and divided into equal‐sized groups (n = 10 per group) not predetermined by a statistical method. Animals were treated with vehicle, DFL23693 (10 mg·kg−1, p.o.) and DFL23448 (10 mg·kg−1, i.v.). The volume of liquid administered was 0.5 mL for p.o. administration and 0.3 mL for i.v. Details of the anaesthesia are provided below. At the end of the procedures, the animals were killed by cervical dislocation.
Induction of orofacial and neuropathic pain
Orofacial pain hypersensitivity was induced by http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4196 according to the method described by Raboisson and Dallel (2004) with some modifications. Briefly, 2.5% formalin (0.1 mL) was injected into the upper lip, just lateral to the nose using a 27G sterile needle mounted on a 1 mL syringe; the needle was inserted into the lip close to the border between hairy and glabrous skin and advanced up to the hilt towards the vibrissal pad. Thus, the formalin solution spread out within the upper lip and the rostral half of the vibrissal pad inducing a stereotypical response characterized by two distinct phases: an initial neurogenic phase (0–3 min) and a second inflammatory phase (10–30 min). Differently from Raboisson and Dallel's method, we evaluated pain hypersensitivity by mechanical and cold stimuli developed 24 h after formalin injection using the Orofacial Stimulation Test (Ugo Basile, Italy).
Neuropathic pain behaviour was induced by ligation of the sciatic nerve according to the method described by Bennett and Xie (1988). Rats were anaesthetized (100 mg·kg−1 http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4233 and 10 mg·kg‐1 http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=523 i.p.), and the left sciatic nerve was exposed at the level of the thigh by blunt dissection through the biceps femoris. We assessed the depth of anesthesia through absent of reflex in response to noxious stimuli such as pinch of paw. Proximal to the sciatic's trifurcation, about 12 mm of nerve was freed of adhering tissue, and four ligatures were loosely tied around it with about 1 mm spacing so that the epineural circulation was preserved. The length of nerve thus affected was 6–8 mm long. The animals were allowed to recover and used the day after the surgery. Sham rats underwent the same surgical procedure to expose but not ligate the left sciatic nerve.
Orofacial stimulation test
The orofacial stimulation test system measures hypersensitivity of the trigeminal area. Animals initially underwent 5 days of training utilizing the orofacial stimulation test after a 12 h period of food deprivation (fasting period). Rats were placed in a standard rat cage with a plastic divider to create two rooms. In the anterior room of the cage, there was an Ugo Basile apparatus with a drinking window for the rat's head to enter and acquire a reward (milk and chocolate) located on the opposing aspect of the drinking window. During the training period, each rat was individually placed in the posterior room for 10 min to familiarize it to its environment, then the drinking window was opened, and the test rat was subsequently monitored for 10 min to allow it to drink the milk and chocolate (Zuo et al., 2013). At the day of behavioural assessment, mechanical or thermal stimulation was induced by the insertion of an appropriate module in the drinking window (not present during the training period). During this time, rats received approximately 3–5 stimulations prior to formalin (basal) and 15–20 stimulations 24 h after formalin injection, for both mechanical and thermal stimulator. Similar to the adaptation training, all thermal or mechanical module experiments were preceded by a 12 h fasting period, 10 min for the rats to be familiarized with testing environment and a subsequent 10 min for behavioural assessment.
Mechanical stimulator
Experiments were performed utilizing the mechanical model that consisted of a cassette with 10 tungsten wires placed 3 mm apart from each other and 8 mm from the opening hole to the cassette held to produce a bending force from the drinking window of the apparatus. The animal's face contacted the tungsten wires of the mechanical module as the rat projected its head through the hole in the apparatus in order to drink milk and chocolate located on the exterior aspect of the drinking window. In each experiment, a blind observer activated a stopwatch every time the test rat drank milk and chocolate (also defined as ‘contact time’); when the test rat moved away from the drinking window, the stopwatch was stopped. Data were generated using the total contact time(s) to mechanical stimulus reached in a total testing time of 600 s. Basal data were obtained from the total contact time (reached in a total testing time of 600 s) 24 h before formalin injection.
Thermal stimulator
In the thermal model, there was a surrounding metal tubing at the opening; the temperature of the circulating solution (water) was controlled by a thermal circulating bath unit (Ugo Basile, Italy). The distance between the metal tube and the nipple of the milk/chocolate bottle was 14 mm. For thermal stimulation, the thermal module was set at 5°C; the animal's orofacial region was shaved 3 days before formalin injection and subjected to cold stimuli when the rat contacted the metal tube as it poked its head through the hole to obtain the milk/chocolate. In each experiment, a blind observer activated a stopwatch every time that the test rat drank milk and chocolate (also defined as ‘contact time’); when the test rat moved away from the drinking window, the stopwatch was stopped. Data were generated from the total contact time(s) to thermal stimulus reached in a total testing time of 600 s. Basal data were obtained from the total contact time (reached in a total testing time of 600 s) 24 h before formalin injection.
Mechanical allodynia
To assess the changes in sensation or in the development of mechanical allodynia, sensitivity to tactile stimulation was measured using the Dynamic Plantar Aesthesiometer (DPA, Ugo Basile, Italy). Individual animals were placed in plexiglas boxes (30 × 30 × 25 cm) with a mesh metal floor covered by a plastic dome that enabled the animal to walk freely, but not to jump, for approximately 30 min thus allowing behavioural acclimatization to the environment. When both paws were resting on the floor, the experimenter started the test. The mechanical stimulus was then delivered in the mid‐plantar skin of the hind paw. The cut‐off was fixed at 50 g. Ipsilateral and contralateral paws were tested twice per session, before ligation (day 0) and then 7 and 14 days after surgery at 1, 3 and 5 h post dose. The means of the paws withdrawal (express in g) were calculated from an average of two separate measures.
Cold allodynia
Cold sensitivity was measured as the number of foot withdrawal responses after application of acetone to the dorsal surface of the paw (Choi et al., 1994). Individual animals were placed in Plexiglas boxes (30 × 30 × 25 cm). A drop of acetone (25 μL) was applied over the dorsal surface of the paws with a syringe connected to a thin polyethylene tube while the rats were standing on a metal mesh. A brisk foot withdrawal response, after the spread of acetone, was considered as a sign of cold allodynia. No familiarization period was required, because it was not necessary for the animal's paw to be on the floor. Cold responses were measured on ipsilateral (ligated) and contralateral paws 7 and 14 days after surgery at 1, 3 and 5 h post dose. The procedure was repeated 3 times at 5 min intervals. In results are reported as the mean of the paw withdrawal (expressed as a number) determined from an average of three separate measures.
Experimental design
The effect of DFL23693 was evaluated 3 h following p.o. administration, while the effect of DFL23448 was evaluated 1 h following i.v. administration, in agreement with their pharmacokinetic characteristics. Antinociceptive activity by mechanical and thermal stimuli was measured 24 h after formalin treatment; two separate experiments were done.
In the neuropathic pain experiment, the antiallodynic effect of the compounds was evaluated on days 7 and 14 after nerve ligation.
Body temperature
Rats were placed into an environmental room maintained at a constant temperature of 21 ± 0.3°C and relative humidity of 55 ± 2%. The animals were allowed to acclimatize for 24 h before taking the first temperature reading. Body temperature (Tb) was recorded using a rectal probe for rats (Model RAT2, Ugo Basile, Italy), which was inserted approximately 5 cm into the colon. A digital thermometer (Model BAT‐10, Ugo Basile, Italy) was used. Vehicle (p.o. or i.v.), DFL23693 (10 mg·kg−1 p.o.) and DFL23488 (10 mg·kg−1 i.v.) were administered for four consecutive days; each day; Tb was measured before treatment (baseline) and at 1, 3, 5 and 7 h following DFL23693 administration and at 0.5, 1, 2 and 3 h following DFL23448 administration, according to their different pharmacokinetic profiles.
Icilin‐induced “wet‐dog” shaking in rats
Icilin, a TRPM8 agonist, was used to induce shaking in rats (Wei, 1983). Animals were first habituated to the testing room for 30 min; after that, they were randomized into treatment groups and treated with vehicle (p.o. or i.v.) or DFL23693 (10 mg·kg−1 p.o.) or with DFL23488 (10 mg·kg−1 i.v.). Icilin was administered i.p. at 1 mg·kg−1 dissolved in 1% Tween80/H2O. The number of intermittent but rhythmic ‘wet dog‐like’ shakes (WDS) of neck, head and trunk in each animal was counted for a period of 30 min following icilin administration. DFL23693 and DFL 23448 were, respectively, administered 3 and 1 h before icilin injection.
Blinding and randomization
For most in vitro protocols, blinding was not feasible as experiments were conducted by an individual experimenter, whereas in in vivo experiments, drug treatments were conducted in a blinded fashion. In neuropathic pain experiments, we excluded the sham group from blinding conditions since all rats subjected to chronic constriction injury (CCI) had a unique posture as they tended to retract the ipsilateral paw and to hold it up from the floor, in a guarding posture, whereas sham rats did not show this behaviour. In all in vivo tests, randomization of animals and treatments were performed by different experimenters. Animals were allocated to experimental groups based on their baseline responses to behavioural tests. In order to reduce the effect of extraneous variables on behavioural assessment, concurrent vehicle‐treated groups were present throughout all experiments. All treatments were performed in the morning, between 08:00 and 09:00 h; behavioural testing was conducted between 10:00 and 15:00 h. No animal was excluded from statistical analysis.
Statistical analysis
Data and statistical analysis in this study comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Statistical analyses were performed on raw data using Prism 5 Graphpad software (GraphPad Software Inc., San Diego, CA, USA).
In vitro data are presented as mean ± SD.
In vivo data are presented as mean ± SEM. In the orofacial test, data are presented as cumulative contact time (s). In CCI experiments, DPA data are presented as paw withdrawal threshold (g); cold allodynia data are presented as number of paw withdrawals. The significance of differences between groups was determined by two‐way repeated measurements ANOVA followed by post hoc Bonferroni's test. For the body temperatures, the data are expressed as differences between temperature obtained after injection (Tpost dose) and temperature before treatment (Tbasal); results are presented as ΔTb ± SEM. The significance of differences between groups was determined by one‐way or two‐way repeated measurements ANOVA followed by post hoc Bonferroni's test.
In icilin test, data are presented as number of WDS; the significance of differences between groups was determined by Student's t‐test.
P < 0.05 was considered statistically significant for all tests.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/2018 (Alexander et al., 2017a,b,c).
Results
In vitro compounds characterization
DFL23448 was tested in an in vitro hTRPM8 calcium influx assay using recombinant HEK‐293 cell line. DFL23448 resulted a potent TRPM8 blocker on icilin and Cooling Agent 10 (menthol analogue)‐induced TRPM8 activation showing an IC50 ± SD of 75 ± 9 and 55 ± 6 nM respectively (Figure 1A). Following our screening grid, the compound was also tested in the Cold Stimulation Test in which the intracellular calcium concentration was measured by detecting the fluorescence changes in response to the temperature change from 25 to 14°C, showing an IC50 ± SD of 21.4 ± 5 nM (Figure 1B). Despite the high potency and selectivity, the oral use of DFL23448 was limited by poor exposure in rats attributable to its elimination through glucuronidation mechanisms. With the aim to improve the metabolic stability of DFL23448, Structure–Activity Relationship (SAR) studies were performed starting from the knowledge of the pharmacophore features of the first series of TRPM8 blockers from which DFL23448 derived.
Figure 1.

Dose‐response curves for effect of DFL23448 on responses induced by icilin (green line) and cooling agent 10 (red line) (A) and in the cold stimulation test (B).
A novel class of small MW of TRPM8 blockers has been identified, and among the active compounds, DFL23693 was selected as the best molecule. DFL23693 is a moderately less potent inhibitor comparing with its cognate DFL23448, when evaluated through icilin and Cooling Agent 10‐induced TRPM8 activation, with an IC50 ± SD of 64 ± 6 and 81 ± 4 nM respectively (Figure 2A). On the other hand, DFL23693 showed the same high potency of DF23448 in the cold physiological activation assay (IC50 ± SD = 22.7 ± 6 nM; Figure 2B).
Figure 2.

Dose‐response curves for effect of DFL23693 on responses induced by icilin (green line) and cooling agent 10 (red line) (A) and in the cold stimulation test (B).
DFL23693 and DFL23448 physicochemical and in vitro absorption, distribution, metabolism and excretion (ADME) properties were obtained to provide the means for a full comparison of the compounds (Table 1). The compounds have the pKa values close to that of the carboxylic acid (5.89 ± 0.02 for DFL23448 and 4.34 ± 0.01 for DFL23693); this explains the high protein binding in rat plasma that is 97.68 ± 0.01% for DFL23448 and 98.99 ± 0.06% for DFL23693. Both compounds have an excellent chemical stability (>98.5%) when heated at 37°C for 5 days. The aqueous solubility of DFL23448 and DFL23693 at pH 7.4 buffered solution was 0.631 ± 0.02 and 15.2 ± 0.11 mM respectively (Table 1).
Table 1.
Physicochemical and in vitro ADME properties of DFL23448 and DFL23693
| Assay | DFL23448 | DFL23693 |
|---|---|---|
| Aqueous solubility at pH 7.4 (mM) | 0.631 ± 0.02 | 15.2 ± 0.11 |
| % chemical stability (PBS, 37°C, 5 days) | 98.9 ± 4.2 | 98.7 ± 5.1 |
| pKa | 5.89 ± 0.02 | 4.34 ± 0.01 |
| Rat plasma protein binding (%) | 97.68 ± 0.01 | 98.99 ± 0.06 |
| hERG inhibition IC50 (μM) | 410 ± 5 | 20 ± 0.9 |
| CYP P450 inhibition IC50 (μM) | >30 for all tested isoforms | >30 for all tested isoforms |
Data are reported as mean ± SD.
Further characterization of the compounds showed the absence of inhibition of hERG potassium channel. The concentration–response curve of the test items on IKr ion channel current resulted in an IC50 value of 410 ± 5 μM for DFL23448 and 20 ± 0.9 μM for DFL23693 (Table 1). The compounds also demonstrated no inhibition of selected CYP enzymes implicated in drug–drug interactions with an IC50 value >30 μM (Table 1).
In vitro selectivity of DFL23448 and DFL23693 versus other TRP channels or GPCRs
DFL23448 and DFL23693 showed a very good selectivity towards a panel of http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=694 when tested at 10 μM in the radioligand binding assay. DFL23448 and DFL23693 exhibited an inhibition lower than 25% compared with the radiolabelled reference compound specific on these targets: α1A, α2A, β1 and β2 adrenoceptors; H1, H2, M2, M3, NK1, δ, κ, μ, NOP (ORL1), 5‐HT1A, CB2, D2, D3 and B1 receptors. In addition, DFL23448 and DFL23693 did not inhibit the calcium efflux in TRPV1, TRPV4, TRPA1 and Nav1.7 ion channels triggered by capsaicin, GSK1016790A, isothiocyanate (AITC) and veratridine respectively. The recorded IC50s were >10 μM for all the ion channels tested (Table 2).
Table 2.
In vitro selectivity of DFL23448 and DFL23693 versus other TRP channels or GPCRs
| Receptor | % inhibition of control specific binding [10 μM] (n = 3) | |
|---|---|---|
| DFL23448 | DFL23693 | |
| α1A | 12.50 ± 4.58 | 13.90 ± 4.11 |
| α2A | 2.58 ± 3.84 | 1.41 ± 2.12 |
| β1 | 4.66 ± 1.46 | 9.42 ± 1.32 |
| β2 | 7.98 ± 0.99 | 5.15 ± 2.22 |
| H1 | 10.82 ± 2.01 | 7.42 ± 0.91 |
| H2 | 5.82 ± 2.52 | 0.41 ± 0.21 |
| M2 | 17.65 ± 3.45 | 3.24 ± 1.25 |
| M3 | 11.67 ± 2.42 | 15.76 ± 1.03 |
| NK1 | 2.02 ± 0.81 | 3.12 ± 0.91 |
| μ | 21.85 ± 3.23 | 3.61 ± 1.27 |
| κ | 21.57 ± 6.53 | 17.62 ± 4.18 |
| δ | 1.86 ± 1.50 | 2.96 ± 0.17 |
| NOP | 14.47 ± 4.55 | 11.3 ± 3.93 |
| 5‐HT1A | 4.05 ± 1.71 | 5.9 ± 1.81 |
| CB2 | 10.58 ± 3.56 | 12.34 ± 4.09 |
| D2 | 7.57 ± 1.68 | 8.90 ± 3.23 |
| D3 | 2.51 ± 1.09 | 11.34 ± 3.98 |
| B1 | 21.54 ± 4.51 | 4.67 ± 2.65 |
| IC50 towards ion channels | ||
| TRPV1 | IC50 > 10 μM | IC50 > 10 μM |
| TRPV4 | IC50 > 10 μM | IC50 > 10 μM |
| TRPA1 | IC50 > 10 μM | IC50 > 10 μM |
| Nav1.7 | IC50 > 10 μM | IC50 > 10 μM |
Pharmacokinetics in the rats
The pharmacokinetic parameters of DFL23448 and DFL23693 following single i.v. and p.o. administration to male rats were evaluated (Table 3). I.v. administration of DFL23448 (3 mg·kg−1) showed a clearance of 3.11 ± 0.11 mL·min−1·kg−1and a t 1/2 of 0.74 ± 0.05 h. The distribution volume was 0.082 ± 0.012 L·kg−1 P.o. administration of 10 mg·kg−1 of DFL23448 caused a maximum plasma concentration (Cmax) of 47 ± 6 ng·mL−1 and the tmax occurred at 4.51 ± 0.32 h after dosing. The p.o. t 1/2 was 5.11 ± 0.43 h. Intravenous administration of DFL23693 at 5 mg·kg−1 showed a low clearance (5.31 ± 0.31 mL·min−1·kg−1) with a t 1/2 of 3.15 ± 0.10 h and a distribution volume of 1.69 ± 0.11 L·kg−1. Urine analysis data showed that a very small quantity of DFL23693 (1.51 ± 0.08%) was excreted through urine over a period of 72 h. P.o. administration at 10 mg·kg−1 of DFL23693 showed a Cmax of 10 673 ± 805 ng·mL−1 and the tmax occurred at 15 min after dosing. The p.o. t 1/2 was 3.13 ± 0.32 h. DFL23693 showed a high p.o. absolute bioavailability (F%) of 98.8%. Table 3 summarizes the pharmacokinetic parameters of the compounds.
Table 3.
PK Parameters of DFL23448 and DFL23693 in rats
| Compound | I.v. route | Oral route | |||||||
|---|---|---|---|---|---|---|---|---|---|
| CL (mL·min−1·kg−1) | Vss (L·kg−1) | t 1/2 (h) | AUC (0‐inf) (ng·h·mL−1) | Cmax (ng·mL−1) | tmax (h) | t 1/2 (h) | AUC (0‐inf) (ng·h·mL−1) | F% | |
| DFL23448 | 3.11 ± 0.11 | 0.082 ± 0.012 | 0.74 ± 0.05 | 20 125 ± 7014 | 47 ± 6 | 4.51 ± 0.32 | 5.11 ± 0.43 | 670 ± 87 | 0.99 |
| DFL23693 | 5.31 ± 0.31 | 1.69 ± 0.11 | 3.15 ± 0.10 | 12 890 ± 1161 | 10 673 ± 805 | 0.25 ± 0.03 | 3.13 ± 0.32 | 25 467 ± 1329 | 98.8 |
DFL23448 was administered at a dose of 3 mg·kg−1 i.v. and 10 mg·kg−1 p.o. DFL23693 was administered at a dose of 5 mg·kg−1 i.v. and 10 mg·kg−1 p.o. Values shown are mean ± SD; CL, clearance; Vss, volume of distribution at steady state; t 1/2, elimination t 1/2; AUC (0‐inf) area under the plasma concentration‐time curve extrapolated from zero to infinity; Cmax, maximum plasma concentration; tmax, time to reach peak or maximum plasma drug concentration; F, oral bioavailability. Preclinical pharmacokinetic parameters of DFL23448 and DFL23693. Values shown are mean ± SD (n = 5).
In vivo results
Effect of DFL23693 and DFL23448 in formalin‐induced orofacial pain hypersensitivity
The formalin test is an important means of assessing pain‐evoked behaviours induced by moderately intense and long‐lasting noxious stimuli. As recently reported (Cervantes‐Durán et al., 2016), intraplantar injection of formalin produces a long‐lasting secondary allodynia and hyperalgesia in rats. In agreement with these results, we observed that upper lip formalin injection in vehicle treated‐group produced a significant reduction of contact time 24 h after challenge both in mechanical (Figure 3A, B) and cold (Figure 3C, D) stimulation test (white bars; #P < 0.05 vs. the respective basal group). Oral pretreatment with DFL23693 (10 mg·kg−1; administrated 3 h before orofacial test, in agreement with its pharmacokinetic characteristics) significantly decreased formalin‐induced pain hypersensibility; indeed, 24 h following formalin challenge, the compound increased the contact time in both mechanical and cold allodynia tests (Figure 3A, C, grey‐white columns; *P < 0.05 vs. vehicle + 24 h group). A very similar activity was also obtained with i.v. DFL23448 treatment (10 mg·kg−1); on the basis of its pharmacokinetic profile, the administration of the drug 1 h before orofacial test produced a significant antinociceptive effect on both mechanical and cold stimuli (Figure 3B, D, respectively; grey‐black columns; *P < 0.05 vs. vehicle + 24 h group).
Figure 3.

Effect of vehicle, DFL23693 and DFL23448 on formalin‐induced mechanical (A and B) and thermal (C and D) allodynia. After 24 h from formalin, rats were treated with vehicle, DFL23693 (10 mg·kg−1 p.o.) and DFL23448 (10 mg·kg−1 i.v.); the test was done 3 h following p.o. administration (DFL23693) or 1 h following i.v. administration (DFL23448). The cumulative contact time was counted for 10 min. Data are shown as mean ± SEM of 10 animals per group. #P < 0.05 versus the respective basal response; *P < 0.05 versus vehicle + 24 h group.
Effect of DFL23693 and DFL23448 in CCI‐induced cold and mechanical allodynia
CCI model is one of the most commonly employed animal model of neuropathic pain. In this model, we evaluated the efficacy of the compounds 7 and 14 days after sciatic nerve ligation, time in which allodynia and hyperalgesia are well evident (Bennett and Xie, 1988; Xing et al., 2007). As illustrated in Figures 4 and 5, CCI‐vehicle treated animals showed a significant cold and mechanical allodynia if compared with sham rats. In particular, in cold allodynia experiments, the number of paw withdrawal threshold on ipsilateral paw of vehicle treated groups resulted significantly increased 7 and 14 days after surgery, due to neuropathy (Figure 4A, B, white bars; #P < 0.05 vs. sham rats); no effect was observed on the contralateral (unligated) paws (data not shown). Oral DFL23693 administration (10 mg·kg−1) significantly reduced the number of ipsilateral paw withdrawal at 3 and 5 h post dose (Figure 4A, grey‐white columns, *P < 0.05 vs. CCI‐vehicle treated rats and Figure 4B, grey‐white columns, *P < 0.05 vs. CCI‐vehicle treated rats); no effect was observed at 1 h. Intravenous DFL23448 administration (10 mg·kg−1) showed a significant reduction of cold allodynia only 1 h post dose on days 7 and 14 following CCI (Figure 4A, B, grey‐black columns, *P < 0.05 vs. CCI‐vehicle rats), whereas no activity was determinate 3 and 5 h post dose (Figure 4A, B, grey‐black columns). In DPA test, paw withdrawal threshold on ipsilateral paw resulted significantly reduced at days 7 and 14, due to neuropathy (Figure 5A, B, white columns; #P < 0.05 vs. sham rats); no effect was observed on the contralateral (unligated) paws (data not shown). At 7 and 14 days from sciatic nerve ligation, DFL23693 (10 mg·kg−1 p.o.) showed a significant reduction of mechanical allodynia at 3 and 5 h post treatment (Figure 5A, B, grey‐white columns; *P < 0.05 vs. CCI‐vehicle treated rats). In agreement with the results obtained in cold allodynia, DFL23448 (10 mg·kg−1 i.v.) significantly increased paw withdrawal latency only 1 h post dose (Figure 5A, B, grey‐black columns; *P < 0.05 vs. CCI‐vehicle treated rats), whereas no activity was observed at 3 and 5 h (Figure 5A, B, grey‐black columns).
Figure 4.
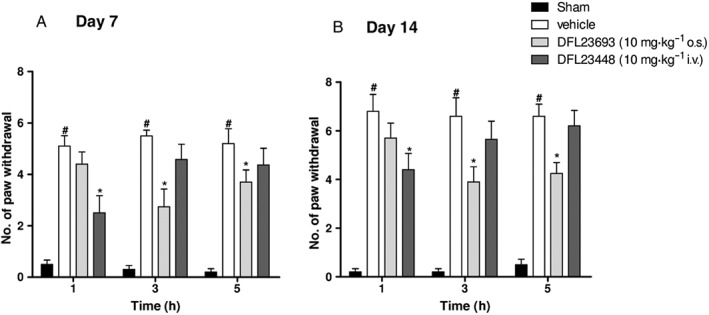
Effect of vehicle, DFL23693 and DFL23448 on CCI‐induced cold allodynia 7 and 14 days after injury. On day 7 following sciatic nerve ligation, rats were treated with vehicle, p.o. DFL23693 (10 mg·kg−1 p.o.) and i.v. DFL23448 (10 mg·kg−1 i.v.). Allodynia induced by cold stimulus was evaluated 1, 3 and 5 h post dose on ipsilateral paw. Data are shown as mean ± SEM of 10 animals per group. #P < 0.05 versus Sham; *P < 0.05 versus vehicle group.
Figure 5.

Effect of vehicle, DFL23693 and DFL23448 on CCI‐induced mechanical allodynia 7 and 14 days after injury. On day 7 following sciatic nerve ligation, rats were treated with vehicle, p.o. DFL23693 (10 mg·kg−1 p.o.) and i.v. DFL23448 (10 mg·kg−1 i.v.). Allodynia induced by mechanical stimulus was evaluated 1, 3 and 5 h post dose on ipsilateral paw. Data are shown as mean ± SEM of 10 animals per group. #P < 0.05 versus Sham; *P < 0.05 versus vehicle group.
Effect of DFL23693 and DFL23448 on body temperature
We evaluated the effects of the compounds on Tb. During the first day of treatment, DFL23693 (10 mg·kg−1 p.o.) produced a significant decrease of Tb by 1.91°C at 3 h and 1.4°C at 5 h (Figure 6A, black square; *P < 0.05 vs. vehicle group), while no significant effect was detectable at 1 and 7 h following p.o. administration. I.v. administration of DFL23448 (10 mg·kg−1) produced a significant effect with a Tb decrease of 0.8°C at 0.5 h and 1.08°C at 1 h (*P < 0.05 vs. vehicle group), while no effect was detectable at 2 and 3 h (Figure 6B, black square). As expected, intramuscular administration of icilin (7.5 mg·kg−1) increased Tb values with a maximum of 1.1°C 2 h after treatment (Figure 6B, black triangle; *P < 0.05 vs. vehicle group). Icilin was used as negative control, and it was administered only on the first day of treatment.
Figure 6.

Effect of vehicle, DFL23693 (A), DFL23448 (B) and icilin (B) on body temperature. Rats were treated with vehicle, DFL23693 (10 mg·kg−1 p.o.) and DFL23448 (10 mg·kg−1 i.v.) for four consecutive days; each day Tb was measured 1–3 to 5–7 h post DFL23693 dose and 0.5–1 to 2–3 h post DFL23448 dose. Icilin (7.5 mg·kg−1 i.m.) was used as negative control, and it was administered only on the first day of treatment. Tb data collected n the first day of treatment (A and B) and at 3 h post dose for DFL23693 (C) and 1 h post dose for DFL23448 (D), for 4 days of treatment. Data are shown as mean [differences between temperature obtained after injection (Tpost dose) and temperature before treatment (Tbasal)] ± SEM of 10 animals per group. *P < 0.05 versus vehicle group.
A marked reduction of magnitude in Tb decrease starting from day 2 was visible for both the compounds at each of the considered experimental time. In particular, DFL23693 3 h post dose induced a decrease of 0.96°C on day 2, 0.63°C on day 3 and 0.27°C on day 4 respectively (Figure 6C). DFL23448 1 h post dose induced a decrease of 0.53, 0.34 and 0.38°C on days 2, 3 and 4 respectively (Figure 6D).
Effect of DFL23693 and DFL23448 on icilin‐induced WDS
To evaluate the compounds' ability to block the spontaneous wet‐dog shake induced by icilin, we administered DFL23693 or DFL23448 before the challenge with icilin (1 mg·kg−1 i.p.) and recorded WDS for a period of 30 min. In the vehicle treated group, a mean of about 170 shakes was counted during the 30 min interval (Figure 7, white column). Pretreatment with DFL23693 (10 mg·kg−1 p.o.) and DFL23448 (10 mg·kg−1 i.v.) significantly reduced icilin‐induced WDS. In particular, in p.o. DFL23693‐pretreated (3 h) animals, about 77 shakes were counted, demonstrating a reduction of 51% (Figure 7A, grey‐white column; *P < 0.05). In i.v. DFL23448‐pretreated (1 h) rats, about 109 shakes were counted throughout the 30 min observation period with a reduction of 37% (Figure 7B, grey‐black column; *P < 0.05).
Figure 7.
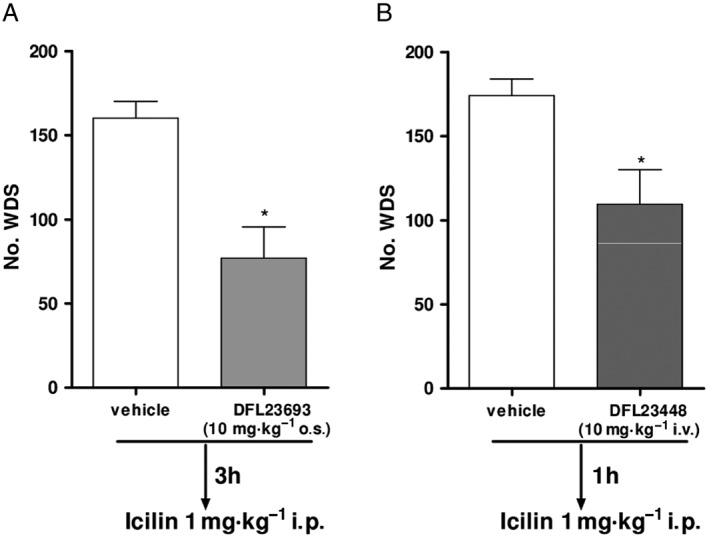
Effect of vehicle, DFL23693 (A) and DFL23448 (B) on icilin‐induced WDS. Rats were pretreated with vehicle, DFL23693 (10 mg·kg−1 p.o.) and DFL23448 (10 mg·kg−1 i.v.). The test was done 3 h following p.o. administration and 1 h following i.v. administration. The number of WDS were counted for 30 min following icilin injection (1 mg·kg−1 i.p.). Data are shown as mean ± SEM of 10 animals per group. *P < 0.05 versus vehicle group.
Discussion
In nearly two decades of research, numerous TRP channels, including TRPA1, TRPV1 and TRPV4, have been clearly demonstrated to be key mediators of pain process and modulation of these channels that have been proven effective in many preclinical models of pain and in some cases useful treatments for human diseases.
Among the pain‐related TRPs, TRPM8 has been progressively recognized for its role in the modulation of pain and nociception (Fernández‐Peña and Viana, 2013). In recent years, several manuscripts documented an increase of TRPM8 expression in different pain models including chemotherapy‐induced neuropathy (Gauchan et al., 2009), experimental colitis (Hosoya et al., 2014) and diabetic neuropathy (Nam et al., 2014), confirming its role in injury‐evoked allodynia in the somatic sensory nervous system (Harrington et al., 2011; Sarria et al., 2012) and underlining the key role of this channel in pain management (Dai, 2016). Notably, in animal models of neuropathic pain, the expression of TRPM8 increases in DRGs in response to sciatic or spinal nerve damage, and this increase correlates closely with the development of neuropathic pain (Frederick et al., 2007; Su et al., 2011; Koh et al., 2016). Blocking the activity of the channel by the administration of TRPM8 antagonists or by genetic ablation using mice lacking TRPM8, significantly attenuated thermal responses in nerve injury‐induced pain (Knowlton et al., 2011; Patel et al., 2014). This evidence suggests that the in vivo antagonism of TRPM8 holds promise for the successful treatment of neuropathic cold hypersensitivity (Marwaha et al., 2016; Weyer and Letho, 2017). A key challenge in the discovery of TRPM8 blockers is the identification of highly selective molecules with a good pharmacokinetic profile suitable to provide an efficacious dose response. In this paper, we describe two novel small MW TRPM8 inhibitors that exhibited a clear efficacy in the reduction of cold and mechanical allodynia in acute and chronic pain models. In vitro data showed that both DFL23693 and DFL23448 potently inhibit human TRPM8 activation by chemical and physiological stimulation and display a high selectivity. In vivo, the compounds were assessed for their TRPM8 antagonist activity under both normal and pathological conditions. In the in vivo experiments, DFL23693 was given by p.o. administration since it showed complete p.o. bioavailability (98.8%) in rats, whereas compound DFL23448 was administered i.v. due to its poor oral bioavailability (0.99%).
In icilin‐induced WDS, a model of skeletal muscle‐mediated heat generation in which the role of TRPM8 has been widely characterized (Werkheiser et al., 2006; Rossi et al., 2009), systemic administration of the drugs was able to reduce the numbers of WDS produced by icilin, in line with previous literature (Tamayo et al., 2012; Lehto et al., 2015). Gavva et al. (2012) showed that the administration of selective TRPM8 antagonists produces a small but significant decrease in Tb, impossible to be reproduced in TRPM8 knockout mice, suggesting that the body temperature decrease is entirely mediated by TRPM8. In agreement with these findings and other published data (Ito et al., 2016), our results show that a single administration of DFL23693 or DFL23448 resulted in transient hypothermia that appeared attenuated after repeated dosing of the compounds. It has been widely demonstrated that TRPM8 is involved in thermoregulation (Gavva et al., 2012), a role that is not entirely unexpected given that other temperature sensitive ion channels, particularly the heat‐gated TRPV1, have also been implicated in the regulation of body temperature (Gavva et al., 2007; Gavva et al., 2008). The precise role of TRPM8 in thermoregulation and energetic homeostasis is still unclear. In fact, although only a modest defect in thermoregulation under specific environmental conditions has been observed in TRPM8 null mice, TRPM8 activation on the skin has been proposed to act as a thermostat to prevent further cooling (Tajino et al., 2011), and subcutaneous injection of icilin results in a clear hyperthermic effect that is absent in TRPM8 knockout mice (Knowlton et al., 2010). In contrast with the above preclinical results, in the first clinical trial conducted in healthy volunteers using a TRPM8 antagonist, PF‐05105679, the compound showed a clear pharmacological activity on cold pain in the absence of hypothermia at any dose tested (Winchester et al., 2014), paving the way to further clinical investigation on this class of molecules. Several new TRPM8 antagonists have been reported to be effective in vitro in the nanomolar range and investigated for their therapeutic potential in the treatment of cold hypersensitivity in different neuropathic pain models and in the management of urological disorders (Patel et al., 2014; Lehto et al., 2015; Mistretta et al., 2016). To confirm the involvement of this channel in pain, it has been demonstrated that TRPM8 is necessary for the detection of chemical and thermal cooling stimuli in some sensory neurons and that TRPM8 knockout CCI‐mice did not exhibit significant increase in acetone‐induced cold allodynia (Colburn et al., 2007). In coherence with this observation, Xing et al. (2007) showed that TRPM8‐immunoreactive positive neurons in the L5 DRGs was increased in CCI animal model and that a non‐selective TRPM8 blocker attenuated cold allodynic responses in these mice. In a recent paper, Su et al. (2011) showed that intrathecal administration of menthol induced cold sensation and that the injection of a TRPM8 antisense oligonucleotide attenuated cold hyperalgesia in CCI rats. Our data showed that in cold allodynia experiments, systemic administration of DFL23693 and DFL23448 reduced the number of paw withdrawal threshold produced by acetone, and surprisingly, the antiallodynic effect was also observed in response to mechanical stimulation, thus suggesting an involvement of TRPM8 also in mechanical allodynia. Our data are also in agreement with recent findings by Sałat and Filipek (2015), which showed the effect of a specific TRPM8 antagonist, AMTB, on mechanical allodynia in a mouse model of paclitaxel‐induced neuropathic pain. These results are also supported by findings demonstrating that neuropathic pain models, including CCI, present spontaneous discharge in myelinated and unmyelinated fibres (Zimmermann, 2001; Seal et al., 2009), a hallmark that strongly contributes to the development and maintenance of neuropathic pain, and that TRPM8 is expressed on both fibres (Wasner et al., 2004; Dhaka et al., 2008). The higher efficacy in the in vivo models of DFL23693 compared with DFL23448 is coherent with the improved PK‐ADME properties of the compound. Indeed, the rapid onset and short duration of the pharmacological effect of DFL23448 perfectly reflect its pK profile (tmax and t 1/2). Oral administration of DFL23693 results in plasma concentrations higher than in vitro IC50 both at 3 and 5 h post treatment (97 and 63 nM, respectively) thus explaining the sustained efficacy in cold and mechanical allodynia models. Furthermore, the low volume of distribution (0.082 ± 0.012 L·kg−1) of DFL23448, associated with its high protein binding accounts for a limited distribution to peripheral terminals of sensory neurons that strongly impacts on its in vivo efficacy. It is worth mentioning that also the activation of TRPM8 has been associated with an analgesic effect and that TRPM8 agonists such as menthol or icilin can alleviate pain in neuropathic animals models (Proudfoot et al., 2006). This goes with the well‐known fact that cooling the skin produces a soothing sensation on the background of inflammatory pain. The nervous circuitries involved in these analgesic effects of cold temperature are currently unclear, and in this complex scenario, translating novel potent and selective TRPM8 modulators into the clinic will allow a better comprehension of the therapeutic potential and safety profile of these agents for pain management. While the role of TRPM8 in chronic pain (such as chronic inflammation or neuropathic pain) has been thoroughly described, there is little information on its contribution in acute pain. Capsaicin‐ or formalin‐induced neurogenic inflammation are two classical acute pain models abundantly used to evaluate the antinociceptive properties of drugs; even if behavioural symptoms (oedema licking or biting responses) are similar regardless of the kind of chemical used, they have distinct mechanisms of action. Indeed, capsaicin is an agonist of TRPV1 channels, whereas formalin not only stimulates TRPA1 channels (McNamara et al., 2007), but it has also others pronociceptive mechanisms (Coste et al., 2012; Sasso et al., 2012). Precisely for its characteristic to develop a more articulated pain, we used formalin‐induced orofacial pain. Following formalin injection, rats displayed orofacial rubbing and rapid head flinching due to direct activation of primary afferent neurons (phase I); then a persistent pain induction (phase II) was associated with the central sensitization and release of multiple inflammatory factors (Luccarini et al., 2006). As recently reported by http://www.ncbi.nlm.nih.gov/pubmed/?term=Garc%C3%ADa%20G%5BAuthor%5D&cauthor=true&cauthor_uid=25036442 (2014), ionic channels participate in the acute nociception induced by formalin as well as in the development and maintenance of secondary mechanical allodynia and hyperalgesia. Specifically, several published data have highlighted a role for the thermo‐channel TRPA1 in formalin‐induced pain as well as in other different pathological pain conditions including neuropathic pain models (del Camino et al., 2010; Wei et al., 2011). Here, we hypothesized that TRPM8 could play a key regulatory role in the development of symptoms closely correlated to hypersensitivity. TRPM8 channels are expressed in many trigeminal neurons (McKemy et al., 2002) and trigeminal nerve endings (Takashima et al., 2007), but their specific involvement in trigeminal pain is still unknown. The novel selective TRPM8 inhibitors that have been fully characterized in this study are able to reduce orofacial allodynia induced by formalin at 24 h. Indeed, both compounds, DFL23693 and DFL23448, increased significantly the contact time to both mechanical and cold stimuli suggesting a key role of this channel in formalin‐induced long‐lasting mechanical and termal allodynia in rats. These findings demonstrate for the first time that TRPM8 blockers are able to reduce pain hypersensitivity induced by formalin in the orofacial test reinforcing the hypothesis that TRPM8 antagonists may have a role in the therapeutic treatment of orofacial pain conditions.
In conclusion, in this paper, the authors, having identified a novel class of selective TRPM8 antagonists with unique binding properties to the ion channel, report their full in vitro and in vivo characterization and the evaluation of their effect on cold and mechanical allodynia in relevant inflammatory and neuropathic pain models. Overall, these findings encourage the use of TRPM8 modulators in the therapeutic management of cold and tactile‐evoked pain, preferably in the orofacial region.
Author contributions
R.R. designed, conceptualized, conducted and analysed the experiments and drafted the manuscript; C.D.C, C.A. and C.C. conducted and analysed the experiments; L.B. conceptualized and designed the experiments and drafted the manuscript; G.B. and A.A. conducted and analysed the experiments and drafted the manuscript; M.A. and A.C. drafted the manuscript and revised it for critically important intellectual content, and approved the final version of the manuscript.
Conflict of interest
LB, MA, GB and AA are employees of Dompé Farmaceutici S.p.A., Italy. The company has interests in the development of TRPM8 antagonists for the treatment of pain conditions. The other authors declare that they have no conflicts of interest.
Declaration of transparency and scientific rigour
This http://onlinelibrary.wiley.com/doi/10.1111/bph.13405/abstract acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
We thank Mr Giovanni Esposito and Mr Angelo Russo for animal care and assistance. This research did not receive any specific grant from funding agencies in the public, commercial or not‐for‐profit sectors.
De Caro, C. , Russo, R. , Avagliano, C. , Cristiano, C. , Calignano, A. , Aramini, A. , Bianchini, G. , Allegretti, M. , and Brandolini, L. (2018) Antinociceptive effect of two novel transient receptor potential melastatin 8 antagonists in acute and chronic pain models in rat. British Journal of Pharmacology, 175: 1691–1706. doi: 10.1111/bph.14177.
References
- Abe J, Hosokawa H, Okazawa M, Kandachi M, Sawada Y, Yamanaka K et al (2005). TRPM8 protein localization in trigeminal ganglion and taste papillae. Brain Res Mol Brain Res 136: 91–98. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Striessnig J, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Voltage‐gated ion channels. Br J Pharmacol 174: S160–S194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Peters JA, Kelly E, Marrion NV, Faccenda E, Harding SD et al (2017c). The Concise Guide to PHARMACOLOGY 2017/18: Ligand‐gated ion channels. Br J Pharmacol 174: S130–S159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aneiros E, Dabrowski M (2009). Novel temperature activation cell‐based assay on thermo‐TRP ion channels. J Biomol Screen 14: 662–667. [DOI] [PubMed] [Google Scholar]
- Bennett GJ, Xie YK (1988). A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 33: 87–107. [DOI] [PubMed] [Google Scholar]
- Campero M, Baumann TK, Bostock H, Ochoa JL (2009). Human cutaneous C fibres activated by cooling, heating and menthol. J Physiol 587: 5633–5652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervantes‐Durán C, Vidal‐Cantú GC, Godínez‐Chaparro B, Granados‐Soto V (2016). Role of spinal 5‐HT2 receptors subtypes in formalin‐induced long‐lasting hypersensitivity. Pharmacol Rep 68: 434–442. [DOI] [PubMed] [Google Scholar]
- Choi Y, Yoon YW, Na HS, Kim SH, Chung JM (1994). Behavioral signs of ongoing pain and cold allodynia in a rat model of neuropathic pain. Pain 59: 369–376. [DOI] [PubMed] [Google Scholar]
- Colburn RW, Lubin ML, Stone DJ Jr, Wang Y, Lawrence D, D'Andrea MR et al (2007). Attenuated cold sensitivity in TRPM8 null mice. Neuron 54: 379–386. [DOI] [PubMed] [Google Scholar]
- Coste O, Möser CV, Sisignano M, Kynast KL, Minden A, Geisslinger G et al (2012). The p21‐activated kinase PAK 5 is involved in formalin‐induced nociception through regulation of MAP‐kinase signaling and formalin‐specific receptors. Behav Brain Res 234: 121–128. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Y (2016). TRPs and pain. Semin Immunopathol 38: 277–291. [DOI] [PubMed] [Google Scholar]
- del Camino D, Murphy S, Heiry M, Barrett LB, Earley TJ, Cook CA et al (2010). TRPA1 contributes to cold hypersensitivity. J Neurosci 30: 15165–15174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Descoeur J, Pereira V, Pizzoccaro A, Francois A, Ling B, Maffre V et al (2011). Oxaliplatin‐induced cold hypersensitivity is due to remodelling of ion channel expression in nociceptors. EMBO Mol Med 3: 266–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhaka A, Earley TJ, Watson J, Patapoutian A (2008). Visualizing cold spots: TRPM8‐expressing sensory neurons and their projections. J Neurosci 28: 566–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández‐Peña C, Viana F (2013). Targeting TRPM8 for pain relief. Open Pain J 6: 154–164. [Google Scholar]
- Frederick J, Buck ME, Matson DJ, Cortright DN (2007). Increased TRPA1, TRPM8, and TRPV2 expression in dorsal root ganglia by nerve injury. Biochem Biophys Res Commun 358: 1058–1064. [DOI] [PubMed] [Google Scholar]
- García G, Martínez‐Rojas VA, Rocha‐González HI, Granados‐Soto V, Murbartián J (2014). Evidence for the participation of Ca(2+)‐activated chloride channels in formalin‐induced acute and chronic nociception. Brain Res 1579: 35–44. [DOI] [PubMed] [Google Scholar]
- Gauchan P, Andoh T, Kato A, Kuraishi Y (2009). Involvement of increased expression of transient receptor potential melastatin 8 in oxaliplatin‐induced cold allodynia in mice. Neurosci Lett 458: 93–95. [DOI] [PubMed] [Google Scholar]
- Gavva NR, Bannon AW, Surapaneni S, Hovland DN Jr, Lehto SG, Gore A et al (2007). The vanilloid receptor TRPV1 is tonically activated in vivo and involved in body temperature regulation. J Neurosci 27: 3366–3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavva NR, Davis C, Letho SG, Rao S, Wang W, Zhu DX (2012). Transient receptor potential melastatin 8 (TRPM8) channels are involved in body temperature regulation. Mol Pain 8: 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavva NR, Treanor JJ, Garami A, Fang L, Surapaneni S, Akrami A (2008). Pharmacological blockade of the vanilloid receptor TRPV1 elicits marked hyperthermia in humans. Pain 136: 202–210. [DOI] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington AM, Hughes PA, Martin CM, Yang J, Castro J, Isaacs NJ et al (2011). A novel role for TRPM8 in visceral afferent function. Pain 152: 1459–1468. [DOI] [PubMed] [Google Scholar]
- Hatem S, Attal N, Willer JC, Bouhassira D (2006). Psychophysical study of the effect of topical application of menthol in healthy volunteers. Pain 122: 190–196. [DOI] [PubMed] [Google Scholar]
- Hosoya T, Matsumoto K, Tashima K, Nakamura H, Fujino H, Murayama T et al (2014). TRPM8 has a key role in experimental colitis‐induced visceral hyperalgesia in mice. Neurogastroenterol Motil 26: 1112–1121. [DOI] [PubMed] [Google Scholar]
- Ito H, Aizawa N, Sugiyama R, Watanabe S, Takahashi N, Tajimi M et al (2016). Functional role of the transient receptor potential melastatin 8 (TRPM8) ion channel in the urinary bladder assessed by conscious cystometry and ex vivo measurements of single‐unit mechanosensitive bladder afferent activities in the rat. BJU Int 117: 484–494. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowlton WM, Bifolck‐Fisher A, Bautista DM, McKemy DD (2010). TRPM8, but not TRPA1, is required for neural and behavioral responses to acute noxious cold temperatures and cold‐mimetics in vivo. Pain 150: 340–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowlton WM, Daniels RL, Palkar R, McCoy DD, McKemy DD (2011). Pharmacological blockade of TRPM8 ion channels alters cold and cold pain response in mice. PloS One 6: e25894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh WU, Choi SS, Kim JH, Yoon HJ, Ahn HS, Lee SK et al (2016). The preventive effect of resiniferatoxin on the development of cold hypersensitivity induced by spinal nerve ligation: involvement of TRPM8. BMC Neurosci 17: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehto SG, Weyer AD, Zhang M, Youngblood BD, Wang J, Wang W et al (2015). AMG2850, a potent and selective TRPM8 antagonist, is not effective in rat models of inflammatory mechanical hypersensitivity and neuropathic tactile allodynia. Naunyn Schmiedebergs Arch Pharmacol 388: 465–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luccarini P, Childeric A, Gaydier AM, Voisin D, Dallel R (2006). The orofacial formalin test in the mouse: a behavioural model for studying physiology and modulation of trigeminal nociception. J Pain 7: 908–914. [DOI] [PubMed] [Google Scholar]
- Marwaha L, Bansal Y, Singh R, Saroj P, Bhandari R, Kuhad A (2016). TRP channels: potential drug target for neuropathic pain. Inflammopharmacology 24: 305–317. [DOI] [PubMed] [Google Scholar]
- McKemy DD, Neuhausser WM, Julius D (2002). Identification of a cold receptor reveals a general role for TRP channels in thermosensation. Nature 416: 52–58. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara CR, Mandel‐Brehm J, Bautista DM, Siemens J, Deranian KL, Zhao M et al (2007). TRPA1 mediates formalin‐induced pain. Proc Natl Acad Sci U S A 104: 13525–13530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistretta FA, Russo A, Castiglione F, Bettiga A, Colciago G, Montorsi F et al (2016). DFL23448, a novel transient receptor potential melastin 8‐selective ion channel antagonist, modifies bladder function and reduces bladder overactivity in awake rats. J Pharmacol Exp Ther 356: 200–211. [DOI] [PubMed] [Google Scholar]
- Nam JS, Cheong YS, Karm MH, Ahn HS, Sim JH, Kim JS (2014). Effects of nefopam on streptozotocin‐induced diabetic neuropathic pain in rats. Korean J Pain 27: 326–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namer B, Seifert F, Handwerker HO, Maihöfner C (2005). TRPA1 and TRPM8 activation in humans: effects of cinnamaldehyde and menthol. Neuroreport 16: 955–959. [DOI] [PubMed] [Google Scholar]
- Parks DJ, Parsons WH, Colburn RW, Meegalla SK, Ballentine SK, Illig CR et al (2011). Design and optimization of benzimidazole‐containing transient receptor potential melastatin 8 (TRPM8) antagonists. J Med Chem 54: 233–247. [DOI] [PubMed] [Google Scholar]
- Parra A, Madrid R, Echevarria D, Olmo S, Morenilla‐Palao C, Acosta MC et al (2010). Ocular surface wetness is regulated by TRPM8‐dependent cold thermoreceptors of the cornea. Nat Med 16: 1396–1399. [DOI] [PubMed] [Google Scholar]
- Patel R, Goncalves L, Newman R, Jiang FL, Goldby A, Reeve J (2014). Novel TRPM8 antagonist attenuates cold hypersensitivity after peripheral nerve injury in rats. J Pharmacol Exp Ther 349: 47–55. [DOI] [PubMed] [Google Scholar]
- Peier AM, Moqrich A, Hergarden AC, Reeve AJ, Andersson DA, Story GM et al (2002). A TRP channel that senses cold stimuli and menthol. Cell 108: 705–715. [DOI] [PubMed] [Google Scholar]
- Proudfoot CJ, Garry EM, Cottrell DF, Rosie R, Anderson H, Robertson DC et al (2006). Analgesia mediated by the TRPM8 cold receptor in chronic neuropathic pain. Curr Biol 16: 1591–1605. [DOI] [PubMed] [Google Scholar]
- Raboisson P, Dallel R (2004). The orofacial formalin test. Neurosci Biobehav Rev 28: 219–226. [DOI] [PubMed] [Google Scholar]
- Rossi HL, Vierck CJ Jr, Caudle RM, Yezierski RP, Neubert JK (2009). Dose‐dependent effects of icilin on thermal preference in the hindpaw and face of rats. J Pain 10: 646–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sałat K, Filipek B (2015). Antinociceptive activity of transient receptor potential channel TRPV1, TRPA1 and TRPM8 antagonists in neurogenic and neuropathic pain models in mice. J Zhejiang Univ Sci B 16: 167–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarria I, Ling J, Xu GY, Gu JG (2012). Sensory discrimination between innocuous and noxious cold by TRPM8‐expressing DRG neurons of rats. Mol Pain 8: 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasso O, Russo R, Vitiello S, Raso GM, D'Agostino G, Iacono A et al (2012). Implication of allopregnanolone in the antinociceptive effect of N‐palmitoylethanolamide in acute or persistent pain. Pain 153: 33–41. [DOI] [PubMed] [Google Scholar]
- Seal RP, Wang X, Guan Y, Raja SN, Woodbury CJ, Basbaum AI et al (2009). Injury‐induced mechanical hypersensitivity requires C‐low threshold mechanoreceptors. Nature 462: 651–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su L, Wang C, Yu YH, Ren YY, Xie KL, Wang GL (2011). Role of TRPM8 in dorsal root ganglion in nerve injury‐induced chronic pain. BMC Neurosci 12: 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajino K, Hosokawa H, Maeqawa S, Matsumura K, Dhaka A, Kobayashi S (2011). Cooling‐sensitive TRPM8 is thermostat of skin temperature against cooling. PLoS One 6: e17504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takashima Y, Daniels RL, Knowlton W, Teng J, Liman ER, McKemy DD (2007). Diversity in the neural circuitry of cold sensing revealed by genetic axonal labeling of transient receptor potential melastatin 8 neurons. J Neurosci 27: 14147–14157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamayo NA, Bo Y, Gore V, Ma V, Nishimura N, Tang P et al (2012). Fused piperidines as a novel class of potent and orally available transient receptor potential melastatin type 8 (TRPM8) antagonists. J Med Chem 55: 1593–1611. [DOI] [PubMed] [Google Scholar]
- Wasner G, Schattschneider J, Binder A, Baron R (2004). Topical menthol – a human model for cold pain by activation and sensitization of C nociceptors. Brain 127: 1159–1171. [DOI] [PubMed] [Google Scholar]
- Wei ET (1983). Inhibition of shaking movements in rats by central administration of cholinergic and adrenergic agents. Psychopharmacology (Berl) 81: 111–114. [DOI] [PubMed] [Google Scholar]
- Wei H, Koivisto A, Saarnilehto M, Chapman H, Kuokkanen K, Hao B et al (2011). Spinal transient receptor potential ankyrin 1 channel contributes to central pain hypersensitivity in various pathophysiological conditions in the rat. Pain 152: 582–591. [DOI] [PubMed] [Google Scholar]
- Werkheiser JL, Rawls SM, Cowan A (2006). Mu and kappa opioid receptor agonists antagonize icilin‐induced wet‐dog shaking in rats. Eur J Pharmacol 547: 101–105. [DOI] [PubMed] [Google Scholar]
- Weyer AD, Letho SG (2017). Development of TRPM8 antagonists to treat chronic pain and migraine. Pharmaceuticals 10: 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winchester WJ, Gore K, Glatt S, Petit W, Gardiner JC, Conlon K (2014). Inhibition of TRPM8 channels reduces pain in the cold pressor test in humans. J Pharmacol Exp Ther 351: 259–269. [DOI] [PubMed] [Google Scholar]
- Xing H, Chen M, Ling J, Tan W, Gu JG (2007). TRPM8 mechanism of cold allodynia after chronic nerve injury. J Neurosci 27: 13680–13690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M (2001). Pathobiology of neuropathic pain. Eur J Pharmacol 429 (1–3): 23–37. [DOI] [PubMed] [Google Scholar]
- Zuo X, Ling JX, Xu GY, Gu JG (2013). Operant behavioral responses to orofacial cold stimuli in rats with chronic constrictive trigeminal nerve injury: effects of menthol and capsazepine. Mol Pain 9: 28. [DOI] [PMC free article] [PubMed] [Google Scholar]