

Abstract
Background
To identify predictive markers for responders in lapatinib-treated patients and to demonstrate molecular changes during lapatinib treatment via cell-free genomics.
Patients and methods
We prospectively evaluated the efficacy of combining lapatinib with capecitabine and oxaliplatin as first line neoadjuvant therapy in patients with previously untreated, HER2-overexpressing advanced gastric cancer. A parallel biomarker study was conducted by simultaneously performing immunohistochemistry and next-generation sequencing (NGS) with tumor and blood samples.
Results
Complete response was confirmed in 7/32 patients (21.8%), 2 of whom received radical surgery with pathologic-confirmed complete response. Fifteen partial responses (46.8%) were observed, resulting in a 68.6% overall response rate. NGS of the 16 tumor specimens demonstrated that the most common co-occurring copy number alteration was CCNE1 amplification, which was present in 40% of HER2+ tumors. The relationship between CCNE1 amplification and lack of response to HER2-targeted therapy trended toward statistical significance (66.7% of non-responders versus 22.2% of responders harbored CCNE1 amplification; P = 0.08). Patients with high level ERBB2 amplification by NGS were more likely to respond to therapy, compared with patients with low level ERBB2 amplification (P = 0.02). Analysis of cfDNA showed that detectable ERBB2 copy number amplification in plasma was predictive to the response (100%, response rate) and changes in plasma-detected genomic alterations were associated with lapatinib sensitivity and/or resistance. The follow-up cfDNA genomics at disease progression demonstrated that there are emergences of other genomic aberrations such as MYC, EGFR, FGFR2 and MET amplifications.
Conclusions
The present study showed that HER2+ GC patients respond differently according to concomitant genomic aberrations beyond ERBB2, high ERBB2 amplification by NGS or cfDNA can be a positive predictor for patient selection, and tumor genomic alterations change significantly during targeted agent therapy.
Keywords: gastric cancer, genomics, next-generation sequencing, liquid biopsy, Guardant360, ERBB2
Key Message
Our study showed that HER2+ GC patients respond differently according to concomitant genomic aberrations beyond ERBB2. High ERBB2 amplification by NGS or cfDNA can be a positive predictor for patient selection, and tumor genomic alterations may change significantly during targeted agent therapy.
Introduction
Human epidermal growth factor 2 (HER2) is the first validated novel target in HER2-overexpressed gastric cancer (GC). In the ToGA trial, it was reported that the greatest overall survival (OS) benefit with trastuzumab was seen in patients who had tumors with high HER2 expression, as determined by immunohistochemistry (IHC) (hazard ratio 0.65; median OS, 16.0 versus 11.8 months) [1]. Lapatinib (Tykerb), a dual EGFR1 and HER2 tyrosine kinase inhibitor (TKI), was tested in the Lapatinib Optimization Study in HER2+ Gastric Cancer (LOGiC) trial as first-line treatment in combination with capecitabine/oxaliplatin (CapeOx) and in the Tykerb with taxol in Asian ErbB2+ GC (TyTAN) trial as combination with paclitaxel in second-line treatment [2, 3]. Unfortunately, both trials failed to demonstrate OS benefit with the addition of lapatinib, with the exception of Asian and younger patients in subgroup analysis of the LOGiC study [2, 4]. Unlike the ToGA trial, the TyTAN and LOGiC trials employed HER2 amplification by fluorescence in situ hybridization (FISH) as the biomarker, regardless of the HER2 IHC results, which may have affected the outcomes of the TyTAN and LOGiC trials due to inclusion of patients without marked HER2 protein expression. The subset of patients in the TyTAN trial who received lapatinib and whose tumors showed high HER2 expression by IHC had improved OS (hazard ratio, 0.59; P = 0.176), similar to the ToGA trial [5]. Hence, overall negative results from LOGiC or TyTAN trials do not preclude potential benefit from lapatinib in an optimally selected HER2+ GC patient population. Nevertheless, it is essential to achieve better understanding through further molecular dissection of HER2+ GC patients: (i) who achieved long-term response to lapatinib, (ii) who achieved initial response but do not sustain durable response, and (iii) who did not respond to lapatinib initially.
GCs are molecularly heterogeneous diseases. As the understanding of the genomic subtypes of GC deepens, it is becoming clear that HER2-overexpressed GC is associated with concomitant genomic alterations. Based on The Cancer Genome Atlas (TCGA) study, concomitant and recurrent focal amplifications were identified in ERBB2+ GC at the CCNE1, CDK6, EGFR, MET, and MYC loci [6]. Previously, our group also reported the status of various concomitant genomic alterations in HER2-overexpressed patients treated with trastuzumab through proteomic and high-through sequencing technologies [7–9]. However, it is not yet clear whether these concomitant genomic alterations influence responsiveness or resistance with respect to HER2-targeted agents in GC.
Herein, we report data from a phase II trial of combination therapy with CapeOx and lapatinib in HER2-overexpressed GC, as first-line therapy. A parallel-biomarker study was conducted by analyzing patient tumor and blood samples, using IHC, tissue-based next-generation sequencing (NGS), and serial plasma-based cell-free DNA (cfDNA) NGS testing.
Patients and methods
Patients
Patients enrolled in this study had measurable, histologically confirmed metastatic and/or recurrent gastric adenocarcinoma that is potentially resectable as defined below. Either the primary or metastatic tumor tissues had to have HER2 considered positive by IHC 3+, or IHC 2+ with ERBB2 gene amplification by silver in situ hybridization (SISH). The trial was conducted in accordance with the Declaration of Helsinki and the Guidelines for Good Clinical Practice (ClinicalTrial.gov.Identifier: NCT# 02015169). The trial protocol was approved by the institutional review board of Samsung Medical Center (Seoul, Korea), and all patients provided written informed consent before enrollment. GlaxoSmithKline (GSK) and Novartis (Seoul, Korea) provided lapatinib but were not involved in patient accrual, data analysis, or manuscript preparation.
To be eligible to participate in this study, patients were required to be at least 18 years old, have at least one measurable lesion according to Response Evaluation Criteria in Solid Tumors (RECIST 1.1), and Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1. Exclusion criteria included any other prior radiotherapy, palliative chemotherapy, or investigational therapies to treat the disease. Adequate hematologic function, hepatic function, and renal function were required. Potentially resectable patients were defined as the following: patients with liver metastases were limited to those with two to five liver metastases. Cases suspicious of peritoneal seeding by imaging without definite evidence of ascites and/or peritoneal enhancement were allowed to enter the study based on investigator’s decision. Isolated metastases were also allowed for study entry.
Study design and treatment
This prospective open-label trial was designed as a single-arm phase II study at an academic cancer center. Enrolled patients received CapeOx (capecitabine 1, 700 mg/m2/day, and oxaliplatin 130 mg/m2) plus lapatinib 1, 250 mg/day. Treatment was administered in 21-day cycles, consisting of intravenous oxaliplatin on day 1 (for up to 8 cycles) and oral capecitabine in two daily doses from day 1 to day 14. Lapatinib was daily administered continuously and treated until up to 8 cycles. In cases where any study drug was discontinued, patients could continue receiving the remaining drugs. When patients achieved partial response (PR) or complete response (CR) during treatment, they were assessed for the possibility of curative resection. If possible, patients were guided to curative resection; otherwise, they were continuously treated for up to 8 cycles.
Assessment
At baseline, the medical history, physical examination, blood tests, urinalysis, electrocardiography, echocardiogram, chest X-ray, and abdomen and pelvis computed-tomography (CT) scan results of the patients were reviewed. Physical examinations, chest X-rays, blood tests were repeated before beginning each cycle of chemotherapy. Tumor responses were evaluated every two cycles according to the RECIST 1.1 criteria. Toxicities were graded based on the National Cancer Institute Common Terminology Criteria for Adverse Events 4.0.
Online only method (supplementary method, available at Annals of Oncology online)
Sample size and statistical analysis
According to single-arm binomial design, a sample size of 32 patients with 10% unassessable rate was needed to accept the hypothesis that the true CR is greater than 20% with 90% power and to reject the hypothesis that the CR is <5% with a type I error rate of 5% [11, 12]. The primary end point of the trial was the CR rate per RECIST 1.1 including both radiologic and pathologic CR. The secondary end points included response rate (RR), disease control rate (DCR), progression-free survival (PFS), OS, safety profile, and exploratory biomarker analysis. The PFS was defined as the time from the start of treatment until the date of disease progression or death resulting from any cause. The OS was measured from the start of treatment to the date of death from any causes. The RR was calculated as the percentage of patients experiencing a confirmed CR or PR, and DCR as RR + stable disease (SD), per the RECIST 1.1 guidelines. Statistical associations between continuous variables were evaluated using Spearman’s correlations and associations between continuous and categorical variables were evaluated using rank-sum statistics.
For biomarker-experiments, comparisons between responder and non-responders were carried out by two-tailed Student’s t tests with P values of <0.05 considered significant.
Results
Patients
Thirty-two patients were enrolled in this study between May 2013 and November 2015. The clinicopathologic characteristics of the 32 patients enrolled in this study are summarized in Table 1. The median age of the patients was 64 years (range 23–80 years), and the majority of the patients were male (81.3%). An 81.3% of the patients had poorly differentiated or moderately differentiated adenocarcinoma and 3.1% of the patients had signet ring cell carcinoma. Two patients had EBV+ tumors and no patients had an MSI-high tumor. Twenty-eight patients had HER2-overexpressed GC with IHC 3+, and four patients (patient #29, 13, 22, 5) were IHC2+ and SISH+.
Table 1.
Baseline characteristics
| N = 32 | % | ||
|---|---|---|---|
| Sex | Male | 26 | 81.3 |
| Female | 6 | 18.8 | |
| Age | Range | 23–80 | |
| Median | 64 | ||
| ECOG PS | 0 | 1 | 3.1 |
| 1 | 31 | 96.9 | |
| Surgery | Total | 20 | 62.5 |
| Curative intent | 14 | 43.8 | |
| TG (total gastrectomy) | 7 | ||
| STG (subtotal gastrectomy) | 6 | ||
| ESD (endoscopic submucosal dissection) | 1 | ||
| Palliative | 4 | 12.5 | |
| TG | 1 | ||
| STG | 1 | ||
| Gastrojejunostomy | 2 | ||
| O and C (open and close) | 2 | 6.3 | |
| Pathologic subtype | Adenocarcinoma | 28 | 87.5 |
| Papillary adenocarcinoma | 3 | 9.4 | |
| Signet ring cell | 1 | 3.1 | |
| Differentiation | W/D (well differentiated) | 1 | 3.1 |
| M/D (modestly differentiated) | 16 | 46.9 | |
| P/D (poorly differentiated) | 13 | 34.4 | |
| Unknown | 2 | 6.3 | |
| Lauren | Diffuse | 3 | 9.4 |
| Intestinal | 6 | 18.8 | |
| Mixed | 1 | 3.1 | |
| Not determined | 22 | 68.8 | |
| EBV | |||
| Positive | 2 | 6.3 | |
| Negative | 18 | 56.3 | |
| Not determined | 12 | 37.5 | |
| MSS | |||
| MSS | 29 | 90.6 | |
| MSI-high | 0 | – | |
| Not determined | 3 | 9.4 | |
| HER2 overexpression | |||
| 2+* | 4 | 12.5 | |
| 3+ | 28 | 87.5 | |
| Prior palliative chemotherapy | 0 | 0.0 |
Drug efficacy
The cut-off date for outcome analysis was 1 April 2017, and the median follow-up time was 22.9 months. Response evaluations were available for 29 of all enrolled patients. CR was achieved in seven patients (21.8%; 95% CI 7.5–36.1) including two pathologic CRs; thus, the CR rate met the primary end point of the study. Fifteen PRs (46.8%) and four patients with stable disease were observed, revealing an overall RR of 68.6% (95% CI 49.9–83.8) and DCR of 81.3 (95% CI 63.6–92.8). Among 22 patients with CR or PR, 2 patients with CR received R0 resection with complete removal of tumor, 1 patient underwent radiofrequency ablation of liver metastasis and 2 patients refused surgery for resectable lesion following chemotherapy. Pathologic CR was confirmed in postoperative specimens from these 2 patients. The median PFS was 9.0 months (95% CI 4.4–13.6 months) and the median OS was 14.2 months (95% CI 12.3–16.1 months). The waterfall plot and swimmer’s plot of responders are provided in Figure 1A and B. Of note, all 7 patients with CR had durable responses ranging from 6.2 to 45.9 months. Of 14 PR patients, 9 patients experience >50% reduction in tumor burden per RECIST 1.1.
Figure 1.

(A) Waterfall plot; (B) swimmer plot; (C) H index (upper panel), discordant HER2 IHC between primary and metastatic tissue specimen (lower panel); (D) H index in correlation to % tumor reduction.
Patient inter-tumor heterogeneity, heterogeneity of HER2 staining, H-score and response to lapatinib
Of 32 patients, we were able to concurrently obtain 10 primary-metastasis paired samples. Of 10 paired samples, 6 patients had concordant HER2 results and 4 patients had discordant HER2 positivity between primary and metastasis (Figure 1C and Table 2). Of 6 concordant patients, 4 achieved PR, 1 CR and 1 PD (Table 2). All of the concordant patients had synchronous metastases. Of discordant patients, patient #22 had gastric HER2 2+ but synchronous SCN LN HER2 0 and had progressive disease after 1 cycle. Patients #13 and #3 had HER2 negative primary gastric tumors but at recurrence 2–3 years post-gastrectomy, had HER2+ liver metastases: #13, with heterogeneous HER2 2+ liver metastasis, had de novo resistance but #3, with homogeneous HER2 3+ liver metastasis, achieved PR with >80% tumor reduction. Of 32 patients, we were able to analyze 29 baseline tumor specimens for HER2 heterogeneity staining, H-score, and correlated these variables with response to lapatinib. Of 7 patients with PD and SD, 5 patients demonstrated heterogeneity in HER2 staining and 2 patients had homogeneous HER2 staining. In contrast, of 7 patients with CR, 5 patients had homogeneous HER2 staining in their tumor. All but two patients (metachronous liver metastases specimen) were based on HER2 staining in primary gastric tumor. The % tumor reduction by CapeOx/lapatinib was significantly correlated with high H-score (Figure 1D) with a cut-off of 200.
Table 2.
Concomitant genomic alterations
| Pt# | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 10 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | 22 | 23 | 24 | 25 | 26 | 27 | 28 | 29 | 30 | 31 | 32 |
| Response | CR | CR | PR | PR | SD | CR | CR | CR | PR | PD | PR | PR | PR | PR | PR | PR | SD | PD | PD | PR | PR | CR | CR | PR | SD | PR | PR | SD | PR |
| HER2 IHC | 3+ | 3+ | 2+ | 3+ | 2+ | 3+ | 3+ | 3+ | 3+ | 3+ | 2+ | 3+ | 3+ | 3+ | 3+ | 3+ | 3+ | 3+ | 2+ | 3+ | 3+ | 3+ | 3+ | 3+ | 3+ | 2+ | 3+ | 3+ | 3+ |
| Site of HER2 test | G | G | G | G | G | G | G | G | G | G | M | G | G | G | G | G | G | G | G | G | G | G | G | G | G | G | G | G | G |
| Tissue type | E | E | E | S | E | S | S | E | E | S | Bx | E | E | S | E | E | E | S | E | E | E | E | E | E | E | E | E | E | E |
| Heterogeneity of HER2 | HO | HO | HO | HE | HE | HO | HE | HO | HE | HE | HO | HO | HO | HO | HO | HO | HO | HO | HO | HO | HO | HE | HO | HO | HE | HO | HE | HE | HE |
| Primary-Meta (or Recurrent) matched biopsy | NA | + | + | + | NA | + | + | + | + | NA | + | NA | NA | NA | + | + | NA | NA | + | NA | + | NA | + | NA | NA | NA | + | NA | NA |
| Discrepancy between primary and metastasis | NA | NA | G (0) L (3+) | NA | NA | G (3+) L (3+) | NA | NA | G (3+) S (0) | NA | G (0) L (2+) | NA | NA | NA | G (3+) S (3+) | G (3+) L (3+) | NA | NA | G (2+) LN (0) | NA | G (3+) Sk (3+) | NA | G (3+) L (3+) | NA | NA | NA | G (3+) LN (3+) | NA | NA |
| FU biopsy during lapatinib | NA | 3+ 0 | 2+ 2+ | 3+ 3+ | NA | NA | 3+ 0 | 3+ 0 | 3+ 3+ | NA | NA | NA | NA | NA | NA | 3+ 3+ | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| H-index | 300 | 270 | 300 | 100 | 55 | 300 | 200 | 280 | 80 | 190 | 240 | 270 | 290 | 300 | 250 | 290 | 100 | 30 | 180 | 270 | 300 | 200 | 250 | 270 | 80 | 180 | 70 | 60 | 210 |
G, gastric; M, metastasis; E, endoscopic biopsy; S, surgical biopsy; HO, homogeneous; HE, heterogeneous; NA, not available.
Concomitant genomic alteration in HER2-overexpressed GC
Among 32 enrolled patients, 16 patients had sufficient tissue for NGS. The majority of patients’ tumors (86.7%) harbored TP53 mutations. In addition, 10 (62.5%) had at least 1 concomitant genomic alteration besides TP53 mutation (Table 3). The most common co-occurring copy number alteration was CCNE1 amplification, which was present in 40% of HER2+ tumors. Interestingly, patients whose tumors harbored CCNE1 amplification were less likely to respond to HER2-targeted therapy, compared with patients without CCNE1 amplification (66.7% of non-responders versus 22.2% of responders harbored CCNE1 amplification; P = 0.08), suggesting that CCNE1 amplification may be a negative predictor of response to HER2-directed therapy (Figure 2A). In contrast, patients with high level ERBB2 amplification were more likely to respond to therapy, compared with patients with low level ERBB2 amplification (P = 0.02; Figure 2B). Additionally, there was a trend towards CCNE1 amplified tumors having lower HER2 log2 ratio, compared with the CCNE1 non-amplified tumors (P = 0.055; supplementary Figure S1, available at Annals of Oncology online). Of the patients who experienced a PR to therapy, two were found to have low level ERBB2 amplification based on NGS. However, the ERBB2 log2 ratio of these two cases was most likely artificially low due to low tumor purity, as evidenced by the low TP53 allele fraction of 8.4% and 4.9%, respectively. Based on the ERBB2 log2 ratio, we estimated the ERBB2 copy number for each case. The mean ERBB2 copy number of patients who responded to HER2 directed therapy was 24.17, while the ERBB2 copy number of non-responders was 3.3 (supplementary Figure S2, available at Annals of Oncology online). We also found a high correlation between ERBB2 log2 ratio and HER2 IHC H-index (R2 = 0.378; Figure 2C).
Table 3.
Concomitant genomic alterations
| Pt# | Best response | CCNE1 IHC | ERBB2 log2 | ERBB2 estimated copy number | H-index | Heterogeneity of HER2 staining | TP53 allele fraction | Other alterations |
|---|---|---|---|---|---|---|---|---|
| 004 | PD | CCNE1 negative | −0.0333 | 1.95 | 100 | Hetero | 0.62963 | NOTCH3 amp, BRD4 amp, TERT loss, APC frameshift |
| 005 | SD | CCNE1 positive (100%) | 0.428 | 2.69 | 55 | Hetero | 0.2962 | CCNE1 amp, |
| MYC amp, APC loss, TP53 loss, ARID1A | ||||||||
| 006 | CR | NA | 0.281 | 2.43 | 300 | Homo | 0.049 | ERBB3mutation, ARID1B inframe del |
| 010 | PR | CCNE1 negative | – | 80 | Hetero | – | BCL9 frameshift | |
| 012 | PD | CCNE1 positive (100%) | 0.679 | 3.20 | 190 | Hetero | 0.2213 | PTEN loss, NOTCH2 missense |
| CCNE1 amp | ||||||||
| 013 | PD | CCNE1 positive (100%) | 0.12 | 2.17 | Hetero | 0.2 | CCNE1 amp | |
| 014 | PR | CCNE1 negative | 2.22 | 9.32 | 270 | Homo | 0.54 | loss, APC nonsense, ARID1A |
| nonsense, CTNNB1 missense, RHOA | ||||||||
| 015 | PR | CCNE1 negative | 4.31 | 39.7 | 290 | Homo | – | – |
| 016 | PR | CCNE1 negative | 3.74 | 26.72 | 300 | Homo | 0.409 | SMAD2/4, |
| BCL2, KEAP1 loss, RNF43 mut, | ||||||||
| CDK8 mut | ||||||||
| 017 | PR | CCNE1 positive (100%) | 0.307 | 2.47 | 250 | Homo | 0.084 | FGFR2 amp, IDH2 amp, |
| PIK3CG missense | ||||||||
| 018 | PR | NA | 2.37 | 10.34 | 290 | Homo | 0.3259 | CCNE1 amp, |
| VEGFA amp, CDK1NA amp, CCND3 amp | ||||||||
| 019 | SD | CCNE1 positive (80%) | 1.69 | 6.45 | 100 | Homo | 0.1185 | CCNE1 amp, |
| EGFR amp, SMAD3/4 loss | ||||||||
| 021 | N.E. | CCNE1 negative | 2.16 | 8.94 | 300 | Homo | 0.3789 | CDKN2A loss |
| 022 | PD | CCNE1 negative | – | 180 | Homo | NA | CRKL amp | |
| 023 | PR | CCNE1 negative | 5.02 | 64.89 | 270 | Homo | 0.4375 | ETV4 amp, |
| CCND3 frameshift | ||||||||
| 024 | PR | CCNE1 negative | 4.90 | 59.71 | 300 | Homo | 0.4262 | CRKL amp, |
| APC nonsense |
Figure 2.
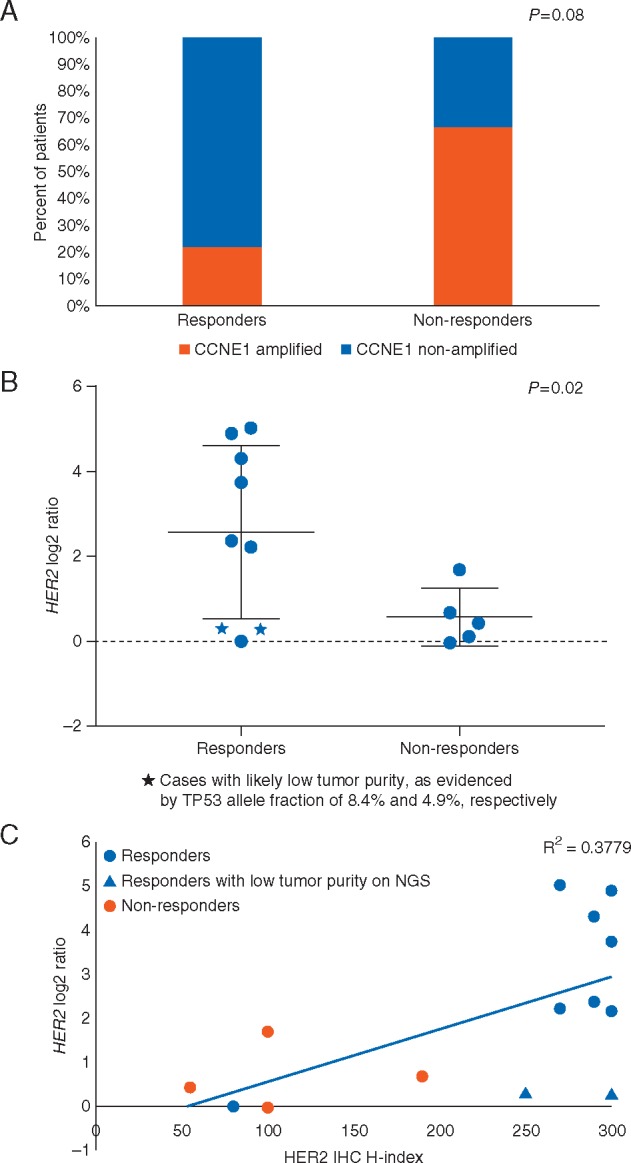
(A) CCNE1 amplification by NGS between responders and non-responders; (B) HER2 log ratio by NGS between responder and non-responders; (C) correlation between HER2 log ratio by NGS and HER2 IHC H-index.
One patient, a 25-year old male with no family history of GC was diagnosed of metastatic, ERBB2 amplified and CRLK amplified GC. IHC showed HER2 2+ and HER2 SISH (4.9 copies by SISH) in primary gastric tumor. The patient rapidly progressed after one cycle of CapeOx/lapatinib to brain and abdomen (Figure 3A). The patient failed to subsequent second- and third-line chemotherapy and died of disease. In contrast, another patient whose cancer harbored both ERBB2 and CRLK amplifications achieved PR. However, that patient’s tumor had a high level amplification of ERBB2 (log2 ratio 4.9) and low level gain of CRLK (log2 ratio 2.05).
Figure 3.

(A) ERBB2 and CRKL co-amplified patient; (B) expression of HER2 and CRKL by IHC in tumor specimen; (C) short-hairpin RNA (shRNA)-mediated CRKL knockdown in HER2-amplified PDCs effectively suppressed CRKL expression in CRKL-amplified/overexpressed PDCs. The CRKL knockdown in an HER2-amplified PDC line rescued lapatinib resistance in cell-viability assays with statistical significance (P = 0.0003). The inhibition of both HER2 and CRKL with lapatinib and shCRKL, respectively, demonstrated substantial ERK and AKT downregulation by western blotting.
CRKL co-amplification drives de novo lapatinib resistance (supplementary data, available at Annals of Oncology online)
Changes of HER2 status in tumor upon disease progression
Post-progression biopsies were available from the primary tumor sites of seven patients who were treated by CapeOx/lapatinib. IHC testing for HER2 was carried out for post-progression specimens from these 7 patients. Four patients (57%) had tumors that showed persistent HER2 overexpression at the time of progression following CapeOx/lapatinib chemotherapy (Table 2), whereas 3 (43%) patients converted to an HER2-negative tumor after progression. Hence, 43% of the patients showed changes in HER2 positivity after receiving targeted agents underscoring the importance of rebiopsy at progression, especially after failure to targeted agents in line with previous study [13].
Plasma ERBB2 status as a predictor and molecular correlate of response based on cfDNA NGS
Several cases illustrate a correlative response of plasma ERBB2 copy number assessment and response or progression on treatment (Figure 4A and B). Patient #8 was diagnosed with HER2+ GC with several liver metastases. At baseline, this patient had ERBB2 amplification (orange), PIK3CA E545K mutation, NRAS G12S mutation and TP53 R273C mutation with somatic alteration burden (highest variant allele fraction at a given time point) of 27.8% (Pt#8). After eight cycles of CapeOx/lapatinib, somatic alteration burden decreased to 0.1% when the patient achieved near CR (PR per RECIST1.1). The patient received surgery after eight cycles of CapeOx/lapatinib. The patient’s cancer recurred as a soft tissue mass around the celiac axis and cfDNA genomic profiling at that time showed re-emergence of PIK3CA E545K and TP53 R273C mutation but no ERBB2 amplification. For Pt#17, ERBB2 amplification level decreased when the patient responded to CapeOx/laptinib. After six cycles, patient developed peritoneal seeding with ascites and follow-up cfDNA demonstrated newly emerged FGFR2 amplification and CCNE1 amplification. When pt#23 progressed to lapatinib, there was newly emerged EGFR amplification in addition to ERBB2 amplification. In Pt#24, patient progressed after seven cycles of CapeOx/lapatinib and ERBB2 amplification level increased upon progression. Pt#29 demonstrated multiple cfDNA genomic aberration such as ERBB2 amplification, TP53 R175H mutation, MYC amplification. At the time of response, somatic alteration burden decreased from 34.2% to 0.1% and achieved CR. After long-term maintenance response to lapatinib, patient developed recurrence to peritoneal seeding when cfDNA demonstrated no ERBB2 amplification but MYC amplification, TP53 R175H mutation and newly emerged MET amplification. This patient had rapidly progressive disease on subsequent chemotherapy. Pt#32 showed ERBB2 amplification, TP53A144P mutation, and TP53R196Q mutation at baseline. The patient achieved PR after eight cycles of CapeOx/lapatinib. After prolonged duration of response, patient progressed to liver and primary tumor and cfDNA demonstrated newly emerged MYC amplification, SMAD4 R361H mutation and FGFR1 R54C mutation. The patient failed to all of subsequent salvage chemotherapy which correlated well with increase in somatic alteration burden by cfDNA. Interestingly, both Pt#31 and Pt#19 did not have ERBB2 amplification detected by cfDNA and both demonstrated progression to CapeOX chemotherapy.
Figure 4.



Correlation between ctDNA ERBB2 amplification and lapatinib response (A) Waterfall plot for subset of patients with ctDNA assessment at baseline; (B) CtDNA follow-up in lapatinib treated GC patients in correlation to radiologic assessment.
Discussion
HER2-directed therapy, such as trastuzumab, has conferred improved survival in patients with HER2+ GC; thus, is currently considered the standard approach [1]. Nevertheless, not all patients with HER2+ GC benefit from this approach due to innate or acquired resistance. To date, many studies designed to identify optimal candidates for anti-HER2 therapy, such as an appropriate HER2 evaluation, have been conducted. However, these strategies are not sufficient to enable precision medicine for HER2-directed GC therapy. In the analysis of genomic subtyping of GC in TCGA [6] and ACRG [14], HER2+ tumors were found to be interlinked with other complex genomic alterations. Previously, our NGS analysis of trastuzumab-treated HER2+ GC patients revealed similar findings [8]. However, to date, it remains unclear which kinds of concomitant genomic alterations in HER2+ GC can affect the outcomes of HER2-directed therapy. In addition, recent studies have strongly suggested that high copy number detected by cfDNA can be predictive to targeted agents such as MET inhibitor or FGFR inhibitor in GC [15, 16]. Thus, we conducted this NGS-based biomarker study with matched tumor and blood samples as part of this phase II clinical trial.
First, by performing IHC and NGS, we comprehensively interrogated the existence of concomitant genomic alterations in HER2-overexpressed GC patients treated with lapatinib. TCGA study reported concomitant and recurrent amplifications of CCNE1, CDK6, EGFR, MET, and MYC in HER2+ GC. Results from our previous study also showed that five genes (CCNE1, PIK3CA, KRAS, CDK4, and CDK6) were concomitantly co-amplified and that some genes such as TP53, CDKN2A, KRAS, KIT, and PIK3CA were concomitantly co-mutated in HER2+ GC [8]. Data generated in this study revealed several examples in patients where concomitant genes were co-amplified (CCNE1, SRC, FGFR2, VEGFA, MYC, CRKL) or co-mutated (TP53, ARID1A, APC, ERBB3). Specifically, almost all patients had TP53 mutations and 62.5% of patients had two or more concomitant genomic alterations besides HER2 overexpression. Despite the small number of cases and diverse genomic profiles of the tumors, there was a correlation between CCNE1 amplification and lack of response to HER2-directed therapy, which trended towards significance. Even though CCNE1 amplification has been shown to promote resistance to HER2-targeted therapy in breast cancer, there are only preclinical data and anecdotal clinical evidence that CCNE1 amplification may play a role in resistance to HER2-targeted therapy in GC [9]. Our study is the first to demonstrate its potential role as a biomarker of resistance in a larger series of patients with ERBB2 amplified GC. Furthermore, in concordance with previous studies including the ToGA and TRIO-013/LOGiC trial [3–5, 17], we found that high-level HER2 amplification was associated with response to HER2-directed therapy. This study revealed the significant correlation between estimated ERBB2 copy number based on ERBB2 log2 and the treatment response (P = 0.037). Unfortunately, estimated ERBB2 copy number data were only one of the seven patients with CR. Thus, the relation between CR and the level of estimated ERBB2 copy number must be further investigated. ERBB2 amplification by NGS was strongly correlated with HER2 protein expression by IHC. The level of ERBB2 amplification was the strongest predictor of response to lapatinib-containing therapy. However, according to the PFS in patients with the same responses to lapatinib, concomitant genomic profiles were distributed very differently. These findings suggested that additional concomitant genomic alterations beyond HER2 overexpression might affect the response and the response duration for HER2-directed therapy in HER2+ GC. Due to the diverse biological networks associated with the altered genes and the complexity of mechanisms underlying HER2 signaling, it is difficult to discover a single indicator to predict the sensitivity to HER2-directed therapy beyond HER2 overexpression.
Second, based on high plasma-to-tissue concordance for key driver alterations in an earlier prospective validation study conducted by our group, we concluded that plasma-based NGS (Guardant360) could serve as a surrogate for tissue-based sequencing of solid tumor driver mutations [10]. We extended our validation of the same cfDNA NGS assay by validating plasma-detected ERBB2 and MET copy number amplifications (CNAs) as robust predictors of treatment response in the NEXT-2 study, although the sample size was modest (ClinicalTrial.gov.Identifier: NCT#02140463). In the current study, we again find plasma-based ERBB2 amplification as a robust predictor of response to lapatinib plus chemotherapy in line with previous studies on MET and FGFR2 inhibitor in GC [15, 16]. As an exploratory analysis, peripheral blood for plasma cfDNA analysis (Guardant360®) was collected from 9 of the 32 tissue HER2+ enrolled patients, before initiating treatment and throughout treatment, at every CT evaluation. One pre-treatment sample had insufficient DNA for analysis (no ctDNA detected), and the other eight samples were adequate. Six of the eight had plasma ERBB2 amplification with 100% RR (95% CI 54–100), including one CR (Figure 4A). There was no correlation between plasma ERBB2 copy number and depth of response (P-value 0.46). In the two of eight adequate samples where no ERBB2 amplification was detected, both had stable disease. Progression-free survival for the six plasma ERBB2 amplified cases was median 9.0 months (95% CI 1.8–16.2).
Third, we found that serial cfDNA sequencing demonstrated tumor evolution and change in genomic profile through lapatinib treatment of the first time (Figure 4). Cell-free DNA NGS may mitigate needle biopsy sampling errors related to intra- or inter-tumor heterogeneity, particularly at progression after treatment pressure has driven emergence of multiple acquired resistance mutations and multiple subclones. For these reasons, sequencing of circulating cfDNA has been suggested as a reasonable alternative to tumor tissue-based genomic testing in NSCLC [18]. Advanced GC may be an ideal candidate for cfDNA NGS in the first line, even before treatment-driven multiclonality, because of the relatively extreme heterogeneity of HER2 status within and between tumors found in this study and others [19]. Significant inter-tumor heterogeneity in GC has also been reported for MET and FGFR amplifications [15, 16, 20]. In this study, through serial cfDNA sequencing, we affirmed the association of changes in plasma-detected ERBB2 copy number with tumor responses to lapatinib plus CapeOx. This finding was consistent with our previous analysis of samples from colorectal cancer patients with an anti-EGFR monoclonal antibody [10]. Interestingly, we found that patients with detectable cfDNA ERBB2 CNA achieved response while the two patients with other cfDNA alterations detected but ERBB2 CNA not detected, achieved stable disease as best response. This finding raises the question as to whether presence or absence of ERBB2 CNA may be more predictive of response than tissue HER2 status but needs to be validated in a larger dataset. Overall, our data suggested the potential utility of analyzing cfDNA as an alternative genomic test in gastrointestinal cancers at baseline and recurrence, reducing the need for repeated tumor biopsies and to identify mechanisms of drug resistance.
CRKL overexpression/amplification is known as a key mechanism conferring resistance to EGFR inhibitors in EGFR-mutant non-small cell lung cancer. We found one patient with co-amplified CRKL through genomic analysis for the enrolled patients. This patient showed de novo resistance to CapeOX plus lapatinib. We confirmed the overexpression of CRKL using IHC in both tumor tissue and the matching PDC line established in this study. By performing MTT assays with a CRKL co-amplified PDC line and a shRNA-mediated CRKL-knockdown PDC line, we demonstrated the inhibitory effect of CRKL amplification/overexpression on HER2-directed therapy against HER2+ GC. However, this finding of CRKL was found in only one case. Hence we need to concretize the role of CRKL in ERBB2 direct therapies in further study.
This study has several limitations. It is a single-arm phase II trial, with limited sample size, patient selection, heterogeneous disease, and possible enrollment bias. Furthermore, this study included exploratory analyses for many biomarkers with diverse methods. This multiplicity and/or complexity of analyses with the limited sample size might influence the outcomes. Hence, the finding of the exploratory biomarker-analyses cannot be entirely conclusive. Nevertheless, the treatment regimen (lapatinib plus CapeOx) showed moderate activity and an acceptable toxicity profile as a first-line treatment in tissue HER2-overexpressed (IHC 3+ or IHC2+ and FISH+) as well as plasma ERBB2 copy number amplified GC patients. Moreover, our parallel biomarker study revealed that HER2-overexpressed GC patients had a diverse pattern of concomitant genomic alterations. Serial genomic analysis using cfDNA should also help predict responses to treatment and to understand the mechanism of treatment resistance. Our study showed that HER2+ GC patients may respond differently according to concomitant genomic aberrations beyond HER2, high ERBB2 amplification by NGS or cfDNA can be a positive predictor for patient selection, and tumor genomic alterations change significantly during targeted agent therapy. Based on our findings, we recommend that GC patients with high ERBB2 copy number detected either by NGS or cfDNA may benefit from lapatinib and this should be further validated in larger scale clinical trials using these biomarkers as selection criteria.
Funding
This work was supported by funding from the Korean Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (HI14C3418, HI16C1990). Support was also provided by a grant from the 20 by 20 Project of Samsung Medical Center (GF01140111). This investigator-initiated trial was also funded by a study-drug donation from Novartis.
Disclosure
None declared except for KB, RL, and AT who are employees of Guardant Health, USA.
References
- 1. Bang YJ, Van Cutsem E, Feyereislova A. et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet 2010; 376(9742): 687–697. [DOI] [PubMed] [Google Scholar]
- 2. Satoh T, Xu RH, Chung HC. et al. Lapatinib plus paclitaxel versus paclitaxel alone in the second-line treatment of HER2-amplified advanced gastric cancer in Asian populations: tyTAN–a randomized, phase III study. JCO 2014; 32(19): 2039–2049. [DOI] [PubMed] [Google Scholar]
- 3. Press MF, Ellis CE, Gagnon RC. et al. HER2 status in advanced or metastatic gastric, esophageal, or gastroesophageal adenocarcinoma for entry to the TRIO-013/LOGiC trial of lapatinib. Mol Cancer Ther 2017; 16(1): 228–238. [DOI] [PubMed] [Google Scholar]
- 4. Hecht JR, Bang YJ, Qin SK. et al. Lapatinib in combination with capecitabine plus oxaliplatin in human epidermal growth factor receptor 2-positive advanced or metastatic gastric, esophageal, or gastroesophageal adenocarcinoma: tRIO-013/LOGiC—a randomized phase III trial. JCO 2016; 34(5): 443–451. [DOI] [PubMed] [Google Scholar]
- 5. Kwak EL, Bang YJ, Camidge DR. et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med 2010; 363(18): 1693–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bass AJ, Thorsson V, Shmulevich I. et al. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014; 513(7517): 202–209., [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lee J, Kim S, Kim P. et al. A novel proteomics-based clinical diagnostics technology identifies heterogeneity in activated signaling pathways in gastric cancers. PLoS One 2013; 8(1): e54644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lee JY, Hong M, Kim ST. et al. The impact of concomitant genomic alterations on treatment outcome for trastuzumab therapy in HER2-positive gastric cancer. Sci Rep. 2015; 5(1): 9289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kim J, Fox C, Peng S. et al. Preexisting oncogenic events impact trastuzumab sensitivity in ERBB2-amplified gastroesophageal adenocarcinoma. J Clin Invest 2014; 124(12): 5145–5158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kim ST, Lee WS, Lanman RB. et al. Prospective blinded study of somatic mutation detection in cell-free DNA utilizing a targeted 54-gene next generation sequencing panel in metastatic solid tumor patients. Oncotarget 2015; 6(37): 40360–40369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kang YK, Kang WK, Shin DB. et al. Capecitabine/cisplatin versus 5-fluorouracil/cisplatin as first-line therapy in patients with advanced gastric cancer: a randomised phase III noninferiority trial. Ann Oncol 2009; 20(4): 666–673. [DOI] [PubMed] [Google Scholar]
- 12. Cunningham D, Starling N, Rao S. et al. Capecitabine and oxaliplatin for advanced esophagogastric cancer. N Engl J Med 2008; 358(1): 36–46. [DOI] [PubMed] [Google Scholar]
- 13. Park SR, Park YS, Ryu MH. et al. Extra-gain of HER2-positive cases through HER2 reassessment in primary and metastatic sites in advanced gastric cancer with initially HER2-negative primary tumours: results of GASTric cancer HER2 reassessment study 1 (GASTHER1). Eur J Cancer 2016; 53: 42–50. [DOI] [PubMed] [Google Scholar]
- 14. Cristescu R, Lee J, Nebozhyn M. et al. Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat Med 2015; 21(5): 449–456. [DOI] [PubMed] [Google Scholar]
- 15. Pearson A, Smyth E, Babina IS. et al. High-level clonal FGFR amplification and response to FGFR inhibition in a translational clinical trial. Cancer Discov 2016; 6(8): 838–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kwak EL, Ahronian LG, Siravegna G. et al. Molecular heterogeneity and receptor coamplification drive resistance to targeted therapy in MET-amplified esophagogastric cancer. Cancer Discov 2015; 5(12): 1271–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gomez-Martin C, Plaza JC, Pazo-Cid R. et al. Level of HER2 gene amplification predicts response and overall survival in HER2-positive advanced gastric cancer treated with trastuzumab. JCO 2013; 31(35): 4445–4452. [DOI] [PubMed] [Google Scholar]
- 18. Ettinger DS, Wood DE, Aisner DL. et al. Non-Small Cell Lung Cancer Version 42017. NCCN Non-Small Cell Lung Cancer Guidelines (Version4.2017) 2017; http://www.nccn.org/professionals/physician_gls/pdf/nscl.pdf (4 March 2017, date last accessed).
- 19. Fusco N, Bosari S.. HER2 aberrations and heterogeneity in cancers of the digestive system: implications for pathologists and gastroenterologists. World J Gastroenterol 2016; 22(35): 7926–7937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Van Cutsem E, Bang YJ, Mansoor W. et al. A randomized, open-label study of the efficacy and safety of AZD4547 monotherapy versus paclitaxel for the treatment of advanced gastric adenocarcinoma with FGFR2 polysomy or gene amplification. Ann Oncol 2017; 28(6): 1316–1324. [DOI] [PubMed] [Google Scholar]