

ABSTRACT
The epithelial layer of the gastrointestinal tract contains invaginations, called glands or crypts, which are colonized by symbiotic and pathogenic microorganisms and may function as designated niches for certain species. Factors that control gland colonization are poorly understood, but bacterial chemotaxis aids occupation of these sites. We report here that a Helicobacter pylori cytoplasmic chemoreceptor, TlpD, is required for gland colonization in the stomach. tlpD mutants demonstrate gland colonization defects characterized by a reduction in the percentage of glands colonized but not in the number of bacteria per gland. Consistent with TlpD's reported role in reactive oxygen species (ROS) avoidance, tlpD mutants showed hallmarks of exposure to high ROS. To assess the role of host-generated ROS in TlpD-dependent gland colonization, we utilized mice that lack either the ability to generate epithelial hydrogen peroxide or immune cell superoxide. tlpD gland colonization defects were rescued to wild-type H. pylori levels in both of these mutants. These results suggest that multiple types of innate immune-generated ROS production limit gland colonization and that bacteria have evolved specific mechanisms to sense and direct their motility in response to this signal and thus spread throughout tissue.
KEYWORDS: MCP, biogeography, chemoreceptors, crypts, glands, reactive oxygen species, signal transduction, stomach
INTRODUCTION
The epithelium of the gastrointestinal (GI) tract contains invaginations, called glands in the stomach and crypts in the intestine, which are thought to protect GI stem cells and also serve as a niche for particular microbes to promote chronic colonization by these microbial species. Our knowledge of the factors that control the colonization of these structures is incomplete. Host factors that have been implicated in controlling gland colonization include the production of mucus (1), the production of antimicrobial peptides (2), and the presence of resident immune cells in the lamina propria (3). Gland colonization, therefore, requires microbes to bypass these defensive strategies.
Bacteria appear to have special adaptations to the gland niche. These include the abilities to use certain carbohydrates (4) and to perform chemotaxis (5–7). The chronically colonizing gastric pathogen Helicobacter pylori is one such microbe that requires chemotaxis for gland colonization (5–7). Chemotaxis permits bacteria to sample their environment via chemoreceptors that use ligand-binding signals to alter the autophosphorylation of a complexed histidine kinase, CheA. Ultimately, this pathway alters flagellar motility to allow bacteria to follow, or repel themselves from, gradients of specific signals (8). Chemotaxis is not critical for initial colonization when high doses of bacteria are given (5). At 1 day postinfection, wild-type and nonchemotactic H. pylori bacteria both colonize ∼5 to 10% of gastric glands (5). At 3 days postinfection, however, wild-type (WT) H. pylori colonization increases to up to ∼30% of glands but the nonchemotactic mutant (Che−) is unable to carry out this increase and instead stays at <10% of glands colonized (5). This trend continues throughout the first month of infection, with colonization by the wild type spreading and that of Che− H. pylori maintaining, such that at 2 weeks postinfection, the wild type has colonized ∼50% of glands and the Che− strain has colonized <10%. These data suggest that chemotaxis plays a key role in H. pylori's ability to expand into new glands and that 2 weeks of infection is a good single time point to assess colonization patterns.
Chemoreceptors are the proteins that sense the environment and guide chemotaxis. H. pylori expresses four chemoreceptors, three of which (TlpA, TlpB, and TlpC) are embedded in the inner membrane and one (TlpD) that is fully cytoplasmic. The relevance of individual chemoreceptors on overall gastric colonization has been gauged previously by the level of colonization defect that a particular mutant displays. Among individual H. pylori chemoreceptor mutants, tlpD mutants have displayed the most severe colonization attenuation in two animal models of infection (9, 10). The exact nature of the tlpD mutant colonization deficit, however, has remained unclear, as has the role of specific signals and chemoreceptors in gland colonization.
TlpD is a cytoplasmic chemoreceptor that contains a C-terminal CZB domain that binds zinc (11, 12). TlpD has been linked to a chemotactic response to multiple stress-related signals, including electron transport chain inhibitors (13), acid (7), and reactive oxygen species (ROS), including hydrogen peroxide (H2O2) or the superoxide generators metronidazole and paraquat (14). One hypothesis is that these signals are connected because they all affect oxidative stress experienced in the cytoplasm (14). Gastric Helicobacter bacteria are known to encounter host-generated ROS derived from both epithelial and immune cells during infection and must cope with this stress to successfully colonize (15, 16).
One key host response to H. pylori is the upregulation of host ROS. Data supporting this include numerous studies that have documented the influx of immune cells that produce ROS, a measured increase in gastric ROS, and an upregulation of H2O2-generating epithelial enzymes (15, 16). ROS are produced by both gastric epithelial cells and innate immune cells and include hydrogen peroxide (H2O2), superoxide (O2−), and hypochlorous acid (HOCl) (15). To counter these stresses, H. pylori possesses a suite of ROS detoxification systems, including catalase, superoxide dismutase, and peroxiredoxins (17). H. pylori mutants lacking these systems are sensitive to ROS and are also attenuated in the host (17). ROS production limits colonization of several microbes at epithelial surfaces in the stomach and intestine (16, 18); in agreement with this idea, mouse mutants that lack the epithelial DUOX enzyme produce less H2O2 and allow elevated colonization by a relative of H. pylori, Helicobacter felis (16). ROS production may serve to drive microbes away from the epithelial surface, as microbial adherence to intestinal epithelial cells promotes H2O2 production and hosts respond to H. pylori infection with elevated ROS (17, 18). However, it is not clear how ROS affects colonization within microniches, including the glands.
To define the contribution of TlpD in gastric colonization, we determined its effect on the colonization of the bulk epithelia and gastric glands at a fixed time point of 2 weeks postinfection. We chose a 2-week time point because chemotaxis plays a significant role in the spread of H. pylori between glands that is highly apparent at this time point (5). Furthermore, tlpD mutants have colonization defects of the bulk epithelia at this time, suggesting that this receptor's contribution toward H. pylori distribution at the gastric epithelium should be evident (9). We report here that tlpD mutants show specific deficits in colonizing a broad swathe of gastric glands and display hallmarks of exposure to elevated ROS. This result raised the possibility that gland colonization defects could be due to an inability of tlpD mutants to successfully navigate in response to ROS, an idea that was further supported by the observation that tlpD mutants colonized fewer glands than WT H. pylori to elevated densities within these glands. To assess whether host-generated ROS impacted H. pylori colonization, we compared the colonization and distribution levels of the WT and tlpD and nonchemotactic cheY mutant strains in mice deficient in either epithelial dual oxidases (Duoxa−/−) or phagocytic NOX2 NADPH oxidase (Cybb−/−). Infection of either Duoxa−/− or Cybb−/− mutant mice rescued the gland colonization defects of the tlpD mutants noted in WT hosts. Our results suggest that ROS production impacts H. pylori gland transit and that TlpD-mediated chemotactic responses are needed to expand to new glands.
RESULTS
tlpD mutants have minor colonization defects but achieve normal per gland loads.
To study the contribution of TlpD in gland colonization, we planned to use the bacterial localization in isolated glands (BLIG) approach, which requires fluorescent bacteria. We thus transformed H. pylori ΔtlpD with the pTM115 plasmid that drives constitutive green fluorescent protein (GFP) expression. This plasmid has been shown to be stably maintained and to express GFP for mouse infection durations of up to 6 months (5). To begin our analysis of TlpD's role in colonization, we orally infected WT C57BL6 mice with WT or tlpD or cheY mutant variants of H. pylori that each expressed GFP. cheY encodes the central chemotaxis signaling proteins, so mutants that lack it are fully nonchemotactic (Che−), while mutants that lack tlpD lose only responses sensed by that receptor and thus are partially chemotactic. After 2 weeks of infection, a time when chemotaxis is needed for gland-gland spread (5), the colonization levels in the total tissue of the stomach corpus and antrum were determined. tlpD mutants showed colonization defects in the antrum and corpus of WT mice (Fig. 1A), similar to that previously reported (9), although these differences were not statistically significant. These results suggested that tlpD GFP-positive (GFP+) H. pylori infections behaved similarly to tlpD infections lacking GFP described previously (9) and encouraged the analysis of gland colonization by the mutant.
FIG 1.
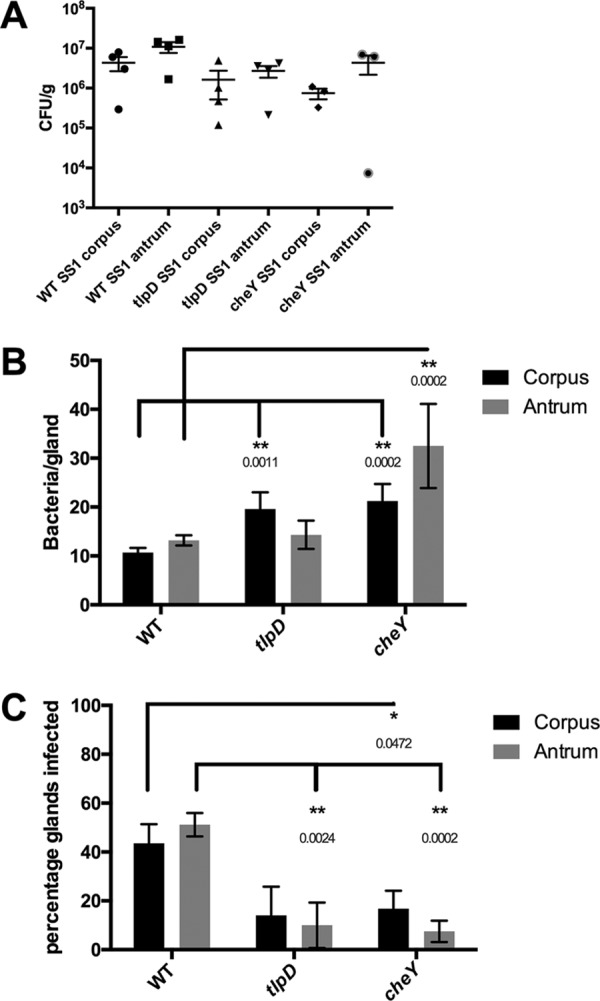
tlpD mutants have deficits in gland occupancy in WT mice but not in colonization of total tissue or individual glands. A comparison of colonization levels of WT mice by H. pylori GFP+ SS1 WT, tlpD mutant, and cheY mutant strains is shown. Mice were orally infected, and stomachs were collected and analyzed for tissue and gland colonization after 2 weeks of infection. (A) Numbers of CFU/g for corpus and antrum regions for mice infected with H. pylori GFP+ SS1 WT (n = 4), tlpD mutant (n = 4), and cheY mutant (n = 3) strains. Differences between groups were not statistically significant. (B) Gland loads in the isolated corpus and antral glands. These numbers are the average number of bacteria counted per gland, excluding uninfected glands. Numbers of infected glands were as follows: WT-infected corpus, 436 glands from six mice; WT-infected antrum, 508 glands from six mice; tlpD mutant-infected corpus, 67 glands from five mice; tlpD mutant-infected antrum, 48 glands from five mice; cheY mutant-infected corpus, 58 glands from four mice; cheY mutant-infected antrum, 24 glands from four mice. (C) Gland occupancy in the isolated corpus and antral glands, representing the percentage of glands infected with the indicated H. pylori strain. Error bars represent standard errors of the mean (SEM) for all panels. Numbers of mice infected are the same as described for gland loads. Statistical differences are indicated by * (P < 0.05) and ** (P < 0.01) as analyzed by Student's t test.
We next examined TlpD's role in gland colonization. To monitor gland colonization, we employed the BLIG approach, in which gastric glands are isolated from infected corpus or antrum tissue, epithelial cells are labeled with Hoechst DNA stain, and glands are examined for the presence of GFP+ H. pylori by fluorescence microscopy (5). The bacteria within glands were counted manually, and two parameters of gland colonization were compared between H. pylori strains. The first parameter was gland bacterial load, the number of bacteria per infected gland. Our calculation of gland bacterial load excludes noninfected glands. The second parameter was gland occupancy, the percentage of glands infected.
In WT mice, WT H. pylori colonized the glands of the corpus and the antrum to similar levels, averaging 10 bacteria/infected gland as reported previously (Fig. 1B) (5). Loss of TlpD did not affect gland load in the antrum but caused an ∼1.8-fold increase in gland load in the corpus compared with that of the WT (Fig. 1B). Full loss of chemotaxis (cheY mutants) also resulted in elevated gland loads of 2- to 3-fold in both the corpus and the antrum relative to those of WT H. pylori (Fig. 1B). These results suggest that chemotactic defects did not impair growth within glands and, if anything, resulted in elevations in bacterial gland load.
The loss of tlpD or chemotaxis results in a reduction in gland occupancy throughout the stomach in WT hosts.
Because tlpD and cheY mutants appeared to have altered gland phenotypes, we next analyzed gland occupancy to determine the percentage of glands infected by H. pylori. This frequency likely reflects both the initial population of glands infected by H. pylori and the ability to spread and colonize new glands. In WT mice, WT H. pylori colonized 40 to 50% of corpus and antral glands by 2 weeks of infection and were found in similar proportions in both regions (Fig. 1C). tlpD mutants showed an ∼3-fold reduced occupancy in both the corpus and antrum relative to that of WT H. pylori (Fig. 1C). cheY mutant gland occupancy was also decreased relative to that of WT H. pylori, with significant reductions in both the corpus and the antrum (Fig. 1C). These results suggest that chemotaxis generally, and TlpD specifically, is required for H. pylori to occupy new glands.
tlpD mutants show hallmarks of elevated ROS exposure relative to WT H. pylori.
We reported recently that TlpD mediates chemotactic repellent responses to multiple ROS (14). Combining this information with our data above suggested that tlpD mutant gland colonization defects could be due to an inability of these mutants to sense and repel themselves away appropriately from ROS. We therefore asked whether tlpD mutants experienced differential oxidative stress in vivo. For this approach, we used quantitative real-time PCR of mRNA isolated from infected mouse tissue. We examined the expression of the catalase gene (katA) mRNA by H. pylori strains, whose expression has been shown to be sensitive to several oxidative stresses (19, 20). We determined that this gene was modestly upregulated in vitro in our strains following exposure to 1 mM H2O2 for 20 min, although the differences were not statistically significant (Fig. 2A). This result suggested that katA mRNA could serve as a reasonable proxy for H2O2 exposure in vivo. We next assessed whether the expression of katA mRNA differed between WT and tlpD, and cheY mutant H. pylori bacteria during infection of WT mice. tlpD mutants expressed significantly more katA mRNA than WT H. pylori in the antrum and modestly more in the corpus (Fig. 2B). These results suggest that tlpD mutants experience elevated oxidative stress during infection. Conversely, cheY mutants did not express elevated catalase mRNA (Fig. 2B). This outcome suggests that the loss of TlpD specifically leads H. pylori to be exposed to conditions that are different from those encountered by the WT, consistent with high exposure to oxidative stress.
FIG 2.
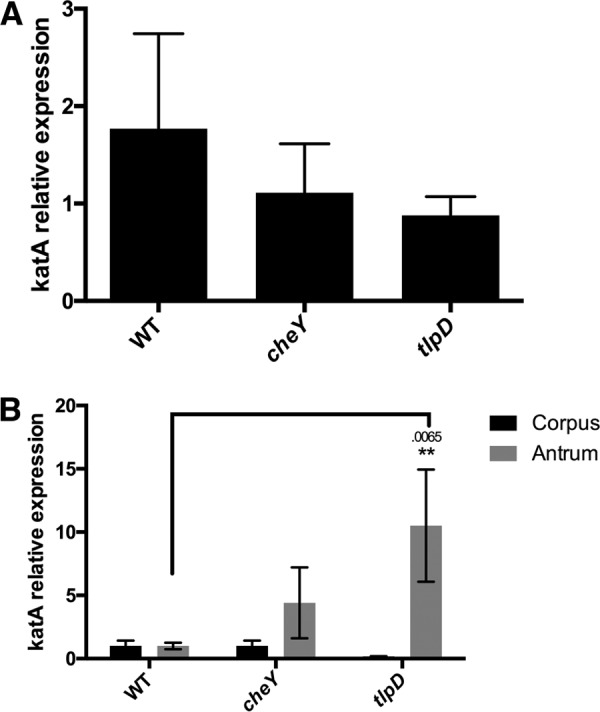
tlpD mutants show evidence of ROS exposure in vivo. A comparison of catalase mRNA expression levels in vitro and in vivo between H. pylori strains is shown. (A) Mean ± SEM of fold change increases in katA mRNA of H. pylori strains exposed to 1 mM H2O2 for 20 min, normalized to gapB. Differences between groups were not significant. (B) Comparison of mean ± SEM of katA expression levels by H. pylori strains in three WT mice, normalized to gapB. Statistical differences are indicated by * (P < 0.05) and ** (P < 0.01) as analyzed by Student's t test, with actual P values indicated above the bar. gapB expression was insensitive to H2O2 exposure based on comparison to 16S rRNA.
Gland colonization defects of tlpD are rescued in hosts deficient in H2O2 production by gastric epithelial cells.
The results presented above suggest that TlpD helps to mitigate the exposure of H. pylori to oxidative stress in the mouse. In order to follow up on oxidative stress and its role in TlpD-mediated colonization, we next infected two mutant mouse hosts that were deficient in the production of H2O2 and O2− production. The first of these lacks the dual oxidase (DUOX) heterodimeric enzyme complex by virtue of loss of the Duoxa-encoded subunit (16). DUOX is expressed by gastric epithelial cells and generates extracellular H2O2, which may serve to limit physical interactions between microbes and the epithelial surface (18). DUOX has been implicated in limiting the colonization of a related Helicobacter species in the stomachs of mice (16).
To examine whether DUOX impacted H. pylori colonization, Duoxa−/− mice were infected as done with WT mice for 2 weeks, at which point the mice were sacrificed and the colonization levels of WT and cheY and tlpD mutant GFP+ H. pylori bacteria were compared. All H. pylori strains colonized the Duoxa−/− mutants to levels that were a bit elevated but not significantly different from those in WT mouse hosts (Fig. 3A). Gland loads were also generally similar between WT and Duoxa−/− mouse glands, across WT and tlpD mutant H. pylori bacteria in both locations and in cheY mutants in the corpus (Fig. 3B). There was a modest increase in gland load in the antrum of the tlpD mutant and a very large decrease in loads of the cheY mutant, suggesting that the effect of DUOX was greatest in the antrum.
FIG 3.
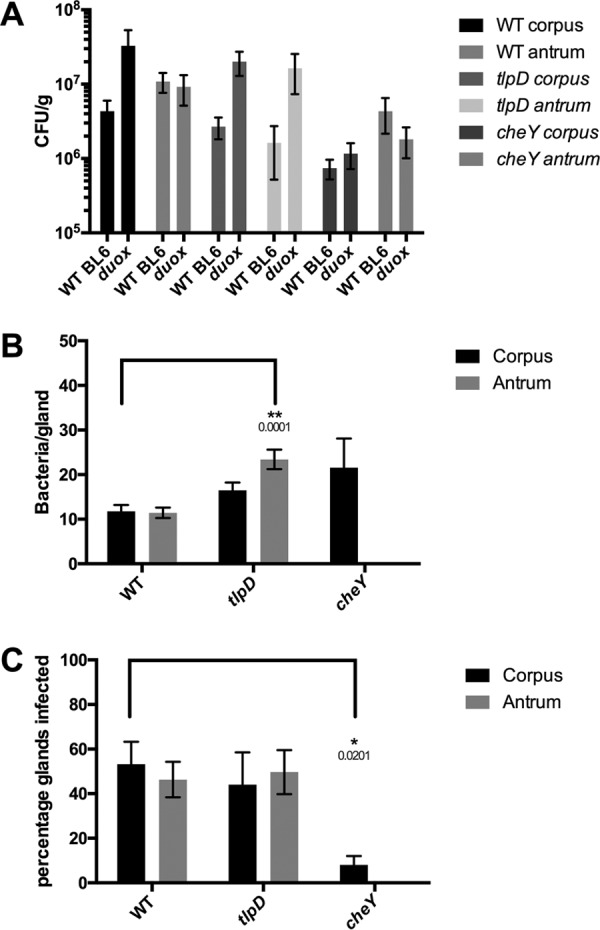
Loss of epithelial H2O2 rescues tlpD mutant gland defects. Colonization of Duoxa−/− mice by WT, tlpD mutant, or cheY mutant GFP+ H. pylori SS1 strains at 2 weeks postinfection is shown. Mice were orally infected, and stomachs were collected and analyzed for tissue and gland colonization. (A) Numbers of CFU/g at 2 weeks postinfection for corpus and antrum regions infected with WT (n = 4), tlpD mutant (n = 5), and cheY mutant (n = 5) GFP+ H. pylori SS1 strains. Data for WT mice (referred to as WT BL6 in the figure) are the same as those in Fig. 1 and are displayed here for comparison. Duox refers to Duoxa−/− mice in the figure. (B) Gland loads in the isolated corpus and antral glands, representing the average number of bacteria counted per gland, excluding uninfected glands. Numbers of infected glands were as follows: WT-infected corpus, 313 glands from six mice; WT-infected antrum, 472 glands from six mice; tlpD mutant-infected corpus, 132 from six mice; tlpD mutant-infected antrum, 149 glands from three mice; cheY mutant-infected corpus, 24 glands from three mice. (C) Gland occupancy in the isolated corpus and antral glands, representing the percentage of glands infected with the indicated H. pylori strain. Error bars represent SEM for all panels. Numbers of mice infected are the same as described for gland loads. Statistical differences are indicated by * (P < 0.05) and ** (P < 0.01) as analyzed by Student's t test.
We next assessed how the loss of Duoxa−/− would alter gland occupancy. WT H. pylori gland occupancy was seemingly unaffected by the loss of Duoxa−/−, as ∼50% of the glands were infected in this background as well as in WT mice (Fig. 3C). Interestingly, the tlpD mutant showed an increase in gland occupancy compared to its levels in a WT mouse, rising from <15% occupied to over 40% (Fig. 3C). Indeed, the tlpD mutant achieved gland occupancy levels in the corpus and antrum that were not statistically different from those of WT H. pylori (Fig. 3C). In contrast, the cheY mutant was not rescued, suggesting that the loss of Duoxa−/− rescue is specific to signals sensed by TlpD and requires chemotaxis. This apparent rescue in tlpD gland occupancy suggests that the loss of H2O2 production by gastric epithelial cells allows for tlpD mutants to move more readily into new gastric glands in both the corpus and the antrum.
Gland colonization defects of tlpD are rescued in hosts deficient in O2− production by phagocytes.
We next assessed the contribution of phagocyte ROS production to H. pylori gland colonization. Phagocyte ROS production was assessed in Cybb−/− mice that lack the catalytic subunit of phagocyte oxidase (Phox). Cybb−/− mice were infected as described above, and the same colonization parameters were compared between WT and cheY and tlpD mutant GFP+ H. pylori strains.
The overall colonization of the corpus and antrum was seemingly unaffected by the loss of Cybb for all three H. pylori strains, showing no significant differences from WT mouse infections (Fig. 4A). Gland loads, on the other hand, were affected in Cybb−/− hosts. Both WT and tlpD H. pylori strains showed significantly elevated gland loads in the corpus relative to those of WT BL6 infections (P = 0.0413 and P < 0.0001), achieving 20 to 30 bacteria/gland in both regions (Fig. 4B). Loads in the antrum followed similar trends, with the WT showing significant elevations relative to those of WT BL6 infections (P = 0.0102), while tlpD changes were enhanced but not significantly so. cheY mutants did not follow this trend in Cybb−/− hosts and instead showed reduced gland loads in the corpus (P = 0.0027) and the antrum (P = 0.0118) relative to those of WT BL6 infections. tlpD mutants, but not the WT, showed significantly elevated gland loads relative to those of cheY mutants in the antrum (P = 0.001) (Fig. 4B). This outcome suggests that superoxide may normally limit H. pylori numbers per gland in a chemotaxis-dependent way. Lastly, we compared gland occupancies in Cybb−/− hosts. Strikingly, the tlpD mutant gland occupancy in both the corpus and the antrum climbed to levels that were not different from those of WT H. pylori. This finding suggests that as with Duoxa−/− infections, gland occupancy defects of tlpD mutants were rescued by the loss of host ROS. As seen with Duoxa−/− infections, cheY mutant gland occupancy did not appear to benefit from the loss of Cybb (Fig. 4C). Interestingly, the gland occupancy by WT and tlpD mutant H. pylori strains was ∼ 2-fold lower in Cybb−/− mice than in WT BL6 hosts. These results suggest that the loss of superoxide production by phagocytes rescues gland colonization defects of tlpD mutant H. pylori, as was observed in Duoxa−/− hosts. Chemotaxis appears necessary for this rescue, as cheY mutants showed gland colonization values similar to those observed in WT mice. Taken together, these results suggest that host-generated ROS serves as a barrier for gland colonization by H. pylori, which the bacteria overcome via TlpD-mediated chemotactic responses. Furthermore, tlpD colonization defects can be attributed to low gland occupancy in the corpus and the antrum, which can be rescued to WT levels by disrupting host ROS production.
FIG 4.
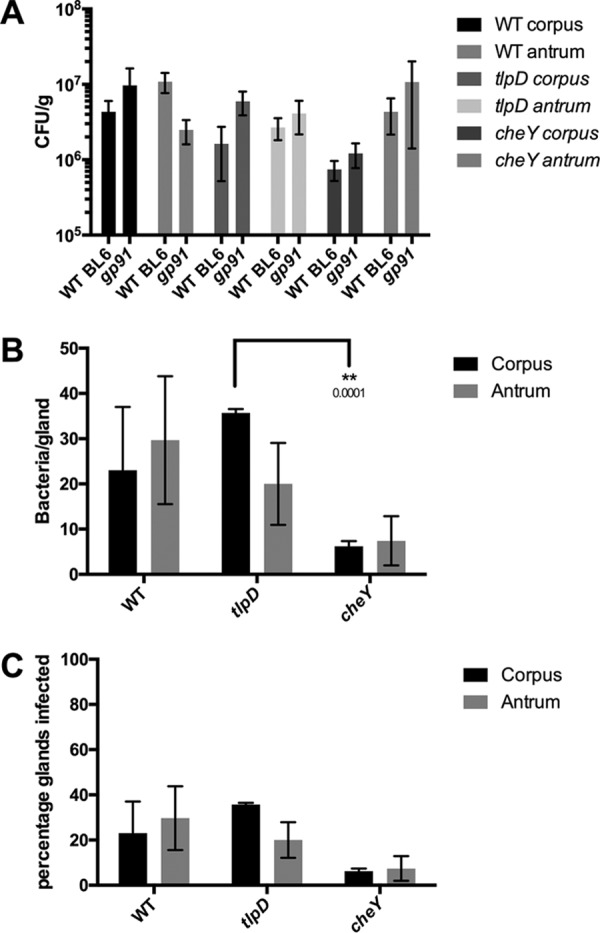
Loss of immune superoxide rescues tlpD mutant gland defects. Colonization of Cybb−/− mice by WT, tlpD mutant, or cheY mutant GFP+ H. pylori SS1 strains at 2 weeks postinfection is shown. Mice were orally infected, and stomachs were collected and analyzed for tissue and gland colonization. (A) Numbers of CFU/g at 2 weeks postinfection for corpus and antrum regions using WT (n = 6), tlpD mutant (n = 14), and cheY mutant (n = 6) GFP+ H. pylori SS1 strains. Data for WT mice are the same as those in Fig. 1 and are displayed here for comparison. WT BL6 refers to wild type and gp91 refers to Cybb−/− mice. (B) Gland loads in the isolated corpus and antral glands, representing the average number of bacteria counted per gland, excluding uninfected glands. Numbers of infected glands are as follows: WT-infected corpus, 69 glands from three mice; WT-infected antrum, 89 glands from three mice; tlpD mutant-infected corpus, 107 glands from three mice; tlpD mutant-infected antrum, 60 glands from three mice; cheY mutant-infected corpus, 31 glands from three mice; cheY mutant-infected antrum, 37 glands from three mice. (C) Gland occupancy in the isolated corpus and antral glands, representing the percentage of glands infected with the indicated H. pylori strain. Error bars represent SEM for all panels. Numbers of mice infected are the same as described for gland loads. Statistical differences are indicated by * (P < 0.05) and ** (P < 0.01) as analyzed by Student t test.
DISCUSSION
We report here that H. pylori requires repellent ROS chemotaxis to be able to successfully colonize glands. Factors that control gland colonization throughout the GI tract are poorly understood, although it appears that an interplay exists between host and microbe to regulate gland access. Host factors known to limit gland colonization include mucus production (1), oxygen gradients emanating from the epithelial surface (21), and antimicrobial peptide production (2). Microbial adaptations that have been reported to aid gland colonization include chemotaxis (5–7), sugar transport systems (4), and the ability to dampen host immune responses (22). Therefore, it seems reasonable to posit that glands represent a desired but protected niche for some microbes in the GI tract. Our work demonstrates that ROS limits gland access and that chemotaxis helps overcome this barrier.
TlpD orthologous chemoreceptors similarly contain the CZB domain and are quite common; the CZB domain is the second most prevalent identifiable domain found in cytoplasmic chemoreceptors (12). These TlpD orthologs are found in many bacterial species, including multiple pathogens that colonize mucosal surfaces, such as most Campylobacter spp., Helicobacter spp., Salmonella enterica serovar Typhimurium, Proteus mirabilis, and Vibrio anguillarum (11). The common occurrence of CZB domain chemoreceptors raises the possibility that chemotactic responses to ROS at the mucosal surface may be a generalizable phenomenon and drive outcomes of multiple infections.
Host ROS generation has previously been implicated in limiting microbial adhesion in the intestine, and Duoxa−/− mice showed elevated mucosal penetrance by a subset of the microbiota (18). Our data suggest that host ROS play an important role in restricting gland access in the stomach and that bacteria can use chemotaxis to overcome this barrier. tlpD and cheY mutants both colonized a low percentage of the total glands in WT mice at 2 weeks postinfection (Fig. 1). This finding is consistent with that of Keilberg et al., which showed that wild-type and Che− H. pylori strains both initially colonize ∼5% of glands, with chemotaxis needed to expand to new glands over the following days (5). This discrepancy results in wild-type H. pylori occupying ∼50% of glands at 2 weeks postinfection, and Che− strains occupying <10%. A similar pattern was observed for the tlpD mutant, in that it also occupied only 5 to 10% of glands after 2 weeks of infection. This phenotype is consistent with the idea that WT, ΔtlpD, and Che− H. pylori strains all colonize a small number of glands initially and multiply within the glands they colonize (Fig. 1B). Only wild-type bacteria, however, are able to expand to a high number of glands, suggesting that both chemotaxis in general and TlpD specifically are needed for this expansion (Fig. 1C). Indeed, both ΔtlpD and Che− strains were found in higher numbers per infected gland, suggesting that they may be unable to leave glands as effectively as the WT (Fig. 1B). Overall, these phenotypes suggest that TlpD and chemotaxis are critically important for occupation of a high number of glands.
Based on this observation, we sought to understand the host factors that limit gland expansion. Previous work had shown that TlpD responds to ROS in vitro (14), so we first asked whether tlpD mutants were exposed to elevated ROS in the host. Indeed, we found evidence of high ROS exposure for tlpD and cheY mutants in the gastric antrum (Fig. 2). We therefore employed two mouse mutants that lack distinct types of ROS. The first of these lacked the DUOX enzymes, which produce H2O2 from the apical side of gastric epithelial cells that both line gastric glands and are at the mucosal surface (16). DUOXA−/− mice had a striking phenotype: tlpD mutants now colonized ∼50% of glands, the same percentage as that of the wild type at 2 weeks of infection (Fig. 3C). tlpD mutants, interestingly, still had somewhat elevated numbers per gland (Fig. 3B), suggesting that they might have difficulty getting out of the glands or possibly not be able to sense a dispersive signal. However, the increased ability of these mutants to occupy multiple glands suggested that DUOX enzymes play a major role in limiting H. pylori from entering new glands, a host attribute that TlpD helps overcome. DUOX enzymes are substantially upregulated by Helicobacter infection (16), suggesting that it may be more challenging for H. pylori to enter glands after an infection has begun.
We gained more insight into bacterial navigation using a Cybb−/− mouse mutant, which lacks the ability to make immune-cell-produced ROS. Cybb−/− mice also had a striking phenotype. In these mice, WT H. pylori colonized fewer glands than it did in WT mice, consistent with a previous but less quantitative report (23), but WT H. pylori achieved higher numbers of bacteria per infected gland (Fig. 4B). These two phenotypes are consistent with H. pylori having increased difficulty leaving a gland without the Cybb phagocyte oxidase and thus fewer glands colonized. ΔtlpD mutants behaved similarly to WT H. pylori, colonizing the same percentage of glands also to high levels. This outcome agrees with the idea that the signal lost in Cybb−/− mutants is sensed via TlpD. Cybb−/− mutants display a complicated phenotype for Helicobacter infections (23, 24). Surprisingly, they have an elevated inflammatory score for lymphocytes, possibly due to an elevated Th1 response, and have been speculated to upregulate alternate antibacterial pathways that could keep H. pylori in the gland (23). Overall, one interpretation of the Cybb−/− data is that Cybb normally creates an ROS dispersive signal that helps to drive H. pylori out of the glands, but the elevated total inflammation present limits full colonization.
Our work additionally suggests that chemotaxis is not required for growth once bacteria are in glands, because we observed that nonchemotactic and tlpD mutants obtained high numbers per gland in WT mice. These results match those in an earlier report, which presented the average number of bacteria per gland in a manner that included uninfected glands in that calculation (5). Excluding uninfected glands from the data reported by Keilberg et al. concerning gland loads for WT and cheY H. pylori produced similar values as those described in this report (5). It is not yet known what sets gland load, but it has been observed that this number varies over the course of a mouse infection, climbing to an average of ∼15 to 25 bacteria/gland within the first month and then dropping to less than 5 by 6 months of infection (5). Our results show that chemotaxis can affect these within-gland levels, somewhat surprisingly playing a role to limit bacterial numbers. Our data suggest the possibility that chemotaxis plays a critical role in gland exit, such that without chemotaxis or TlpD specifically, bacterial numbers rise in the glands but bacteria cannot effectively leave. This phenotype in turn creates poor gland occupancy.
Previous work showed that TlpD drives chemotaxis repellent responses in vitro (7, 13, 14), and our data are consistent with the idea that it also mediates chemotaxis repellent responses in the host. Specifically, we found that tlpD mutants display hallmarks of high ROS exposure, in agreement with the idea that these mutants cannot avoid ROS. One role for this response in vivo comes from the observation that hosts upregulate defensive ROS production upon H. pylori infection (17). Thus, H. pylori may experience a delay from initially colonizing glands to experiencing stress imparted by the host. Therefore, a repellent response mediated by TlpD could limit the detrimental effect of these stresses.
In conclusion, our data support a model in which host ROS generation is sensed by the cytoplasmic chemoreceptor TlpD, which initiates a chemotaxis repellent response to drive H. pylori out of glands and promote the gland colonization in new regions, possibly with decreased ROS production. TlpD homologs are found in other pathogens that colonize mucosal surfaces, which raises the possibility that chemotactic responses to ROS at the mucosal surface may drive colonization dynamics in other infections as well.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
WT and cheY SS1 GFP+ H. pylori strains described previously were employed for mouse infections (5). H. pylori was cultured on either Columbia horse blood agar (CHBA) or brucella broth with 10% fetal bovine serum (FBS; Life Technologies) (BB10). CHBA consisted of Columbia agar (BD) with 5% defibrinated horse blood (Hemostat Labs, Davis, CA), 50 μg/ml cycloheximide, 10 μg/ml vancomycin, 5 μg/ml cefsulodin, 2.5 U/ml polymyxin B, and 0.2% (wt/vol) β-cyclodextrin. All chemicals were from Thermo Fisher or Gold Biotech. Cultures were incubated at 37°C under 5 to 7% O2, 10% CO2, and balance N2.
Creation of tlpD GFP+ H. pylori mutants.
tlpD GFP+ H. pylori strain SS1 (KO1614) was constructed by transformation of ΔtlpD::cat SS1 (KO914) (14, 25) with the plasmid pTM115 (5, 26) isolated from H. pylori strain SS1 and was selected on CHBA plates containing 15 μg/ml kanamycin (5, 9).
Animal infections and H. pylori colonization calculations.
The University of California, Santa Cruz, Institutional Animal Care and Use Committee approved all animal protocols and experiments. Cybb−/−-targeted homozygous null mice in a B6.129S background were obtained from Jackson Laboratory (Bar Harbor, ME; JAX stock no. 002365) (27); Duoxa−/− mice lacking functional dual oxidase enzymes by virtue of loss of the duoxa1-duoxa2 maturation subunits (28) were obtained as heterozygotes on the B6 background from the University of Michigan. All mice were obtained as breeding pairs and bred at UC Santa Cruz. Duoxa−/− mice were generated, screened, and maintained as previously described (16). In brief, Duoxa genotyping was performed by isolating genomic DNA from tail tissue with the Qiagen DNeasy blood and tissue kit, followed by PCR with a common primer (DA-WT/KO), a WT allele-specific primer (DA-WT-R), and a knockout allele-specific primer (DA-KO-R) (16). Genotypes were judged by the presence of the WT allele as a 381-bp fragment and the knockout allele as a 568-bp fragment (28).
Six- to 8-week-old mice (male and female) were infected intraorally by allowing the animals to drink a 50-μl suspension from a pipette tip containing H. pylori grown to mid-exponential phase and concentrated to an optical density at 600 nm of 3.0 (∼5 × 107/50 μl) in BB10 medium, as done previously (6). At the end of an infection period, mice were sacrificed by CO2 narcosis. The stomach was removed, opened along the lesser curvature, and washed in phosphate-buffered saline (PBS) to remove food. The corpus and antrum were divided based on tissue coloration and cut into pieces, which were then processed to analyze total bacterial colonization or gland isolation or for RNA extraction. For total bacterial colonization, corpus and antral tissue was weighed, homogenized with the Bullet Blender (Next Advance) with 1.0-mm zirconium silicate beads, and then plated to determine the number of CFU per gram of stomach tissue on CHBA with the addition of 20 μg/ml bacitracin, 10 μg/ml nalidixic acid, and 15 μg/ml kanamycin.
Gland isolation and microscopy.
Glands were isolated by incubating dissected gastric tissue in Dulbecco's phosphate-buffered saline (DPBS) (Millipore) plus 5 mM EDTA at 4°C for 2 h with agitation, as described previously (5, 29). The tissue was subsequently transferred to DPBS containing 1% sucrose and 1.5% sorbitol and shaken for 30 s. Glands were labeled with 10 μg/ml Hoechst DNA stain (Life Technologies). Glands were kept on ice and examined as soon as possible. Ten microliters of shaken tissue was placed on glass slides and visualized with a Nikon Eclipse E600 microscope with fluorescence filters for 4′,6′-diamidino-2-phenylindole (DAPI), GFP, and red fluorescent protein (RFP). For each time point and infection, 100 glands each were imaged for the corpus and antrum, and the number of H. pylori cells inside the gland was counted manually for each gland. Gland load levels were calculated by averaging the number of bacteria observed in colonized glands per mouse and H. pylori strain. Gland occupancy was calculated as the frequency of glands occupied per mouse host and averaged over at least three mice. Gland colonization comparisons were made for at least three mice per genotype and H. pylori strain.
RNA isolation and qPCR.
Gastric tissue was flash frozen in liquid nitrogen and homogenized in TRIzol (Invitrogen), and RNA was isolated by following the TRIzol RNA isolation protocol (GIBCO). DNA was removed by following the TURBO DNA-free kit protocol (Life Technologies). cDNA was produced with a high-capacity cDNA reverse transcription kit (Life Technologies) using random primers. Quantitative PCR (qPCR) was performed using the SensiFAST SYBR No-ROX kit (Bioline) with the primers listed below. Primer efficiency was calculated by amplifying serial dilutions of WT H. pylori genomic DNA, plotting the threshold cycle (CT) values obtained per dilution, and calculating the slope. Efficiencies were derived from the slope using the following equation: efficiency = −1 + 10(−1/slope) (30). Relative fold changes were calculated using the ΔΔCT method with Pfaffl correction for PCR amplification efficiency, using 16S and gapB as reference genes with the following primers, listed 5′-3′ (30): 16S forward, GGAGGATGAAGGTTTTAGGATTG; 16S reverse, TCGTTTAGGGCGTGGACT; katA forward, AGAGGTTTTGCGATGAAGT; katA reverse, CGTTTTTGAGTGTGGATGAA; gapB forward, GCCTCTTGCACGACTAACGC; gapB reverse, CTTTGCTCACGCCGGTGCTT.
In vitro treatment of H. pylori with H2O2.
Overnight cultures of H. pylori strains were adjusted to an optical density at 600 nm (OD600) of 0.2 and split into two cultures, with one receiving treatment with 1 mM H2O2 for 20 min. RNA isolation and qPCR protocols were identical to those described above.
ACKNOWLEDGMENTS
We thank Christina Yang for contributing references and discussion about factors that affect gland colonization. We also thank the reviewers for their thoughtful comments.
The described project was supported by National Institute of Allergy and Infectious Diseases (NIAID, NIH) grants R21AI117345 (to K.M.O.) and RO1DK087708-01 (to J.Y.K.) and by UCSC Committee on Research funds (to K.M.O.).
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Millet YA, Alvarez D, Ringgaard S, von Andrian UH, Davis BM, Waldor MK. 2014. Insights into Vibrio cholerae intestinal colonization from monitoring fluorescently labeled bacteria. PLoS Pathog 10:e1004405. doi: 10.1371/journal.ppat.1004405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Petnicki-Ocwieja T, Hrncir T, Liu Y-J, Biswas A, Hudcovic T, Tlaskalova-Hogenova H, Kobayashi KS. 2009. Nod2 is required for the regulation of commensal microbiota in the intestine. Proc Natl Acad Sci U S A 106:15813–15818. doi: 10.1073/pnas.0907722106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Macpherson AJ, Slack E, Geuking MB, McCoy KD. 2009. The mucosal firewalls against commensal intestinal microbes. Semin Immunopathol 31:145–149. doi: 10.1007/s00281-009-0174-3. [DOI] [PubMed] [Google Scholar]
- 4.Lee SM, Donaldson GP, Mikulski Z, Boyajian S, Ley K, Mazmanian SK. 2013. Bacterial colonization factors control specificity and stability of the gut microbiota. Nature 501:426–429. doi: 10.1038/nature12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Keilberg D, Zavros Y, Shepherd B, Salama NR, Ottemann KM. 2016. Spatial and temporal shifts in bacterial biogeography and gland occupation during the development of a chronic infection. mBio 7:e01705-16. doi: 10.1128/mBio.01705-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Howitt MR, Lee JY, Lertsethtakarn P, Vogelmann R, Joubert L-M, Ottemann KM, Amieva MR. 2011. ChePep controls Helicobacter pylori infection of the gastric glands and chemotaxis in the Epsilonproteobacteria. mBio 2:e00098-11. doi: 10.1128/mBio.00098-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang JY, Goers Sweeney E, Guillemin K, Amieva MR. 2017. Multiple acid sensors control Helicobacter pylori colonization of the stomach. PLoS Pathog 13:e1006118. doi: 10.1371/journal.ppat.1006118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lertsethtakarn P, Ottemann KM, Hendrixson DR. 2011. Motility and chemotaxis in Campylobacter and Helicobacter. Annu Rev Microbiol 65:389–410. doi: 10.1146/annurev-micro-090110-102908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rolig AS, Shanks J, Carter JE, Ottemann KM. 2012. Helicobacter pylori requires TlpD-driven chemotaxis to proliferate in the antrum. Infect Immun 80:3713–3720. doi: 10.1128/IAI.00407-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Behrens W, Schweinitzer T, Bal J, Dorsch M, Bleich A, Kops F, Brenneke B, Didelot X, Suerbaum S, Josenhans C. 2013. Role of energy sensor TlpD of Helicobacter pylori in gerbil colonization and genome analyses after adaptation in the gerbil. Infect Immun 81:3534–3551. doi: 10.1128/IAI.00750-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Draper J, Karplus K, Ottemann KM. 2011. Identification of a chemoreceptor zinc-binding domain common to cytoplasmic bacterial chemoreceptors. J Bacteriol 193:4338–4345. doi: 10.1128/JB.05140-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Collins KD, Lacal J, Ottemann KM. 2014. Internal sense of direction: sensing and signaling from cytoplasmic chemoreceptors. Microbiol Mol Biol Rev 78:672–684. doi: 10.1128/MMBR.00033-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schweinitzer T, Mizote T, Ishikawa N, Dudnik A, Inatsu S, Schreiber S, Suerbaum S, Aizawa S-I, Josenhans C. 2008. Functional characterization and mutagenesis of the proposed behavioral sensor TlpD of Helicobacter pylori. J Bacteriol 190:3244–3255. doi: 10.1128/JB.01940-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Collins KD, Andermann TM, Draper J, Sanders L, Williams SM, Araghi C, Ottemann KM. 2016. The Helicobacter pylori CZB cytoplasmic chemoreceptor TlpD forms an autonomous polar chemotaxis signaling complex that mediates a tactic response to oxidative stress. J Bacteriol 198:1563–1575. doi: 10.1128/JB.00071-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Handa O, Naito Y, Yoshikawa T. 2010. Helicobacter pylori: a ROS-inducing bacterial species in the stomach. Inflamm Res 59:997–1003. doi: 10.1007/s00011-010-0245-x. [DOI] [PubMed] [Google Scholar]
- 16.Grasberger H, Zaatari El M, Dang DT, Merchant JL. 2013. Dual oxidases control release of hydrogen peroxide by the gastric epithelium to prevent Helicobacter felis infection and inflammation in mice. Gastroenterology 145:1045–1054. doi: 10.1053/j.gastro.2013.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Flint A, Stintzi A, Saraiva LM. 2016. Oxidative and nitrosative stress defences of Helicobacter and Campylobacter species that counteract mammalian immunity. FEMS Microbiol Rev 40:938–960. doi: 10.1093/femsre/fuw025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grasberger H, Gao J, Nagao-Kitamoto H, Kitamoto S, Zhang M, Kamada N, Eaton KA, Zaatari El M, Shreiner AB, Merchant JL, Owyang C, Kao JY. 2015. Increased expression of DUOX2 is an epithelial response to mucosal dysbiosis required for immune homeostasis in mouse intestine. Gastroenterology 149:1849–1859. doi: 10.1053/j.gastro.2015.07.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Olczak AA, Wang G, Maier RJ. 2005. Up-expression of NapA and other oxidative stress proteins is a compensatory response to loss of major Helicobacter pylori stress resistance factors. Free Radic Res 39:1173–1182. doi: 10.1080/10715760500306729. [DOI] [PubMed] [Google Scholar]
- 20.Ernst FD, Bereswill S, Waidner B, Stoof J, Mäder U, Kusters JG, Kuipers EJ, Kist M, van Vliet AHM, Homuth G. 2005. Transcriptional profiling of Helicobacter pylori Fur- and iron-regulated gene expression. Microbiology 151:533–546. doi: 10.1099/mic.0.27404-0. [DOI] [PubMed] [Google Scholar]
- 21.Pédron T, Nigro G, Sansonetti PJ. 2016. From homeostasis to pathology: decrypting microbe-host symbiotic signals in the intestinal crypt. Philos Trans R Soc Lond B Biol Sci 371:20150500. doi: 10.1098/rstb.2015.0500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Round JL, Lee SM, Li J, Tran G, Jabri B, Chatila TA, Mazmanian SK. 2011. The Toll-like receptor 2 pathway establishes colonization by a commensal of the human microbiota. Science 332:974–977. doi: 10.1126/science.1206095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blanchard TG, Yu F, Hsieh CL. 2003. Severe inflammation and reduced bacteria load in murine helicobacter infection caused by lack of phagocyte oxidase activity. J Infect Dis 187:1609–1615. doi: 10.1086/374780. [DOI] [PubMed] [Google Scholar]
- 24.Keenan JI, Peterson RA, Hampton MB. 2005. NADPH oxidase involvement in the pathology of Helicobacter pylori infection. Free Radic Biol Med 38:1188–1196. doi: 10.1016/j.freeradbiomed.2004.12.025. [DOI] [PubMed] [Google Scholar]
- 25.Williams SM, Chen YT, Andermann TM, Carter JE, McGee DJ, Ottemann KM. 2007. Helicobacter pylori chemotaxis modulates inflammation and bacterium-gastric epithelium interactions in infected mice. Infect Immun 75:3747–3757. doi: 10.1128/IAI.00082-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Amieva MR, Vogelmann R, Covacci A, Tompkins LS, Nelson WJ, Falkow S. 2003. Disruption of the epithelial apical-junctional complex by Helicobacter pylori CagA. Science 300:1430–1434. doi: 10.1126/science.1081919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pollock JD, Williams DA, Gifford MAC, Li LL, Du X, Fisherman J, Orkin SH, Doerschuk CM, Dinauer MC. 1995. Mouse model of X-linked chronic granulomatous disease, an inherited defect in phagocyte superoxide production. Nat Genet 9:202–209. doi: 10.1038/ng0295-202. [DOI] [PubMed] [Google Scholar]
- 28.Grasberger H, De Deken X, Mayo OB, Raad H, Weiss M, Liao X-H, Refetoff S. 2012. Mice deficient in dual oxidase maturation factors are severely hypothyroid. Mol Endocrinol 26:481–492. doi: 10.1210/me.2011-1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mahe MM, Aihara E, Schumacher MA, Zavros Y, Montrose MH, Helmrath MA, Sato T, Shroyer NF. 2013. Establishment of gastrointestinal epithelial organoids. Curr Protoc Mouse Biol 3:217–240. doi: 10.1002/9780470942390.mo130179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]