

Abstract
Hyenas (family Hyaenidae), as the sister group to cats (family Felidae), represent a deeply diverging branch within the cat-like carnivores (Feliformia). With an estimated population size of <10,000 individuals worldwide, the brown hyena (Parahyaena brunnea) represents the rarest of the four extant hyena species and has been listed as Near Threatened by the IUCN. Here, we report a high-coverage genome from a captive bred brown hyena and both mitochondrial and low-coverage nuclear genomes of 14 wild-caught brown hyena individuals from across southern Africa. We find that brown hyena harbor extremely low genetic diversity on both the mitochondrial and nuclear level, most likely resulting from a continuous and ongoing decline in effective population size that started ∼1 Ma and dramatically accelerated towards the end of the Pleistocene. Despite the strikingly low genetic diversity, we find no evidence of inbreeding within the captive bred individual and reveal phylogeographic structure, suggesting the existence of several potential subpopulations within the species.
Keywords: evolution, hyena, genomics, population genomics, diversity
Introduction
Hyaenidae occupy a major, albeit species-poor branch within Feliformia. The family has a rich fossil history but is now restricted to only four extant species (Koepfli et al. 2006). Members of Hyaenidae occupy a variety of different niches, including predatory, scavenging, and termite-feeding. The most notable, and arguably most important, of these niches is that of the scavenger (Gusset and Burgener 2005; Watts and Holekamp 2007). Scavengers are known to be important for maintaining ecosystem function with profound roles in nutrient cycling and in influencing disease dynamics (Benbow et al. 2015). However, despite the close relationship of Hyaenidae to the well-studied Felidae family, of which a number of genomes have already been sequenced (Cho et al. 2013; Dobrynin et al. 2015; Abascal et al. 2016), and its ecological importance, no genomic studies have been carried out on the Hyaenidae family. Genomic data sets can help to more accurately gauge population structure and connectivity, taxonomic relationships, genetic diversity, and the demographic history of a species than more traditional methods (e.g., mitochondrial markers or microsatellites) (Allendorf et al. 2010; Steiner et al. 2013; Shafer et al. 2015).
The brown hyena (Parahyaena brunnea) is predominantly a scavenger, mainly feeding on large vertebrate carrion (Watts and Holekamp 2007). Brown hyena are generally found in arid and semiarid areas across southern Africa, in small clans ranging in size from a single female and her cubs to extended families that include a female, her adult offspring of both sexes, and an immigrant male. Females, but not males, breed in the natal clan, necessitating males to leave their natal area by either immigrating into a new clan or by adopting a nomadic lifestyle in order to have reproductive success (Watts and Holekamp 2007). The brown hyena is the rarest of all extant hyena species with estimates of population size being <10,000 individuals worldwide and are listed as Near Threatened by the International Union for Conservation of Nature (IUCN) (Wiesel 2015). Despite its listing as Near Threatened, brown hyenas continue to be persecuted, often considered as problem animals by farmers or killed for trophy hunting. Incidental and often deliberate poisoning, shooting, and trapping of these animals all hamper the survival of this ecologically important species (Kent and Hill 2013).
Previous genetic studies of the brown hyena had limited results in regard to population structure but have hinted towards very low genetic diversity within the species (Rohland et al. 2005; Knowles et al. 2009). Species-wide genetic comparisons using a short fragment of the mitochondrial cytochrome b gene found no variability regardless of sample origin (Rohland et al. 2005). Moreover, a study investigating population structure within Namibian brown hyenas, using microsatellites, also found no detectable population structure (Knowles et al. 2009). One possible explanation for the inability of both of these studies to find population structure could be very low levels of genetic diversity within the brown hyena. Even though extant genetic diversity does not necessarily correlate with current day population sizes (Bazin et al. 2006; Leffler et al. 2012), it represents an important parameter for understanding past evolutionary events. Moreover, knowledge of the evolutionary processes affecting a species is critical to inform conservation plans aimed at the long-term management of its evolutionary potential (Romiguier et al. 2014). Thus, these early indications of low diversity are important to investigate further.
Here, we present both complete mitochondrial genomes and low-coverage nuclear genomes of brown hyena from populations across its range in southern Africa (fig. 1), as well as a single high-coverage nuclear genome from a captive individual bred from wild-caught parents. We find both mitochondrial and nuclear genomic diversity to be extremely low, yet we find no signs of inbreeding in the high-coverage genome. Furthermore, demographic analyses suggest that this low diversity results from a continuous decrease in effective population size over the last million years. Finally, we further reveal a number of potential subpopulations across the brown hyena’s range revealed by low-coverage complete nuclear genomes.
Fig. 1.
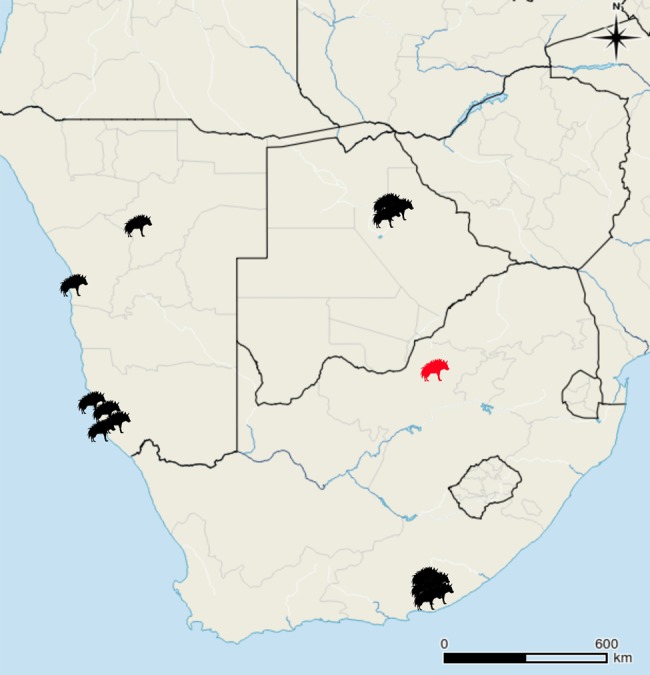
Map of the sampling locations of the wild-caught brown hyena included in this study. The red hyena indicates the original area of the South African population prior to translocations in 2003. Map generated using QGIS 2.0.1-Dufour (QGIS Development Team, 2016. QGIS Geographic Information System. Open Source Geospatial Foundation Project. http://www.qgis.org/).
Results
Genome Reconstructions
Using ∼56× coverage data from Illumina 2 ×150 bp PE shotgun sequencing, we obtained a de novo assembly of a striped hyena (Hyaena hyaena) nuclear genome with a scaffold N50 of ∼2 Mb using Allpaths LG (Butler et al. 2008), default parameters, and an additional gap closing step using Gapcloser (Luo et al. 2012) (supplementary table S1, Supplementary Material online). Benchmarking Universal Single-Copy Orthologs (BUSCO) analyses (Simão et al. 2015) using both the eukaryotic and mammalian databases show high levels of complete BUSCOs (89.4% and 93.4%, respectively) (supplementary table S2, Supplementary Material online), giving us confidence that our assembly is of both good quality and completeness. With this as reference, we successfully mapped low-coverage nuclear genomes (2.1–3.7×) from 14 wild-caught brown hyena individuals originating from Namibia, South Africa, and Botswana and a high-coverage nuclear genome from a single captive individual (supplementary tables S3 and S4, Supplementary Material online) using Burrows-Wheeler Alignment Tool (BWA) (Li and Durbin 2009).
Due to the lack of a published brown hyena mitochondrial genome, we assembled the complete mitochondrial genome de novo from the captive individual using MITObim (Hahn et al. 2013) using a similar approach to Westbury et al. (2017). We used default parameters apart from mismatch value, where we used zero. We ran three independent assemblies using a different bait reference sequence for each (domestic cat [U20753.1], spotted hyena [JF894377.1], and striped hyena [NC_020669.1]). All three independent MITObim runs produced identical brown hyena mitochondrial sequences, providing strong evidence that our reconstructed mitochondrial genome is correct. We then mapped the wild individuals to this sequence using BWA (supplementary table S4, Supplementary Material online).
Genetic Diversity
Mitochondrial DNA diversity estimates of the 14 wild-caught brown hyena individuals show an average of only four mutational mismatches (k) among them (fig. 2). In order to investigate the relative level of and to put this k value into context, we compared this value against mitochondrial diversity estimates calculated from a number of other mammalian species of different IUCN conservation statuses. This comparison showed the brown hyena to have a very low level of diversity relative to other species used in the comparison. Brown hyena mitochondrial diversity is two times lower than the species with the next lowest level of mitochondrial diversity, the tasmanian devil (Sarcophilus harrisii), with a k value of 8, and 47.5 times lower than the chimpanzee (Pan troglodytes), which has a k value of 190. We then calculated genome-wide autosomal heterozygosity estimates of our high-coverage captive individual to be 0.000121 (SD = 0.000022, calculated using 20-Mb nonoverlapping windows constituting only covered bases). We compared this result with the heterozygosity of a number of species for which nuclear genomes of at least 20× coverage were available (fig. 3A and supplementary table S5, Supplementary Material online). This showed the brown hyena to have extremely low levels of autosomal heterozygosity, consistent with the very low levels of diversity we found within the mitochondrial genome. The captive brown hyena individual is known to be the offspring of wild-caught parents, albeit in a captive environment, and should therefore not display a large extent of inbreeding as can be found in some captive bred populations. We therefore measured levels of heterozygosity in various window sizes across the brown hyena nuclear genome in order to test for inbreeding in this brown hyena. Visual inspections show no considerable stretches of homozygosity ≥5 Mb (supplementary fig. S1, Supplementary Material online), suggesting no obvious or substantial signs of inbreeding on the genome of this individual that would differ from what is expected in an out-bred individual (McQuillan et al. 2008). However, while it is believed that the parents of the captive bred individual came from the same population, it is unknown whether the relatedness of the parents and therefore the level of inbreeding within this individual represents those found in nature.
Fig. 2.
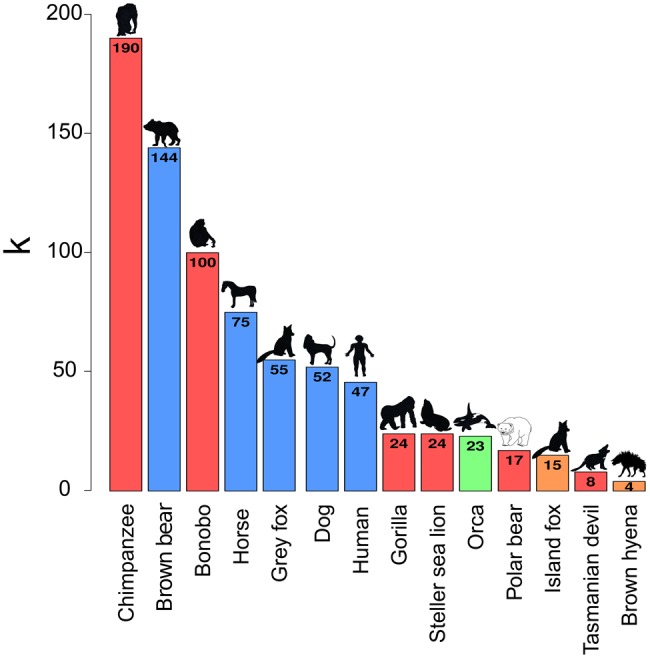
Mitochondrial diversity comparisons of the brown hyena to a number of other mammalian species for which such data are available. k represents the average number of substitutions expected between the mitochondrial genomes of two randomly selected individuals of the same species, bar colors represent conservation status (red—endangered, orange—near threatened, blue—least concern, green—unavailable) according to the IUCN.
Fig. 3.

Estimated nuclear heterozygosity levels in the brown hyena and comparisons to other mammalian species. (A) Average genome-wide autosomal heterozygosity comparisons. Y axis represents the average proportion of sites within the autosomes to be heterozygous. Bar colors represent conservation status (red—endangered, orange—near threatened, blue—least concern, green—insufficient data) according to the IUCN. (B) Heterozygosity density comparisons between the four species with the lowest estimated average genome-wide heterozygosity levels in this study. Y axis represents the number of heterozygous sites within the 1-Mb window. X axis represents the window. Colors represent species (blue—brown hyena, black—Iberian lynx, magenta—orca, red—cheetah, green—Channel Island fox; San Miguel Island).
We further analyzed the distribution of heterozygosity across the autosomes by randomly selecting 1,000 nonoverlapping windows consisting of 1 Mb of covered sites and comparing the results to the four other species with the lowest heterozygosity included in this study, the orca (Orcinus orca), cheetah (Acinonyx jubatus), San Miguel Channel Island fox (Urocyon littoralis littoralis), and Iberian lynx (Lynx pardinus) (fig. 3B). The brown hyena autosomes have consistently lower levels of heterozygosity across the genome when compared with the Iberian lynx, orca, and cheetah, excluding the windows with the lowest levels of heterozygosity which may indicate higher levels of inbreeding in the other species. However, while the brown hyena has a lower level of average genome-wide diversity, for the majority of the windows, the San Miguel Channel Island fox has lower heterozygosity than the brown hyena.
Demographic History
As sex chromosomes may have a different demographic history than autosomes, we identified and removed scaffolds in our assembly likely to originate from the X chromosome before running demographic analyses. We aligned our striped hyena assembly by synteny (Grabherr et al. 2010) to the cat X chromosome (126,427,096 bp) using Satsuma (Grabherr et al. 2010) and found 195 scaffolds (supplementary table S6, Supplementary Material online), totaling 117,479,157 bp. The alignments were visualized using Circos (Krzywinski et al. 2009) (supplementary fig. S2, Supplementary Material online). The Circos alignment showed that the scaffolds found using Satsuma cover the complete cat X chromosome. These scaffolds were then removed, along with all scaffolds under 1 Mb in length, before running a Pairwise Sequentially Markovian Coalescent (PSMC) model analysis (Li and Durbin 2011). We estimated the mutation rate per generation to be 7.5 ×10−9 by carrying out a pairwise distance analysis between the autosomes of the striped and brown hyena using a consensus base identity by state (IBS) approach in ANGSD. Using this result (pairwise distance = 0.00525) we then calculated the average per generation mutation rate assuming the divergence date of the two species to be 4.2Ma (Koepfli et al. 2006), a genome-wide strict molecular clock and a generation time of 6 years. This mutation rate and a generation time of 6 years was used to calibrate and plot the PSMC analysis (fig. 4). Additional PSMC plots using alternative mutation rates representing the minimum (2.6Ma) and maximum (6.4Ma) values within the 95% credibility interval of the brown and striped hyena divergence from Koepfli et al. (2006) (mutation rates = 12 ×10−9 and 5.0×10−9, respectively) can be seen in supplementary figure S3, Supplementary Material online.
Fig. 4.

Pairwise Sequentially Markovian Coalescent (PSMC) model plot of the autosomes of one high-coverage brown hyena. The Y axis represents effective population size and the X axis represents time in years. Light red bars show bootstrap support values. Calibrated using a generation time (g) of 6 years and a mutation rate (μ) of 7.5×10−9 per generation. Blue line presents δ18O levels based on data found in Zachos et al. (2001).
PSMC analyses using the brown hyena autosomes were consistent with its low genomic diversity, and revealed a continuous gradual decrease in effective population size from ∼44,000 to ∼11,000 over the last 1 My. This was followed by a more rapid recent decrease towards the end of the Late Pleistocene (∼40 kya), culminating in an effective population size of ∼2,600 for the present day (fig. 4). As PSMC uses linkage to infer demographic history, we also ran the analysis using all autosomal scaffolds regardless of length to investigate the influence shorter scaffolds would have on the result. This analysis also produced the same pattern and result.
Population Structure
To investigate population structure we constructed a haplotype network using the mitochondrial genomes of the wild-caught individuals. This revealed some phylogeographic structure with all but one haplotype being geographically restricted. This single haplotype was, however, shared among individuals from all three sampled countries (fig. 5A).
Fig. 5.

Population structure analyses. (A) Median joining mitochondrial haplotype network of the 14 wild-caught brown hyena individuals included in this study. Lines on the connecting branches represent single base differences, size of the circle represents number of individuals belonging to a single haplotype and colors represent sampling country (black—South Africa, blue—Namibia, red—Botswana). (B) Principal components analysis produced using genotype likelihoods for the low-coverage genomes of the 14 wild-caught brown hyena individuals in this study. Colors represent country of origin (black—South Africa, blue—Namibia, red—Botswana). Percentages on the X and Y axis represent the percentage of variance explained by each respective component. (C) Densitree phylogenetic tree. Light gray lines represent phylogenetic trees produced from single scaffolds. The dark black line represents the root canal as defined by Densitree.
We then used the mapped low-coverage nuclear genomes of the wild-caught individuals to infer nuclear population structure by carrying out genome wide principal component analyses (PCA) using both single base identity by state (IBS) and genotype likelihoods (GL), in addition to maximum likelihood (ML) phylogenetic analyses of pseudohaploidized consensus sequences. The results clustered individuals consistent with their geographical origin in all cases (fig. 5B andsupplementary fig. S4, Supplementary Material online). PCA using both single base IBS and GL produced similar results (fig. 5B andsupplementary fig. S5, Supplementary Material online).
In contrast to whole nuclear genomic data, both PCA and phylogenetic analyses using single scaffolds did not support a unanimous phylogeographic pattern. Independent PCA results for the nine longest scaffolds are shown in supplementary figure S6, Supplementary Material online. Although they do generally support some phylogeographic structure, individuals from different regions partially intermingle in these plots. When running independent per-scaffold ML phylogenetic analyses with scaffolds over 2 MB, we find that in 49 out of 333 trees (14.7%), the South African samples form a monophyletic clade, in 72 out of 333 trees (21.6%), the Botswana samples form a monophyletic clade, and in 17 out of 333 trees (5.1%), the Namibian samples form a monophyletic clade. This nonunanimous pattern shows up as clouds surrounding all nodes within the brown hyena lineage when visualizing all independent ML trees simultaneously using Densitree (Bouckaert 2010) (fig. 5C). Although comparatively few individual trees support monophyly of either of the three geographical groups, the root canal (consensus tree with highest clade support), defined by Densitree, nevertheless shows a similar topology as a tree constructed using the complete nuclear genome (fig. 5C andsupplementary fig. S4, Supplementary Material online). This consensus tree is again consistent with the geographic origin of the individuals.
Admixture and population structure analyses using NGSadmix (Skotte et al. 2013) reached convergence for K values (i.e., assumed number of populations) of 2–5, providing an estimate of the number of populations currently inhabiting southern Africa (fig. 6). When considering a K value congruent with the number of countries of origin, K3 (fig. 6B), individuals NamBH13 and NamBH21 stand out as being admixed with all three populations, although this may be an artifact resulting from incorrect population assignment. A K value of four, however, clusters these individuals as an independent population (fig. 6C). The latter is consistent with their geographic origins (northern Namibia while all other Namibian samples are from the south) and previous findings (Wiesel 2015 unpublished data) investigating the distribution patterns of individuals across Namibia (supplementary fig. S7, Supplementary Material online).
Fig. 6.

Admixture plots produced using different K values in NGSAdmix. (A)K = 2, (B)K = 3, (C)K = 4, (D)K = 5. Y axis represents the admixture proportion and X axis represents the individual.
To further test this, we carried out D-statistic comparisons testing for population structure (fig. 7). A high D value could either represent differential levels of admixture or more recent common ancestry and therefore an incorrect predefined topology. Taking the latter into account, we placed individuals into one of three (defined as: Namibia, Botswana, or South Africa) or one of four (defined as: northern Namibia, southern Namibia, Botswana, or South Africa) predefined populations as hypothesized by the PCA and NGSadmix results and compared the D values produced from “correct” topologies, that is, branches H1 and H2 belong within the same population (fig. 7A), and “incorrect” topologies, that is, branches H2 and H3 belong within the same population (fig. 7B). When considering four predefined populations (fig. 7C and supplementary table S7, Supplementary Material online), it can consistently be seen that when we break the predefined population structure and therefore topology, we find higher D values than when individuals from the same predefined population are in the H1 and H2 positions. Assuming that the correct topology is that with the lowest D value, and as there is clear separation between D values recovered when comparing “correct” and “incorrect” topologies when using four predefined populations, we conclude that there are indeed four populations within our data set. This pattern is not seen when only three predefined populations (fig. 7D and supplementary table S8, Supplementary Material online) are considered as there are many overlapping D values (seen as an overlap in SDs) within the Namibian population when “correct” (mean D score = 0.0130, SD = 0.00838) and “incorrect” topologies (mean D score = 0.0907, SD = 0.0827) are tested. This led us to reject the possibility of one single population within Namibia. Taken together, our results suggest four populations, with a split between northern and southern Namibia. This analysis revealed the same population structure as the NGSadmix analyses, thus corroborating the observation that this data set consists of four populations rather than three.
Fig. 7.

Topology test using D-statistics. (A) D-statistic analysis demonstrating the correct predetermined population structure. (B) D-statistic analysis demonstrating the incorrect predetermined population structure. (C) D-statistics comparisons when four populations are determined a priori. (D) D-statistics comparisons when three populations are determined a priori. Red colored circles represent comparisons when the predefined population structure is not broken within the comparison. Blue colored circles represent comparisons when the predefined population structure is broken within the comparison. X axis represents the D value.
Finally, we carried out IBS analyses on a number of other mammalian species for which species-wide low-coverage data were available in order to compare these with the population structure within the brown hyena. Each species, including the brown hyena, had 10 representatives randomly selected and pairwise distance comparisons were carried out within each species (supplementary fig. S8, Supplementary Material online). For the brown hyena, most pairwise distance values are closely overlaid (mean value = 0.467, SD = 0.032), representing a relatively high level of shared diversity between different putative populations and suggesting extensive gene flow among these populations. This low variability in pairwise distances, as shown by the SD, is not as small as in some species, such as domesticated sheep (mean value = 0.471, SD = 0.011), which are known to be approaching panmixia (Peter et al. 2007) but still smaller than within other species included in this analysis such as the orca (mean value = 0.461, SD = 0.147) and the chimpanzee (mean value = 0.481, SD = 0.074). This result still suggests some, albeit low, level of differentiation within the brown hyena. We also found no abnormalities, that is, potential subspecies, large differences between populations, or indications that a number of the individuals were extraordinarily related, which could be driving the population signal we find in the brown hyena.
Discussion
Using complete mitochondrial genomes of 14 wild-caught brown hyena originating from across southern Africa and a single high-coverage nuclear genome from an individual bred in captivity from wild-caught parents, we find that brown hyena display very low levels of both mitochondrial and nuclear genomic diversity, lower than that of many other mammalian species, including several listed as endangered (figs. 2 and 3A). Genomic diversity was even lower than in individuals from species famously known to have extremely low levels of genomic diversity, such as the cheetah (Dobrynin et al. 2015) and the Iberian lynx (Abascal et al. 2016), both of which have gained considerable research attention because of this characteristic. Interestingly, many other species with low levels of genomic diversity (e.g., Iberian lynx and Channel Island foxes) have restricted ranges, which is thought to increase the likelihood of inbreeding due to low population connectivity hence a bigger impact of genetic drift and ultimately culminating in low diversity. The brown hyena, comparatively, has a range of ∼2,400,000 km2 (Wiesel 2015) and as we show, high levels of population connectivity, making its low genetic diversity both a unique and unexpected finding. Furthermore, despite very low levels of genetic diversity within both the nuclear and mitochondrial genomes, we found no strong signs of inbreeding in the nuclear genome of a brown hyena born in captivity from wild-caught parents (fig. 3B andsupplementary fig. S1, Supplementary Material online). This was a somewhat unexpected result as low genomic diversity is generally expected to arise within very small or recently bottlenecked populations within which, due to the limited number of alleles in the gene pool, one would expect to find signs of inbreeding. This finding is, however, in accordance with the large geographical range of the brown hyena.
Independent from inbreeding, low genomic diversity has commonly been associated with an increased likelihood of genetic disease and a decreased adaptive potential in a changing environment, both of which can lead to increased extinction rates (Reed and Frankham 2003; Spielman et al. 2004). However, in contrast to the Iberian lynx (Peña et al. 2006; Jiménez et al. 2008; Martínez et al. 2013) or the Florida panther (Roelke, Forrester, et al. 1993; Roelke, Martenson, et al. 1993; Johnson et al. 2010), currently, there is no apparent sign of severe genetic diseases in the brown hyena, suggesting that brown hyenas are not negatively affected by their low genetic diversity. However, further investigations would be required to unravel the true influence that low genetic diversity may have on the survivability of the brown hyena. A possible explanation for this phenomenon, low genetic diversity but a lack of detrimental consequences, has previously been described within mountain and eastern lowland gorillas (Xue et al. 2015). This study hypothesized that long-term low effective population sizes may have prevented the accumulation of severely deleterious mutations in these gorillas due to the more frequent exposure of these alleles in a homozygous state. This would allow the mutation to be more efficiently purged from the gene pool than in populations with larger effective sizes which can allow the deleterious allele to persist in a heterozygous state.
Interestingly, different populations of Channel Island foxes represent another set of examples where extremely low genetic diversity has not resulted in obvious genetic defects (Robinson et al. 2016). The only island fox population with >20× genome coverage, that is, the one from San Miguel Island, has higher overall genomic diversity than the brown hyena. However, another island fox population, from San Nicolas Island, although not sequenced to sufficient coverage for our analyses, has been shown to display even lower levels of genomic diversity than the San Miguel Island population when compared using allozymes and hypervariable minisatellite DNA (Wayne et al. 1991) as well as medium coverage genomes (Robinson et al. 2016). Thus, while a direct comparison to our brown hyena data is not (yet) possible, there are likely mammalian island populations with even lower genomic diversity than the brown hyena. Independent of their absolute levels of genomic diversity, Channel Island fox populations show heterozygosity hotspots in a number of genes with high levels of ancestral variation, despite low levels of genome-wide heterozygosity. These heterozygosity hotspots have been interpreted as likely important adaptive variation allowing for the retention of evolutionary potential in Channel Island foxes (Robinson et al. 2016). In contrast, heterozygosity appears to be fairly evenly distributed across the genome in the brown hyena (fig. 3B and supplementary fig. S1, Supplementary Material online) suggesting this may not the case for the brown hyena. However, as each estimate in this study only shows the nuclear genomic diversity of a single individual and as levels are expected to vary between and even within natural populations, some caution must be taken when extrapolating these results to a species wide level.
Nevertheless, given these differences in the distribution of heterozygosity between brown hyena and Channel Island foxes, it is likely that different demographic processes may have led to the observed patterns. The PSMC analyses on our high-coverage brown hyena genome show that the very low levels of genomic diversity are associated with a gradual, yet steady decline in effective population size over the last million years, with a more rapid decline towards the end of the Late Pleistocene (fig. 4). In contrast, Channel Island Fox populations are believed to have first reached the northern Channel Islands at the end of the Pleistocene, ∼40–25 kya, became isolated at the end of the Wisconsin glacial epoch ∼17 kya, and were subsequently transported to the southern islands by Native Americans ∼3,000 years ago (Collins 1991). Thus, it is likely, based on the small size of these islands and the small population size of the individual island populations, that these populations underwent rapid and dramatic bottlenecks caused by multiple founder effects which may have led to the low diversity seen today. Such different population demographics, together with other lineage specific factors such as mutation rates and mating systems, can be expected to result in different distributions of heterozygosity along the genome. However, it is not immediately clear why the latter would result in heterozygosity islands within a sea of almost no diversity while the former resulted in low diversity across the entire genome with a lack of such heterozygosity islands. Even though some island fox populations seem to have lower genetic diversity than the brown hyena, considering the much larger geographic distribution of the latter (by several orders of magnitude) and the much higher census population size, the low genomic diversity of the brown hyena is not only remarkable but also begs for an explanation. These findings also suggest that the correlation between genetic diversity and the perceived risk of extinction may not be as strong as previously thought, since many species with higher genetic diversity are considered to be at greater risk of extinction. However, further research will be needed to uncover the direct relationships of low genetic diversity, fitness, and extinction risk.
The brown hyena is known to have had a more extensive range, with Middle Pleistocene fossils having been found in Kenya (Werdelin and Barthelme 1997) and other putative brown hyena fossils found as far north as Ethiopia (Werdelin and Solounias 1991). The continuous decrease in effective population size and low levels of genomic diversity seen today may have occurred due to a variety of factors. These could include the shrinking of suitable habitats during the Pleistocene (deMenocal 2004), the decrease in carcass availability caused by a number of extinctions within large-bodied carnivore guilds from 2 to 1 Ma (Lewis 1997; Werdelin and Lewis 2005), the migration of new competitors in the form of canids from Eurasia (Koepfli et al. 2015), or a combination of these factors. Interestingly, a number of the brown hyena’s larger bodied bone cracking relatives went extinct during the Pleistocene (Lewis 1997). Thus, although it experienced a drastic population decline, the versatility, and omnivorous diet of the brown hyena may have allowed it to survive until today.
Through the use of a combination of different population structure analyses on the entire nuclear genome we were able to define, for the first time, population structure within the brown hyena (figs. 5–7). We found structuring in concordance with the geographic origins of the individuals, which represented four different populations. Furthermore, by comparing these results to those produced using mitochondrial genomes and single scaffolds, we show that even when using millions of base pairs (i.e., single scaffolds >2 Mb; fig. 5C andsupplementary fig. S6, Supplementary Material online) difficulties with accurately defining population structure can arise. This may be due to the presence of a continually falling effective population size and a large recent bottleneck within the species. The recent bottleneck may have limited the variability within the species leading to the observed low level of differentiation between populations. This effect is especially highlighted when inferring population structure using whole nuclear genomes as opposed to single scaffolds. One possible explanation for these fine scale differences could be the rise of pastoralism in Southern Africa ∼2.5kya (Smith 1992). Pastoralism may have acted as a barrier between populations, rapidly reducing population connectivity and thus allowing for the accumulation of genetic differences, most likely through drift, between populations even when species-wide diversity is low.
All things considered, these findings reinforce the value of using whole nuclear genomes in population analyses over other approaches such as microsatellites and even reduced genomic representation techniques, especially in species suspected of having low genomic diversity such as the brown hyena.
Materials and Methods
Samples
15 brown hyena samples were used for this study; five from South Africa, six from Namibia, three from Botswana (fig. 1), and one from Tierpark Berlin (supplementary table S3, Supplementary Material online). One female striped hyena (Hyaena hyaena) from Tierpark Berlin was also included for use as a mapping reference.
Striped Hyena De Novo Assembly
We extracted DNA from Hyena2069, the striped hyena sample from the Tierpark Berlin, on a KingFisher Duo robot using the blood DNA extraction kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. The extract was then built into two PCR free Truseq Illumina sequencing libraries, one with 180 bp and one with 670 bp short inserts. Two Nextera mate-pair libraries were also constructed with sizes of 3 and 6 kb. All of these libraries were constructed at the National Genomics Infrastructure (NGI) in Stockholm. All libraries were then sequenced on an Illumina HiSeqX using 2 × 150 bp paired-end sequencing at the NGI in Stockholm. The 180- and 670-bp insert libraries were sequenced on one lane each, and the mate-pair libraries were multiplexed and sequenced together on a single lane.
The NGI trimmed Illumina adapter sequences from the raw Illumina reads using Trimmomatic (Bolger et al. 2014) and performed a de novo assembly using Allpaths LG (Butler et al. 2008) with default parameters. We then performed an additional gap closing step using Gapcloser (Luo et al. 2012). Assembly quality and completeness were assessed using BUSCOv2 (Simão et al. 2015) using both the eukaryote and mammalian BUSCO databases (supplementary table S2, Supplementary Material online).
Captive Brown Hyena Sample
We extracted DNA from a single female brown hyena sample from Tierpark Berlin on a KingFisher Duo robot using the cell and tissue DNA extraction kit (Thermo Fisher Scientific). The extract was then built into a PCR free Truseq Illumina sequencing library using a 350-bp insert size by the NGI in Stockholm. This library was then sequenced on a single lane on an Illumina Highseq X using 2 × 150 bp paired-end sequencing at the NGI in Stockholm.
Wild-Caught Brown Hyena Samples
We extracted DNA from six blood and three tissue samples using a Qiagen DNeasy blood and tissue extraction kit, following the manufacturer’s protocol. We also extracted five hair samples using the DY04 user modified version of the Qiagen DNeasy kit and protocol. We fragmented DNA extracts using a Covaris sonicator into ∼500-bp fragments. Fragmented extracts were then constructed into Illumina sequencing libraries using a modified version of the protocol set out by Meyer and Kircher (2010) with modifications (Fortes and Paijmans 2015). Library molecules from 400 to 900 bp were selected using a Pippin Prep Instrument (Sage Science) and sequenced on an Illumina Nextseq 500 at Potsdam University, Germany.
Raw Brown Hyena Data Treatment
We trimmed Illumina adapter sequences and removed reads shorter than 30 bp from the raw reads of the 15 brown hyena samples using Cutadapt v1.8.1 (Martin 2011) and merged overlapping reads using FLASH v1.2.1 (Magoč and Salzberg 2011).
Mitochondrial Genome Assembly
As no brown hyena mitochondrial sequence was available, we reconstructed one using the shotgun data from the high-coverage individual. We assembled the mitochondrial genome through iterative mapping using MITObimv1.8 (Hahn et al. 2013) on 40 million trimmed and merged reads, subsampled using seqtk (Li 2012). We removed duplicate reads using prinseq (Schmieder and Edwards 2011). MITObim was performed in three independent runs using three different starting bait reference sequences. The references included the domestic cat (U20753.1), spotted hyena (JF894377.1), and striped hyena (NC_020669.1). We implemented MITObim using default parameters apart from mismatch value where we used zero. Output maf files were converted to sam files and visualized using Geneious v9.0.5 (Kearse et al. 2012). Consensus sequences were constructed in Geneious using a 75% base call consensus threshold, and only sites with over 20× coverage were considered.
The reconstructed mitochondrial genome served as a reference sequence for subsequent mitochondrial DNA mapping analyses. We mapped the trimmed and merged reads from our 14 wild-brown hyenas to the reconstructed reference sequence using BWAv0.7.15 (Li and Durbin 2009), using the mem algorithm and default parameters and parsed the mapped files using Samtools v1.3.1 (Li et al. 2009). The consensus sequences were constructed using ANGSDv0.913 (Korneliussen et al. 2014), only considering read mapping and phred base quality scores >25.
Mitochondrial Analyses
The mitochondrial genomes from the 14 wild-caught brown hyenas were aligned using Mafftv7.271 (Katoh and Standley 2013). We constructed a median joining haplotype network of the alignment using Popart (Leigh and Bryant 2015). In order to compare the species-wide mitochondrial diversity of the brown hyena to other previously published data, we calculated average pairwise distances (k) for the brown hyena and a number of other mammalian species. To do this, we randomly selected individuals to represent each species from Genbank (supplementary table S9, Supplementary Material online), performed a multiple sequence alignment using Mafft and calculated the overall mean pairwise distances, treating gaps, and missing data as complete deletions using MEGA6 (Tamura et al. 2013) (fig. 2B). We then further separated some of these species into either populations or subspecies (supplementary table S10, Supplementary Material online).
Low-Coverage Nuclear Genome Analyses
Trimmed and merged data were mapped to the striped hyena de novo assembly using BWA v0.7.15 (Li and Durbin 2009) and parsed using Samtools v1.3.1 (Li and Durbin 2009).
We applied the following filtering options for all analyses involving ANGSD (Korneliussen et al. 2014): we only considered sites where at least 10 individuals had coverage (-minInd 10) and only included sites for which the per-site coverage across all individuals was <75. We implemented quality filtering by setting a minimum base quality score of 25 (-minQ 25), minimum mapping quality score of 25 (-minMapQ 25), and only allowed reads that mapped uniquely to one location (-unique_only 1). We also adjusted quality scores around indels (-baq 1) (Li 2011).
Brown Hyena Population Structure
Principal component analyses (PCA) were carried out using both single read IBS analyses and GL analyses in ANGSDv0.913 (Korneliussen et al. 2014). IBS analyses were restricted to single nucleotide polymorphisms (SNPs) occurring in at least two individuals. This was done to remove singletons, which could represent sequencing errors. We computed genotype likelihoods in ANGSD and converted outputs to a covariance matrix using ngsTools (Fumagalli et al. 2014). Covariance matrices were converted into PCA outputs and visualized using R (R Development Core Team 2008). For the phylogenetic analyses, we performed Maximum likelihood analyses with RAxML v8.2.10 (Stamatakis 2014), specifying the striped hyena as outgroup and using the GTR + GAMMA substitution model. We prepared the infile for this by computing consensus sequences using ANSGD with the above-mentioned filters. We then performed genome-wide alignments, removed sites with missing data in three or more individuals, sites where singletons occurred within the brown hyena ingroup, and invariant site positions using a custom Perl script.
We then repeated the phylogenetic and IBS PCA analyses using single scaffolds. PCA analyses were carried out using nine independent analyses on the nine largest scaffolds (scaffolds 0–8) and maximum likelihood analyses were carried out independently for single scaffolds >2 Mb. We calculated admixture proportions using NGSadmix (Skotte et al. 2013) setting K values from 2 to 7. We used ANGSD genotype likelihood values as input, only including SNPs with a P value of <1 ×10−6. NGSadmix analyses were repeated a maximum of 100 times per K. Only those that converged (produced a consistently identical likelihood score) within these 100 analyses were considered as meaningful. D-statistic analyses were implemented in ANGSDv0.913, sampling a single base per site while specifying the striped hyena as outgroup with default parameters.
Comparative Population Structures
In order to compare the population structure within the brown hyena to those of other species, 10 individuals per species were randomly selected from a number of different mammals for which such data were publicly available (supplementary table S11, Supplementary Material online). Comparisons between individuals were performed using single base IBS, only considering sites where at least seven individuals had coverage and SNPs that occurred in at least two individuals. Other filtering options were: a minimum base quality score of 25 (-minQ 25), minimum mapping quality score of 25 (-minMapQ 25), and reads that mapped uniquely to one location (-unique_only 1). Quality scores around indels were also adjusted for (-baq 1).
Species Heterozygosity Estimates
High coverage, single individual representatives for a number of species were assessed for autosomal heterozygosity levels to be compared against that of our high coverage, captive bred brown hyena individual. Raw data were selected from a range of different species (supplementary table S12, Supplementary Material online). Raw reads were all treated comparably, using Cutadapt v1.8.1 (Martin 2011) to trim Illumina adapter sequences and FLASH v1.2.1 (Magoč and Salzberg 2011) to merge overlapping reads. We mapped each species to its respective reference sequence using BWAv0.7.15 (Li and Durbin 2009) and processed the mapped reads further using Samtools v1.3.1 (Li et al. 2009). To adjust for biases that could be introduced by unequal levels of coverage, the resulting bam files were all subsampled to an average of 20× using Samtools (Li et al. 2009). The autosomal heterozygosity was estimated from sample allele frequencies, taking genotype likelihoods into account for each species representative using ANGSDv0.913 (Korneliussen et al. 2014). The software ANGSD was chosen as it can overcome the biases that may arise due to differential coverage across the genome. Instead of other methods that rely on direct SNP/genotype calling from the data, ANGSD uses genotype likelihoods in downstream analyses. This allows for the incorporation of statistical uncertainties into the analysis, which in turn reduce the biases caused by differential coverage across the genome. SDs were calculated by estimating the heterozygosity in various window sizes across the genome. These windows were constituted of either 20, 50, or 100 Mb of covered sites (supplementary table S5, Supplementary Material online). For the 1-Mb window analysis, we used the same analysis to estimate heterozygosity as before but performed the analysis in nonoverlapping windows consisting of 1 Mb of covered sites. To investigate potential signs of inbreeding, we investigated the genome of the captive bred brown hyena for runs of homozygosity (supplementary fig. S1, Supplementary Material online). We performed this analysis by estimating heterozygosity across the genome using three different sized windows, 2, 1, and 0.5 Mb. Heterozygosity proportions for each window were then plotted and visually investigated for large numbers of consecutive windows with relatively low levels of heterozygosity using a 5-Mb run of homozygosity as the threshold to indicate recent inbreeding, as has been previously suggested (McQuillan et al. 2008).
Demographic Inference
The demographic history of the brown hyena was calculated using only the autosomal chromosomes in PSMC (Li and Durbin 2011). Scaffolds representing the X chromosome of the striped hyena were determined through a synteny analysis to the cat X chromosome (CM001396.2) using Satsuma synteny (Grabherr et al. 2010). These scaffolds were then removed along with any scaffold shorter than 1 Mb. A consensus diploid sequence was constructed using Samtools (Li et al. 2009) to be used as input for PSMC. PSMC was implemented using parameters previously shown to be meaningful when considering human data. One hundred bootstrap analyses were undertaken. When plotting, we assumed a generation time of 6 years and a mutation rate of 7.5 ×10−9 per generation for autosomes. Generation time was estimated based off of the estimated breeding ages of a number of Namibian brown hyena based on tooth wear. The ages of these individuals being ∼12, 14, 9–11, 4–5, 3–4, and 6 years of age.
In order to estimate the mutation rate, we carried out a pairwise distance analysis on the striped and brown hyena’s autosomes using a consensus base IBS approach in ANGSDv0.913. We then calculated the average per generation mutation rate assuming the divergence date of the two species to be 4.2Ma (Koepfli et al. 2006), a genome-wide strict molecular clock and a generation time of 6 years. Additional analyses utilizing different mutation rates based on the 95% confidence interval of the brown and striped hyena divergence from Koepfli et al. 2006 (2.6 and 6.4 Ma) can be seen in supplementary figure S3, Supplementary Material online.
Supplementary Material
Supplementary data are available at Molecular Biology and Evolution online.
Supplementary Material
Acknowledgments
This work was supported by the European Research Council (consolidator grant GeneFlow # 310763 to M.H.). The authors also acknowledge support from Science for Life Laboratory, the Knut and Alice Wallenberg Foundation, the National Genomics Infrastructure funded by the Swedish Research Council, and Uppsala Multidisciplinary Center for Advanced Computational Science for assistance with massively parallel sequencing, as well as de novo assembly of the striped hyena and access to the UPPMAX computational infrastructure. L.D. acknowledges support from the Swedish Research Council and FORMAS. We would like to thank Prof. Yoshan Moodley for his suggestions on the manuscript. We would finally like to thank Binia De Cahsan for producing the animal icons found in figures 1–3.
Author Contributions
The project was conceived by M.V.W. and M.H. M.V.W. and L.D. performed lab work; M.V.W., S.H., and A.B. performed DNA analyses and interpretation of results. I.W., V.L., R.W., D.M.P., F.S., and A.L. assisted with locating and sampling of specimens. Final editing and manuscript preparation was coordinated by M.V.W. All contributing authors read and agreed to the final manuscript.
References
- Abascal F, Corvelo A, Cruz F, Villanueva-Cañas JL, Vlasova A, Marcet-Houben M, Martínez-Cruz B, Cheng JY, Prieto P, Quesada V.. 2016. Extreme genomic erosion after recurrent demographic bottlenecks in the highly endangered Iberian lynx. Genome Biol. 171:251.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allendorf FW, Hohenlohe PA, Luikart G.. 2010. Genomics and the future of conservation genetics. Nat Rev Genet. 1110:697–709. [DOI] [PubMed] [Google Scholar]
- Bazin E, Glémin S, Galtier N.. 2006. Population size does not influence mitochondrial genetic diversity in animals. Science 3125773:570–572. [DOI] [PubMed] [Google Scholar]
- Benbow ME, Tomberlin JK, Tarone AM.. 2015. Carrion ecology, evolution, and their applications. Boca Raton FL: CRC Press. [Google Scholar]
- Bolger AM, Lohse M, Usadel B.. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 3015:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouckaert RR. 2010. DensiTree: making sense of sets of phylogenetic trees. Bioinformatics 2610:1372–1373. [DOI] [PubMed] [Google Scholar]
- Butler J, MacCallum I, Kleber M, Shlyakhter IA, Belmonte MK, Lander ES, Nusbaum C, Jaffe DB.. 2008. ALLPATHS: de novo assembly of whole-genome shotgun microreads. Genome Res. 185:810–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho YS, Hu L, Hou H, Lee H, Xu J, Kwon S, Oh S, Kim H-M, Jho S, Kim S.. 2013. The tiger genome and comparative analysis with lion and snow leopard genomes. Nat Commun. 4:2433.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins PW. 1991. Interaction between island foxes (Urocyon littoralis) and Indians on islands off the coast of southern California: i. Morphologic and archaeological evidence of human assisted dispersal. J Ethnobiol. 11:51–81. [Google Scholar]
- deMenocal PB. 2004. African climate change and faunal evolution during the Pliocene–Pleistocene. Earth Planet Sci Lett. 220(1–2):3–24. [Google Scholar]
- Dobrynin P, Liu S, Tamazian G, Xiong Z, Yurchenko AA, Krasheninnikova K, Kliver S, Schmidt-Küntzel A, Koepfli K-P, Johnson W et al. , 2015. Genomic legacy of the African cheetah, Acinonyx jubatus. Genome Biol. 16:277.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortes GG, Paijmans JLA.. 2015. Analysis of whole mitogenomes from ancient samples. Methods Mol Biol. 1347:179–195. [DOI] [PubMed] [Google Scholar]
- Fumagalli M, Vieira FG, Linderoth T, Nielsen R.. 2014. ngsTools: methods for population genetics analyses from next-generation sequencing data. Bioinformatics 3010:1486–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabherr MG, Russell P, Meyer M, Mauceli E, Alfoldi J, Di Palma F, Lindblad-Toh K.. 2010. Genome-wide synteny through highly sensitive sequence alignment: satsuma. Bioinformatics 269:1145–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusset M, Burgener N.. 2005. Estimating larger carnivore numbers from track counts and measurements. Afr J Ecol. 434:320–324. [Google Scholar]
- Hahn C, Bachmann L, Chevreux B.. 2013. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads—a baiting and iterative mapping approach. Nucleic Acids Res. 4113:e129.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiménez A, Sánchez B, Pérez Alenza D, García P, López JV, Rodriguez A, Muñoz A, Martínez F, Vargas A, Peña L.. 2008. Membranous glomerulonephritis in the Iberian lynx (Lynx pardinus). Vet Immunol Immunopathol. 121(1–2):34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson WE, Onorato DP, Roelke ME, Land ED, Cunningham M, Belden RC, McBride R, Jansen D, Lotz M, Shindle D et al. , 2010. Genetic restoration of the Florida panther. Science 3295999:1641–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM.. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 304:772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C et al. , 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2812:1647–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent VT, Hill RA.. 2013. The importance of farmland for the conservation of the brown hyaena Parahyaena brunnea. Oryx 4703:431–440. [Google Scholar]
- Knowles JC, Van Coeverden de Groot PJ, Wiesel I, Boag PT.. 2009. Microsatellite variation in namibian brown hyenas (Hyaena brunnea): population structure and mating system implications. J Mammal. 906:1381–1391. [Google Scholar]
- Koepfli K-P, Jenks SM, Eizirik E, Zahirpour T, Van Valkenburgh B, Wayne RK.. 2006. Molecular systematics of the Hyaenidae: relationships of a relictual lineage resolved by a molecular supermatrix. Mol Phylogenet Evol. 383:603–620. [DOI] [PubMed] [Google Scholar]
- Koepfli K-P, Pollinger J, Godinho R, Robinson J, Lea A, Hendricks S, Schweizer RM, Thalmann O, Silva P, Fan Z et al. , 2015. Genome-wide evidence reveals that African and Eurasian golden jackals are distinct species. Curr Biol. 2516:2158–2165. [DOI] [PubMed] [Google Scholar]
- Korneliussen TS, Albrechtsen A, Nielsen R.. 2014. ANGSD: analysis of next generation sequencing data. BMC Bioinformatics 15:356.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA.. 2009. Circos: an information aesthetic for comparative genomics. Genome Res. 199:1639–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leffler EM, Bullaughey K, Matute DR, Meyer WK, Segurel L, Venkat A, Andolfatto P, Przeworski M.. 2012. Revisiting an old riddle: what determines genetic diversity levels within species? PLoS Biol. 109:e1001388.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leigh JW, Bryant D.. 2015. popart: full-feature software for haplotype network construction. Methods Ecol Evol. 69:1110–1116. [Google Scholar]
- Lewis ME. 1997. Carnivoran paleoguilds of Africa: implications for hominid food procurement strategies. J Hum Evol. 32(2–3):257–288. [DOI] [PubMed] [Google Scholar]
- Li H. 2011. Improving SNP discovery by base alignment quality. Bioinformatics 278:1157–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. 2012. seqtk Toolkit for processing sequences in FASTA/Q formats. Available from: https://github.com/lh3/seqtk, last accessed November 17, 2016.
- Li H, Durbin R.. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2514:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R.. 2011. Inference of human population history from individual whole-genome sequences. Nature 4757357:493–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2516:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, He G, Chen Y, Pan Q, Liu Y et al. , 2012. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 11:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magoč T, Salzberg SL.. 2011. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2721:2957–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 171:10–12. [Google Scholar]
- Martínez F, Manteca X, Pastor J.. 2013. Retrospective study of morbidity and mortality of captive Iberian lynx (Lynx pardinus) in the ex situ conservation programme (2004-June 2010). J Zoo Wildl Med. 444:845–852. [DOI] [PubMed] [Google Scholar]
- McQuillan R, Leutenegger A-L, Abdel-Rahman R, Franklin CS, Pericic M, Barac-Lauc L, Smolej-Narancic N, Janicijevic B, Polasek O, Tenesa A et al. , 2008. Runs of homozygosity in European populations. Am J Hum Genet. 833:359–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer M, Kircher M.. 2010. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb Protoc. 20106:pdb.prot5448, db.prot5448. [DOI] [PubMed] [Google Scholar]
- Peña L, Garcia P, Jiménez MA, Benito A, Pérez Alenza MD, Sánchez B.. 2006. Histopathological and immunohistochemical findings in lymphoid tissues of the endangered Iberian lynx (Lynx pardinus). Comp Immunol Microbiol Infect Dis. 29(2–3):114–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter C, Bruford M, Perez T, Dalamitra S, Hewitt G, Erhardt G.. 2007. Genetic diversity and subdivision of 57 European and Middle-Eastern sheep breeds. Anim Genet. 381:37–44. [DOI] [PubMed] [Google Scholar]
- R Development Core Team. 2008. R: a language and environment for statistical computing. Available from: http://www.R-project.org, last accessed December 10, 2015. [Google Scholar]
- Reed DH, Frankham R.. 2003. Correlation between fitness and genetic diversity. Conserv Biol. 171:230–237. [Google Scholar]
- Robinson JA, Ortega-Del Vecchyo D, Fan Z, Kim BY, vonHoldt BM, Marsden CD, Lohmueller KE, Wayne RK.. 2016. Genomic flatlining in the endangered island fox. Curr Biol. 269:1183–1189. [DOI] [PubMed] [Google Scholar]
- Roelke ME, Forrester DJ, Jacobson ER, Kollias GV, Scott FW, Barr MC, Evermann JF, Pirtle EC.. 1993. Seroprevalence of infectious disease agents in free-ranging Florida panthers (Felis concolor coryi). J Wildl Dis. 291:36–49. [DOI] [PubMed] [Google Scholar]
- Roelke ME, Martenson JS, O’Brien SJ.. 1993. The consequences of demographic reduction and genetic depletion in the endangered Florida panther. Curr Biol. 36:340–350. [DOI] [PubMed] [Google Scholar]
- Rohland N, Pollack JL, Nagel D, Beauval C, Airvaux J, Pääbo S, Hofreiter M.. 2005. The population history of extant and extinct hyenas. Mol Biol Evol. 2212:2435–2443. [DOI] [PubMed] [Google Scholar]
- Romiguier J, Gayral P, Ballenghien M, Bernard A, Cahais V, Chenuil A, Chiari Y, Dernat R, Duret L, Faivre N et al. , 2014. Comparative population genomics in animals uncovers the determinants of genetic diversity. Nature 5157526:261.. [DOI] [PubMed] [Google Scholar]
- Schmieder R, Edwards R.. 2011. Quality control and preprocessing of metagenomic datasets. Bioinformatics 276:863–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafer ABA, Wolf JBW, Alves PC, Bergström L, Bruford MW, Brännström I, Colling G, Dalén L, De Meester L, Ekblom R et al. , 2015. Genomics and the challenging translation into conservation practice. Trends Ecol Evol. 302:78–87. [DOI] [PubMed] [Google Scholar]
- Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM.. 2015. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 3119:3210–3212. [DOI] [PubMed] [Google Scholar]
- Skotte L, Korneliussen TS, Albrechtsen A.. 2013. Estimating individual admixture proportions from next generation sequencing data. Genetics 1953:693–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AB. 1992. Origins and spread of pastoralism in Africa. Annu Rev Anthropol. 211:125–141. [Google Scholar]
- Spielman D, Brook BW, Frankham R.. 2004. Most species are not driven to extinction before genetic factors impact them. Proc Natl Acad Sci U S A. 10142:15261–15264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 309:1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner CC, Putnam AS, Hoeck PEA, Ryder OA.. 2013. Conservation genomics of threatened animal species. Annu Rev Anim Biosci. 1:261–281. [DOI] [PubMed] [Google Scholar]
- Tamura K, Stecher G, Peterson D, Filipski A, Kumar S.. 2013. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 3012:2725–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts HE, Holekamp KE.. 2007. Hyena societies. Curr Biol. 1716:R657–R660. [DOI] [PubMed] [Google Scholar]
- Wayne RK, George SB, Gilbert D, Collins PW, Kovach SD, Girman D, Lehman N.. 1991. A morphologic and genetic study of the island fox, Urocyon littoralis. Evolution 458:1849–1868. [DOI] [PubMed] [Google Scholar]
- Werdelin L, Barthelme J.. 1997. Brown hyena (Parahyaena brunnea) from the Pleistocene of Kenya. J Vert Paleontol. 174:758–761. [Google Scholar]
- Werdelin L, Lewis ME.. 2005. Plio-Pleistocene Carnivora of eastern Africa: species richness and turnover patterns. Zool J Linn Soc. 1442:121–144. [Google Scholar]
- Werdelin L, Solounias N.. 1991. The Hyaenidae: taxonomy, systematics and evolution. Fossils Strata 30:1–104. [Google Scholar]
- Westbury M, Baleka S, Barlow A, Hartmann S, Paijmans JLA, Kramarz A, Forasiepi AM, Bond M, Gelfo JN, Reguero MA et al. , 2017. A mitogenomic timetree for Darwin’s enigmatic South American mammal Macrauchenia patachonica. Nat Commun. 8:15951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesel I. 2015. Parahyaena brunnea The IUCN red list of threatened species 2015: e.T10276A82344448. Available from: http://dx.doi.org/10.2305/IUCN.UK.2015-4.RLTS.T10276A82344448.en, last accessed April 14, 2017.
- Xue Y, Prado-Martinez J, Sudmant PH, Narasimhan V, Ayub Q, Szpak M, Frandsen P, Chen Y, Yngvadottir B, Cooper DN et al. , 2015. Mountain gorilla genomes reveal the impact of long-term population decline and inbreeding. Science 3486231:242–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachos J, Pagani M, Sloan L, Thomas E, Billups K.. 2001. Trends, rhythms, and aberrations in global climate 65 Ma to present. Science 2925517:686–693. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.