

Abstract
The evolution of asexual organisms is driven not only by the inheritance of genetic modification but also by the acquisition of foreign DNA. The contribution of vertical and horizontal processes to genome evolution depends on their rates per year and is quantified by the ratio of recombination to mutation. These rates have been estimated for bacteria; however, no estimates have been reported for phages. Here, we delineate the contribution of mutation and recombination to dsDNA phage genome evolution. We analyzed 34 isolates of the 936 group of Siphoviridae phages using a Lactococcus lactis strain from a single dairy over 29 years. We estimate a constant substitution rate of 1.9 × 10−4 substitutions per site per year due to mutation that is within the range of estimates for eukaryotic RNA and DNA viruses. The reconstruction of recombination events reveals a constant rate of five recombination events per year and 4.5 × 10−3 nucleotide alterations due to recombination per site per year. Thus, the recombination rate exceeds the substitution rate, resulting in a relative effect of recombination to mutation (r/m) of ∼24 that is homogenous over time. Especially in the early transcriptional region, we detect frequent gene loss and regain due to recombination with phages of the 936 group, demonstrating the role of the 936 group pangenome as a reservoir of genetic variation. The observed substitution rate homogeneity conforms to the neutral theory of evolution; hence, the neutral theory can be applied to phage genome evolution and also to genetic variation brought about by recombination.
Keywords: phage evolution, substitution rate, recombination rate, bacteriophage
Introduction
Viruses that infect bacteria—termed bacteriophages—are ubiquitous in nature (Cobián Güemes et al. 2016). Phage interaction with bacteria occurs by two different routes; in the lytic cycle, phages replicate and lyse the host, while in the lysogenic cycle, phage DNA is integrated into the host chromosome and is replicated with the host genome. Differential phage predation modulates bacterial population structure (Bouvier and Del Giorgio 2007), while phage-mediated gene transfer can facilitate bacterial adaptation to specific habitats or lifestyles (Waldor and Mekalanos 1996; Coleman et al. 2006). Thus, phage interaction with bacteria is a major contributor to bacterial evolution (Pal et al. 2007) and ecology, for example, bacterial cell lysis during phage infection impacts marine biogeochemical cycles (Suttle 2007; Jover et al. 2014). Furthermore, the impact of phage predation on the biodiversity of bacterial populations is of utmost importance in biotechnological applications such as dairy fermentation (Samson and Moineau 2013).
Phage–bacteria interaction is characterized by antagonistic coevolution where the rate of evolution of both partners has direct consequences for bacterial resilience and phage virulence (Gomez and Buckling 2011; Schwartz and Lindell 2017). The rate of mutation as estimated by fluctuation experiments ranges between ∼10−10 mutations per nucleotide per replication in bacteria, and a rate of 1–8 × 10−7 mutations per nucleotide per infection for dsDNA bacteriophages (Sanjuán et al. 2010). The rate of nucleotide substitution measures the number of mutations that persist in the population over time. It is typically estimated by calibrating the number of substitutions with dated fossils (Thorne and Kishino 2002) or with heterochronous samples from a measurably evolving population (Drummond et al. 2003). Current estimates of the substitution rate per site per year range between 10−8 and 10−5 in bacteria and have been reported as ∼10−5 in eukaryotic dsDNA viruses and ∼10−3 in eukaryotic RNA viruses, respectively (Biek et al. 2015). There is generally a strong temporal signal in bacterial genomes (Duchêne et al. 2016). Variation of the substitution rate over time can be attributed to epidemics resulting in shorter generation times in Yersinia pestis (Cui et al. 2013). An assessment of temporal signal and estimates for substitution rates in phages are currently lacking.
Genome evolution of both phages and bacteria includes the acquisition of genetic material via genetic recombination, that is, DNA acquisition within the lineage. Genetic recombination occurs on two different scales. On the micro-scale, replacing recombination has the potential to alter multiple nucleotides in a single event (termed here recombination). On the macro-scale, genetic recombination can result in the acquisition or the deletion of genes and leads to gene content variation over time (termed here gene gain and loss). Phages encode various proteins that facilitate recombination and footprints of genetic recombination have been observed in phage genomes (Martinsohn et al. 2008; Szczepańska 2009). Recombination between phages occurs mainly through coinfection, which has been shown to be prevalent in bacterial populations (Flores et al. 2011; Roux et al. 2014; Díaz-Muñoz 2017). During coinfection, temperate phages can recombine with prophages in the host genome (De Paepe et al. 2014), whereas lytic phages can recombine with other lytic types (Shcherbakov et al. 1992) or with prophages (Bouchard and Moineau 2000). The high gene content variation observed in natural phage populations suggests that gene gain and loss is frequent (Hendrix et al. 1999). Nonetheless, the rates of recombination and of gene gain and loss in phage genome evolution are yet unknown.
The dairy environment is characterized by the presence of multiple phage types for Lactococcus lactis (Mahony et al. 2012), with dsDNA Siphoviridae phages of the 936 group as the most prevalent type (Murphy et al. 2016). Industrial dairy fermentation is based on actively growing starter cultures of lactic acid bacteria in the production vessels (Parente et al. 2017). This industrial dairy environment implements numerous phage control measures on a regular basis, for example, sanitation and disinfection, selection of starter cultures with numerous phage defense systems (Hayes et al. 2017). Nevertheless, lytic phages that are capable of infecting actively growing starter culture strains in the production vessels persist in the dairy environment (Parente et al. 2017). Thus, phage evolution is supposed to occur in dairy production due to permanent phage–host interactions (Mahony et al. 2017). Notably, it has recently been shown by qPCR methodology that these undefined starter cultures may also contain bacteriophages (Muhammed et al. 2017).
Here, we study genome evolution of one lineage of strictly lytic phages that we sampled repeatedly from a dairy production line over 29 years. The availability of heterochronous phage isolates supplies a unique opportunity to estimate the rates of the different processes during phage genome evolution.
Results
Lactococcus lactis Phage Genomes
Bacteriophages were sampled from a single German dairy that used one distinct, undefined, multiple-strain starter culture over three decades (termed here culture A, 1985–2014, fig. 1). The phage-sensitive L. lactis subsp. cremoris strain CA-49 was isolated from culture A and used for isolation and propagation of 34 lytic phages (termed here culture A phages). We confirmed by electron microscopy that all phages belong to the group of strictly lytic 936 Siphoviridae phages (supplementary fig. S1, Supplementary Material online). All phages were sequenced, yielding genomes of length between 29,097 and 31,049 base pairs (bp) (average 30,099 bp), that encode between 51 and 56 protein-coding sequences (CDSs) (table 1). The inferred protein sequences were clustered into 74 homologous protein families that comprise 40 core families and 34 variable families (supplementary table S1, Supplementary Material online).
Fig. 1.

Timeline of culture A phage isolates. Bar height is the number of samples taken in each quarter. Electron micrographs of representative isolates are shown (see also table 1, supplementary fig. S1, Supplementary Material online).
Table 1.
Data Set Summary.
| Phage | Isolation Time | Reads | Genome Length | cos Site | %GC | Number of CDSs | % Coding | Accession |
|---|---|---|---|---|---|---|---|---|
| LP8511 | November 1985 | 272,016 | 29,953 | CACAAAGGACT | 35.11 | 52 | 91.29 | MF775669 |
| LP9104 | April 1991 | 291,287 | 29,157 | CACAAAGGACT | 35.15 | 52 | 91.16 | MF775670 |
| LP9205a | May 1992 | 295,658 | 29,098 | CACAAAGGACT | 34.94 | 51 | 90.57 | MF775671 |
| LP9205b | May 1992 | 278,048 | 30,296 | CACAAAGGACT | 34.82 | 53 | 91.20 | MF775672 |
| LP9206a | June 1992 | 272,071 | 29,097 | CACAAAGGACT | 34.95 | 52 | 91.15 | MF775673 |
| LP9206b | June 1992 | 338,562 | 30,309 | CACAAAGGACT | 34.85 | 53 | 91.16 | MF775674 |
| LP9206c | June 1992 | 496,242 | 29,291 | not detected | 34.96 | 53 | 90.53 | MF775675 |
| LP9207 | July 1992 | 342,030 | 29,203 | CACAAAGGACT | 35.01 | 54 | 90.93 | MF775676 |
| LP9210 | October 1992 | 263,126 | 29,525 | CACAAAGGACT | 35.14 | 52 | 90.98 | MF775677 |
| LP9404 | April 1994 | 281,524 | 30,072 | CACAAAGGACT | 34.95 | 54 | 91.35 | MF775678 |
| LP9405a | May 1994 | 322,642 | 30,083 | CACAAAGGACT | 34.97 | 53 | 91.52 | MF775679 |
| LP9405b | May 1994 | 336,096 | 29,294 | CACAAAGGACT | 34.93 | 52 | 90.95 | MF775680 |
| LP9406 | June 1994 | 300,558 | 30,237 | CACAAAGGACT | 34.84 | 54 | 91.60 | MF775681 |
| LP9609 | September 1996 | 339,049 | 30,945 | CACAAAGGACT | 34.76 | 55 | 91.36 | MF775682 |
| LP9701 | January 1997 | 336,018 | 30,545 | CACAAAGGACT | 34.92 | 54 | 91.21 | MF775683 |
| LP9801 | January 1998 | 341,504 | 30,204 | CACAAAGGACT | 34.86 | 55 | 91.64 | MF775684 |
| LP9903 | March 1999 | 335,635 | 29,557 | CACAAAGGGCT | 34.96 | 54 | 91.78 | MF775685 |
| LP9908 | August 1999 | 312,195 | 29,668 | CACAAAGGACT | 34.83 | 53 | 91.55 | MF775686 |
| LP0004a | April 2000 | 292,061 | 30,093 | CACAAAGGACT | 34.82 | 55 | 92.52 | MF775687 |
| LP0004b | April 2000 | 235,063 | 30,953 | CACAAAGGACT | 34.73 | 56 | 92.71 | MF775688 |
| LP0004c | April 2000 | 311,085 | 30,044 | CACAAAGGACT | 34.80 | 55 | 92.27 | MF775689 |
| LP0004d | April 2000 | 426,489 | 30,104 | CACAAAGGACT | 34.82 | 55 | 92.20 | MF775690 |
| LP0109 | September 2001 | 307,620 | 30,537 | CACAAAGGACT | 34.83 | 54 | 91.87 | MF775691 |
| LP0202 | February 2002 | 351,377 | 30,236 | CACAAAGGACT | 34.93 | 54 | 92.15 | MF775692 |
| LP0209 | September 2002 | 339,677 | 30,339 | CACAAAGGACT | 34.76 | 53 | 91.78 | MF775693 |
| LP0212 | December 2002 | 98,163 | 29,916 | CACAAAGGACT | 34.77 | 52 | 91.61 | MF775694 |
| LP0304 | April 2003 | 374,920 | 30,344 | CACAAAGGACT | 34.88 | 54 | 91.65 | MF775695 |
| LP0509 | September 2005 | 357,592 | 30,547 | CACAAAGGACT | 34.94 | 54 | 91.07 | MF775696 |
| LP0604 | April 2006 | 390,026 | 30,547 | CACAAAGGACT | 34.93 | 54 | 91.07 | MF775697 |
| LP0903 | March 2009 | 363,869 | 31,049 | CACAAAGGACT | 34.90 | 54 | 91.24 | MF775698 |
| LP1005 | May 2010 | 350,641 | 30,541 | CACAAAGGACT | 34.89 | 56 | 90.77 | MF775699 |
| LP1011 | November 2010 | 332,613 | 30,542 | CACAAAGGAAT | 34.87 | 56 | 90.29 | MF775700 |
| LP1110 | October 2011 | 363,558 | 30,528 | CACAAAGGACT | 34.89 | 55 | 90.86 | MF775701 |
| LP1407 | July 2014 | 311,560 | 30,508 | CACAAAGGACT | 34.91 | 55 | 90.69 | MF775702 |
| LP1502a | February 2015 | 355,385 | 30,247 | CACAAAGGGCT | 35.12 | 52 | 90.65 | MF775703 |
| LP1502b | February 2015 | 331,478 | 30,227 | CACAAAGGACT | 35.11 | 52 | 90.71 | MF775704 |
| LP1502c | February 2015 | 309,510 | 30,247 | CACAAAGGACT | 35.11 | 52 | 90.73 | MF775705 |
Note.—Phages from 1985 to 2014 (culture A phages) were propagated on host L. lactis subsp. cremoris CA-49. Phages from 2015 (culture B phages) were propagated on host L. lactis subsp. lactis IL1403. Phage names indicate isolation time (in YYMM format), that is, the first isolate LP8511 is from November 1985 and the last culture A isolate LP1407 is from July 2014.
Substitution Rate of a Measurably Evolving Phage Population
To estimate the rate of nucleotide substitutions, we aligned the 34 genomes. This yielded a 35,111-bp long alignment, where 4,713 (13%) of the sites are variable, of which 289 (6.1%) are multimorphic. The high sequence similarity between the phage genomes indicates that the phage isolates indeed document a single phage lineage. Reconstructing a maximum likelihood (ML) tree from the aligned genomes shows a weak correlation between the root to tip distance and the sampling time (r2 = 0.43, using TempEst; fig. 2A), hence, there is a weak temporal signal in the data. In order to delineate between mutational processes (i.e., vertical evolution) and recombination (i.e., horizontal evolution), we tested for the presence of recombination. Since a signal of recombination was detected in the alignment (P < 10−6, phi test for recombination), we further identified sites affected by recombination using ClonalFrameML (Didelot and Wilson 2015). These sites were masked (i.e., excluded) from the current analysis, retaining 16,073 (46%) alignment positions. In the masked alignment, 91 (0.57%) of the sites are variable, of which 3 (3.3%) are multimorphic. Recombination signal was eliminated in the masked alignment (P = 0.0625, phi test for recombination). Estimating the phylogeny from the masked alignment (termed here masked phylogeny) revealed a strong temporal signal (r2 = 0.97, fig. 2B). This indicates that the rate of nucleotide substitution resulting from mutations is homogeneous over the sampling time. The strong temporal signal allowed for a reliable estimation of nucleotide substitution rates from the masked alignment. A dated phylogeny estimated using BEAST (Bouckaert et al. 2014) revealed a caterpillar-like tree topology, where one main lineage persists through time (fig. 3). We estimated the substitution rate in this dated phylogeny as 1.888 × 10−4 substitutions per site per year, with a 95% highest posterior density (HPD) interval of 1.470 × 10−4–2.316 × 10−4 (fig. 3, supplementary table S2, Supplementary Material online). Testing alternative models for population size dynamics supports a constant population size over time (supplementary table S2, Supplementary Material online). Furthermore, we validated that the observed substitution pattern is best explained by a strict molecular clock model, which also supports that the substitution rate is homogeneous over the sampling time (supplementary table S2, Supplementary Material online). Based on the average genome length of 30,099 bp, we estimate the genome-wide number of substitutions as 1.888 × 10−4 substitutions/site/year × 30,099 sites, that is, 5.683 substitutions/year.
Fig. 2.

Temporal signal in whole-genome alignment as estimated by TempEst (Rambaut et al. 2016). Genetic distance from the root in substitutions per site is calculated either from (A) the phylogeny reconstructed from the whole alignment (supplementary fig. S2A, Supplementary Material online), or (B) the phylogeny reconstructed from the masked alignment (supplementary fig. S2B, Supplementary Material online).
Fig. 3.

Dated phylogeny and substitution rates. (A) Densitree plot constructed using BEAST with strict clock and constant population size model (supplementary table S2, model 1, Supplementary Material online). All trees from the posterior distribution are displayed in green. The root canal, that is, the phylogeny with the highest clade support, is displayed in blue. Phylogenetic conflict is present for relationships between samples isolated close in time, nonetheless, the bifurcating root canal topology is well represented among the sampled trees. (B) Substitution rate estimates. Vertical lines denote mean, boxes denote 95% HPD intervals and whiskers denote the range of the posterior distributions.
To further validate the genome-wide nucleotide substitution rate, we calculated the substitution rate independently for the 40 core protein families. Alignments of the core protein families show a varying extent of recombination up to 100% (supplementary table S1, Supplementary Material online). The gene alignments were masked for the signal of recombination and were concatenated into a 13,149 bp-long alignment, where 77 (0.59%) of the sites are variable. Estimating the substitution rate from the core families alignment yielded 1.737 × 10−4 substitutions per site per year, which is within the 95% HPD substitution rate calculated from the whole-genome alignment (fig. 3, supplementary table S2, Supplementary Material online). Further comparison of the rate among the three codon positions shows that the second position is the slowest, while the third position is the fastest evolving (fig. 3, supplementary table S2, Supplementary Material online). This is expected from the genetic code structure and serves as a confirmation for our approach. Notably, every codon position follows a strict molecular clock model (supplementary table S2, Supplementary Material online).
Our results reveal a strong molecular clock signal in the vertical evolution of a lineage of the 936 group of phages over 29 years. The strength of temporal signal in our data is comparable to recent estimates from measurably evolving bacteria populations (e.g., r2 = 0.93 for Staphylococcus aureus ST239) (Duchêne et al. 2016). The substitution rate per site per year that we estimated here for lactococcal phages is within the range of eukaryotic dsDNA viruses (∼10−5) and eukaryotic RNA viruses (∼10−3) (Biek et al. 2015). For Siphoviridae phages residing in the human gut, a previous short-term estimate based on a sampling time span of 2.5 years revealed rates between 10−3 and 10−4.5 substitutions per site per year (Minot et al. 2013). We note that estimates from short-term data sets are known to exceed those from long-term data sets due to the presence of transient polymorphic sites (Aiewsakun and Katzourakis 2016). This is consistent with the observation of high rates in a previous short-term data set (Minot et al. 2013). In addition, the rate reported here might still be slightly overestimated.
The latency period in phages of the 936 group is about 30 min and the adsorption time is up to 10 min (Müller-Merbach et al. 2007). Thus, these phages can—in principal—complete up to 36 generations per day or ∼13,000 generations per year. The actual number of generations per year is expected to be lower than this theoretical maximum as it depends on the encounter rate of a free host and, in particular, on the availability of actively growing host cells during dairy fermentation in the production vessels (Parente et al. 2017). Nonetheless, our results reveal that a lineage of the 936 group of phages accumulated about 6 genome-wide substitutions per year within this number of generations.
Taken together, the variation in the complete alignment is high with 13% of sites being variable. In contrast, the masked alignment has only 0.57% of variable sites. The high level of variation in the complete alignment might have been introduced by horizontal processes (i.e., recombination) or by mutational hotspots. In the presence of mutational hotspots, mutations happen multiple times at the same sites and generate a high amount of multimorphic sites. Thus, the low frequency of multimorphic sites (6.1% in the complete alignment and 3.3% in the masked alignment) refutes the presence of mutational hotspots in the phage genomes. In addition, the magnitude of the substitution rate cannot explain the diversity observed in the complete genomes. Hence, point mutations are unlikely to constitute the major driver of phage genome evolution.
The Extent of Recombination in Phage Evolution
To estimate the rate of recombination, we reconstructed recombination events based on the whole-genome alignment and the masked phylogeny. In total, 345 recombination events were detected; those are evenly distributed over terminal and internal branches (supplementary fig. S3, Supplementary Material online). The relative contribution of recombination and mutation to nucleotide alterations was estimated by ClonalFrameML as r/m = 23.50 (supplementary fig. S3, Supplementary Material online). To further validate the detection of recombination, we excluded 471 (1.34%) positions showing alignment uncertainty. Applying ClonalFrameML to this alignment resulted in r/m = 23.24. Consequently, our r/m estimate is robust to sequence alignment errors.
The relative contribution of recombination and mutation to nucleotide alterations has been so far estimated only for prokaryotic genomes. The analysis of species-wide data resulted in generally low r/m values, for example, 0.283 in S. aureus (Didelot and Wilson 2015) and 3.4 in Bacillus cereus (Ansari and Didelot 2014). Higher values were observed in studies restricted to a particular lineage, for example, Sulfolobus islandicus isolated from a single location showed r/m estimates between 1.8 and 13 (Cadillo-Quiroz et al. 2012); an r/m of 7.2 was estimated for a Streptococcus pneumoniae lineage isolated over 20 years (Croucher et al. 2011). An extreme ratio of r/m between 12 and 62 was observed in Helicobacter pylori (Kennemann et al. 2011), which might be attributable to the low sampling density and the short evolutionary time scale of 3 years in that study. Based on our estimate of r/m for a distinct dairy Siphoviridae phage lineage, we conclude that the effect of recombination relative to mutation can be elevated in phage genome evolution in comparison to bacteria.
Temporal Signal in Phage Recombination Rates
Mapping the recombination events on the masked phylogeny revealed a strong correlation between the number of recombination events and time (r2 = 0.90, fig. 4A). Strong temporal signals are observed also for the total recombination event length (r2 = 0.85) and for the number of nucleotide alterations as a result of recombination (r2 = 0.86, fig. 4). The strong temporal signal of recombination over the sampling time allowed us to estimate the rate of recombination from the linear regression fit. This revealed a rate of 5.091 recombination events per year. The total recombination event length is estimated as 809 nucleotides transferred per year, whereas the effect of recombination is inferred as 135 genome-wide nucleotide alterations per year that correspond to 135/30,099 = 4.485 × 10−3 nucleotide alterations per site per year (fig. 4). The relative contribution of recombination to mutation, r/m, is not correlated with time (fig. 4D), which demonstrates a constant r/m ratio over time. These results show that the estimated recombination rates are homogeneous over time in phage short-term evolution.
Fig. 4.

Recombination rate estimation. Estimation of recombination rate in (A) number of events: 5.091 [4.063–6.810] recombination events per year, (B) total recombination event length: 809.0 [654.4–1290] nucleotides transferred per year, (C) effect of recombination: 135.0 [91.22–170.9] nucleotides altered per year. Numbers in square brackets give the range for estimates based on 20 bootstrap phylogenies. (D) r/m per branch with r being the number of nucleotide alterations mapped to the branch and m being the branch length in genome-wide substitutions. r/m is independent of the node height, that is, distance from the root in substitutions per site. Due to the strong temporal signal in substitutions, this demonstrates a constant r/m over time.
Altogether, we observe homogeneous rates of nucleotide alterations due to mutation and recombination in our data set. This enabled us to estimate the relative contribution of recombination to mutation directly from the rates as r/m = 135/5.683 = 23.76. Note that the r/m ratio calculated from the rates per year is not different from the ClonalFrameML estimate that does not take the temporal signal into account; this provides further support for a homogeneous r/m ratio over time.
Gene Content Evolution
To study gene content evolution, we focused on the variable homologous protein families. Of the 34 variable protein families, nine families are variable due to nonsense mutations. Six of the nonsense families were generated by the presence of a premature stop codon in the upstream CDSs. Four of the premature stop codon substitutions were independent events in four different isolates and resulted in a truncation of the neck passage structure protein (NPS, family 11, fig. 5C). The truncation of this protein results in a phenotypic change, where the NPS is absent in the virion (Murphy et al. 2016) (supplementary fig. S1, Supplementary Material online). The remaining three nonsense families have a substitution that either changes the start codon or introduces a frame shift mutation (supplementary fig. S4, Supplementary Material online).
Fig. 5.

Distribution of protein families along the genome. (A) Genome annotation. (B) Presence–absence matrix of variable protein families. Based on maximum parsimony, 25 gain and 42 loss events are inferred for gene indel families. (C) Consensus genome annotation based on the whole-genome alignment. Protein family numbers of core clusters are denoted below the genes, which are colored yellow (late region), orange (early region), and red (middle region). Protein family number of variable clusters is denoted above the gene. Singleton clusters are colored in gray. Overlapping variable genes are plotted in parallel. Several properties are shown aligned to the consensus genome annotation: Variable sites—sliding window analysis (width 500 bp, offset 100 bp) of number of columns having multiple nucleotides; Recombination—sliding window analysis (width 500 bp, offset 100 bp) of the number of recombination events in each window; presence in masked alignment—positions included in the masked alignment are colored in black. Diversity varies along the genomes. The early region is most diverse in terms of gene content and nucleotide differences. This region is also most affected by recombination. In contrast, the late region is very conserved. In accordance with previous observations (Mahony et al. 2012), we also found hotspots of diversity among the structural genes in regions that overlap with the genes for the tail length tape measure protein (family 16) and the phage lysin (family 22).
The remaining 25 variable protein families are not caused by nonsense mutations, but vary most likely due to horizontal processes (termed here gene indel families). The presence–absence pattern of those families shows frequent gene gain and loss, where each protein family was both gained and lost at least once (fig. 5B). An extreme case of gene indel dynamics is observed for DNA polymerase, where two protein families were identified (gene families 64 and 66, fig. 5). Exactly one DNA polymerase protein family is present in each genome at a conserved genomic location. Those two protein families are alternating frequently over time due to their recurrent replacement by recombination. Notably, since the substitution rate is homogeneous in the genomes of all 34 phage isolates, the substitution rate does not vary between phages having a different DNA polymerase. Furthermore, we observe that no gene indel family was gained or lost between 2005 and 2014 (fig. 5B). The correlation between pairwise gene indel content distance and time is weak (r2 = 0.19, supplementary fig. S5, Supplementary Material online). This demonstrates the absence of temporal signal in gene gain and loss evolution.
Gene Content Evolution in the Pangenome of the 936 Group of Phages
The frequent gene loss and regain suggests the existence of a pangenome (i.e., genetic reservoir) that is accessible by genetic recombination. To examine the genetic connectivity between culture A phages and other phages of the 936 group, we reconstructed protein families for all phages of the 936 group. This includes 90 publicly available genomes (supplementary table S3, Supplementary Material online) and 3 phages of the 936 group isolated in 2015 from the same dairy as the previous 34 culture A phages, after switching to a different undefined starter culture (termed here culture B phages, table 1). A comparative genomics analysis of the 127 genomes yielded 203 homologous protein families. Of these, 24 families are core families (supplementary table S1, Supplementary Material online).
In the presence of homogeneous substitution rates and homogeneous gene gain and loss rates, an association between genetic distance and gene content distance is expected. In the 936 group of phages, a comparison between the gene content distance and genetic distance revealed two clusters (fig. 6). The first cluster comprises closely related strains, including pairs with a genetic distance smaller than 0.07 substitutions/site and similar gene content (average distance 0.1270). Distantly related strains are clustered separately; these have a higher gene content distance (0.3773). Genetic distance and gene content distance are weakly correlated within each cluster (fig. 6). The clustering pattern resembles ecotypes observed in marine phage populations (Marston and Martiny 2016). The biased sampling of dairy phages from just a few factories can also mimic a clustering pattern, thus, the sampling density in our data is insufficient to determine the presence of different ecotypes for the 936 group of phages.
Fig. 6.
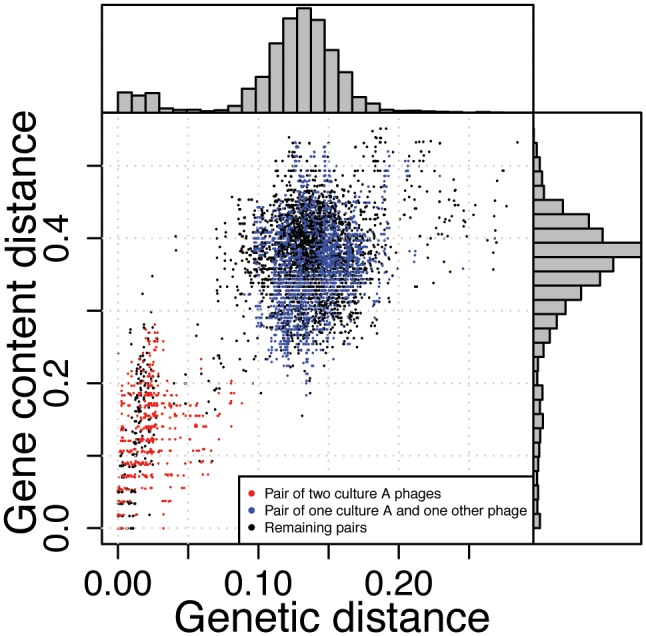
Association between pairwise gene content distance and genetic distance for all phages of the 936 group. Pairwise genetic distances in substitutions per site are estimated from the codon alignments of the 24 core genes. Gene content distances are based on Jaccard index. There is a clear correlation between the genetic distance of a pair of strains and its protein content similarity (r2=0.5352, n = 8001, P < 10−6). This association is, however, the result of two clusters in the data. There is a weak linear relationship for closely related strains that show a genetic distance smaller than 0.07 substitutions/site (r2=0.1798, n = 866, P < 10−6) and a very weak relationship for distantly related strains with larger genetic distances (r2=0.02276, n = 7,135, P < 10−6). Phages of the 936 group form discrete clusters that are characterized by a low genetic distance inside clusters and a high genetic distance between clusters (supplementary fig. S6, Supplementary Material online). The clustering pattern remains when using alternative distance measures (as in Mavrich and Hatfull 2017, supplementary fig. S7, Supplementary Material online).
To quantify the extent of genetic connectivity between culture A phages and other phages of the 936 group, we performed a comparative phylogenetic analysis of protein families. A phylogenetic network reconstructed from the concatenated alignment of 24 core protein families shows many conflicting splits, yet there is a strong signal supporting the monophyly of culture A phages (supplementary fig. S8, Supplementary Material online). In 7 out of 63 protein families (having at least 2 members, excluding nonsense families), culture A phages are not monophyletic. This indicates that genetic recombination with other phages of the 936 group has occurred in these families (supplementary fig. S9, Supplementary Material online). These include structural proteins, phage lysin, HNH homing endonuclease, and DNA polymerase. An example is the Sak3 (sensitivity to AbiK protein 3) phylogeny, that shows a clear signal of recurrent replacing recombination with various donors of the 936 group of phages. This protein is a target for the bacterial abortive infection system (Abi) (Bouchard and Moineau 2004); hence, the frequent recombination events in sak3 are an indication for rapid phage evolution due to antagonistic coevolution with the host.
Culture A phage genomes encode three protein families that have no homologs in other phages of the 936 group (protein families 13, 67, and 68). Protein family 67 is observed in all isolates from 2005 and later (fig. 5). It is a shortened version of the phage antirepressor protein (Ant) found in prophage regions of L. lactis genomes. Phage antirepressor proteins function as inactivators of prophage repressors and are usually associated with temperate phages. A premature stop codon is observed in phage isolates from 2010 and later, resulting in a shorter version of Ant and in the evolution of nonsense family 68 (fig. 5). In addition, the tail protein extension tpeX gene (family 13, fig. 5) encodes an extension of the major tail protein (family 12, fig. 5) that is expressed by translational read-through and that is visible as a thin spiral structure on the phage tails (Murphy et al. 2016). The TpeX protein sequence of culture A phages is highly diverged from TpeX in other phages of the 936 group (supplementary fig. S11, Supplementary Material online) indicating the existence of a culture A specific TpeX variant.
While protein families specific to culture A phages are rare, most variable protein families of culture A phages are also present in other phage genomes of the 936 group. Our results thus demonstrate recurrent recombination events between culture A phages and the pangenome of the 936 group of phages. The observed recombination events are best explained by host coinfection by related, yet genetically different, phages. The genetic connectivity within the pangenome indicates that different phages of the 936 group share an environment where frequent coinfection events occur. Hence, different phage types can propagate and evolve in an undefined starter culture that comprises diverse host strains.
Discussion
Our results reveal a strong temporal signal in phage genome evolution including a homogeneous rate of nucleotide substitution and a homogeneous rate of recombination. Homogeneous nucleotide substitution rates, which have been termed “molecular clock” (Zuckerkandl and Pauling 1965), are an important outcome of the neutral theory of molecular evolution (Kimura 1969; Bromham and Penny 2003). The theory posits that most mutations are neutral or deleterious such that polymorphisms observed in genomic data are neutral or nearly neutral (Ohta and Kimura 1971). Indeed, site-directed mutagenesis of ssDNA phage f1 revealed a majority of neutral mutations in the distribution of fitness effects (Peris et al. 2010). Furthermore, assuming homogeneous generation times and a constant mutation rate, the neutral theory predicts that genome evolution follows a molecular clock (Ohta and Kimura 1971). Our study demonstrates the presence of a molecular clock for substitutions originating from mutational processes in phage short-term evolution. Notably, the higher substitution rate observed for the third codon position indicates that synonymous substitutions are preferred. Thus, the application of the neutral theory to phage evolution is supported by the presence of the molecular clock and the prevalence of synonymous substitutions, which has been shown before for eukaryotic RNA viruses (Gojobori et al. 1990). As the neutral theory states homogeneous generation times, the strong molecular clock in phage evolution implies that the phage infection cycle length is homogeneous over the sampling time period in this biotechnological environment. In addition, we show that the molecular clock holds for recombination events. We hypothesize that this stems from a constant coinfection rate that adjusts the pace of the recombination process. Thus, we show that the neutral theory is also applicable to phage genome evolution. The homogeneity of recombination rate further supports the applicability of the neutral theory to genetic variation brought about by horizontal processes.
Our analysis does not reveal a molecular clock signal in gene content evolution. Variable protein families are enriched in the early transcriptional region (fig. 5) that typically encodes nonessential proteins involved in phage–host interaction (Roucourt and Lavigne 2009). Clustering of host interaction genes in phages has been previously observed, for example, for antiCRISPR genes in Pseudomonas aeruginosa phages (Bondy-Denomy et al. 2013), in hypervariable regions in T4-like phages (Comeau et al. 2007), and by the presence of genomic islands in viral metagenomes (Mizuno et al. 2014). This suggests that gene gain and loss in this region is driven by phage–bacteria antagonistic coevolution, which is characterized by a strong selection pressure. The lack of a molecular clock signal for gene content evolution may thus be explained by deviation from the assumption of the neutral theory, indicating that selection is at play.
Here, we show that the effect of recombination relative to mutation is elevated in a lineage of the 936 group of phages in comparison to bacteria. The prevalence of recombination in the 936 group of phages is consistent with the occurrence of the recombination proteins single-stranded binding protein (SSB) and sensitivity to AbiK protein 3 (Sak3) in the core genome of this group. SSB from a phage of the 936 group can stimulate RecA (Scaltriti et al. 2009) and Sak3 is a DNA–single strand annealing protein involved in homologous recombination due to interaction with RecA (Bouchard and Moineau 2004; Scaltriti et al. 2011) (see also supplementary figs. S9D and S10, Supplementary Material online). Here, we show evidence that recombination is an important evolutionary process that shapes the genomes of phages of the 936 group and we suggest that these proteins are major contributors to this process.
Materials and Methods
Sampling and DNA Extraction
Whey samples were collected from a German dairy from 1985 to 2014. Within this 29-year period, a mesophilic undefined (multiple-strain) starter culture “A” was constantly used for milk fermentation. The phage-sensitive L. lactis subsp. cremoris strain CA-49 was isolated from culture A and used for isolation and propagation of all 34 virulent phages. In 2014, milk fermentation with culture A was replaced in the dairy by an alternative undefined starter culture B. Since (i) strain CA-49 from culture A could not propagate 936 phages from culture B, and (ii) phage-sensitive single-colony isolates could not be isolated from culture B, laboratory strains L. lactis subsp. lactis IL1403 (Bolotin et al. 2001) and L. lactis subsp. cremoris MG1363 (Gasson 1983) were used for phage screening. This resulted in the isolation and propagation of three 936 culture B phages on strain IL1403.
Lactococcal strains were grown at 30 °C in M17 broth (Terzaghi and Sandine 1975) supplemented with 1% lactose (LM17). 10 mM CaCl2 was added for phage propagation (LM17-Ca). Plaque assays were done according to the double agar layer protocol (Adams 1959) from serial dilutions of these whey samples and also from phage lysates in LM17-broth. Phage isolates were obtained by picking individual plaques from the host bacterial lawns in LM17-Ca soft agar. To remove bacterial cells and debris, whey samples were centrifuged (17,500 × g, 20 min, 4 °C) and the supernatants were filtered with 0.45 µm membrane filters (minisart, Sartorius, Göttingen, Germany). For each phage isolate, three successive plaque isolations were performed. Lysates of the last purified phage isolates propagated on LM17-broth were fortified with 10% glycerin and stored at −80 °C. Lactococcal strains were grown in reconstituted skim milk and early log phase cultures were stored at −80 °C.
For DNA extraction, phages were propagated in 500 ml of an exponentially grown host culture. Phage concentration and purification by CsCl density gradient ultracentrifugation were done as described elsewere (Sambrook and Russel 2001). Purity and quantity of the phage DNA was checked by standard agarose (1.2%) gel electrophoresis and in a Nanodrop 2000c spectrophotometer (Thermo Fisher Scientific, Dreieich, Germany). Phage DNA was subsequently concentrated using disposable spin columns (Genomic DNA Clean & Concentrator, Zymo Research Europe, Freiburg), and their purity and concentration for sequencing was finally determined with a Qubit 3.0 Fluorometer (Thermo Fisher Scientific).
Transmission Electron Microscopy
For electron microscopy, phages purified by CsCl gradient ultracentrifugation were used. Negative staining with 2% uranyl acetate was done as described earlier (Vegge et al. 2006; Murphy et al. 2016). Specimens were viewed in a Tecnai 10 transmission electron microscope (FEI Thermo Fisher Scientific, Eindhoven, the Netherlands) at an acceleration voltage of 80 kV equipped with a Megaview G2 CCD camera (Emsis, Muenster, Germany).
Sequencing and Assembly
Phage DNA was sequenced with Illumina MiSeq after NexteraXT library preparation. This resulted in 98,163–496,242 paired-end reads of length 300 bp per sample (table 1). A first assembly was obtained by SPAdes v3.9.0 (Bankevich et al. 2012). In the case of highly fragmented assemblies, reads were enriched for phage origin as follows. Reads were mapped onto contigs with BWA MEM v0.7.5 (Li and Durbin 2009). Then two different coverage cutoffs were applied, 100 and 1,000. Contigs with an average coverage above the cutoff were filtered using samtools v1.3.1 (Li et al. 2009) and paired-end reads were extracted from highly-covered contigs using bedtools v2.25.0 (Quinlan and Hall 2010). These subsets of paired-end reads were reassembled with SPAdes. Thus, up to three assemblies were obtained per sample. Their assembly graphs were inspected by bandage v0.8.0 for circular contigs (Wick et al. 2015). Although the packaged DNA is linear, circular assemblies are expected to result from the concatemer step of DNA replication (Casjens and Gilcrease 2009). Thus, a circular assembly was assessed as a confirmation of a completely assembled phage contig. For samples where no circular contig was detected, assembly with PlasmidSPAdes v3.9.0 (Antipov et al. 2016) was attempted and Recycler v0.6 (Rozov et al. 2017) was used with the precomputed assemblies. This resulted in circular assemblies for all except four samples (LP9206c, LP9406, LP0109, and LP0903, table 1). For circular assemblies, in all except one sample (LP1407), a direct repeat was detected with Tandem repeats finder v4.09 (Benson 1999). A cohesive (cos) site was present before the tandem repeat and circular genomes were consistently cut at the start of the cos site. The completeness of the four linear assemblies was confirmed by gene content and in all but one sample (LP9206c, table 1) by the presence of the cos site at the beginning of the contig. In the four linear assemblies, the tandem repeat was already located at the end of the contig. Mappings were visualized by Tablet v1.16.09.06 (Milne et al. 2010) for quality control.
Genome annotation was predicted by the RAST server (Aziz et al. 2008). Additional functional annotations for predicted CDSs were obtained by blastp against the nr database at NCBI. For culture A phages, the annotation was extended by hmmsearch (Finn et al. 2015), or by obtaining the Phagonaute (Delattre et al. 2016) annotation of a related protein. Raw reads and annotated assemblies are deposited in GenBank under BioProject PRJNA398675.
Phylogenetic Analysis and Rate Estimation
The following evolutionary analysis was performed for culture A phages. ProgressiveMauve v2.4.0 (Darling et al. 2010) with the option –seed-family found only one block confirming that the genomes are collinear. MAFFT v7.123 b (Katoh and Standley 2013) and the E-INS-i algorithm was used to obtain a whole-genome nucleotide alignment. Pairwise genetic distances were estimated from the whole-genome nucleotide alignments using RAxML v8.2.9 (Stamatakis 2014). Average nucleotide identity (ANI) was estimated using pyani v0.2.4 (Pritchard 2017) based on BLASTN+ v2.4.0 (Altschul et al. 1990). ANI distance is 1-ANI. Alignment column uncertainty was measured by the Heads or Tails (HoT) method (Landan and Graur 2007) as the columns that are different in the forward and reverse alignment. Maximum likelihood (ML) phylogenies were estimated with RAxML v8.2.9 (Stamatakis 2014) using the GTRGAMMA model and 100 bootstrap replicates. TempEst v1.5 (Rambaut et al. 2016) was used to estimate the strength of the association between divergence and sampling time for a given phylogeny with branch lengths and dated tips based on the best-fitting root found by correlation. The presence of recombination in an alignment was indicated by the phi test (Bruen et al. 2006) implemented in SplitsTree4 (Huson and Bryant 2006).
Recombination events were detected with ClonalFrameML v1.25 (Didelot and Wilson 2015) and the respective kappa estimated under the HKY model with RAxML. The ClonalFrameML model detects recombination from an external source but is also applicable to recombination inside the sampled population (Didelot and Wilson 2015). Recombined segments detected with ClonalFrameML are characterized by the start and end position in the alignment and the branch in the phylogeny where the segment is introduced. Alternatively, recombination was detected using gubbins v2.2.0 (Croucher et al. 2015), once with default parameters and once by setting the outgroup to LP8511. We restricted ourselves to a method that detects recombination by stretches of clustered polymorphisms. Alternative phylogenetics methods [e.g., RDP4 (Martin et al. 2015)] only detect recombination inside the sampled lineage and might result in an underestimation of recombination events and thus in an overestimation of the substitution rate. In addition, methods based on the assumption that all isolates were sampled at the same time [e.g., ClonalOrigin (Didelot et al. 2010), BratNextGen (Marttinen et al. 2012)] are not applicable.
Alignments were masked from recombination by masking recombinant stretches detected by ClonalFrameML. Thereby, recombinant stretches on terminal lineages were replaced by gaps and recombinant stretches on internal lineages result in masking of the whole alignment region. The ML phylogeny was reestimated for the masked alignment. The process was repeated once to detect additional recombined segments with the new phylogeny. A third iteration did not result in additional recombined segments with ClonalFrameML. An alternative recombination detection method—gubbins (Croucher et al. 2015)—retained a shorter alignment with less temporal signal (r2 < 78% with different gubbins options) in comparison to the ClonalFrameML analysis. Therefore, we continued the analysis with the recombination results from ClonalFrameML.
ClonalFrameML was also used as the basis to estimate the rate of recombination. The rate of recombination events per year is estimated by TempEst from a phylogeny where the branch lengths represent the number of recombination events detected on each branch by ClonalFrameML. Analogously, the total recombination segment length transferred per year is estimated from a phylogeny where the branch lengths represent the total length of all segments detected on each branch. Finally, the number of nucleotide alterations by recombination per year is estimated from a phylogeny where the branch lengths represent the sum of altered nucleotides for all segments detected on each branch.
Dated phylogenies were estimated with BEAST v2.4.4 (Bouckaert et al. 2014) using the HKY substitution model with a discrete gamma distribution of 16 categories across sites. The strict clock prior was set to an exponential distribution with mean 0.001. Additional rate priors that were tested and showed similar results are Uniform[0, 10], exponential distribution with mean 0.0001, 0.01, 0.1, 1. Deviations from the strict molecular clock were modeled by lognormally autocorrelated rates (Drummond et al. 2006) and a random local clock (Drummond and Suchard 2010). The population size prior was a coalescent constant population. Deviations from a constant population size were modeled by an exponential growth, the Bayesian Skyline Plot (Drummond et al. 2005), or the Extended Bayesian Skyline Plot (Heled and Drummond 2008). Tracer v1.6 (Rambaut et al. 2014) was used to visualize log files and to calculate the 95% HPD highest posterior density interval. All chains were run for at least 107 generations and until all parameters had effective sample sizes of at least 200, with a burnin of 10% and sampling every 1,000 iterations. Convergence was visually checked by Tracer using a second independent chain. Bayes factors were calculated based on marginal likelihood estimation by stepping-stone sampling (Baele et al. 2012). Posterior distributions of trees were visualized by DensiTree v2.2.5 (Bouckaert and Heled 2014).
Gene Content Analysis
Homologous protein families for culture A phages were calculated as follows. Bidirectional best hits with BLASTP+ 2.4.0 (Altschul et al. 1990) hits (e-value < 10−10) were realigned with needle [package emboss (Rice et al. 2000)]. Sequence pairs having global sequence identity <60% were excluded. The remaining pairs were clustered using MCL v14.137 (Enright et al. 2002). Clusters were manually curated based on the whole-genome alignment by joining six clusters with 50%–60% amino acid identity if they were aligned in the whole-genome alignment. This results in 74 clusters of which 40 are universal (core families). The 34 variable families were manually classified based on the whole-genome alignment into different modes of emergence: gene indel if they are absent in the remaining strains (25 families) and nonsense if they emerged by nonsense mutations (9 families). Nonsense families encompass gene loss by frameshift mutation (one family, supplementary fig. S4, Supplementary Material online), gene gain by start codon evolution (two families, supplementary fig. S4, Supplementary Material online), and potential artefactual CDS due to premature stop codon in the upstream CDS (six families).
The Jaccard index for a pair of genomes is the number of protein families shared by both genomes divided by the number of protein families present in any of the two genomes. This similarity was converted into a gene content distance by one minus Jaccard index. Alternatively, the gene content distance was calculated as one minus the average proportion of shared genes. The number of gain and loss events that is necessary to explain the presence absence data was estimated based on maximum parsimony using GLOOME (Cohen et al. 2010) on the tree of the masked whole-genome alignment.
Protein families were aligned with MAFFT and the E-INS-i algorithm. Codon alignments were generated by pal2nal v14 (Suyama et al. 2006). Recombination was detected on the codon alignments using ClonalFrameML and the phylogeny of the masked whole-genome alignment. BEAST analysis was based on a partition model of codon positions with a common phylogeny.
The publicly available genome sequences of 90 lactococcal phages of the 936 group were downloaded from NCBI (supplementary table S3, Supplementary Material online). Protein families for all available phage isolates of the 936 group were calculated by bidirectional best blastp (e-value < 10−10) with a global sequence identity ≥ 50% and MCL clustering. This resulted in 203 protein families with a core of 24 families present in all 127 phage isolates. Protein families were aligned with MAFFT and the E-INS-i algorithm. A core alignment was generated by concatenating alignments from 24 core protein families after removing duplicates. Alignments were visualized by Jalview (Waterhouse et al. 2009). Phylogenies are computed by RAxML with the PROTGAMMALG option.
Supplementary Material
Supplementary data are available at Molecular Biology and Evolution online.
Supplementary Material
Acknowledgments
We thank Maxime Godfroid (Kiel University) for providing a script to count the alterations introduced by recombination. The manuscript was greatly improved by comments from Bas E. Dutilh (Utrecht University). We thank Giddy Landan, Nils Hülter, Fernando Domingues Kümmel Tria, and Tanita Wein (Kiel University) for critical comments on the manuscript. Inka Lammertz and Angela Back (Max Rubner-Institut, Kiel) are acknowledged for technical assistance in phage isolation and propagation, DNA extraction, and transmission electron microscopy. The study was supported by the European Research Council (Grant No. 281357 to TD) and the Bioinformatics Network at Kiel University.
References
- Adams MH. 1959. Enumeration of bacteriophage particles In: Ross SR, editor. Bacteriophages. London: Interscience Publishers, Ltd; p. 27–34. [Google Scholar]
- Aiewsakun P, Katzourakis A.. 2016. Time-dependent rate phenomenon in viruses. J Virol. 9016:7184–7195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ.. 1990. Basic local alignment search tool. J Mol Biol. 2153:403–410. [DOI] [PubMed] [Google Scholar]
- Ansari MA, Didelot X.. 2014. Inference of the properties of the recombination process from whole bacterial genomes. Genetics 1961:253–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antipov D, Hartwick N, Shen M, Raiko M, Lapidus A, Pevzner PA.. 2016. plasmidSPAdes: assembling plasmids from whole genome sequencing data. Bioinformatics 3222:3380–3387. [DOI] [PubMed] [Google Scholar]
- Aziz RK, Bartels D, Best A, DeJongh M, Disz T, Edwards R, Formsma K, Gerdes S, Glass EM, Kubal M.. 2008. The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9:75.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baele G, Lemey P, Bedford T, Rambaut A, Suchard MA, Alekseyenko AV.. 2012. Improving the accuracy of demographic and molecular clock model comparison while accommodating phylogenetic uncertainty. Mol Biol Evol. 299:2157–2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD et al. , 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 195:455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson G. 1999. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 272:573–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biek R, Pybus OG, Lloyd-Smith JO, Didelot X.. 2015. Measurably evolving pathogens in the genomic era. Trends Ecol Evol. 306:306–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolotin A, Wincker P, Mauger S, Jaillon O, Malarme K, Weissenbach J, Ehrlich SD, Sorokin A.. 2001. The complete genome sequence of the lactic acid bacterium Lactococcus lactis ssp. lactis IL1403. Genome Res. 115:731–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondy-Denomy J, Pawluk A, Maxwell KL, Davidson AR.. 2013. Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature 4937432:429–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard JD, Moineau S.. 2000. Homologous recombination between a lactococcal bacteriophage and the chromosome of its host strain. Virology 2701:65–75. [DOI] [PubMed] [Google Scholar]
- Bouchard JD, Moineau S.. 2004. Lactococcal phage genes involved in sensitivity to AbiK and their relation to single-strand annealing proteins. J Bacteriol. 18611:3649–3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouckaert R, Heled J.. 2014. DensiTree 2: seeing trees through the forest. bioRxiv: 012401. [Google Scholar]
- Bouckaert R, Heled J, Kühnert D, Vaughan T, Wu C-H, Xie D, Suchard M. a, Rambaut A, Drummond AJ.. 2014. BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput Biol. 104:e1003537.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouvier T, Del Giorgio PA.. 2007. Key role of selective viral-induced mortality in determining marine bacterial community composition. Environ Microbiol. 92:287–297. [DOI] [PubMed] [Google Scholar]
- Bromham L, Penny D.. 2003. The modern molecular clock. Nat Rev Genet. 43:216–224. [DOI] [PubMed] [Google Scholar]
- Bruen TC, Philippe H, Bryant D.. 2006. A simple and robust statistical test for detecting the presence of recombination. Genetics 1724:2665–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadillo-Quiroz H, Didelot X, Held NL, Herrera A, Darling A, Reno ML, Krause DJ, Whitaker RJ.. 2012. Patterns of gene flow define species of thermophilic Archaea. PLoS Biol. 102:e1001265.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casjens SR, Gilcrease EB.. 2009. Determining DNA packaging strategy by analysis of the termini of the chromosomes in tailed-bacteriophage virions. Methods Mol Biol. 502:91–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobián Güemes AG, Youle M, Cantú VA, Felts B, Nulton J, Rohwer F.. 2016. Viruses as winners in the game of life. Annu Rev Virol. 31:197–214. [DOI] [PubMed] [Google Scholar]
- Cohen O, Ashkenazy H, Belinky F, Huchon D, Pupko T.. 2010. GLOOME: gain loss mapping engine. Bioinformatics 2622:2914–2915. [DOI] [PubMed] [Google Scholar]
- Coleman ML, Sullivan MB, Martiny AC, Steglich C, Barry K, DeLong EF, Chisholm SW.. 2006. Genomic islands and the ecology and evolution of Prochlorococcus. Science 3115768:1768–1770. [DOI] [PubMed] [Google Scholar]
- Comeau AM, Bertrand C, Letarov A, Tétart F, Krisch HM.. 2007. Modular architecture of the T4 phage superfamily: a conserved core genome and a plastic periphery. Virology 3622:384–396. [DOI] [PubMed] [Google Scholar]
- Croucher NJ, Harris SR, Fraser C, Quail M. a, Burton J, van der Linden M, McGee L, von Gottberg A, Song JH, Ko KS et al. , 2011. Rapid pneumococcal evolution in response to clinical interventions. Science 3316016:430–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croucher NJ, Page AJ, Connor TR, Delaney AJ, Keane JA, Bentley SD, Parkhill J, Harris SR.. 2015. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 433:e15.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y, Yu C, Yan Y, Li D, Li Y, Jombart T, Weinert LA, Wang Z, Guo Z, Xu L et al. , 2013. Historical variations in mutation rate in an epidemic pathogen, Yersinia pestis. Proc Natl Acad Sci. 1102:577–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling AE, Mau B, Perna NT.. 2010. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 56:e11147.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Paepe M, Hutinet G, Son O, Amarir-Bouhram J, Schbath S, Petit M-A.. 2014. Temperate phages acquire DNA from defective prophages by relaxed homologous recombination: the role of Rad52-like recombinases. PLoS Genet. 103:e1004181.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delattre H, Souiai O, Fagoonee K, Guerois R, Petit M-A.. 2016. Phagonaute: a web-based interface for phage synteny browsing and protein function prediction. Virology 496:42–50. [DOI] [PubMed] [Google Scholar]
- Díaz-Muñoz SL. 2017. Viral coinfection is shaped by host ecology and virus–virus interactions across diverse microbial taxa and environments. Virus Evol. 31:vex011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didelot X, Lawson D, Darling A, Falush D.. 2010. Inference of homologous recombination in bacteria using whole-genome sequences. Genetics 1864:1435–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didelot X, Wilson DJ.. 2015. ClonalFrameML: efficient inference of recombination in whole bacterial genomes. PLoS Comput Biol. 112:e1004041.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond AJ, Ho SYW, Phillips MJ, Rambaut A.. 2006. Relaxed phylogenetics and dating with confidence. PLoS Biol. 45:e88.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond AJ, Pybus OG, Rambaut A, Forsberg R, Rodrigo AG.. 2003. Measurably evolving populations. Trends Ecol Evol. 189:481–488. [Google Scholar]
- Drummond AJ, Rambaut A, Shapiro B, Pybus OG.. 2005. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol Biol Evol. 225:1185–1192. [DOI] [PubMed] [Google Scholar]
- Drummond AJ, Suchard MA.. 2010. Bayesian random local clocks, or one rate to rule them all. BMC Biology 8:114.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchêne S, Holt KE, Weill F-X, Le Hello S, Hawkey J, Edwards DJ, Fourment M, Holmes EC.. 2016. Genome-scale rates of evolutionary change in bacteria. Microb Genomics 211:e000094.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enright AJ, Van Dongen S, Ouzounis CA.. 2002. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 307:1575–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RD, Clements J, Arndt W, Miller BL, Wheeler TJ, Schreiber F, Bateman A, Eddy SR.. 2015. HMMER web server: 2015 update. Nucleic Acids Res. 43(W1):W30–W38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores CO, Meyer JR, Valverde S, Farr L, Weitz JS.. 2011. Statistical structure of host-phage interactions. Proc Natl Acad Sci U S A. 10828:E288–E297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasson MJ. 1983. Plasmid complements of Streptococcus lactis NCDO 712 and other lactic streptococci after protoplast-induced curing. J Bacteriol. 1541:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gojobori T, Moriyama EN, Kimura M.. 1990. Molecular clock of viral evolution, and the neutral theory. Proc Natl Acad Sci. 8724:10015–10018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez P, Buckling A.. 2011. Bacteria-phage antagonistic coevolution in soil. Science 3326025:106–109. [DOI] [PubMed] [Google Scholar]
- Hayes S, Murphy J, Mahony J, Lugli GA, Ventura M, Noben J-P, Franz CMAP, Neve H, Nauta A, van Sinderen D.. 2017. Biocidal inactivation of Lactococcus lactis bacteriophages: efficacy and targets of commonly used sanitizers. Front Microbiol. 8:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heled J, Drummond AJ.. 2008. Bayesian inference of population size history from multiple loci. BMC Evol Biol. 8:289.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrix RW, Smith MC, Burns RN, Ford ME, Hatfull GF.. 1999. Evolutionary relationships among diverse bacteriophages and prophages: all the world’s a phage. Proc Natl Acad Sci U S A. 965:2192–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson DH, Bryant D.. 2006. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol. 232:254–267. [DOI] [PubMed] [Google Scholar]
- Jover LF, Effler TC, Buchan A, Wilhelm SW, Weitz JS.. 2014. The elemental composition of virus particles: implications for marine biogeochemical cycles. Nat Rev Microbiol. 127:519–528. [DOI] [PubMed] [Google Scholar]
- Katoh K, Standley DM.. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 304:772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennemann L, Didelot X, Aebischer T, Kuhn S, Drescher B, Droege M, Reinhardt R, Correa P, Meyer TF, Josenhans C et al. , 2011. Helicobacter pylori genome evolution during human infection. Proc Natl Acad Sci. 10812:5033–5038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M. 1969. The rate of molecular evolution considered from the standpoint of population genetics. Proc Natl Acad Sci. 634:1181–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landan G, Graur D.. 2007. Heads or Tails: a simple reliability check for multiple sequence alignments. Mol Biol Evol. 246:1380–1383. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R.. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2514:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R.. 1000 Genome Project Data Processing Subgroup 2009. The sequence alignment/map format and SAMtools. Bioinformatics 2516:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahony J, Cambillau C, van Sinderen D.. 2017. Host recognition by lactic acid bacterial phages. FEMS Microbiol Rev. 41(Supp_1):S16–S26. [DOI] [PubMed] [Google Scholar]
- Mahony J, Murphy J, van Sinderen D.. 2012. Lactococcal 936-type phages and dairy fermentation problems: from detection to evolution and prevention. Front Microbiol. 3:335.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marston MF, Martiny JBH.. 2016. Genomic diversification of marine cyanophages into stable ecotypes: cyanophage diversification into ecotypes. Environ Microbiol. 1811:4240–4253. [DOI] [PubMed] [Google Scholar]
- Martin DP, Murrell B, Golden M, Khoosal A, Muhire B.. 2015. RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evol. 11:vev003.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinsohn JT, Radman M, Petit M-A.. 2008. The lambda red proteins promote efficient recombination between diverged sequences: implications for bacteriophage genome mosaicism. PLoS Genet. 45:e1000065.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marttinen P, Hanage WP, Croucher NJ, Connor TR, Harris SR, Bentley SD, Corander J.. 2012. Detection of recombination events in bacterial genomes from large population samples. Nucleic Acids Res. 401:e6.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavrich TN, Hatfull GF.. 2017. Bacteriophage evolution differs by host, lifestyle and genome. Nat Microbiol. 2:17112.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne I, Bayer M, Cardle L, Shaw P, Stephen G, Wright F, Marshall D.. 2010. Tablet—next generation sequence assembly visualization. Bioinformatics 263:401–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minot S, Bryson A, Chehoud C, Wu GD, Lewis JD, Bushman FD.. 2013. Rapid evolution of the human gut virome. Proc Natl Acad Sci U S A. 11030:12450–12455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno CM, Ghai R, Rodriguez-Valera F.. 2014. Evidence for metaviromic islands in marine phages. Front Microbiol. 5:27.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhammed MK, Krych L, Nielsen DS, Vogensen FK.. 2017. A high-throughput qPCR system for simultaneous quantitative detection of dairy Lactococcus lactis and Leuconostoc bacteriophages. PLoS ONE 123:e0174223.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller-Merbach M, Kohler K, Hinrichs J.. 2007. Environmental factors for phage-induced fermentation problems: replication and adsorption of the Lactococcus lactis phage P008 as influenced by temperature and pH. Food Microbiol. 24(7–8):695–702. [DOI] [PubMed] [Google Scholar]
- Murphy J, Bottacini F, Mahony J, Kelleher P, Neve H, Zomer A, Nauta A, van Sinderen D.. 2016. Comparative genomics and functional analysis of the 936 group of lactococcal Siphoviridae phages. Sci Rep. 6:21345.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ota T, Kimura M.. 1971. On the constancy of the evolutionary rate of cistrons. J Mol Evol. 11:18–25. [DOI] [PubMed] [Google Scholar]
- Pal C, Maciá MD, Oliver A, Schachar I, Buckling A.. 2007. Coevolution with viruses drives the evolution of bacterial mutation rates. Nature 4507172:1079–1081. [DOI] [PubMed] [Google Scholar]
- Parente E, Cogan TM, Powell IB.. 2017. Starter cultures: general aspects In: Cheese, 4th ed Elsevier; p. 201–226. Available from http://linkinghub.elsevier.com/retrieve/pii/B9780124170124000089 [Google Scholar]
- Peris JB, Davis P, Cuevas JM, Nebot MR, Sanjuán R.. 2010. Distribution of fitness effects caused by single-nucleotide substitutions in bacteriophage f1. Genetics 1852:603–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard L. 2017. pyani: Python module for average nucleotide identity analyses. Available from https://github.com/widdowquinn/pyani
- Quinlan AR, Hall IM.. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 266:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A, Lam TT, Max Carvalho L, Pybus OG.. 2016. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 21:vew007.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A, Suchard MA, Drummond AJ.. 2014. Tracer v1.6. Available from: http://beast.bio.ed.ac.uk/Tracer
- Rice P, Longden I, Bleasby A.. 2000. EMBOSS : the European Molecular Biology Open Software Suite. Trends Genet. 166:276–277. [DOI] [PubMed] [Google Scholar]
- Roucourt B, Lavigne R.. 2009. The role of interactions between phage and bacterial proteins within the infected cell: a diverse and puzzling interactome. Environ Microbiol. 1111:2789–2805. [DOI] [PubMed] [Google Scholar]
- Roux S, Hawley AK, Torres Beltran M, Scofield M, Schwientek P, Stepanauskas R, Woyke T, Hallam SJ, Sullivan MB.. 2014. Ecology and evolution of viruses infecting uncultivated SUP05 bacteria as revealed by single-cell- and meta-genomics. eLife 3:e03125.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozov R, Brown Kav A, Bogumil D, Shterzer N, Halperin E, Mizrahi I, Shamir R.. 2017. Recycler: an algorithm for detecting plasmids from de novo assembly graphs. Bioinformatics 334:475–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Russel DW.. 2001. Molecular cloning: a laboratory manual, 3rd ed Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press [Google Scholar]
- Samson JE, Moineau S.. 2013. Bacteriophages in food fermentations: new frontiers in a continuous arms race. Annu Rev Food Sci Technol. 4:347–368. [DOI] [PubMed] [Google Scholar]
- Sanjuán R, Nebot MR, Chirico N, Mansky LM, Belshaw R.. 2010. Viral mutation rates. J Virol. 8419:9733–9748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaltriti E, Launay H, Genois M-M, Bron P, Rivetti C, Grolli S, Ploquin M, Campanacci V, Tegoni M, Cambillau C et al. , 2011. Lactococcal phage p2 ORF35‐Sak3 is an ATPase involved in DNA recombination and AbiK mechanism. Mol Microbiol. 801:102–116. [DOI] [PubMed] [Google Scholar]
- Scaltriti E, Tegoni M, Rivetti C, Launay H, Masson J-Y, Magadan AH, Tremblay D, Moineau S, Ramoni R, Lichière J et al. , 2009. Structure and function of phage p2 ORF34p2, a new type of single-stranded DNA binding protein. Mol Microbiol. 736:1156–1170. [DOI] [PubMed] [Google Scholar]
- Schwartz DA, Lindell D.. 2017. Genetic hurdles limit the arms race between Prochlorococcus and the T7-like podoviruses infecting them. ISME J. 118:1836–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcherbakov VP, Plugina LA, Nesheva MA.. 1992. Genetic recombination in bacteriophage T4: single-burst analysis of cosegregants and evidence in favor of a splice/patch coupling model. Genetics 1314:769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 309:1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suttle C. 2007. a. 2007. Marine viruses–major players in the global ecosystem. Nat Rev Microbiol. 510:801–812. [DOI] [PubMed] [Google Scholar]
- Suyama M, Torrents D, Bork P.. 2006. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 34(Web Server):W609–W612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczepańska AK. 2009. Bacteriophage-encoded functions engaged in initiation of homologous recombination events. Crit Rev Microbiol. 353:197–220. [DOI] [PubMed] [Google Scholar]
- Terzaghi BE, Sandine WE.. 1975. Improved medium for lactic streptococci and their bacteriophages. Appl Microbiol. 296:807.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorne JL, Kishino H.. 2002. Divergence time and evolutionary rate estimation with multilocus data. Syst Biol. 515:689–702. [DOI] [PubMed] [Google Scholar]
- Vegge CS, Vogensen FK, Grath SM, Neve H, van Sinderen D, Brøndsted L.. 2006. Identification of the lower baseplate protein as the antireceptor of the temperate lactococcal bacteriophages TP901-1 and Tuc2009. J Bacteriol. 1881:55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldor MK, Mekalanos JJ.. 1996. Lysogenic conversion by a filamentous phage encoding cholera toxin. Science 2725270:1910–1914. [DOI] [PubMed] [Google Scholar]
- Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ.. 2009. Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 259:1189–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wick RR, Schultz MB, Zobel J, Holt KE.. 2015. Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 3120:3350–3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuckerkandl E, Pauling L.. 1965. Evolutionary divergence and convergence in proteins Evolving genes and proteins. Elsevier; p. 97–166. Available from http://linkinghub.elsevier.com/retrieve/pii/B9781483227344500176 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.