

Abstract
Animal bodies comprise diverse arrays of cells. To characterize cellular identities across an entire body, we have compared the transcriptomes of single cells randomly picked from dissociated whole larvae of the marine annelid Platynereis dumerilii. We identify five transcriptionally distinct groups of differentiated cells, each expressing a unique set of transcription factors and effector genes that implement cellular phenotypes. Spatial mapping of cells into a cellular expression atlas, and wholemount in situ hybridization of group-specific genes reveals spatially coherent transcriptional domains in the larval body, comprising, for example, apical sensory-neurosecretory cells versus neural/epidermal surface cells. These domains represent new, basic subdivisions of the annelid body based entirely on differential gene expression, and are composed of multiple, transcriptionally similar cell types. They do not represent clonal domains, as revealed by developmental lineage analysis. We propose that the transcriptional domains that subdivide the annelid larval body represent families of related cell types that have arisen by evolutionary diversification. Their possible evolutionary conservation makes them a promising tool for evo–devo research.
Keywords: evo-devo, single-cell transcriptomics, expression atlas, transcriptional domain, cell type family, Platynereis dumerilii
Introduction
The organization of cells into tissues and organs represents a fundamental feature of animal bodies. Recently, a number of studies have applied single-cell transcriptomics to assess cellular diversity within tissues, such as the vertebrate pancreas (Muraro et al. 2016), intestine (Grun et al. 2015), and different parts of the brain, including cortex, basal ganglia, and hypothalamus (Zeisel et al. 2015; Tasic et al. 2016; Romanov et al. 2017). This approach allows the molecular characterization of cell types within a given tissue, as well as an assessment of their heterogeneity. However, the sheer number of cells in conventional vertebrate or insect model systems has so far limited the comparison of cellular identities to single tissues or organs, and hampered comparisons across entire bodies.
Other laboratories have focused on the molecular comparison of different tissues, referred to as tissue transcriptomics. This approach determines and compares the expression profiles of entire tissues via bulk RNAseq (Henrichsen et al. 2009; Ramskold et al. 2009; Brawand et al. 2011; Breschi et al. 2016), with the potential to compare across the entire body. However, this approach requires defining tissues a priori by morphology or marker gene expression. This is problematic because a given tissue may be composed of very different cell types; and similar cell types can be found in distinct tissues as is the case for vertebrate ciliary photoreceptors, which populate both the retina and pineal organ (Arendt 2008). Therefore, this approach lacks the resolution necessary for understanding how cell types are distributed across entire organisms, and how this relates to morphologically defined tissues.
To advance on this, we have established the marine annelid Platynereis dumerilii, a molecular model species for development, evolution, and neurobiology (Fischer and Dorresteijn 2004; Williams and Jekely 2016), as an experimental paradigm to explore how cellular identities compare and relate to each other across an entire animal body. The Platynereis larval body is ideally suited for investigating the transcriptional organization of an entire organism as it harbors a variety of morphologically distinct cell types, but has a relatively low number of cells (Fischer et al. 2010), making it amenable for applying single-cell RNAseq to the whole organism. Further, Platynereis develops highly stereotypically, which allows for the construction of a cellular atlas onto which single-cell transcriptomes can be spatially mapped (Tomer et al. 2010; Asadulina et al. 2012; Vergara et al. 2016).
Here, we apply single-cell RNAseq to randomly sampled cells from the dissociated whole larvae at 48-h postfertilization (hpf). Our whole-body analysis reveals that, at this stage, the larval annelid body comprises five well-defined groups of differentiated cells with distinctive expression profiles. Cells in each group share expression of a unique set of transcription factors together with effector genes encoding group-specific cellular structures and functions. To correlate these groups with larval morphology, we establish a gene expression atlas for 48 hpf Platynereis larvae using the recent “Profiling by Signal Probability mapping” (ProSPr) pipeline (Vergara et al. 2016). For each group, we then locate individual cells in this atlas using an established algorithm for spatial mapping of single cells (Achim et al. 2015). The spatial distribution of each group was further validated by conducting wholemount in situ hybridization of selected group-specific genes. We thus reveal that the five distinct groups of differentiated cells spatially subdivide the larval body into coherent and nonoverlapping transcriptional domains that comprise (1) sensory-neurosecretory cells located around the apical tip of the larva, (2) peptidergic prospective midgut cells, (3) somatic myocytes, (4) cells with motile cilia constituting the larval ciliary bands, and (5) larval surface cells with epidermal and neural characteristics. We also show that these domains do not reflect developmental lineage, as they unite cells of distinct clonal origin. We propose that the five transcriptional domains represent evolutionarily related cell types that share fundamental characteristics at the regulatory and effector gene level (so-called cell type families) and discuss their possible evolutionary conservation across larger phylogenetic distances.
Results
Single-Cell RNA-Seq Identifies Five Groups of Differentiated Cells
To explore cell type diversity on the whole organism level, we dissociated whole larvae of a marine annelid, P.dumerilii at 48 hpf, and randomly captured cells for single-cell RNA-sequencing (scRNA-seq) (fig. 1). At this stage of development, the larva is comprised of relatively few cells (∼5000), but has many differentiated cell types, including different ciliated cells, neurons, and myocytes. The collected cells were optically inspected to exclude doublets, multiple cells, or cell debris. Sequenced samples were further filtered computationally to remove low complexity transcriptomes, lowly expressed genes, and transcriptomic doublets (supplementary fig. 1, Supplementary Material online and see Materials and Methods). A total of 373 cells and 31300 transcripts passed filtering steps and were used for downstream analysis. To group the cells into distinct clusters, we used a sparse clustering strategy, which identified seven groups of cells. We used the scran package to find group specific marker genes and discovered that in pairwise comparisons across all groups, two clusters were consistently highly similar to one another. Therefore, we merged these two closely related groups (fig. 1 and supplementary fig. 2, Supplementary Material online, and see further details and justification in Materials and Methods).
Fig. 1.

Single-cell transcriptomics of Platynereis 48 hpf larvae. Cells of the 48 hpf larvae were dissociated and randomly selected for single-cell RNA-sequencing using the Fluidigm C1 Single-cell AutoPrep system. Combining sparse clustering with spatial positioning of single cells allows the identification of robust cell groups within the data. The clustering approach enables identification of genes that characterize each cell type. Finally, we used hierarchical clustering to investigate the similarity between the identified cell clusters.
To characterize the remaining six groups further, we identified differentially expressed genes (see Materials and Methods). The largest group of cells, which resulted from combining the two closely related groups, was characterized by the specific expression of genes known to be active in developmental precursors, such as DNA replication (DNA topoisomerase, Rfc3, Rfc5, Mcm3, Mcm4, Mcm7), cell proliferation (Pcna, Md2l1, Apc7, supplementary fig. 2i, Supplementary Material online), or chromatin remodeling (Nucleoplasmin, Bptf), suggesting this group was primarily composed of undifferentiated cells. Cells in the other five groups showed significantly lower expression of these markers (supplementary table 1, Supplementary Material online, FDR < 0.1, Materials and Methods), suggesting these are differentiated cells. For each of these five groups, pairwise differential expression analysis revealed distinct sets of group-specific effector genes (i.e., differentially expressed genes that encode the particular structural and/or functional properties of cells) and group-specific transcription factors, many of which are known to act as terminal selectors in cell type differentiation (Hobert 2011; Arendt, Musser, et al. 2016) (supplementary table 2, Supplementary Material online).
We next applied hierarchical clustering to validate group membership and explore higher level transcriptional relationships among groups. First, we validated the clustering of differentiated cells into the above groups by constructing a tree and performing bootstrap resampling using all single-cell transcriptomes (Materials and Methods) (supplementary fig. 3, Supplementary Material online). This tree confirmed each group’s integrity, albeit with variable support. Second, we explored relationships among the different groups by conducting hierarchical clustering with bootstrapping using an average gene expression vector computed for each cell group (see tree in fig. 4 for support values; Materials and Methods).
Fig. 4.

Identification and validation of tissue-specific genes. On top, Distance tree showing the hierarchical relationships between the differentiated cell groups. (a–e) For each identified cell group, the expression of a group-specific marker gene is shown in a bar plot, the respective tissue is shown schematically in the ventral view of Platynereis larva, and visualized by WMISH with respective probes: (a) Pde9 expression in the apical ectoderm (red); (b) Hnf4 expression in the midgut (cyan); (c) St-mhc expression in striated muscle (green); (d) Rsph4 expression in ciliated cells (yellow); and (e) Grm7 expression characterizes the nonapical surface cells (gray). Note that Pde9, Hnf4, Rsph4 and Grm7 are novel markers for the respective cell groups. Each ISH pattern was replicated in at least six animals. Scale bar, 50 μm. Apo, apical organ; pt, prototroch, Z factor=, , expression of gene i in cell j, , mean expression of gene i, , SD of gene i.
Mapping Sequenced Cells to a Cellular Expression Atlas
As a prerequisite for localizing the five distinct groups of differentiated cells in the larval body, we built a gene expression atlas with cellular resolution for the 48 hpf larva, using our recently established Profiling by Signal Probability mapping (ProSPr) pipeline (Vergara et al. 2016). This involved whole-mount in situ hybridization for 176 genes, automated acquisition of up to 40 expression images per gene, and registration of these images to a reference larva to construct an expression average for each gene (fig. 2 and supplementary fig. 4, Supplementary Material online).
Fig. 2.

The ProSPr cellular expression atlas for 48 hpf Platynereis larvae. ProSPr generates a standardized expression pattern for each gene (1–6 are examples) that are registered onto the same averaged larva. As a result, for any bilateral pair of cells in the body (red rings) the expression profile can be determined in a binary fashion (white: not expressed; black: expressed).
Taking advantage of this atlas, we used a dual strategy to investigate the spatial distribution of sequenced cells. First, we located individual cells in the atlas via the comparison of complete, specificity-weighted mRNA profiles for each cell with the positional gene expression vectors derived from the atlas (Achim et al. 2015). Following this strategy, the vast majority (95%) of cells could be mapped to distinct positions within the larva (fig. 3; left panels show the single most likely position for each cell in each group of differentiated cells, right panels show the highest scoring mapping positions for one selected cell in each group). Notably, cells belonging to the same group showed spatially correlated mapping to distinct body regions, as apparent from the overlap with the expression of one marker gene specific for each group. Remarkably, looking at the highest scoring mapping positions for an individual cell, we consistently detected pairs of bilateral coordinates, reflecting the bilateral symmetry of the animal. We also noted different degrees of spatial acuity: While some cells mapped to unique (or closely adjacent) left/right pairs of coordinates (fig. 3b and d, right panels), other cells mapped to segmentally iterated positions, reflecting the segmental nature of the larval trunk with iterated occurrence of cell types (fig. 3f and h, and j, right panels).
Fig. 3.
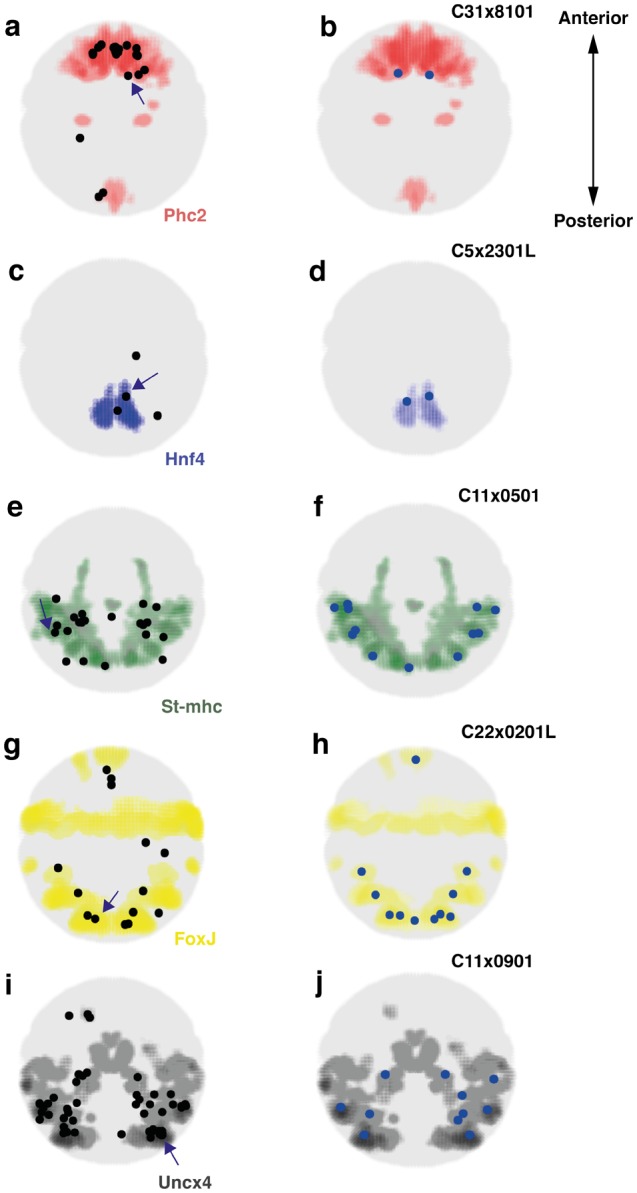
Spatial mapping of single cells to spatially coherent regions characterized by specific marker gene expression. Expression of specific marker gene expression visualized as colored regions within the larva in ventral views. Left panels: Mapping results for all cells belonging to the respective group. Centroids of the voxel clusters to which cells mapped with highest confidence are shown as black dots. Right panels: For each cell group, we show an example of the mapping result for one individual cell (indicated by an arrow on the left panel). (a) Cells of the apical ectoderm group (n=23 cells) map to the Phc2 expressing region in the embryo. (b) Mapping result for the cell C31x8101. (c) Midgut cells (n=4 cells) map to the Hnf4 expressing region. (d) Mapping result for the cell C5x2301L. (e) Striated musculature cells (n=23 cells) map to the St-mhc expressing region. (f) Mapping result for the cell C11x0501. (g) Cells of the ciliary bands (n=14 cells) map to the Foxj expressing region. (h) Mapping result for the cell C22x0201L. (i) Cells of the nonapical ectodermal group (n=55 cells) map to the Uncx4 expression domain. (j) Mapping result for the cell C11x0901.
Transcriptional Domains Subdivide the Larval Body
Complementing the spatial mapping of individual cells, we also determined the overall spatial distribution for each group of differentiated cells in the larval body. To this end, we identified transcripts that showed maximum specificity for each group (fig. 4, left panels) and determined their expression pattern in the larval body using wholemount in situ hybridization (WMISH) (fig. 4, right panels). We reasoned that in combination, the expression patterns of the genes most specific for each group should approximate the overall spatial distribution of cells belonging to that group in the larval body. If not yet included, we fed group-specific genes into the expression atlas to accurately determine expression overlap (and to refine the spatial mapping of individual cells).
The two approaches, that is, the mapping of individual cells (see above) and the mapping of the entire group via marker gene expression, produced entirely consistent results and subdivided the larval body into five spatially coherent, nonoverlapping, and entirely transcriptionally defined regions, which we refer to as transcriptional domains. In particular, group 1 cells mapped to the larval brain around the apical tip of the larva, where Prohormone Convertase 2 (Phc2) is expressed, a marker for this domain (fig. 3a and b). Consistent with this, expression of another domain-specific marker gene, Phosphodiesterase-9 (Pde9), was restricted to the most apical territory of the larva (fig. 4a). Group 2 cells mapped to the midgut primordium alongside the yolk-rich macromeres, as confirmed by the spatially highly restricted expression of Hnf4 (fig. 3c and d and fig. 4b). Cells of group 3 mapped to the differentiating striated myocytes that consistently expressed the Striated muscle myosin heavy chain (St-mhc) gene (fig. 3e and f and fig. 4c); and group 4 cells populated the ciliary bands, matching the expression of Foxj (fig. 3g and h) and of Radial spoke head protein homolog 4 (Rsph4) (fig. 4d) (Fischer et al. 2010). Finally, group 5 cells covered large part of the larval surface, reflected by Uncx4 expression (fig. 3i and j) and by the broad surface expression of the Metabotropic glutamate receptor 7 (Grm7) in the larval episphere and hyposphere (fig. 4e).
Although the transcriptional domains for groups 2, 3, and 4 matched known larval tissues (early midgut, striated myocytes, and ciliary bands), no obvious morphological features or boundaries corresponded to, or delimited groups 1 and 5 transcriptional domains. This is remarkable because the topology of the hierarchical tree (fig. 4 and supplementary fig. 3, Supplementary Material online) supported a deep transcriptional divide between the two groups. Transcriptionally, group 5 cells were more similar to the ciliary bands (group 4) and to striated myocytes (group 3), than to group 1 cells, with high bootstrap support.
To locate the boundary between transcriptional domains 1 and 5 more precisely in the larval body, we investigated the expression of group-specific marker genes taking advantage of our expression atlas. We thus determined that the group 1 transcriptional domain comprised a cruciform patch of differentiated apical cells, deeply embedded in the anterior neuroectoderm and covered by surface cells (fig. 5). In contrast, the expression of group 5-specific genes consistently remained restricted to the larval surface. Coexpression analysis revealed virtually no expression overlap between genes specific for the one or the other group, corroborating their strict separation as distinct transcriptional domains.
Fig. 5.

Spatial extent of the group 1 versus group 5 transcriptional domains as determined by specific marker gene comparison. Gene expression averages at 48 hpf as determined by the ProSPr cellular expression atlas. Group 1 gene Phc2 in red; group 2 genes LaminAC and Pcdh15 in green. (a and b) Apical views; (c and d) ventral views. The inset in (a) and (b) is a scanning electron microscopy apical view in the same orientation, illustrating that the group 1 domains is situated around the apical organ.
The Group 1 Transcriptional Domain Represents the Apical Nervous System
To explore the molecular nature of the groups/transcriptional domains, we investigated their distinctive gene sets. Figure 6 shows the most specific and informative regulatory and effector genes that were found to be specific for each group. Illustrating the power of this approach, the genes specific for groups 2, 3, and 4 cells clearly identify these as differentiated midgut cells, striated myocytes, and the multiciliated cells of the ciliary bands, respectively (fig. 6b, c, and d). Further inspection of the group 1-specific genes (fig. 6a) revealed that many of these encoded neuropeptides, such as the broadly expressed GNXQNpeptide or GGNamide, two lophotrochozoan neuropeptides (Conzelmann et al. 2013), and the neuropeptide-cleaving Prohormone convertase 2 (Phc2) (fig. 5), indicating neurosecretory release from these cells (Tessmar-Raible et al. 2007). In addition, we found noncalcium-binding members of the synaptotagmin family implicated in the generation and fusion of large dense core vesicles for neurosecretion, such as Synaptotagmin17/B/K (Syt17), Sytα, and Syt4 (figs. 6a and 7) (Gustavsson et al. 2012; Moghadam and Jackson 2013; Park et al. 2014). This is in line with electron microscopy data, which show that cells in the dorsomedian Platynereis brain are rich in dense core vesicles while synapses are often sparse or absent (Williams et al. 2017). Group 1 cells also specifically expressed genes associated with photoreception, such as the bistable photopigment Peropsin (Nagata et al. 2010) and components of cGMP-based signal transduction such as Pde9 and the CNG channel components Cngb and Cnga-α (fig. 6a). Individual cells expressed ciliary photoreceptor-specific genes and were thus identified as the brain ciliary photoreceptors (Neuropeptide Y [Npy] in combination with c-opsin or c-opsin2; Arendt et al. 2004), or as photosensory-vasotocinergic cells (expressing Vasotocin–neurophysin, Proenkephalin, and c-opsin; Tessmar-Raible et al. 2007). Interestingly, we also found that neuroglobin was specific for this domain, encoding a monomeric globin evolutionarily older than hemoglobin or myoglobin known to reversibly bind oxygen (Burmester et al. 2000). Taken together, the nature of group 1-specific genes is strong evidence that this domain represents the apical nervous system (ANS) as characterized previously (Tosches and Arendt 2013; Tosches et al. 2014; Williams et al. 2017).
Fig. 6.

Tissue specific marker genes reflect cellular functions. For each of the differentiated tissues, we show a heatmap of tissue specific gene expression: (a) expression profile of group 1 apical nervous system-specific genes; (b) expression profile of group 2 midgut genes; (c) expression profile of group 3 myocyte-specific genes; (d) expression profile of group 4 multiciliated cell-specific genes; and (e) expression profile of group 5 neural/epithelial surface-specific genes. Functionally related groups of genes are highlighted. TF: transcription factor, td: transcriptional domain.
Group 5 Transcriptional Domain Cells Show Epidermal and Neural Characteristics
The ectoderm-derived Group 5 cells expressed genes are indicative of mixed epithelial and neural characteristics (fig. 6e). Expression of some of these genes is shared with both multiciliated cells and striated myocytes, consistent with the close relationship we found between these groups in our hierarchical clustering analysis (compare fig. 4). Among the most broadly expressed genes shared with multiciliated cells and myocytes were LaminA/C, a fibrous protein of the nuclear lamina (Kim et al. 2012) (supplementary fig. 5, Supplementary Material online), and the related Neurofilament-70 (Nf70), a neural intermediate filament found in Lophotrochozoans; Extended Synaptotagmin 2 (Esyt2) has a reported function in membrane lipid composition dynamics, extracellular signal transduction, and cell adhesion (Herdman and Moss 2016); and P4ha2 encodes a component of prolyl 4-hydroxylase, a key enzyme in collagen synthesis. The genes specifically shared between group 5 and the ciliary bands included Anoctamin, a transepithelial chloride channel (Pedemonte and Galietta 2014), Protocadherin-15 (Pcdh15), an atypical cadherin mediating structural integrity of ciliated sensory receptors (Seiler et al. 2005) (supplementary fig. 5, Supplementary Material online), and Semaphorin 2 (Sema2), a secreted extracellular guidance molecule involved in axon pathfinding (Bates and Whitington 2007). In line with an overall epithelial identity of these cells, they expressed the epidermis-specific transcription factor Grainyhead, known for its conserved role in maintaining the epidermal barrier (Boglev et al. 2011), and the ETS-homologous factor that likewise specifies epithelial cell type identities (Feldman et al. 2003).
Most noticeably, however, we detected a set of genes representing an extracellular matrix-remodeling module (fig. 8). These genes encoded matrix-modifying proteases such as a serine protease Plasminogen related to vertebrate Plasmin (Sonderegger and Matsumoto-Miyai 2014) and Hepatocyte Growth Factor (HGF) (Tyndall and Walikonis 2006) (supplementary table 2, Supplementary Material online); metalloproteinases such as Matrix Metalloproteinase (Mmp9) (Michaluk et al. 2011) together with its specific substrate Cd109 (Vadon-Le Goff et al. 2015); and the cysteine proteinase Cathepsin L (Felbor et al. 2002) (fig. 8b). Many of these protease systems have been implicated in the modification of dendritic spines (Michaluk et al. 2011) and synapses (Sonderegger and Matsumoto-Miyai 2014), suggesting nervous system-related functions of this module.
Fig. 8.
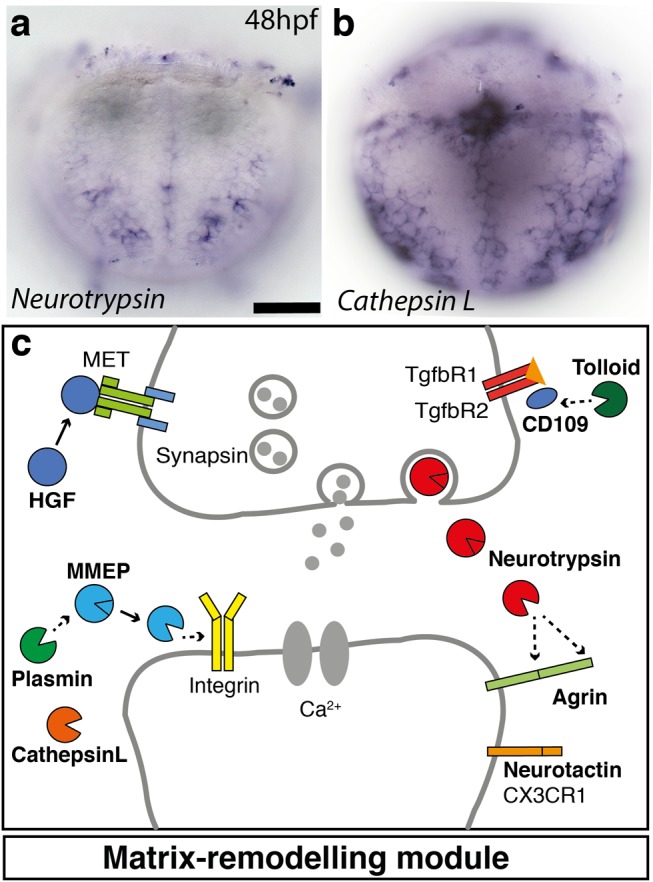
Extracellular matrix remodeling module is specific to group 5 neural/epithelial surface cells. (a and b) WMISH analysis of Neurotrypsin and Cathepsin L expression in Platynereis larvae at 48 hpf. Ventral view focused at the surface. Scale bar, 50 μm c, Schematic of the synapse formation cellular module. Neurotrypsin cleaves agrin locally at the synaptic cleft, triggering the formation of new synapses (Sonderegger and Matsumoto-Miyai 2014). HGF and its receptor MET enhance clustering of synaptic proteins at excitatory synapses (Tyndall and Walikonis 2006). Plasmin cleaves selected synaptic target proteins such as NMDA receptor and matrix metalloproteinases (Sonderegger and Matsumoto-Miyai 2014). Several members of the family of matrix-metalloproteinases such as MMP-9 have been implicated in synapse formation and remodeling (Michaluk et al. 2011). Tolloid-like (Tll) and its substrate CD109 have been implicated in the control of TGFβ signaling and extracellular matrix synthesis (Vadon-Le Goff et al. 2015). Cathepsin L is likewise implicated in extracellular matrix remodeling and neuronal survival (Felbor et al. 2002). The genes specifically expressed in nonapical ectodermal cells are marked in bold.
In line with this, the genes most specific for group 5 (not shared with the ciliary bands or myocytes) abundantly encoded proteins with reported roles in neurite outgrowth and synaptic plasticity. For example, we identified Neurotactin, which encodes a cell surface glycoprotein of the serine esterase superfamily (Speicher et al. 1998), and its specific binding partner Amalgam (Zeev-Ben-Mordehai et al. 2009); both involved in axonal pathfinding in Drosophila (Speicher et al. 1998; Fremion et al. 2000). We also found two orthologs of Neurotrypsin (fig. 8a), a matrix-modifying serine protease, together with the extracellular proteoglycan Agrin (Stephan et al. 2008), indicating that the Neurotrypsin-Agrin system is active in these cells (Matsumoto-Miyai et al. 2009) (fig. 6e). The Hspg2 gene, encoding a conserved proteoglycan related to Agrin, was also part of this set. Furthermore, the specific expression of the nicotinic acetylcholine receptor alpha-9 is noteworthy. In vertebrates, this unconventional receptor plays restricted roles in the mechanosensory hair cells, where it is involved in the reception of efferent signals (Murthy et al. 2009), and in epidermal keratinocytes, where it triggers epithelialization via local, nonneuronal acetylcholine cytotransmission (Chernyavsky et al. 2007). Finally, smaller subsets of group 5 cells also expressed Wnt ligands such as Wnt4 (Pruitt et al. 2014), and the bHLH HES transcription factors Hes1, Hes2, Hes6, and Hes11; and homeodomain transcription factors with conserved roles in neuronal specification such as Uncx4, Tbx20, or Phox2 (Denes et al. 2007; Rabe et al. 2012; Nomaksteinsky et al. 2013) (fig. 6e).
In conclusion, a closer inspection of group 5-specific genes suggests both epithelial and neural properties. However, since none of the group 5 cells showed a clear and distinct neuronal profile as observed in the apical nervous system (supplementary fig. 6, Supplementary Material online), the possible classification of (subsets of) group 5 cells as neuronal remains preliminary. We therefore used our ProSPr 48hpf cellular expression atlas to test for co-expression of pan-neuronal markers with goup/domain-specific genes. Supporting neural characteristics of group 1 (apical nervous system, fig. 9a) and of at least some group 5 cells (figure 9b), our cellular expression atlas revealed extensive coexpression of group 1 markers (Vat1l, Syt4; compare fig. 6a) and restricted but clear coexpression of group 5 markers (Tbx20, uncx4, LaminAC; compare fig. 6e) with the pan-neuronal marker Rab3 (fig. 9).
Fig. 9.

Coexpression of groups 1 and 5 marker genes with the pan-neuronal marker Rab3 indicates the presence of neural cell types. Averaged gene expression patterns for 48 hpf ProSPr used for in silico analysis of coexpression. (a) Group 1. (b) Group 5.
Incongruence of Transcriptional Domains and Developmental Lineage
Finally, we set out to explore a possible correlation between the five transcriptional domains of the larval body and the Platynereis developmental lineage (Ackermann et al. 2005; Vopalensky et al. 2018), to find out whether there was congruence between transcriptional and clonal domains. We reasoned that common origin from a shared developmental precursor would be one possible explanation for the observed transcriptional similarity between cells (Scialdone et al. 2016; Kee et al. 2017). Facilitating this analysis, the lineage of the Platynereis episphere has recently been solved up to 30 hpf by in vivo imaging, and individual cells can be identified in any embryo up to 15 hpf due to the highly stereotypical embryonic development (Vopalensky et al. 2018). Building on these resources, we investigated the clonal origin of all Phc2+ cells of the apical nervous system (fig. 10). We found them to derive from multiple different sublineages of the Platynereis larval episphere, thus representing a multiclonal assembly. Likewise, the group 5 neural-epithelial surface cells and the motile ciliated cells are developmentally heterogeneous, as they both comprise episphere and hyposphere cells that derive from distinct parts of the Platynereis lineage (Ackermann et al. 2005). The cellular lineage of the midgut and somatic myocytes is yet unknown. We can thus conclude that the groups and transcriptional domains for which cellular lineage information is available are polyclonal; and that developmental lineage cannot account for their expression similarity.
Fig. 10.

The developmental lineage of early differentiating Phc2+ cells. (a–d) Multiclonal origin of the first 10 differentiating Phc2+ cells. For each labeled cell, the upper line indicates the name of the cell and the lower line its lineage progeny. (a and b) Represent 14 hpf and (c and d) represent 16 hpf. (a and c) Confocal images of Phc2+ cells and (b and d) the corresponding cells in the tracked lineage (Vopalensky et al. 2018) indicated by red dots. Note that the 10 phc2+ cells labeled in (d) are of different clonal origin as revealed by their deviating lineage progeny. (e and f) The cells present at 30 hpf also mostly originate from different clonal domains, as revelaed by spatial alignment to the clonal map at 30 hpf. Brown lines separate cells descending from the first four distinct cells A, B, C, D.
To further explore the developmental fate of the transcriptional domains, we performed whole-mount in situ hybridization of apical nervous system and neural/epithelial surface marker genes for stages earlier than 48 hpf and for later life-cycle stages (supplementary fig. 7, Supplementary Material online). In the absence of whole-body single-cell data for these stages, these preliminary data suggest that the transcriptional domains remain stable over time and may thus represent an inherent and constant property of the animal body.
Discussion
A New Molecular Subdivision of the Platynereis Larval Body
For this study, we analyzed the transcriptomes of cells randomly collected from dissociated, entire larvae of the marine annelid Platynereis dumerilii. Previous single-cell studies had captured cells from specific body parts and tissues only (such as the mouse forebrain; Zeisel et al. 2015; Tasic et al. 2016). Our data are unique as they allow single-cell level comparison of differentiated cells across the entire body. We were able to sort all differentiated cells into five major groups, based on similarities and differences in gene expression profiles. Spatial mapping of sequenced cells into the 48 hpf ProSPr cellular-resolution expression atlas, in combination with wholemount in situ hybridization of group-specific marker genes, revealed five coherent transcriptional domains in the 48 hpf Platynereis body. In contrast to an expression domain, which delimits expression in the body for a single gene, a transcriptional domain delimits the expression of many genes to a specific part of the body. For the first time, the discovery of transcriptional domains opens up the possibility to understand animal organization that does not rely on predefined morphological boundaries or a priori gene selection, but instead reflects the molecular boundaries that correspond to the most pronounced differences in overall gene expression.
We probed the molecular nature of these transcriptional domains and found they were united by the shared expression of specific sets of transcription factors with known roles in cell fate selection, such as Hnf4 for midgut (Palanker et al. 2009), or Grainyhead for epithelial cells (Boglev et al. 2011). Although we did not test the function of these transcription factors in this study, it is plausible to hypothesize that they play a role in regulating domain-specific effector genes. In line with this, we found each domain shared a coherent set of effector genes representing distinct cellular modules, as exemplified for the apical nervous system (fig. 7) and for the neural/epithelial surface cells (fig. 8). Beyond that, however, we also observed considerable differences in transcription factor and effector gene expression within a given group (as evident from fig. 6), suggesting the presence of several related but distinct cell types. For example, cells of the apical nervous system differ with regard to opsin expression, neuropeptide content, and signaling components—indicating that each, or almost each, cell might represent a separate type. These differences are unlikely to reflect technical noise, as we find known cell type markers coexpressed within individual cells. It is important to stress that, although our sample size is large enough to resolve transcriptional domains, it is too small for the full characterization of individual cell types, which in some cases may be represented by only a single, bilateral pair of cells (Vergara et al. 2016). It is thus plausible that the number of sequenced cells would have to be increased substantially to fully resolve the individual cell types within each given domain.
Fig. 7.
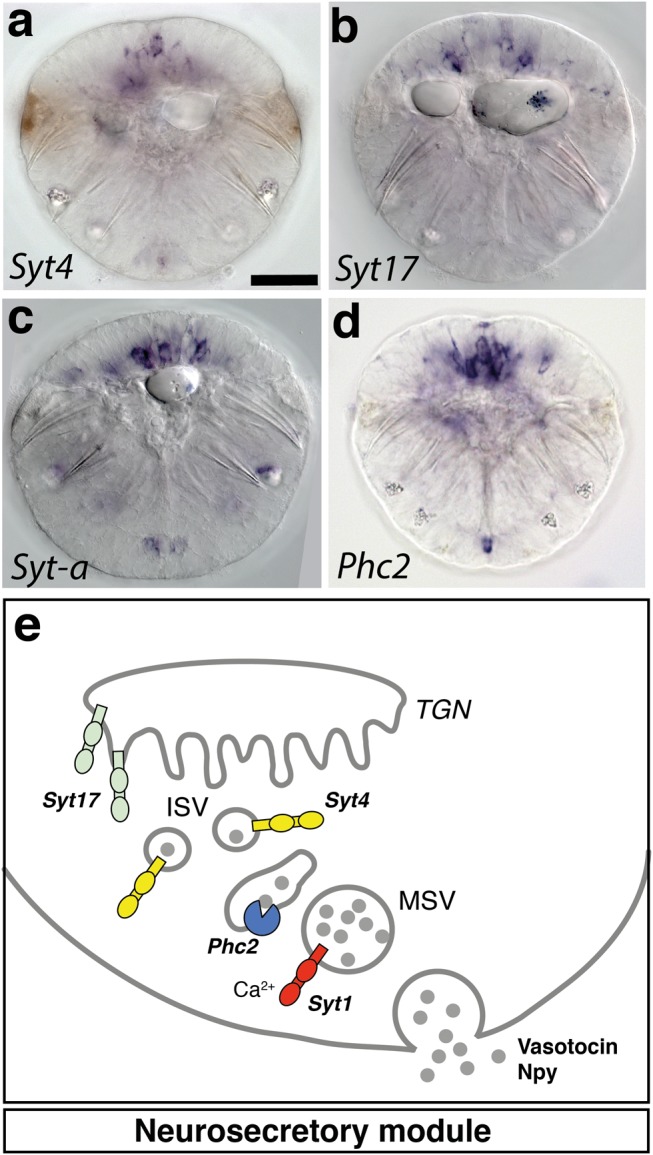
Neurosecretory functional module is specific to group 1 ANS cells. (a–d) WMISH analysis of Syt4, Syt17, Sytα, and Phc2 expression in Platynereis larvae at 48 hpf. Ventral view. Scale bar, 50 μm. (e) Schematic of the neurosecretion cellular module. Syt17 contains an N-terminal cysteine cluster mediating membrane association with the trans-Golgi network (TGN) (Fukuda 2003). Syt4 is involved in the maturation of secretory vesicles (Zhang et al. 2011). Sytα functions in vesicle trafficking of specific subclasses of neuropeptides and/or neuromodulators (Adolfsen et al. 2004). TGN, trans-Golgi network; ISV, immature secretory vesicle, MSV, mature secretory vesicle.
Despite our inability to resolve individual cell types, the comparison of these transcriptional domains to larval morphology and developmental lineage revealed several notable results. First, whereas some domains clearly delineate distinct morphological tissues, such as midgut, striated muscles, and ciliary bands, we also uncovered a fundamental divide between apical nervous system and neural/epithelial surface ectoderm, which was emphasized by the topology of the tree that placed both far apart from each other. This divide was unexpected because the boundary between both domains does not coincide with a clear morphological boundary, and both domains coexpress pan-neuronal markers (fig. 9). Second, we found that the transcriptional domains were largely incongruent with clonal domains (fig. 10). This is clear evidence that their transcriptional similarity is not due to shared developmental lineage, and accords with recent findings for the vertebrate retina (Macosko et al. 2015), which found differentiated rods and cones more closely related to each other transcriptionally than to other retinal cell types, even though they come from different developmental progenitors (Cepko 2015). If not developmental lineage, what then accounts for the similarity of cells and cell types found within the transcriptional domains?
The Concept of Cell Type Families
One plausible explanation for the observed similarities in expression profile between cells belonging to the same group/domain is evolutionary relatedness; or, in other words, that their constituting cell types derive from shared ancestral cell types. We have recently proposed that the diversity of cell types in animals evolved via a sequence of cellular diversification events, producing pairs of sister cell types in diverging lineages. With continued diversification, ancient cell types thus give rise to cell type families. Reflecting their common ancestry, related cell types would share the expression of some transcription factors involved in their combinatorial specification and of (subsets of) cellular effector genes implementing their similar phenotypes (Hobert 2011; Arendt et al. 2016).
The somatic myocytes (fig. 6c) exemplify the concept of cell type families. Although the transcription factor Mef2 is expressed in all cells of the group, the expression of other factors is likewise robust but restricted to few cells in the groups, including MyoD, Hand, Mox, Paraxis, and Twist, consistent with more specific roles as myoblast identity genes as reported, for example, for Drosophila (Dobi et al. 2015). This would suggest that the Platynereis myocyte family contains several distinct myocyte types representing sister cell types (Brunet et al. 2016).
Evolutionary Conservation of the Apical Nervous System
Our hypothesis that the transcriptional domains represent cell type families can be tested via comparison to other species. Given that the Platynereis larval body is composed of five transcriptional domains; and assuming that the cell types of each domain indeed diversified from single founder cell types in animal evolution, then these five founder cell types must have existed early in animal evolution. This would make the transcriptional domains that we have identified in Platynereis very ancient and imply that they should also exist in other remote animal phyla. Formal testing of this hypothesis will require sampling additional species for whole-body single-cell profiling in diverse phyla. However, several initial lines of evidence already support our hypothesis. In particular, it has previously been proposed that the annelid apical nervous system and the vertebrate hypothalamus are homologous. This was based on the conserved developmental expression of transcription factors such as six3 and foxq (Marlow et al. 2014), on the shared presence of a neurosecretory center releasing conserved neuropeptides such as oxytocin-vasopressin (Tessmar-Raible et al. 2007; Williams et al. 2017), on conserved nonvisual ciliary photoreceptors employing ciliary opsin and a vertebrate-type phototransduction cascade (Arendt et al. 2004), and on the shared presence of an active center of melatonin synthesis in control of circadian behavior (Tosches et al. 2014). These similarities have been taken as evidence to propose evolutionary conservation of the apical nervous system as a distinct part of the bilaterian brain (Tosches and Arendt 2013), which may trace back to early metazoan ancestors (Marlow et al. 2014).
Our new data allow us to further challenge and validate this hypothesis, via the comparison of new apical nervous system domain-specific markers identified in Platynereis with expression data available for the vertebrates. And indeed, this comparison is highly supportive of evolutionary conservation. For example, in vertebrates the noncalcium-binding and neurosecretion-specific Syt4 is specifically active in the neuroendocrine hypothalamus, where it is known to regulate the release of the neuropeptide Oxytocin (Zhang et al. 2011). In addition, vertebrate Syt17 has initially been isolated from the vasopressin-secreting supraoptic and paraventricular nuclei and likewise shows strong expression in the hypothalamus (Lee et al. 2001), as does Neuroglobin (Hundahl et al. 2008, 2012; Fabrizius et al. 2016). Finally, the T-box transcription factor T-brain is specifically expressed in the developing para- and periventricular and supraoptic hypothalamic nuclei (Dominguez et al. 2015). These observations suggest that transcriptional domains and the corresponding cell type families are conserved in animal evolution and that their comparative study represents a highly promising new direction in evolutionary research.
Materials and Methods
Cell Dissociation of Platynereis Whole Larvae
Platynereis dumerilii larvae were incubated in filtered natural seawater (FNSW), at 18°C for 47 h to reach the desired stage. For single-cell dissociation, larvae were concentrated using a 40 μm size mesh, picked, washed in Ca, Mg-free artificial sea water (CMF-SW), and incubated for 10 min in 0.5% Pronase (Roche cat # 10165921001), 1% sodium thioglycolate (Sigma T0632) in CMF-SW, at room temperature. Larvae were then rinsed in 1× PBS, and incubated in 500 μl of 150 μg/ml Liberase (Roche, cat # 05401119001) in 1× PBS, for 1 min. Larvae were gently triturated by pipetting, centrifuged at 200×g for 3 min, and reconstituted in 600 μl of 1× PBS. Cells were then dissociated by trituration using a syringe. Dissociated cells were washed twice in 200 μl of 1× PBS and concentrated by centrifuging (1 min, 200×g). Cells were resuspended in 20 μl of 1× PBS, of which 12 μl was loaded on the Fluidigm C1 capture chip.
Cell Capture and Sequencing
Platynereis larval cells were captured, and single-cell cDNA synthesis was performed using the Fluidigm C1 Single-Cell Auto Prep System, and the sequencing libraries were prepared as previously described (Achim et al. 2015). We used Fluidigm IFCs for mRNA-seq with capture sites for 5–10 μm and 10–17 μm to collect the cells, allowing the capture of a full range of cell sizes in 48hf stage of Platynereis. In total, nine IFC-s, each representing an independent collection of cells derived from a different batch of animals, were processed for this study. The information about the samples distribution on chips, the chip sizes, cell number per sample, and the pooling of samples on sequencing lanes is described in supplementary table 3, Supplementary Material online. 100-bp paired-end sequences were generated using the Illumina HiSeq2000 platform. For one library (CN62), 75-bp paired-end sequences were generated using the Illumina NextSeq500 platform. Libraries were sequenced to an average depth of 2.1 million reads.
Data Availability
The raw sequence data for the cells analyzed in this study are available from ArrayExpress, accession numbers E-MTAB-2865 (see supplementary table 5, Supplementary Material online, for matching the cell ID-s between the E-MTAB-2865 and this study) and E-MTAB-5953. The R code used for analysis is available at: https://github.com/MarioniLab/Platynereis2017.
Read Alignment
FastQC (Andrews 2010) was used for the quality trimming of the raw sequencing reads using default settings. For read mapping, the quality-filtered sequencing reads were mapped, using bowtie2, against a Platynereis reference transcriptome combined from the two previously published assemblies (Conzelmann et al. 2013; Achim et al. 2015). Briefly, the transcriptome assemblies were concatenated and contigs showing >94% identities were removed using CD-HIT (Li and Godzik 2006), leaving 44977 transcripts. In the generated reference, each gene should be represented by one transcript, reducing the problem of multiple mapping of the reads. Expression counts for each transcript were obtained using HTSeq (Anders et al. 2015) while only considering uniquely mapped reads.
Gene Annotation
To annotate genes of the two transcriptome assemblies (Conzelmann et al. 2013; Achim et al. 2015), reciprocal BLAST comparison of genes sequences against the Uniref90 protein database (Arendt assembly) or against Swissprot (Jékely assembly) was performed. For each transcript, the BLAST hit with highest E-value was selected for annotation.
Quality Control
Low-quality cells were removed from the data set based on the following filtering criteria. Visual inspection of capture sites on the IFC revealed empty wells, wells with multiple cells or debris captured. Only wells with positive cell capture were further processed for sequencing. For downstream analysis, libraries marked as single cells were chosen. Cells with <1,000 reads mapping to ERCC spike-ins, <100,000 mapped transcriptomic reads or <60% of mapped reads being allocated to the transcriptome were removed. Additionally, cells that express unusually small number of genes (< 2000) were removed. Prior to normalization, genes with >1,000,000 reads or <10 reads across all cells were removed. A tsne plot showing batch labels for each cell is presented in supplementary figure 1l, Supplementary Material online.
Normalization
The BASiCS package (Vallejos et al. 2015) was used to normalize read counts by incorporating ERCC spike-ins for technical noise estimation. Specific ERCC spike-ins were removed if not detected in the data set. Posterior estimates for model parameters were computed by Markov chain Monte Carlo (MCMC) simulation with 40,000 iterations.
The BASiCS_DenoisedCounts function was utilized to compute normalized read counts.
Running the MCMC independently on the different batches confirmed similar small levels of technical noise in the data (supplementary fig. 1h, Supplementary Material online).
Spatial Mapping
Spatial mapping of the sequenced single-cell transcriptomes onto the Platynereis reference atlas was performed as described previously (Achim et al. 2015). For the reference atlas construction, we performed whole-mount in situ hybridization for 176 genes in 48 hpf larvae and produced up to 53 expression images per gene via automated microscopy. We then registered these images to a reference larva and used ProSPr to reconstruct an average expression pattern for each gene that achieves cellular resolution (Vergara et al. 2016). Plotting patterns for all genes onto the same larval average resulted in a cellular resolution atlas (supplementary fig. 4, Supplementary Material online and fig. 3), to which we then correlated the expression vectors of sequenced cells (Achim et al. 2015). The Platynereis reference atlas is provided as supplementary table 4, Supplementary Material online, and the spatial mapping results for all the sequenced cells are provided in supplementary table 5, Supplementary Material online. We defined functional regions within the embryo based on spatial expression patterns of known marker genes. The region in which Phc2 is expressed comprises the apical nervous system. The muscle region of the animal is defined by the expression of St-mhc. Foxj expression defines the ciliated cells and Hnf4 expression the midgut region. The trunk ectoderm can be described by Uncx4 expression. To assign mapped cells to particular regions, we first performed graphical clustering on the voxels for each cell. Second, we identified the voxel cluster with highest mapping confidence for each cell (right hand side of fig. 3). For visualizing the mapping of individual cells, the centroids of all voxel clusters were plotted (left hand side of fig. 3). To compute the overlap of mapped cells to spatial gene expression patterns, we focused on the cluster with highest mapping confidence for each cell. If more than 50% of these voxels also show a particular gene expression (e.g., Syt1), we consider the cell as falling into this gene region.
Detecting Highly Variable Genes
The BASiCS_DetectHVG function in BASiCS allows the detection of highly variable genes by incorporating spike-ins to estimate expected technical variability. Testing was done after MCMC simulation with a variance threshold of 0.98 and an evidence threshold of 0.7.
Clustering
To detect clusters of cells based on the expression of highly variable genes, we used a sparse K-means clustering approach taken from the sparcl package in R (Witten and Tibshirani 2010; Team 2014). We clustered the data using K = 2,…, 10 and chose K = 7 since only small subpopulations emerged at K = 8,…, 10 (supplementary fig. 1j, Supplementary Material online). An elbow plot showing the averaged within-cluster sum of squares is noninformative for the choice of K (supplementary fig. 1i, Supplementary Material online). where is the log10-transformed transcript count of gene i in cell j and cluster k, the log10-transformed mean expression of gene i in cluster k and the number of clusters. The parameters were tuned with 2 < wbounds < 100 using 15 permutations. Genes with weights > 0.03 define the cluster-characteristic features of the data. We compared this clustering strategy to one that does not require an a priori definition of K. The dynamicTreeCut package in R (Langfelder et al. 2008) performs a flexible branch cutting for hierarchical clustering approaches. The mclust package in R (Fraley et al. 2012) performs clustering based on finite mixture modeling of Normals. Comparisons of the cell labels for each K using the sparse clustering approach and the labels detected by dynamicTreeCut and mclust shows a good overlap at K = 7 (supplementary fig. 1k, Supplementary Material online). The following parameters were used for dynamicTreeCut clustering: minClusterSize = 5, deepSplit = 0 and the dendrogram was build using the spearman dissimilarity (see Tree Building) and using ward.D2 as linkage method. The parameters for mclust were chosen by calculating the BIC with the default function mclustBIC. We used the rand index calculated by the cluster_similarity function of the cluster package in R to compare the labels.
Removing Cell Doublets
Possible cell doublets were removed after clustering and marker gene detection. The percentage of group specific marker gene expression (# marker genes expressed/# all genes) was calculated for each cell. Cells with unusually high expression of markers nonspecific for their group were removed from the data set.
Tree Building
To investigate the transcriptional similarity between cell types, we hierarchically clustered cell types before and after averaging gene expression across all cells within each group. The averaging strategy increases stability of the hierarchical clustering algorithm by averaging out dropout events in single-cell data. Distances were calculated based on the Spearman dissimilarity: (Van Dongen and Enright 2012). To compute bootstrapped P values, we used the pvclust package in R (Ryota Suzuki 2006). Hierarchical clustering was performed on the log10-transformed expression counts before and after averaging within each group using the spearman dissimilarity as distance, complete linkage clustering, and 1000 bootstrap iterations to evaluate cluster stability.
Differential Expression
To identify cell group specific enriched marker genes, we used two approaches. First, we used the scde package (Kharchenko et al. 2014) to compare each of the identified cell group against all the remaining samples in order to identify novel group specific genes from the unannotated portion of the Platynereis transcriptome. These genes were then annotated, added to the reference, and the clustering was repeated. Second, differential expression analysis was performed using the findMarkers function from the scran package. This approach uses limma (Ritchie et al. 2015) on the log2-transformed, normalized counts. Group specific marker genes were defined as differentially expressed genes with a FDR < 0.1 and log2FC > 0 for all pairwise comparisons. Initially, differential expression was tested between all seven detected groups. Groups 6 and 7 consistently grouped together when hierarchical clustering (default options from the hclust package) was performed on the log2 fold changes in expression between one group and all others (supplementary fig. 2, Supplementary Material online). We therefore merged groups 6 and 7 and tested differential expression between the now largest group and all other groups (supplementary table 1, Supplementary Material online). In the next step, we tested differential expression pairwise between all differentiated cell groups (supplementary table 2, Supplementary Material online).
In Situ Hybridization
For in situ hybridization (ISH), we collected 48 hpf or 72 hpf Platynereis larvae. Animals were fixed overnight in 4% PFA/1.5× PBS and ISH was performed as described previously (Tessmar-Raible et al. 2005).
Cloning of Platynereis Genes and ISH Probe Synthesis
For the cloning of P. dumerilii cDNA sequences, wild-type Platynereis RNA was isolated by Trizol/phenol/chloroform extraction method, reverse transcribed using SuperScriptIII reverse transcriptase (Life Technologies, cat. # 18080044), and amplified by PCR using TaKaRa ExTaq DNA polymerase (Clontech, cat. # RR001A) and respective gene-specific primers. The resulting PCR fragments were cloned into pCRII-TOPO vector (Life Technologies, cat # K4610-20). The cloned gene sequences and the gene specific primer sequences are available upon request.
For the synthesis of ISH probes, cDNA plasmids were linearized and antisense RNA probes were transcribed using SP6 or T7 RNA polymerase (Roche, cat. #11487671001 and 10881775001, respectively) and DIG RNA-labeling mix (Roche, cat. #11277073910) or Fluorescein RNA labeling mix (Roche, cat. # 11685619910).
Microscopy and Image Processing
For imaging of WMISH samples for ProSPr, samples were mounted in 97% 2,2′-thiodiethanol (Sigma, cat. # 166782) and imaged on a Leica TCS SP8 confocal microscope, using a combination of fluorescence and reflection microscopy (Jekely and Arendt 2007). The colocalization analyses and image postprocessing was performed using Fiji (Schindelin et al. 2012) software. The colorimetric WMISH samples were imaged on Zeiss AXIO Imager M1 microscope. The figure panels were compiled using Adobe Illustrator and Adobe Photoshop software.
Supplementary Material
Supplementary data are available at Molecular Biology and Evolution online.
Supplementary Material
Acknowledgments
We thank Catalina Vallejos for comments on the implementation of BASiCS and all members of the Marioni and Arendt labs for feedback on the project and manuscript. KA and JM were supported by the Marie Curie COFUND programme from the European Commission and by EMBL core funding. NE, PC, VB, and DA were supported by core funding from EMBL. KA, HMV, PYB, PV were supported by the Advanced grant “Brain Evo-Devo” from the European Research Council. JCM was supported by core funding from EMBL and Cancer Research UK.
Author Contributions
K.A. and N.E. planned and conducted experiments, interpreted the data, designed the study, and wrote the manuscript. H.M.V., P.B., and P.C. conducted experiments. T.B. and J.M. interpreted data and edited the manuscript. V.B., J.C.M., and D.A. funded the study. J.C.M. and D.A. designed the study and wrote the manuscript.
References
- Achim K, Pettit J-B, Saraiva LR, Gavriouchkina D, Larsson T, Arendt D, Marioni JC.. 2015. High-throughput spatial mapping of single-cell RNA-seq data to tissue of origin. Nat Biotechnol. 335:503–509. [DOI] [PubMed] [Google Scholar]
- Ackermann C, Dorresteijn A, Fischer A.. 2005. Clonal domains in postlarval Platynereis dumerilii (Annelida: Polychaeta). J Morphol. 2663:258–280. [DOI] [PubMed] [Google Scholar]
- Adolfsen B, Saraswati S, Yoshihara M, Littleton JT.. 2004. Synaptotagmins are trafficked to distinct subcellular domains including the postsynaptic compartment. J Cell Biol. 1662:249–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W.. 2015. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 312:166–169.http://dx.doi.org/10.1093/bioinformatics/btu638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews S. 2010. FastQC: a quality control tool for high throughput sequence data. Available online at:http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
- Arendt D. 2008. The evolution of cell types in animals: emerging principles from molecular studies. Nat Rev Genet. 911:868–882.http://dx.doi.org/10.1038/nrg2416 [DOI] [PubMed] [Google Scholar]
- Arendt D, Musser JM, Baker CVH, Bergman A, Cepko C, Erwin DH, Pavlicev M, Schlosser G, Widder S, Laubichler MD et al. 2016. The origin and evolution of cell types. Nat Rev Genet. 17:744–757. [DOI] [PubMed] [Google Scholar]
- Arendt D, Tessmar-Raible K, Snyman H, Dorresteijn AW, Wittbrodt J.. 2004. Ciliary photoreceptors with a vertebrate-type opsin in an invertebrate brain. Science 3065697:869–871. [DOI] [PubMed] [Google Scholar]
- Arendt D, Tosches MA, Marlow H.. 2016. From nerve net to nerve ring, nerve cord and brain–evolution of the nervous system. Nat Rev Neurosci. 171:61–72.http://dx.doi.org/10.1038/nrn.2015.15 [DOI] [PubMed] [Google Scholar]
- Asadulina A, Panzera A, Verasztó C, Liebig C, Jékely G. 2012. Whole-body gene expression pattern registration in Platynereis larvae. Evodevo 31:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates KE, Whitington PM.. 2007. Semaphorin 2a secreted by oenocytes signals through plexin B and plexin A to guide sensory axons in the Drosophila embryo. Dev Biol. 3022:522–535. [DOI] [PubMed] [Google Scholar]
- Boglev Y, Wilanowski T, Caddy J, Parekh V, Auden A, Darido C, Hislop NR, Cangkrama M, Ting SB, Jane SM et al. , 2011. The unique and cooperative roles of the Grainy head-like transcription factors in epidermal development reflect unexpected target gene specificity. Dev Biol. 3492:512–522. [DOI] [PubMed] [Google Scholar]
- Brawand D, Soumillon M, Necsulea A, Julien P, Csárdi G, Harrigan P, Weier M, Liechti A, Aximu-Petri A, Kircher M et al. 2011. The evolution of gene expression levels in mammalian organs. Nature 478:343–348. [DOI] [PubMed] [Google Scholar]
- Breschi A, Djebali S, Gillis J, Pervouchine DD, Dobin A, Davis CA, Gingeras TR, Guigó R.. 2016. Gene-specific patterns of expression variation across organs and species. Genome Biol. 171:151.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet T, Fischer AH, Steinmetz PR, Lauri A, Bertucci P, Arendt D.. 2016. The evolutionary origin of bilaterian smooth and striated myocytes. Elife 5:e19607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burmester T, Weich B, Reinhardt S, Hankeln T.. 2000. A vertebrate globin expressed in the brain. Nature 4076803:520–523.http://dx.doi.org/10.1038/35035093 [DOI] [PubMed] [Google Scholar]
- Cepko CL. 2015. The Determination of Rod and Cone Photoreceptor Fate. Annu Rev Vis Sci. 1:211–234. [DOI] [PubMed] [Google Scholar]
- Chernyavsky AI, Arredondo J, Vetter DE, Grando SA.. 2007. Central role of alpha9 acetylcholine receptor in coordinating keratinocyte adhesion and motility at the initiation of epithelialization. Exp Cell Res. 31316:3542–3555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conzelmann M, Williams EA, Krug K, Franz-Wachtel M, Macek B, Jekely G.. 2013. The neuropeptide complement of the marine annelid Platynereis dumerilii. BMC Genomics 14:906.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denes AS, Jékely G, Steinmetz PRH, Raible F, Snyman H, Prud’homme B, Ferrier DEK, Balavoine G, Arendt D.. 2007. Molecular architecture of annelid nerve cord supports common origin of nervous system centralization in bilateria. Cell 1292:277–288. [DOI] [PubMed] [Google Scholar]
- Dobi KC, Schulman VK, Baylies MK.. 2015. Specification of the somatic musculature in Drosophila. Wiley Interdiscip Rev Dev Biol. 44:357–375.http://dx.doi.org/10.1002/wdev.182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez L, Gonzalez A, Moreno N.. 2015. Patterns of hypothalamic regionalization in amphibians and reptiles: common traits revealed by a genoarchitectonic approach. Front Neuroanat. 9:3.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabrizius A, Andre D, Laufs T, Bicker A, Reuss S et al. , 2016. Critical re-evaluation of neuroglobin expression reveals conserved patterns among mammals. Neuroscience 337:339–354. [DOI] [PubMed] [Google Scholar]
- Felbor U, Kessler B, Mothes W, Goebel HH, Ploegh HL, Bronson RT, Olsen BR. 2002. Neuronal loss and brain atrophy in mice lacking cathepsins B and L. Proc Natl Acad Sci U S A. 99:7883–7888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman RJ, Sementchenko VI, Watson DK.. 2003. The epithelial-specific Ets factors occupy a unique position in defining epithelial proliferation, differentiation and carcinogenesis. Anticancer Res. 23(3A):2125–2131. [PubMed] [Google Scholar]
- Fischer A, Dorresteijn AWC.. 2004. The polychaete Platynereis dumerilii (Annelida): a laboratory animal with spiralian cleavage, lifelong segment proliferation and a mixed benthic/pelagic life cycle. BioEssays 26:314–325.http://dx.doi.org/10.1002/bies.10409 [DOI] [PubMed] [Google Scholar]
- Fischer AH, Henrich T, Arendt D.. 2010. The normal development of Platynereis dumerilii (Nereididae, Annelida). Front Zool. 7:31.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraley C, Raftery AE, Murphy TB, Scrucca L.. 2012. mclust Version 4 for R: Normal Mixture Modeling for Model-Based Clustering, Classification, and Density Estimation, Technical Report Number 597.
- Fremion F, Darboux I, Diano M, Hipeau-Jacquotte R, Seeger MA, Piovant M.. 2000. Amalgam is a ligand for the transmembrane receptor neurotactin and is required for neurotactin-mediated cell adhesion and axon fasciculation in Drosophila. EMBO J. 19:4463–4472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda M. 2003. Molecular cloning and characterization of human, rat, and mouse synaptotagmin XV. Biochem Biophys Res Commun. 3061:64–71. [DOI] [PubMed] [Google Scholar]
- Grun D, Lyubimova A, Kester L, Wiebrands K, Basak O, Sasaki N, Clevers H, van Oudenaarden A. 2015. Single-cell messenger RNA sequencing reveals rare intestinal cell types. Nature 525:251–255. [DOI] [PubMed] [Google Scholar]
- Gustavsson N, Wu B, Han W.. 2012. Calcium sensing in exocytosis. Adv Exp Med Biol. 740:731–757. [DOI] [PubMed] [Google Scholar]
- Henrichsen CN, Vinckenbosch N, Zollner S, Chaignat E, Pradervand S, Schütz F, Ruedi M, Kaessmann H, Reymond A. 2009. Segmental copy number variation shapes tissue transcriptomes. Nat Genet. 41:424–429. [DOI] [PubMed] [Google Scholar]
- Herdman C, Moss T.. 2016. Extended-Synaptotagmins (E-Syts); the extended story. Pharmacol Res. 107:48–56.http://dx.doi.org/10.1016/j.phrs.2016.01.034 [DOI] [PubMed] [Google Scholar]
- Hobert O. 2011. Regulation of terminal differentiation programs in the nervous system. Annu Rev Cell Dev Biol. 27:681–696.http://dx.doi.org/10.1146/annurev-cellbio-092910-154226 [DOI] [PubMed] [Google Scholar]
- Hundahl CA, Allen GC, Nyengaard JR, Dewilde S, Carter BD, Kelsen J, Hay-Schmidt A.. 2008. Neuroglobin in the rat brain: localization. Neuroendocrinology 883:173–182. [DOI] [PubMed] [Google Scholar]
- Hundahl CA, Fahrenkrug J, Hay-Schmidt A, Georg B, Faltoft B, Hannibal J.. 2012. Circadian behaviour in neuroglobin deficient mice. PLoS One 74:e34462.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jekely G, Arendt D.. 2007. Cellular resolution expression profiling using confocal detection of NBT/BCIP precipitate by reflection microscopy. Biotechniques 42:751–755.http://dx.doi.org/10.2144/000112462 [DOI] [PubMed] [Google Scholar]
- Kee N, Volakakis N, Kirkeby A, Dahl L, Storvall H, Nolbrant S, Lahti L, Björklund ÅK, Gillberg L, Joodmardi E et al. , 2017. Single-Cell Analysis Reveals a Close Relationship between Differentiating Dopamine and Subthalamic Nucleus Neuronal Lineages. Cell Stem Cell. 201:29–40. [DOI] [PubMed] [Google Scholar]
- Kharchenko PV, Silberstein L, Scadden DT.. 2014. Bayesian approach to single-cell differential expression analysis. Nat Methods 11:740–742.http://dx.doi.org/10.1038/nmeth.2967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, McDole K, Zheng Y.. 2012. The function of lamins in the context of tissue building and maintenance. Nucleus 33:256–262.http://dx.doi.org/10.4161/nucl.20392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Zhang B, Horvath S.. 2008. Defining clusters from a hierarchical cluster tree: the Dynamic Tree Cut package for R. Bioinformatics 24:719–720.http://dx.doi.org/10.1093/bioinformatics/btm563 [DOI] [PubMed] [Google Scholar]
- Lee MY, Choi SH, Shin SL, Chin H, Kwon OJ.. 2001. Distribution of B/K protein in rat brain. Cell Tissue Res. 3031:47–56. [DOI] [PubMed] [Google Scholar]
- Li W, Godzik A.. 2006. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22:1658–1659.http://dx.doi.org/10.1093/bioinformatics/btl158 [DOI] [PubMed] [Google Scholar]
- Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM et al. , 2015. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 1615:1202–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marlow H, Tosches MA, Tomer R, Steinmetz PR, Lauri A, Larsson T, Arendt D.. 2014. Larval body patterning and apical organs are conserved in animal evolution. BMC Biol. 12:7.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto-Miyai K, Sokolowska E, Zurlinden A, Gee CE, Lüscher D, Hettwer S, Wölfel J, Ladner AP, Ster J, Gerber U et al. , 2009. Coincident pre- and postsynaptic activation induces dendritic filopodia via neurotrypsin-dependent agrin cleavage. Cell 1366:1161–1171. [DOI] [PubMed] [Google Scholar]
- Michaluk P, Wawrzyniak M, Alot P, Szczot M, Wyrembek P, Mercik K, Medvedev N, Wilczek E, De Roo M, Zuschratter W et al. , 2011. Influence of matrix metalloproteinase MMP-9 on dendritic spine morphology. J Cell Sci. 124(Pt 19):3369–3380. [DOI] [PubMed] [Google Scholar]
- Moghadam PK, Jackson MB.. 2013. The functional significance of synaptotagmin diversity in neuroendocrine secretion. Front Endocrinol (Lausanne) 4:124.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muraro MJ, Dharmadhikari G, Grun D, Groen N, Dielen T, Jansen E, van Gurp L, Engelse MA, Carlotti F, de Koning EJ, et al. 2016. A single-cell transcriptome atlas of the human pancreas. Cell Syst. 3:385–394 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy V, Taranda J, Elgoyhen AB, Vetter DE.. 2009. Activity of nAChRs containing alpha9 subunits modulates synapse stabilization via bidirectional signaling programs. Dev Neurobiol. 6914:931–949.http://dx.doi.org/10.1002/dneu.20753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata T, Koyanagi M, Tsukamoto H, Terakita A.. 2010. Identification and characterization of a protostome homologue of peropsin from a jumping spider. J Comp Physiol A Neuroethol Sens Neural Behav Physiol. 196:51–59.http://dx.doi.org/10.1007/s00359-009-0493-9 [DOI] [PubMed] [Google Scholar]
- Nomaksteinsky M, Kassabov S, Chettouh Z, Stoeklé H-C, Bonnaud L, Fortin G, Kandel ER, Brunet J-F.. 2013. Ancient origin of somatic and visceral neurons. BMC Biol. 11:53.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palanker L, Tennessen JM, Lam G, Thummel CS.. 2009. Drosophila HNF4 regulates lipid mobilization and beta-oxidation. Cell Metab. 93:228–239.http://dx.doi.org/10.1016/j.cmet.2009.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park D, Li P, Dani A, Taghert PH.. 2014. Peptidergic cell-specific synaptotagmins in Drosophila: localization to dense-core granules and regulation by the bHLH protein DIMMED. J Neurosci. 3439:13195–13207.http://dx.doi.org/10.1523/JNEUROSCI.2075-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedemonte N, Galietta LJ.. 2014. Structure and function of TMEM16 proteins (anoctamins). Physiol Rev. 942:419–459.http://dx.doi.org/10.1152/physrev.00039.2011 [DOI] [PubMed] [Google Scholar]
- Pruitt MM, Letcher EJ, Chou HC, Bastin BR, Schneider SQ.. 2014. Expression of the wnt gene complement in a spiral-cleaving embryo and trochophore larva. Int J Dev Biol. 58:563–573. [DOI] [PubMed] [Google Scholar]
- Rabe TI, Griesel G, Blanke S, Kispert A, Leitges M, van der Zwaag B, Burbach JPH, Varoqueaux F, Mansouri A.. 2012. The transcription factor Uncx4.1 acts in a short window of midbrain dopaminergic neuron differentiation. Neural Dev. 71:39.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramskold D, Wang ET, Burge CB, Sandberg R.. 2009. An abundance of ubiquitously expressed genes revealed by tissue transcriptome sequence data. PLoS Comput Biol. 512:e1000598.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK.. 2015. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 437:e47.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanov RA, Zeisel A, Bakker J, Girach F, Hellysaz A, Tomer R, Alpár A, Mulder J, Clotman F, Keimpema E, et al. 2017. Molecular interrogation of hypothalamic organization reveals distinct dopamine neuronal subtypes. Nat Neurosci. 20:176–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryota Suzuki HS. 2006. Pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics 22:1540–1542.http://dx.doi.org/10.1093/bioinformatics/btl117 [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B et al. , 2012. Fiji: an open-source platform for biological-image analysis. Nat Methods 97:676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scialdone A, Tanaka Y, Jawaid W, Moignard V, Wilson NK, Macaulay IC, Marioni JC, Göttgens B. 2016. Resolving early mesoderm diversification through single-cell expression profiling. Nature 5357611:289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiler C, Finger-Baier KC, Rinner O, Makhankov YV, Schwarz H, Neuhauss SCF, Nicolson T.. 2005. Duplicated genes with split functions: independent roles of protocadherin15 orthologues in zebrafish hearing and vision. Development 1323:615–623. [DOI] [PubMed] [Google Scholar]
- Sonderegger P, Matsumoto-Miyai K.. 2014. Activity-controlled proteolytic cleavage at the synapse. Trends Neurosci. 378:413–423.http://dx.doi.org/10.1016/j.tins.2014.05.007 [DOI] [PubMed] [Google Scholar]
- Speicher S, Garcia-Alonso L, Carmena A, Martin-Bermudo MD, de la Escalera S, Jimenez F.. 1998. Neurotactin functions in concert with other identified CAMs in growth cone guidance in Drosophila. Neuron 202:221–233. [DOI] [PubMed] [Google Scholar]
- Stephan A, Mateos JM, Kozlov SV, Cinelli P, Kistler AD, Hettwer S, Rülicke T, Streit P, Kunz B, Sonderegger P et al. , 2008. Neurotrypsin cleaves agrin locally at the synapse. FASEB J. 226:1861–1873. [DOI] [PubMed] [Google Scholar]
- Tasic B, Menon V, Nguyen TN, Kim TK, Jarsky T, Yao Z, Levi B, Gray LT, Sorensen SA, Dolbeare T et al. , 2016. Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nat Neurosci. 192:335–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Team RC. 2014. R: a language and environment for statistical computing. Vienna (Austria): R Foundation for Statistical Computing.
- Tessmar-Raible K, Raible F, Christodoulou F, Guy K, Rembold M, Hausen H, Arendt D.. 2007. Conserved sensory-neurosecretory cell types in annelid and fish forebrain: insights into hypothalamus evolution. Cell 1297:1389–1400. [DOI] [PubMed] [Google Scholar]
- Tessmar-Raible K, Steinmetz PR, Snyman H, Hassel M, Arendt D.. 2005. Fluorescent two-color whole mount in situ hybridization in Platynereis dumerilii (Polychaeta, Annelida), an emerging marine molecular model for evolution and development. Biotechniques 39:460, 62, 64. [DOI] [PubMed] [Google Scholar]
- Tomer R, Denes AS, Tessmar-Raible K, Arendt D. 2010. Profiling by image registration reveals common origin of annelid mushroom bodies and vertebrate pallium. Cell 1425:800-9. [DOI] [PubMed] [Google Scholar]
- Tosches MA, Arendt D.. 2013. The bilaterian forebrain: an evolutionary chimaera. Curr Opin Neurobiol. 236:1080–1089.http://dx.doi.org/10.1016/j.conb.2013.09.005 [DOI] [PubMed] [Google Scholar]
- Tosches MA, Bucher D, Vopalensky P, Arendt D.. 2014. Melatonin signaling controls circadian swimming behavior in marine zooplankton. Cell 1591:46–57.http://dx.doi.org/10.1016/j.cell.2014.07.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyndall SJ, Walikonis RS.. 2006. The receptor tyrosine kinase Met and its ligand hepatocyte growth factor are clustered at excitatory synapses and can enhance clustering of synaptic proteins. Cell Cycle 514:1560–1568.http://dx.doi.org/10.4161/cc.5.14.2918 [DOI] [PubMed] [Google Scholar]
- Vadon-Le Goff S, Hulmes DJ, Moali C.. 2015. BMP-1/tolloid-like proteinases synchronize matrix assembly with growth factor activation to promote morphogenesis and tissue remodeling. Matrix Biol. 44–46:14–23.http://dx.doi.org/10.1016/j.matbio.2015.02.006 [DOI] [PubMed] [Google Scholar]
- Vallejos CA, Marioni JC, Richardson S.. 2015. BASiCS: Bayesian analysis of single-cell sequencing data. PLoS Comput Biol. 116:e1004333.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dongen S, Enright A.. 2012. Metric distances derived from cosine similarity and Pearson and Spearman correlations. arXiv: 1208.3145.
- Vergara HM, Bertucci P, Hantz P, Tosches MA, Vopalensky P, Arendt D.. 2016. A whole-organism cellular gene expression atlas reveals conserved cell type clusters in the ventral nerve cord of Platynereis dumerilii. Proc Natl Acad Sci U S A. 114:5878–5885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vopalensky P, Tosches MA, Achim K, Handberg-Thorsager M, Fischer A, Arendt D.. 2018. From spiral cleavage to bilateral symmetry: the developmental cell lineage of the annelid brain. BIORXIV/2018/268177. [DOI] [PMC free article] [PubMed]
- Williams EA, Jekely G.. 2016. Towards a systems-level understanding of development in the marine annelid Platynereis dumerilii. Curr Opin Genet Dev. 39:175–181.http://dx.doi.org/10.1016/j.gde.2016.07.005 [DOI] [PubMed] [Google Scholar]
- Williams EA, Verasztó C, Jasek S, Conzelmann M, Shahidi R et al. , 2017. Synaptic and peptidergic connectome of a neurosecretory centre in the annelid brain. Elife 6:e26349. [DOI] [PMC free article] [PubMed]
- Witten DM, Tibshirani R.. 2010. A framework for feature selection in clustering. J Am Stat Assoc. 105:713–726.http://dx.doi.org/10.1198/jasa.2010.tm09415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeev-Ben-Mordehai T, Paz A, Peleg Y, Toker L, Wolf SG, Rydberg EH, Sussman JL, Silman I.. 2009. Amalgam, an axon guidance Drosophila adhesion protein belonging to the immunoglobulin superfamily: over-expression, purification and biophysical characterization. Protein Expr Purif. 632:147–157. [DOI] [PubMed] [Google Scholar]
- Zeisel A, Muñoz-Manchado AB, Codeluppi S, Lönnerberg P, La Manno G, Juréus A, Marques S, Munguba H, He L, Betsholtz C et al. , 2015. Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 3476226:1138–1142. [DOI] [PubMed] [Google Scholar]
- Zhang G, Bai H, Zhang H, Dean C, Wu Q, Li J, Guariglia S, Meng Q, Cai D.. 2011. Neuropeptide exocytosis involving synaptotagmin-4 and oxytocin in hypothalamic programming of body weight and energy balance. Neuron 693:523–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw sequence data for the cells analyzed in this study are available from ArrayExpress, accession numbers E-MTAB-2865 (see supplementary table 5, Supplementary Material online, for matching the cell ID-s between the E-MTAB-2865 and this study) and E-MTAB-5953. The R code used for analysis is available at: https://github.com/MarioniLab/Platynereis2017.