

Abstract
Plant homeodomain (PHD) zinc fingers are histone reader domains that are often associated with human diseases. Despite this, they constitute a poorly targeted class of readers, suggesting low ligandability. Here, we describe a successful fragment-based campaign targeting PHD fingers from the proteins BAZ2A and BAZ2B as model systems. We validated a pool of in silico fragments both biophysically and structurally and solved the first crystal structures of PHD zinc fingers in complex with fragments bound to an anchoring pocket at the histone binding site. The best-validated hits were found to displace a histone H3 tail peptide in competition assays. This work identifies new chemical scaffolds that provide suitable starting points for future ligand optimization using structure-guided approaches. The demonstrated ligandability of the PHD reader domains could pave the way for the development of chemical probes to drug this family of epigenetic readers.
The plant homeodomain (PHD) zinc fingers are small reader domains found in several chromatin-binding proteins. They are characterized by the conserved motif Cys4-His-Cys3, which binds two Zn ions important for the structural integrity of the domain. PHDs recognize a diverse set of histone marks,1 as well as DNA sequences;2 the specificity of binding is dictated by the properties of the pocket.3 With more than 170 sequences annotated as PHD finger in the human genome, they are considered one of the largest families among reader domains.4 They are often found located in tandem with other reader domains, suggesting a potential cross-talk among readers. Genetic evidence has linked PHD fingers to disease-related pathways,5 electing them as a new class of potential epigenetic drug targets.1 However, unlike other reader domains (e.g., bromodomains),6 PHD fingers have been proven difficult to target with small molecules, and no chemical probe has been reported to date against them. To this end, only two studies have investigated the ligandability of this class of reader domains. In a first study, the Halo-tag technology was used for screening small molecules against the PHD of JARID1A and identified a chemical scaffold potentially able to disrupt the interaction with the H3K4me3 peptide, although no structural data were provided.7 In a later study, fragment screening was applied to target the PHD finger of Pygo and the best compound identified as binding to a cleft proximal to the histone pocket.8
Here, we describe a fragment-based approach that yielded fragments targeting two ligandable pockets of a PHD domain, one of which is at the PHD/histone interface. As model systems, we chose the PHD fingers of two proteins of the BAZ (bromodomain adjacent to zinc finger) family:9 BAZ2A and BAZ2B. The reason for choosing these targets was their suitability to biophysical and structural investigation, as previously demonstrated by our laboratory.10,11 PHD zinc fingers in BAZ2 lie in proximity of bromodomain readers.9 Several studies already attempted to address the druggability of BAZ2 bromodomains,12−16 whereas, to our knowledge, no study has yet assessed the ligandability of BAZ2 PHD domains. Further motivation came from an ultimate goal to develop chemical probes that could inform on the biological function of these proteins. BAZ2A is the most studied member of the family, and it was previously identified as part of the nucleolar remodelling complex (NoRC), which mediates silencing of rDNA.17 BAZ2A was found to be involved in prostate cancer, and its expression levels were proposed as potential biomarker for the diagnosis of this type of cancer.18 In contrast, BAZ2B is much less characterized. It was found associated with a doubling of the risk of sudden cardiac death (SCD)19 but its biological function remains unknown and information on potential interactions with other macromolecules remains elusive. Therefore, the development of chemical probes able to target BAZ2 reader domains could provide useful tools to shed light on the biological role of these proteins.
We began our ligandability study by considering the known protein–protein interaction of the targeted PHD domains. Previous research has shown that BAZ2 PHDs recognize preferentially unmodified H3 histone tail promoting helicity in the H3 peptide upon binding.10,11 It was also shown that the mutation to alanine of the second and third residues of the H3 histone tail abolishes binding.11,20 Therefore, we hypothesized that the H3 N-terminal 3-mer motif “ART” might be essential to anchor the histone tail to the surface of BAZ2 PHDs. To test this hypothesis, we synthesized and biophysically characterized the binding of ART against both PHD domains. The 3-mer peptide retained binding, as measured by NMR and ITC, with dissociation constants of 1–2 mM (see Figure S1 in the Supporting Information). Next, we solved the crystal structure of the PHD of BAZ2A in complex with ART, which confirmed the expected binding mode with the peptide superposing well with the first three residues of the bound H3 tail crystal structure11 (see Figure 1A, as well as Figure S2 in the Supporting Information). The amidic nitrogen at the C-terminus of the ART peptide forms interactions with the backbone carbonyl of Leu1691 and Asp1688 (Figure 1B). The tripeptide surface covers an area of 610 Å2, which is comparable with the size of small molecules.21 This observation encouraged us toward the possibility to disrupt the PHD–histone interaction with small molecules. Thus, we embarked on a fragment screening campaign to explore the propensity of the protein surface to bind small molecules.
Figure 1.
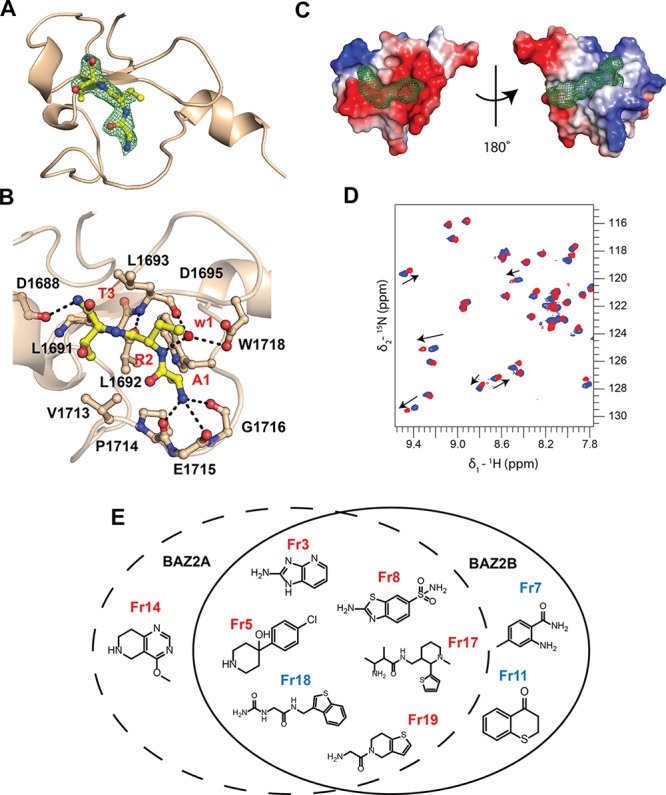
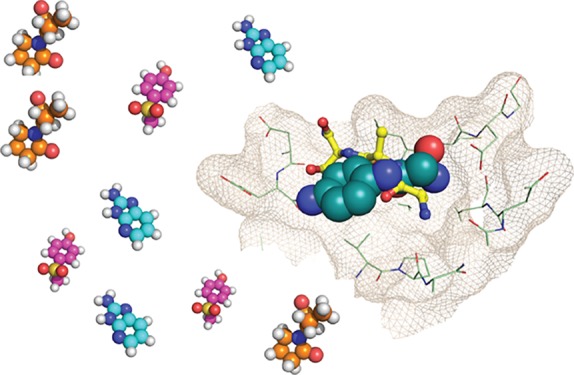
Druggable pockets on BAZ2A/B PHD and validated fragments. (A) Crystal structure of BAZ2A PHD in complex with ART tripeptide. Fo – Fc electron density map of the peptide is contoured at 3σ. The R2 side chain of the peptide is not visible in the electron density. (B) Close-up view of the interactions. (C) Druggable binding sites in BAZ2A PHD (PDB: 4QF2)10 identified by FTMap, shown as green mesh. Protein surface is colored according to the electrostatic potential. (D) Overlay of (15N–1H) HSQC spectra recorded on the apo form of 15N-BAZ2B PHD (blue) and after 5 mM fragment addition (red). Arrows represent the shift direction. (E) Chemical structures of the in silico fragments validated by HSQC. Fragments reporting binding by NMR to the histone pocket are shown in red, and fragments reporting binding by NMR to the back pocket are shown in blue.
First, we assessed the ligandability of the histone pocket of BAZ2A using FTMap22 to probe the apo crystal structure. FTMap readily identified the histone pocket as a hit (Figure 1C), even though it perceived it as a borderline druggable pocket likely requiring charged compounds, in agreement with the salt-bridges present in the BAZ2A–ART complex (Figure 1B). Interestingly, FTMap identified a second druggable binding site in BAZ2A, which is located opposite to the histone pocket and is rather hydrophobic (Figure 1C), which we refer to here as the “back pocket”. Thus, we next assembled a diverse virtual library of a thousand low-molecular-weight compounds and performed in silico docking using Glide 7.0 (Schrödinger, LLC), targeting both pockets in BAZ2 PHDs. Because proposed histone-pocket binders have a tendency to be positively charged, selected compounds were subsequently rescored using MM-GBSA (Prime 3.0, Schrodinger, LLC), which accounts for a better estimation of desolvation penalties upon binding. We normalized the docking rankings by efficiency, visually inspected the top-ranked compounds, and selected 19 fragments (see Table S1 in the Supporting Information). Compounds were purchased and binding to both proteins was interrogated via a biophysical screening cascade.23 First-pass screen was performed using (15N–1H)-heteronuclear single-quantum coherence (HSQC) NMR spectroscopy. Advantages of using HSQC as primary validation step are (a) its sensitivity to low-affinity interactions and (b) the possibility to provide information on the region of binding through chemical shift mapping.24 Furthermore, chemical shift perturbations (CSPs) can be used to estimate the binding affinities24 and, consequently, ligand efficiency of the fragments (see Table S2 in the Supporting Information). Second-pass, thermal shift assay (TSA) was performed to test if the HSQC-validated fragments were able to stabilize or destabilize BAZ2 PHDs in solution. In parallel, an AlphaLISA competition assay25 was developed to assess the ability of these fragments to displace an H3 peptide from the histone pocket. Ultimately, X-ray crystallography was used to investigate the binding mode of the validated fragments.
BAZ2 PHD (15N–1H)-HSQC spectra were suitable for CSP experiments, using resonances previously assigned.11 (15N–1H) HSQC spectra obtained after incubation of each protein with each single fragment were overlaid with the apo form spectrum of the protein; those fragments showing chemical shift for at least one resonance of the spectrum were considered as binders (Figure 1D). Of the 19 compounds tested, nine fragments were confirmed by HSQC as potential binders (∼47% confirmation rate). Most of the hits were common to the two PHDs, with a few fragments selectively binding BAZ2A or BAZ2B (see Figure 1E, as well as Figure S3 in the Supporting Information). Among this pool of validated fragments, it was noted that fragments Fr3 and Fr8 presented a relatively similar scaffold and the poses predicted by docking, which reported binding to the histone pocket, were in agreement with the CSP heat map (see Figure 2A, as well as Figure S3). The predicted binding mode of Fr3 to BAZ2B PHD (Figure 2A) shows how the amino group on the azole derivative is thought to be the driving force of binding. Indeed, it was noted that a similar fragment, Fr15, which carries the −NH2 group on a phenyl ring (Table S1), did not show binding by HSQC. Interestingly, Miller et al. also identified a benzothiazole scaffold (CF4) analogous to Fr8 as a binder of the Pygo PHD.8 In that study, CF4 was shown to bind the protein at a back surface opposite to the histone pocket, which was defined as the benzothiazole cleft.8 Herein, we propose that the benzothiazole scaffold (Fr8), based on our modeling and HSQC experiments, binds, instead, to the histone pocket of BAZ2 PHDs (see Figures S3 and S4 in the Supporting Information). Structural analyses showed no similarities between the benzothiazole cleft of Pygo8 and the histone pocket of BAZ2A PHD (see Figure S4). HSQC experiments also allowed identifying fragments binding to the back pocket, for example, Fr7 for BAZ2B PHD, according to the docking model (see Figure 2B). Binding affinity was not estimated for this fragment, since chemical shift intensities were too low (<0.03 ppm) and resonances did not display relevant changes upon titration, implying binding that was too weak (see Figure S5 in the Supporting Information). The proximity of Fr7 to the Zn and Cys1956 coordinating Zn suggests that it might be possible in the future to increase its binding affinity by growing the fragment with a group able to chelate the Zn or covalently bind, for example, residue Cys1956 (see Figure 2B).7
Figure 2.

Biophysical and structural validation of fragment hits. (A) Docking
pose of BAZ2B PHD and Fr3 showing a set of residues shifted
in HSQC and clustered at the histone pocket. Residues are colored
according to the intensity of the shifts: strong shifts in red (Δδ
> 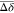 + 2σ), intermediate shifts in orange
(Δδ >
+ 2σ), intermediate shifts in orange
(Δδ > 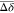 + σ) and lower shifts or no shifts
in green (Figure S3). (B) Docking pose
of BAZ2B PHD and Fr7 with shifts clustered at the back
pocket of BAZ2B and close-up view of in silico predicted
interactions. (C) Crystal structure of BAZ2A PHD in complex with Fr19 (in sticks, with green carbons). Fo – Fc electron density
map is contoured at 3 σ around the bound fragment. The Thr3
methyl hydrophobic pocket is colored in yellow, and the acidic wall
is red. (D) Chemical structures of optimized fragments.
+ σ) and lower shifts or no shifts
in green (Figure S3). (B) Docking pose
of BAZ2B PHD and Fr7 with shifts clustered at the back
pocket of BAZ2B and close-up view of in silico predicted
interactions. (C) Crystal structure of BAZ2A PHD in complex with Fr19 (in sticks, with green carbons). Fo – Fc electron density
map is contoured at 3 σ around the bound fragment. The Thr3
methyl hydrophobic pocket is colored in yellow, and the acidic wall
is red. (D) Chemical structures of optimized fragments.
The nine HSQC-validated hits were tested by TSA, monitoring shifts of the melting temperature of the PHD domains upon the addition of fragments (Figure S6). Some of the fragments showed positive shifts and others showed negative shifts (see Table S3 in the Supporting Information). For example, Fr19, which showed binding to the histone pocket by HSQC, reported negative ΔTm when tested with BAZ2A PHD (Table S3). As previously shown, both positive and negative shifts can yield validated fragment hits.26 We next questioned if these HSQC-validated fragments could affect the PHD–histone interaction. To this extent, an AlphaLISA25 competition assay was developed to measure the displacement of a Flag-tagged peptide.11 We decided to use the peptide ARTAATARKS (referred to as AA mutant), because it binds significantly more tightly than the wild-type sequence (by 4-fold and 14-fold vs BAZ2A and BAZ2B, respectively, as measured by ITC11). Co-crystal structure revealed that the AA mutant peptide retains the same binding mode of the wild-type sequence (see Figure S7 in the Supporting Information), supporting its use as a displacement probe for the assay (further details are described in the Supporting Information). The functionality of the assay was corroborated using the corresponding untagged AA mutant peptide (see Figure S8 in the Supporting Information). We found that some of the tested fragments could displace the histone peptide in the assay in a concentration-dependent manner, for example, Fr3 (see Figure S9 in the Supporting Information). In contrast, other fragments, such as, for example, Fr18, did show interference only at high concentration (>1 mM); this was expected since, by HSQC data, Fr18 binds the back pocket (see Figures S3 and S9).
To investigate the molecular details of the interaction of the BAZ2 PHDs with the nine HSQC-validated fragments, we performed co-crystallization and soaking experiments. The crystal structure of BAZ2A PHD in complex with Fr19 was solved at 1.9 Å. The unbiased Fo – Fc electron density map was observed in two of the four protomers (chains B and C), within the pocket that accommodates Ala1 of the histone peptide (Figure 2C). The 2-amino-N-acetamide moiety of the fragment was fully defined in the electron density, while the density was incomplete for the 4,5,6,7-tetrahydrothieno(3,2-c)pyridine ring of the ligand (see Figure 2C, as well as Figure S10 in the Supporting Information). In the crystal lattice, the fragment binding site from one protein chain packs against one from the neighboring asymmetric unit, and this constrains the 5,6 fused ring of the fragment to adopt two distinct orientations in order to avoid steric clash (see Figure S11 in the Supporting Information). This results in ring–ring stacking interactions from neighboring asymmetric units, and packing against Leu1693 in one chain (see Figure 2C and Figure S11). In both bound conformations, the terminal amino group of the fragment displaces the structural water W1 (Figure 1B) to form three hydrogen bonds: one with the side-chain carboxylate of Asp1695, a second one with the Leu1693 backbone carbonyl, and a third one with a new water molecule (observed bound in three of the four protomers). The amide carbonyl of the fragment forms a hydrogen bond with the Leu1693 backbone NH (see Figure 2C).
The conformations adopted by Fr19 do not allow the 5,6 fused ring to fill the hydrophobic pocket that accommodates the Thr3 methyl group of the H3 peptide, defined by Leu1692, Val1713, and Ile1703 (see Figure 2C and 1B). From these observations, optimization of Fr19 was attempted in silico using molecular docking. The 2-amino-N-acetamide moiety was constrained to the crystallographic conformation, while modeling focused on determining suitable groups to better fill the hydrophobic cavity. Opening of the aliphatic ring showed improvement in the flexibility of the molecule, and allowed one to introduce aromatic substituents attached to the secondary amide. Phenyl and thiazole aryls were identified as suitable groups (Figure 2D). To further stabilize the aromatic ring in the pocket, we also chose phenol and aniline substituents (Figure 2D) to engage in hydrogen bond interactions with Asp1688 on the acidic wall (ref (11), shown in red in Figure 2C). The insertion of a methyl on the 2-amino terminal of the fragment (Figure 2D) aimed to address hydrophobic interactions in the pocket defined by Trp1718 and Pro1714, which accommodate the methyl of Ala1 of the H3 peptide (Figure 1B). Fragments Fr20–Fr23 (Figure 2D) were tested as singleton in HSQC, which confirmed the histone pocket as a region of binding (see Figure S12 in the Supporting Information). Binding affinities, as measured by CSPs, did not remarkably improve (Figure S12).
To gain further insights on the binding mode of these fragments, soaking experiments were performed. Crystal structures of BAZ2A and BAZ2B PHD were solved in complex with Fr23 and of BAZ2B PHD in complex with Fr21 (data collection and refinement statistics are reported in Table 1). Crystal structure of BAZ2B PHD with Fr21 (see Figure 3A, Figure S13D in the Supporting Information, and Table 1) shows that the fragment interacts with residues Leu1947, Leu1948, Asp1950, Gly1971, Ile1968, and Pro1969, which were previously identified as hotspots of binding for the H3 10-mer peptide.11 The secondary amide introduced a higher flexibility able to fill the hydrophobic pocket with the thiazole group (Figure 3A). The ring was modeled with the S atom pointing toward the beta strand of the PHD, in agreement with high-level DFT calculations, which show a strong preference of the amide to remain in the thiazole plane with the S atom pointing toward the amide oxygen (Figure 3D). This conformation is stabilized by an n → σ* electron donation from the lone pair of the amide oxygen (n) to the antibonding S–C orbital (σ*) in the thiazole (En→σ* = 2.7 kcal/mol; see the Supporting Information for more details).
Table 1. Crystallographic Data Collection and Refinement Statisticsa.
| data collection | BAZ2A-ART | BAZ2A-ARTAATARKS | BAZ2A-Fr19 | BAZ2A-Fr23 |
|---|---|---|---|---|
| space group | P43212 | P43212 | P43212 | P43212 |
| cell dimensions | ||||
| a, b, c (Å) | 73.4, 73.4, 99.7 | 72.7, 72.7, 99.9 | 72.1, 72.1, 99.2 | 72.8, 72.8, 99.6 |
| α, β, γ (deg) | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 |
| resolution | 46.03 (2.0) | 45.72 (2.0) | 45.34 (1.9) | 45.2 (2.7) |
| unique observations | 19049 | 18751 | 21069 | 7841 |
| completeness (%) | 99.9 (99.9) | 99.9 (99.9) | 99.3 (98.8) | 100 (100) |
| redundancy | 8.6 (9.0) | 9.7 (9.9) | 3.8 (4.0) | 12 (12.3) |
| CC1/2 | 0.99 (0.78) | 0.99 (0.91) | 0.99 (0.83) | 0.99 (0.95) |
| I/σ(I) | 15.5 (2.1) | 14.0 (4.0) | 14.3 (2.6) | 10.7 (3.7) |
| wavelength (Å) | 0.9282 | 0.9763 | 0.9763 | 0.9686 |
| refinement | ||||
| Rwork/Rfree (%) | 19.3/23.5 | 19.4/22.3 | 18.9/23.1 | 20.8/24.4 |
| rmsd bond (Å) | 0.011 | 0.007 | 0.015 | 0.005 |
| rmsd angle (deg) | 1.6 | 1.26 | 1.9 | 0.75 |
| PDB code | 6FHU | 6FKP | 6FI0 | 6FAP |
| data collection | BAZ2B–Fr21 | BAZ2B–Fr23 |
|---|---|---|
| space group | P212121 | P212121 |
| cell dimensions | ||
| a, b, c (Å) | 38.3, 45.4, 65.0 | 37.8, 45.5, 64.8 |
| α, β, γ (deg) | 90, 90, 90 | 90, 90, 90 |
| resolution | 65.04 (1.95) | 64.83 (2.7) |
| unique observations | 8453 | 3349 |
| completeness | 97.7 (87.1) | 100.0 (100.0) |
| redundancy | 5.8 (4.9) | 5.6 (5.9) |
| CC1/2 | 0.99 (0.91) | 0.96 (0.56) |
| I/σ(I) | 18.1 (1.3) | 5.3 (1.9) |
| wavelength (Å) | 0.9686 | 0.9686 |
| refinement | ||
| Rwork/Rfree (%) | 18.5/23.8 | 22.6/29.9 |
| rmsd bond (Å) | 0.02 | 0.014 |
| rmsd angle (deg) | 1.88 | 1.73 |
| PDB code | 6FHQ | 6FI1 |
Values shown in parentheses are for the highest resolution shell.
Figure 3.
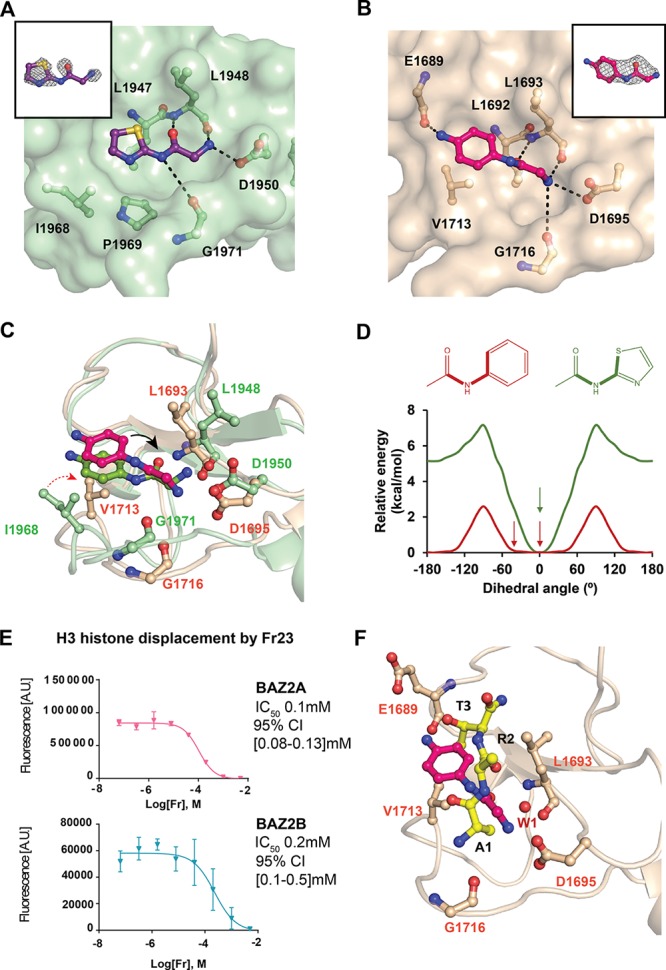
Insights on the binding mode of optimized fragments. (A) Crystal structure of BAZ2B PHD in complex with Fr21 bound to the histone pocket. (B) Crystal structure of BAZ2A PHD in complex with Fr23 bound to the histone pocket. Fo – Fc electron density map of the fragments is in gray and contoured at 2.5σ. (C) Superposition of BAZ2 PHDs in complex with Fr23. The black arrow shows the dihedral angle. The red arrow shows the different orientation of Val1713 in BAZ2A and Ile1968 in BAZ2B. (D) Relative torsion energy of surrogate arylamides. The observed dihedral angles of the parent compounds in complex with BAZ2 are highlighted with an arrow. (E) AlphaLISA dose–response curves of Fr23. The error bars represent the standard deviation of each point (see the Supporting Information for more details). (F) Superposition of BAZ2A in complex with Fr23 and ART peptide.
Crystal structures of both BAZ2A PHD (Figure 3B, as well as Figure S13B in the Supporting Information) and BAZ2B PHD (Figures S13A and S13C) were solved in complex with Fr23. Superposition of the two structures showed that the hotspots of binding are equivalent (Figure 3C). Despite this, some differences were observed in the torsion angle of the acetamide of the ligand. In Fr23 bound to BAZ2B, the torsion angle of the acetamide plane is rotated by only 10° compared to the ring plane, whereas, in BAZ2A, it is rotated clockwise by 46°. The 2-amino-N-acetamide orientation in Fr23 bound to BAZ2A is also retained in Fr19 (Figure S14 in the Supporting Information). From the structure, this rotation is needed to retain the hydrogen bond with Asp1695, despite the aromatic ring being pushed further by Val1713, which is oriented toward the pocket (see Figure 3C). The corresponding residue in BAZ2B is Ile1968, which points outside the pocket, allowing the fragment to be accommodated closer (Figure 3C). The dihedral angle in Fr23 was also studied by DFT calculations using phenylacetamide as model, which shows a flat profile between 0° and 50° (Erel < 0.35 kcal/mol; see Figure 3D). On average, proteins can induce a strain energy of ∼0.6 kcal/mol on a bound ligand;27 thus, the calculated low torsion energy gap is consistent with the hypothesized protein-induced strain of Fr23.
We next interrogated if Fr23 could interfere with the binding of the H3 AA mutant peptide in solution using the AlphaLISA competition assay. Dose–response curves in Figure 3E show displacement of the peptide in both proteins, consistent with the binding mode observed crystallographically. Fr23 binding mode was then compared to the ART binding mode (Figure 3F). The amide carbonyl of Fr23 superposes closely with the peptide R2 carbonyl, making a hydrogen bond with the Leu1693 backbone (Figure 3C). The amino group of Fr23 displaces the structural water W1, which mediates binding of the peptide N-terminal amino group to the protein (Figure 1B). The aromatic ring is accommodated in the Thr3 methyl pocket. Overall, while the fragment occupies the same hot spot pocket as H3, the details of the interaction are different.
In conclusion, this study reports the first fragment screening campaign to identify ligandable pockets of the BAZ2 PHD zinc fingers. In silico ligandability analysis highlighted two potential druggable pockets and targeted virtual screening identified a set of fragments that were validated experimentally for protein binding. From this set, we were able to solve the co-crystal structure of one compound (Fr19) bound to the histone pocket, which guided further in silico optimization of the binding mode, resulting in two more fragments successfully soaked in BAZ2 PHDs. To the best of our knowledge, these are the first fragment-bound crystal structures revealing protein–ligand interactions at the histone pocket of a PHD zinc finger domain. Analysis of these structures highlighted similarities and differences in the molecular recognition between the two proteins, as well as similarities and differences in binding mode between fragments and histone peptide. Co-crystallized fragments recapitulate a conserved anchoring hotspot at the histone binding site, which, however, is not explored by the bound histone tail. Thus, a rational design could take advantage of the importance of the histone tail helicity revealed in previous work,11 by linking our identified fragments with synthetic scaffolds exploring those features of histone binding.28
This work provides a blueprint for the development of future improved ligands of this family of PHDs. The development of chemical probes to disrupt the interaction between BAZ2 PHDs and histone H3 could ultimately provide new chemical tools to interrogate the still elusive activity and biological function of these proteins. Beyond inhibitors of the PHD–histone interaction, a small molecule able to bind the PHD zinc finger could be strategically linked to a selective ligand of the adjacent bromodomain14−16 with the intent to generate bivalent compounds of increased affinity and selectivity for the target protein. Such a bivalent approach targeting tandem domains was demonstrated in recent work, targeting BET bromodomains.29 Ultimately, improved ligands could also be conjugated to E3 ubiquitin ligase ligands, yielding bifunctional chemical degraders to induce proteasomal degradation of the BAZ2 proteins.30,31
Methods
For detailed description of material and methods, see the Experimental Section in the Supporting Information.
Acknowledgments
Funding support is gratefully acknowledged from the European Research Council (No. ERC-2012-StG-311460 DrugE3CRLs Starting Grant, to A.C.); the BBSRC (Ph.D. Studentship, to A.A.); the European Commission (No. H2020-MSCA-IF-2015-806323 Marie Skłodowska-Curie Actions Individual Fellowship, to X.L.); and the Wellcome Trust (Strategic Awards 100476/Z/12/Z for biophysics and drug discovery and 094090/Z/10/Z for structural biology and X-ray crystallography to the Division of Biological Chemistry and Drug Discovery at Dundee). We thank the Diamond Light Source for beamtime (BAG Proposal Nos. MX14980 and MX10071) and support at beamlines i04-1, i03, and i24. We are grateful to P. Fyfe for support with the in-house X-ray facility and to S. Hughes for advice with modeling of the Fr19 co-crystal structure.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.7b01093.
Experimental section and supporting figures (PDF)
Accession Codes
Atomic coordinates and structure factors have been deposited on the Protein Data Bank under accession codes: 6FHU (BAZ2A PHD–H3 3-mer), 6FKP (BAZ2A PHD–H3 AA mutant 10-mer) 6FI0 (BAZ2A PHD–Fr19), 6FAP (BAZ2A PHD–Fr23), 6FHQ (BAZ2B PHD–Fr21), and 6FI1 (BAZ2B PHD–Fr23).
The authors declare no competing financial interest.
Supplementary Material
References
- Musselman C. A.; Kutateladze T. G. (2009) PHD Fingers Epigenetic Effectors and Potential Drug Targets. Mol. Interventions 9, 314–323. 10.1124/mi.9.6.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppikofer M.; Sagolla M.; Haley B.; Zhang H. M.; Kummerfeld S. K.; Sudhamsu J.; Flynn E. M.; Bai T.; Zhang J.; Ciferri C.; Cochran A. G. (2017) Non-canonical reader modules of BAZ1A promote recovery from DNA damage. Nat. Commun. 8, 862. 10.1038/s41467-017-00866-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taverna S. D.; Li H.; Ruthenburg A. J.; Allis C. D.; Patel D. J. (2007) How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat. Struct. Mol. Biol. 14, 1025–1040. 10.1038/nsmb1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L. H.; Zhen X. T.; Denton E.; Marsden B. D.; Schapira M. (2012) ChromoHub: A data hub for navigators of chromatin-mediated signalling. Bioinformatics 28, 2205–2206. 10.1093/bioinformatics/bts340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker L. A.; Allis C. D.; Wang G. G. (2008) PHD fingers in human diseases: disorders arising from misinterpreting epigenetic marks. Mutat. Res., Fundam. Mol. Mech. Mutagen. 647, 3–12. 10.1016/j.mrfmmm.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller S.; Filippakopoulos P.; Knapp S. (2011) Bromodomains as therapeutic targets. Expert Rev. Mol. Med. 13, 1–21. 10.1017/S1462399411001992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner E. K.; Nath N.; Flemming R.; Feltenberger J. B.; Denu J. M. (2012) Identification and characterization of small molecule inhibitors of a plant homeodomain finger. Biochemistry 51, 8293–8306. 10.1021/bi3009278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller T. C.; Rutherford T. J.; Birchall K.; Chugh J.; Fiedler M.; Bienz M. (2014) Competitive binding of a benzimidazole to the histone-binding pocket of the Pygo PHD finger. ACS Chem. Biol. 9, 2864–2874. 10.1021/cb500585s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones M. H.; Hamana N.; Nezu J.; Shimane M. (2000) A novel family of bromodomain genes. Genomics 63, 40–45. 10.1006/geno.1999.6071. [DOI] [PubMed] [Google Scholar]
- Tallant C.; Valentini E.; Fedorov O.; Overvoorde L.; Ferguson F. M.; Filippakopoulos P.; Svergun D. I.; Knapp S.; Ciulli A. (2015) Molecular basis of histone tail recognition by human TIP5 PHD finger and bromodomain of the chromatin remodeling complex NoRC. Structure 23, 80–92. 10.1016/j.str.2014.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bortoluzzi A.; Amato A.; Lucas X.; Blank M.; Ciulli A. (2017) Structural basis of molecular recognition of helical histone H3 tail by PHD finger domains. Biochem. J. 474, 1633–1651. 10.1042/BCJ20161053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson F. M.; Fedorov O.; Chaikuad A.; Philpott M.; Muniz J. R. C.; Felletar I.; von Delft F.; Heightman T.; Knapp S.; Abell C.; Ciulli A. (2013) Targeting Low-Druggability Bromodomains: Fragment Based Screening and Inhibitor Design against the BAZ2B Bromodomain. J. Med. Chem. 56, 10183–10187. 10.1021/jm401582c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiliotopoulos D.; Wamhoff E. C.; Lolli G.; Rademacher C.; Caflisch A. (2017) Discovery of BAZ2A bromodomain ligands. Eur. J. Med. Chem. 139, 564–572. 10.1016/j.ejmech.2017.08.028. [DOI] [PubMed] [Google Scholar]
- Lolli G.; Caflisch A. (2016) High-Throughput Fragment Docking into the BAZ2B Bromodomain: Efficient in Silico Screening for X-Ray Crystallography. ACS Chem. Biol. 11, 800–807. 10.1021/acschembio.5b00914. [DOI] [PubMed] [Google Scholar]
- Chen P.; Chaikuad A.; Bamborough P.; Bantscheff M.; Bountra C.; Chung C. W.; Fedorov O.; Grandi P.; Jung D.; Lesniak R.; Lindon M.; Muller S.; Philpott M.; Prinjha R.; Rogers C.; Selenski C.; Tallant C.; Werner T.; Willson T. M.; Knapp S.; Drewry D. H. (2016) Discovery and Characterization of GSK2801, a Selective Chemical Probe for the Bromodomains BAZ2A and BAZ2B. J. Med. Chem. 59, 1410–1424. 10.1021/acs.jmedchem.5b00209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drouin L.; McGrath S.; Vidler L. R.; Chaikuad A.; Monteiro O.; Tallant C.; Philpott M.; Rogers C.; Fedorov O.; Liu M.; Akhtar W.; Hayes A.; Raynaud F.; Muller S.; Knapp S.; Hoelder S. (2015) Structure enabled design of BAZ2-ICR, a chemical probe targeting the bromodomains of BAZ2A and BAZ2B. J. Med. Chem. 58, 2553–2559. 10.1021/jm501963e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strohner R.; Nemeth A.; Jansa P.; Hofmann-Rohrer U.; Santoro R.; Langst G.; Grummt I. (2001) NoRC—A novel member of mammalian ISWI-containing chromatin remodeling machines. EMBO J. 20, 4892–4900. 10.1093/emboj/20.17.4892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu L., Frommel S. C., Oakes C. C., Simon R., Grupp K., Gerig C. Y., Bar D., Robinson M. D., Baer C., Weiss M., Gu Z., Schapira M., Kuner R., Sultmann H., Provenzano M., ICGC Project on Early Onset Prostate Cancer, Yaspo M. L., Brors B., Korbel J., Schlomm T., Sauter G., Eils R., Plass C., and Santoro R.. BAZ2A (TIP5) is involved in epigenetic alterations in prostate cancer and its overexpression predicts disease recurrence. Nat. Genet. 2015, 47, 22–30 10.1038/ng.3165 [DOI] [PubMed] [Google Scholar]
- Arking D. E.; Junttila M. J.; Goyette P.; Huertas-Vazquez A.; Eijgelsheim M.; Blom M. T.; Newton-Cheh C.; Reinier K.; Teodorescu C.; Uy-Evanado A.; Carter-Monroe N.; Kaikkonen K. S.; Kortelainen M. L.; Boucher G.; Lagace C.; Moes A.; Zhao X.; Kolodgie F.; Rivadeneira F.; Hofman A.; Witteman J. C.; Uitterlinden A. G.; Marsman R. F.; Pazoki R.; Bardai A.; Koster R. W.; Dehghan A.; Hwang S. J.; Bhatnagar P.; Post W.; Hilton G.; Prineas R. J.; Li M.; Kottgen A.; Ehret G.; Boerwinkle E.; Coresh J.; Kao W. H.; Psaty B. M.; Tomaselli G. F.; Sotoodehnia N.; Siscovick D. S.; Burke G. L.; Marban E.; Spooner P. M.; Cupples L. A.; Jui J.; Gunson K.; Kesaniemi Y. A.; Wilde A. A.; Tardif J. C.; O’Donnell C. J.; Bezzina C. R.; Virmani R.; Stricker B. H.; Tan H. L.; Albert C. M.; Chakravarti A.; Rioux J. D.; Huikuri H. V.; Chugh S. S. (2011) Identification of a sudden cardiac death susceptibility locus at 2q24.2 through genome-wide association in European ancestry individuals. PLoS Genet. 7, e1002158. 10.1371/journal.pgen.1002158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarty S.; Essel F.; Lin T.; Zeigler S. (2015) Histone Peptide Recognition by KDM5B-PHD1: A Case Study. Biochemistry 54, 5766–5780. 10.1021/acs.biochem.5b00617. [DOI] [PubMed] [Google Scholar]
- Arkin M. R.; Tang Y. Y.; Wells J. A. (2014) Small-Molecule Inhibitors of Protein-Protein Interactions: Progressing toward the Reality. Chem. Biol. 21, 1102–1114. 10.1016/j.chembiol.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenke R.; Kozakov D.; Chuang G. Y.; Beglov D.; Hall D.; Landon M. R.; Mattos C.; Vajda S. (2009) Fragment-based identification of druggable “hot spots” of proteins using Fourier domain correlation techniques. Bioinformatics 25, 621–627. 10.1093/bioinformatics/btp036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvestre H. L.; Blundell T. L.; Abell C.; Ciulli A. (2013) Integrated biophysical approach to fragment screening and validation for fragment-based lead discovery. Proc. Natl. Acad. Sci. U. S. A. 110, 12984–12989. 10.1073/pnas.1304045110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson M. P. (2014) Using chemical shift perturbation to characterise ligand binding (vol 73, pg 1, 2013). Prog. Nucl. Magn. Reson. Spectrosc. 80, 64–64. 10.1016/j.pnmrs.2014.05.001. [DOI] [PubMed] [Google Scholar]
- Philpott M.; Yang J.; Tumber T.; Fedorov O.; Uttarkar S.; Filippakopoulos P.; Picaud S.; Keates T.; Felletar I.; Ciulli A.; Knapp S.; Heightman T. D. (2011) Bromodomain-peptide displacement assays for interactome mapping and inhibitor discovery. Mol. BioSyst. 7, 2899–2908. 10.1039/c1mb05099k. [DOI] [PubMed] [Google Scholar]
- Dai R.; Geders T. W.; Liu F.; Park S. W.; Schnappinger D.; Aldrich C. C.; Finzel B. C. (2015) Fragment-based exploration of binding site flexibility in Mycobacterium tuberculosis BioA. J. Med. Chem. 58, 5208–5217. 10.1021/acs.jmedchem.5b00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao M. H.; Haq O.; Muegge I. (2007) Torsion angle preference and energetics of small-molecule ligands bound to proteins. J. Chem. Inf. Model. 47, 2242–2252. 10.1021/ci700189s. [DOI] [PubMed] [Google Scholar]
- Yin H.; Lee G. I.; Sedey K. A.; Kutzki O.; Park H. S.; Orner B. P.; Ernst J. T.; Wang H. G.; Sebti S. M.; Hamilton A. D. (2005) Terphenyl-based bak BH3 alpha-helical proteomimetics as low-molecular-weight antagonists of Bcl-X-L. J. Am. Chem. Soc. 127, 10191–10196. 10.1021/ja050122x. [DOI] [PubMed] [Google Scholar]
- Tanaka M.; Roberts J. M.; Seo H. S.; Souza A.; Paulk J.; Scott T. G.; DeAngelo S. L.; Dhe-Paganon S.; Bradner J. E. (2016) Design and characterization of bivalent BET inhibitors. Nat. Chem. Biol. 12, 1089–1096. 10.1038/nchembio.2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zengerle M.; Chan K. H.; Ciulli A. (2015) Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem. Biol. 10, 1770–1777. 10.1021/acschembio.5b00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J.; Qian Y.; Altieri M.; Dong H.; Wang J.; Raina K.; Hines J.; Winkler J. D.; Crew A. P.; Coleman K.; Crews C. M. (2015) Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem. Biol. 22, 755–763. 10.1016/j.chembiol.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.