

ABSTRACT
Coxiella burnetii is the causative agent of human Q fever, a debilitating flu-like illness that can progress to chronic disease presenting as endocarditis. Following inhalation, C. burnetii is phagocytosed by alveolar macrophages and generates a lysosome-like replication compartment termed the parasitophorous vacuole (PV). A type IV secretion system (T4SS) is required for PV generation and is one of the pathogen's few known virulence factors. We previously showed that C. burnetii actively recruits autophagosomes to the PV using the T4SS but does not alter macroautophagy. In the current study, we confirmed that the cargo receptor p62/sequestosome 1 (SQSTM-1) localizes near the PV in primary human alveolar macrophages infected with virulent C. burnetii. p62 and LC3 typically interact to select cargo for autophagy-mediated degradation, resulting in p62 degradation and LC3 recycling. However, in C. burnetii-infected macrophages, p62 was not degraded when cells were starved, suggesting that the pathogen stabilizes the protein. In addition, phosphorylated p62 levels increased, indicative of activation, during infection. Small interfering RNA experiments indicated that p62 is not absolutely required for intracellular growth, suggesting that the protein serves a signaling role during infection. Indeed, the Nrf2-Keap1 cytoprotective pathway was activated during infection, as evidenced by sustained maintenance of Nrf2 levels and translocation of the protein to the nucleus in C. burnetii-infected cells. Collectively, our studies identify a new p62-regulated host signaling pathway exploited by C. burnetii during intramacrophage growth.
KEYWORDS: Coxiella burnetii, Nrf2, autophagy, intracellular pathogen, p62
INTRODUCTION
Human Q fever is an emerging disease caused by the intracellular, Gram-negative bacterium Coxiella burnetii. Inhalation of fewer than 10 organisms is sufficient for infection, contributing to the pathogen's status as a CDC category B select agent (1). Acute disease presents as a debilitating flu-like illness that can progress to chronic disease, commonly manifesting as endocarditis (2). Indeed, C. burnetii is a leading cause of noncultivatable infectious endocarditis (3). Currently, no vaccine is approved for civilian use in the United States and treatment options are limited, with doxycycline being the most effective antibiotic (4). To design improved therapeutics, increased understanding of the pathogen's intracellular lifestyle is needed, as successful cellular infection is required for progression of disease.
The intracellular life cycle of C. burnetii is unique, as the pathogen targets alveolar macrophages for replication within a lysosome-derived parasitophorous vacuole (PV) (5). Two defined C. burnetii virulence determinants are lipopolysaccharide (LPS) and a Dot/Icm type IVB secretion system (T4SS). C. burnetii undergoes LPS phase variation, where the O antigen of full-length LPS (phase I) is truncated (phase II), resulting in reduced virulence (6). Phase I organisms are highly virulent, and full-length LPS serves a shielding function to prevent innate immune detection (7, 8). However, after conversion to phase II, organisms are immunogenic and less virulent. Regardless of the LPS type, PV biogenesis requires a Dot/Icm T4SS that secretes bacterial effector proteins into the host cytoplasm, where they then manipulate numerous processes, including apoptosis and autophagy (9–12). Few effectors have been fully characterized; however, CaeA, CaeB, and AnkG have been implicated in prevention of host apoptosis, while Cig2 promotes PV fusion with autophagosomes (11–13).
Autophagy is a conserved eukaryotic process of degrading and recycling cargo, including defunct organelles and protein aggregates, to acquire nutrients during periods of deprivation. Canonical macroautophagy uses double-membrane autophagosomes that typically originate from the endoplasmic reticulum and fuse with lysosomes (14). Autophagy is tightly regulated, and cargo is identified by specialized receptors (15). Cargo receptors are ubiquitin-interacting proteins that target ubiquitinated substrates for degradation via selective autophagy (16). p62, also known as sequestosome-1 (SQSTM-1), is a cargo receptor that interacts with ubiquitinated cargo and LC3 at the autophagosome membrane to target substrates for autophagic degradation. p62 can also target ubiquitinated bacteria for degradation via selective autophagy (16). Therefore, p62 is often regarded as antibacterial, and the protein's presence on or nearby invading bacteria is typically detrimental to the pathogen. Altered levels of p62 can indicate autophagic flux or autophagy activation (17, 18). Indeed, to determine if cells are undergoing autophagic flux, the levels of both p62 and lipidated LC3-II must be assessed (19). During autophagic flux, the levels of LC3-II increase or remain constant, as the protein is recycled following autophagosome fusion with lysosomes. However, the levels of p62, along with those of selected cargo, decrease during autophagic flux due to degradation within the autophagosome.
p62 also functions as a signaling center in selective autophagy, where selection and degradation of specific proteins alter downstream signaling events (20). p62 contains numerous phosphorylation sites that regulate distinct downstream responses, including S349, which controls the interaction with the Nrf2-Keap1 antioxidant pathway; S403/407, which regulates interaction with ubiquitinated proteins; and T269/S272, which alters cell cycle events (20). Additionally, p62 regulates the inflammasome response by selecting assembled inflammasomes for degradation via autophagy. Inflammasomes are multiprotein complexes required for proinflammatory interleukin-1β (IL-1β) and IL-18 processing and release from the cell upon stimulation (21). p62 recognizes and targets ubiquitinated inflammasome components for degradation, limiting the cellular proinflammatory response (22).
In addition to impacting inflammasome activity, p62 (via S349 phosphorylation) regulates oxidative stress responses by controlling degradation of components of the Nrf2-Keap1 pathway, which is triggered by many factors, including xenobiotics (23–25). Under homeostatic conditions, Keap1 binds and promotes proteasome-dependent degradation of the transcription factor Nrf2. Upon stimulation of this pathway, p62 is phosphorylated, increasing its binding affinity for Keap1 and freeing Nrf2 to translocate to the nucleus and induce transcription of multiple cytoprotective genes (26). Moreover, the Nrf2-Keap1 pathway promotes activation of selective autophagy by inducing transcription of the gene encoding p62 (26).
Previously, we discovered that p62 is recruited to the PV in a T4SS-dependent manner in human macrophages, suggesting an active role for p62 in C. burnetii-host cell interactions. Here, we used our primary human alveolar macrophage platform of infection (27) to assess C. burnetii recruitment and activation of p62. We found that p62 is recruited near the PV of virulent C. burnetii and is stabilized during infection. p62 is recruited in a noncanonical fashion that does not require LC3- or ubiquitin-interacting domains. Additionally, p62 is phosphorylated at S349 during C. burnetii intracellular growth, confirming that the protein is activated. Interestingly, we found that the Nrf2-Keap1 antioxidant pathway is activated in infected cells, suggesting a novel role for this pathway in C. burnetii parasitism of human macrophages.
RESULTS
p62 localizes near the PV formed by C. burnetii in primary hAMs.
We previously showed that LC3 and p62 are recruited to the PV membrane in a T4SS-dependent manner during avirulent C. burnetii infection of THP-1 macrophage-like cells, suggesting that selective autophagy occurs at the PV (10). To address the disease relevance of these findings, we used our primary human alveolar macrophage (hAM) platform of infection. At 72 h postinfection (hpi) by avirulent C. burnetii (Nine Mile II [NMII]), when the PV has expanded and bacterial replication is in progress, we observed p62 near the PV membrane (Fig. 1). These data confirm previous observations in THP-1 cells and indicate that p62 recruitment to the PV occurs in the pathogen's in vivo target cell. Additionally, p62 localized near the PV of virulent (Nine Mile I [NMI]) C. burnetii-infected hAMs (Fig. 1), suggesting that this event occurs during natural infection by disease-causing bacteria and is not an artifact caused by immunogenic avirulent organisms. Moreover, LC3 and p62 colocalized near the PV of NMI-infected hAMs (data not shown). To confirm p62 localization near the NMI PV, infected HeLa cells were assessed using antibody to detect native p62. These results showed p62 puncta near numerous PV (Fig. 1), similar to the results obtained with hAMs. Although fluorescence microscopy alone is not sufficient to conclude that p62 intimately associates with the PV, these results suggest that the protein establishes a proximity to the vacuole that would allow interaction with T4SS effectors, as previously proposed (10).
FIG 1.

p62 localizes near the PV during primary hAM infection with C. burnetii NMI and NMII. (Left and middle) hAMs or HeLa cells were infected for 72 h with virulent (NMI) or avirulent (NMII) C. burnetii or left uninfected and then processed for immunofluorescence confocal microscopy to detect p62 (green), CD63 (red), and C. burnetii (violet). DAPI-stained DNA is blue. The data shown are representative of observations from three individual donors and experiments. (Insets) Magnified regions of p62 localization around the PV. Arrowheads, p62 puncta that colocalize with CD63; asterisks in p62 panels, PV. (Right) Intensity profiles show the limiting membrane of the PV (arrows) and the peaks of p62 intensity (asterisks) near the PV membrane. Native p62 localizes near the CD63-positive PV in primary hAMs and HeLa cells infected with virulent C. burnetii.
p62 ubiquitin-binding and LC3-interacting domains are not required for localization near the PV.
LC3 and p62 colocalization at the PV membrane suggests activation of selective autophagy (10). Generally regarded as an antibacterial protein in the context of intracellular infection, p62 targets ubiquitinated bacteria for degradation via autophagy by directly interacting with LC3. Like most cargo receptors, p62 encodes an ubiquitin-binding domain (UBA) and an LC3-interacting region (LIR) that promote interaction with target proteins (20). Interestingly, p62 recruitment to the PV also requires T4SS function, indicating active recruitment of the protein or interacting proteins by C. burnetii. To assess the mechanism by which p62 is recruited to the PV, we initially transfected HeLa cells as previously described with constructs encoding green fluorescent protein (GFP)-tagged p62 (GFP-p62), GFP-p62 with an LIR deletion (GFP-p62-ΔLIR), or GFP-p62 with a UBA deletion (GFP-p62-ΔUBA) (28) and then infected the transfected cells with C. burnetii (Fig. 2). The goal of these studies was to determine if the LIR or UBA region of p62 is required for recruitment to the PV that forms in macrophage and nonmacrophage cells and if overexpression of GFP-p62 impacts infection. As shown in Fig. 2, GFP-p62-ΔLIR and GFP-p62-ΔUBA were still recruited to the area of the PV, with GFP-p62-ΔUBA also being dispersed in the cytoplasm. Although direct binding of p62 mutants to the PV membrane or associated proteins has not been determined, these results suggest that p62 is recruited near the PV by a noncanonical mechanism.
FIG 2.

p62 LIR and UBA domain mutants localize to the area of the PV. HeLa cells were transfected with GFP constructs encoding wild-type, GFP-tagged-only, or p62 mutant proteins. At 72 hpi with NMII, cells were processed for fluorescence microscopy. CD63 antibody (red) was used to detect the PV, C. burnetii antibody was used to detect bacteria (violet), and DAPI-stained DNA is shown in blue. Experiments were performed in triplicate. (Right) Intensity profiles show the limiting membrane of the PV (arrows) and peaks of p62 intensity (asterisks) at the PV membrane. The absence of an LC3- or ubiquitin-binding domain does not impact p62 localization around the PV.
p62 levels increase during infection of hAMs with virulent C. burnetii.
Previously, we observed that p62 levels were stable during infection of hAMs with avirulent C. burnetii, indicating that infected cells do not undergo normal autophagic flux (17) in response to the pathogen (Fig. 3). We next assessed whether this observation was similar during virulent C. burnetii infection. Interestingly, p62 accumulated in hAMs infected with virulent C. burnetii throughout infection, with levels increasing approximately 2.5-fold above those of uninfected cells at 96 hpi. This finding was surprising, as the only established difference between NMI and NMII is the structure of their respective LPS. These data suggest that neither virulent nor avirulent C. burnetii stimulates normal autophagic flux, as indicated by no decrease in p62 levels in hAMs infected with either isolate, and virulent organisms more effectively stabilize the protein, leading to elevated levels during intracellular growth.
FIG 3.
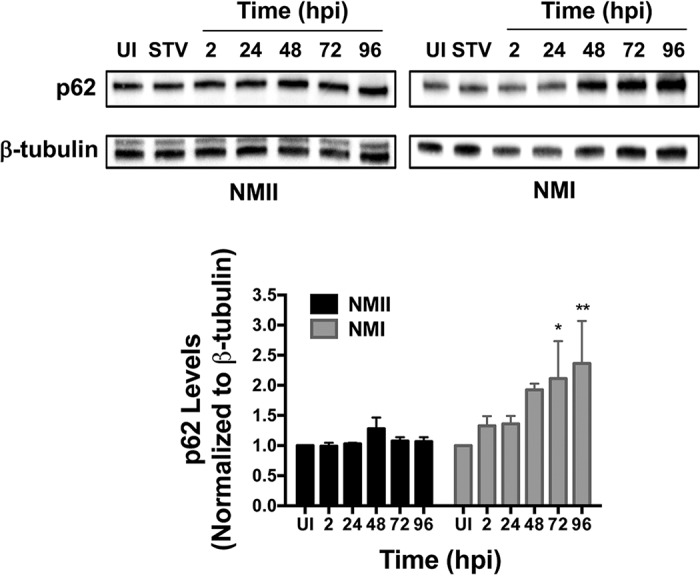
p62 levels increase during virulent C. burnetii infection. hAMs were infected for 2 to 96 h and processed for immunoblot analysis using monoclonal antibodies to detect p62. β-Tubulin served as a loading control, and the blots presented are representative of those for three individual donors and experiments that were used for densitometry analysis. P values were determined using Student's t test. *, P < 0.05; **, P < 0.001. UI, uninfected cells; STV, nutrient-starved uninfected cells (positive control). p62 levels remain stable in cells infected with avirulent bacteria and increase from 72 to 96 hpi in cells infected with virulent C. burnetii.
p62 is stabilized during C. burnetii infection.
The accumulation of p62 suggested that the protein is not susceptible to degradation via autophagy during infection. To address this possibility, hAMs were infected for 72 h and then subjected to nutrient starvation by incubation with Earle's balanced salt solution (EBSS). Nutrient starvation induces macroautophagy, a process that ultimately degrades p62. In both NMII- and NMI-infected hAMs, p62 levels remained stable following EBSS starvation (Fig. 4 and data not shown). As expected, uninfected cells displayed decreased p62 levels during starvation-induced autophagy by 8 h poststarvation. A possible explanation for this observation is that the lysosome-like PV cannot degrade proteins. However, previous studies showed that the PV displays the degradative properties of a lysosome (evidenced by turnover of bovine serum albumin [BSA] and other bacteria) and contains active cathepsin D, a confirmed marker of active lysosomes (29). Together, our data suggest that p62 is specifically stabilized during infection.
FIG 4.
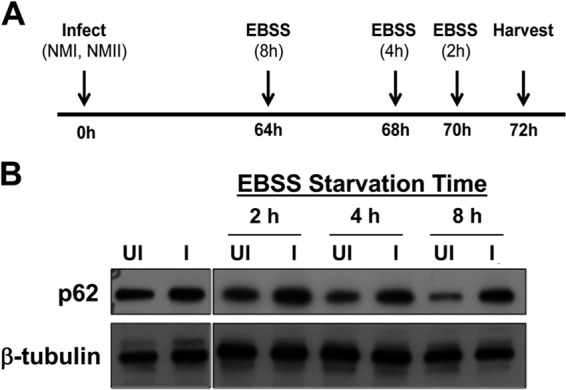
p62 is stabilized in C. burnetii-infected cells. (A) Schematic of experimental design for nutrient starvation. (B) After EBSS starvation, hAM lysates were processed for immunoblot analysis to detect p62. β-Tubulin served as a loading control, and the blots presented are representative of those for three individual donors and experiments. UI, uninfected cells; I, NMII-infected cells. C. burnetii stabilizes p62 that is turned over in nutrient-starved hAMs.
p62 is dispensable for C. burnetii infection and intracellular growth.
We next addressed whether p62 is required for productive infection with C. burnetii. Unfortunately, small interfering RNA (siRNA)-based knockdown in hAMs was insufficient; therefore, we used THP-1 macrophage-like cells, which have been established as a relevant in vitro cell model by multiple laboratories. Using nucleofection, we achieved ∼75% decreased p62 expression (Fig. 5). Using a strain of NMII that constitutively expresses mCherry, we conducted growth curve analysis of C. burnetii in p62-silenced cells. As shown in Fig. 5, decreased p62 expression had no significant effect on bacterial replication, suggesting that p62 is not absolutely required for C. burnetii growth within eukaryotic cells. These results suggest that p62 may play a signaling role in C. burnetii infection that is not required for bacterial growth.
FIG 5.
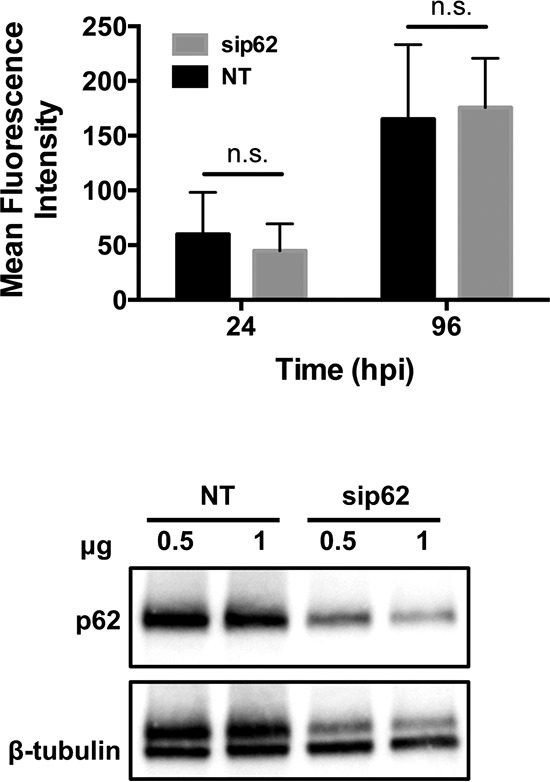
p62 is not required for C. burnetii intracellular replication. (Top) THP-1 cells were transfected with siRNA targeting p62 (sip62) or nontargeting siRNA (NT) as a control. (Bottom) Decreased p62 expression was confirmed by immunoblot analysis, and β-tubulin served as a loading control. Transfected cells were infected with NMII-mCherry for 24 or 96 h. Bacterial replication was monitored at each time point by determination of mCherry fluorescence and normalized to that for uninfected cells with the corresponding treatments. No statistically significant (n.s.) differences were present in the growth curve analysis using Student's t test, indicating that C. burnetii replication is not impacted by decreasing p62 expression.
C. burnetii infection triggers activation of the Nrf2-Keap1 pathway.
Although p62 is not absolutely required for productive C. burnetii infection or replication, the protein may contribute to host signaling in response to the pathogen. p62 is linked to many signaling pathways due to its role in autophagy and ability to act as a scaffolding protein (20). Thus, we searched for p62-regulated signaling pathways that may impact C. burnetii infection processes. The Nrf2-Keap1 pathway promotes p62 stabilization and accumulation (30), similar to our observations during C. burnetii growth in nutrient-starved hAMs. Nrf2 is continually degraded via the proteasome when bound to Keap1. When activated, Keap1 releases Nrf2, stabilizing Nrf2 and allowing translocation to the nucleus. In a bacterial context, p62 targets Salmonella and is subsequently phosphorylated, binding to Keap1 and promoting Nrf2 activation (31). p62 contains multiple residues that are phosphorylated to regulate activation (20), providing a straightforward readout of activation during infection. Here, we focused on S349, as phosphorylation of this residue promotes interaction with Keap1 (26). As shown in Fig. 6A, the levels of phosphorylated p62 (S349) increased substantially throughout infection, indicating that the protein is activated in a manner that promotes interaction with Keap1.
FIG 6.
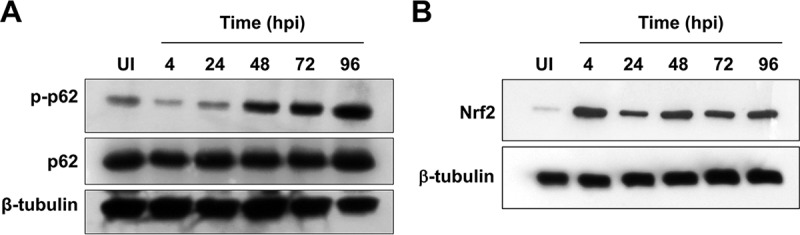
p62 phosphorylation at S349 increases during infection, and Nrf2 levels are stabilized. hAMs were infected with C. burnetii NMII for 4 to 96 h and then harvested and assessed by immunoblot analysis to detect phosphorylated p62 (p-p62) and total p62 (A) or Nrf2 (B). β-Tubulin was probed as a loading control, and the blots presented are representative of those from three individual experiments. The levels of phosphorylated p62 increase during infection and Nrf2 levels remain stable, suggesting activation of both proteins.
To assess whether the Nrf2-Keap1 pathway is activated during infection, we monitored the levels of Nrf2 by immunoblotting, as the protein should be continually degraded if inactive. Immunoblot analysis revealed little detectable Nrf2 in uninfected hAMs; however, when infected with C. burnetii, detectable levels of Nrf2 were present and maintained from 4 to 96 hpi (Fig. 6B). In agreement with these results, a substantial number of infected THP-1 cells compared to uninfected cells contained nuclear Nrf2 (Fig. 7A). To confirm these results, HeLa cells were transfected with constructs encoding GFP-Nrf2, providing quantifiable results. The signal for nuclear GFP-Nrf2 increased in approximately 80% of infected cells, corresponding to the results obtained with THP-1 cells, as opposed to approximately 45% of uninfected cells (Fig. 7B). Combining data from all cells examined under each condition, C. burnetii-infected cells demonstrated nuclear mean fluorescence intensities (MFIs) of 2,265.20, whereas the nuclear MFIs were 1,027.98 for uninfected cells (a ∼2.2-fold increase from that for C. burnetii-infected cells). Furthermore, the nuclear fraction of infected THP-1 cells was isolated and compared to that of uninfected cells. As shown in Fig. 7C, substantial Nrf2 was detected only in the nucleus of C. burnetii-infected cells, indicating activation of the transcription factor. Collectively, these data indicate that Nrf2 signaling is activated during C. burnetii infection coincident with p62 phosphorylation at a Keap1-specific residue.
FIG 7.

Nrf2 translocates to the nucleus during infection. THP-1 cells were infected with NMII for 72 h and then processed for immunofluorescence confocal microscopy to detect C. burnetii and Nrf2 (A) and GFP-Nrf2 expression in HeLa cells (B) or immunoblot analysis to detect Nrf2 and histone H3 (nuclear fraction control) in the nuclear fraction of infected THP-1 cells (C). The results presented are representative of those from three individual experiments. *, P < 0.01, using Student's t test. Arrows indicate regions of intensity profile analysis. Results show that more C. burnetii-infected cells (∼80%) than uninfected cells (∼45%) contain increased nuclear Nrf2, indicating activation of the pathway.
DISCUSSION
In the current study, we identified a potential signaling role for p62 during C. burnetii infection: activation of the cytoprotective Nrf2-Keap1 pathway. Our previous study demonstrated p62 recruitment to the PV in a T4SS-dependent manner, indicating that the pathogen purposely targets this antibacterial protein (10). p62 targets numerous bacteria for destruction, including Salmonella, Listeria, and Legionella (28, 32, 33). For example, p62 is recruited to intracellular Salmonella, targeting the bacterium for destruction in an autophagolysosome. p62 is also required to degrade Listeria lacking the actin-based motility protein ActA. Moreover, the cytokine response to Legionella pneumophila is amplified in the absence of p62, suggesting that the protein is involved in regulation of inflammation in response to bacteria. In contrast, C. burnetii replicates efficiently in a PV surrounded by p62 puncta, suggesting a probacterial function for the protein in this infection system.
Key to C. burnetii virulence is the organism's LPS structure and ability to infect human alveolar macrophages. Here, we used the phase variants NMI and NMII (6) to infect hAMs, our primary cell infection platform. After confirming that p62 and LC3 localization to the area of the PV occurs in hAMs during virulent NMI infection, we obtained perplexing results regarding the levels of p62 during NMI infection, where the levels were significantly higher than those in uninfected cells in the absence of significantly increased p62 expression (not shown). The only defined difference between the NMI and NMII isolates is their respective LPS, the structure of which confers virulence to NMI. Regardless, our data indicate that hAMs do not undergo normal autophagic flux in response to C. burnetii pathotypes, confirming that the pathogen does not activate canonical autophagy. However, p62 is clearly dysregulated during infection, with levels being stabilized during NMII infection and increased during NMI growth in hAMs.
To address how p62 is recruited to the vicinity of the PV, we assessed whether localization was solely due to interaction with the LC3 or ubiquitin present at the PV membrane. Ectopically expressed GFP-p62, GFP-p62-ΔLIR (which does not interact with LC3), and GFP-p62-ΔUBA (which does not bind ubiquitin) are all recruited to the area of the PV, indicating that an interaction with neither LC3 nor ubiquitin is required for p62 localization near the PV. These results indicate a role for p62 outside the realm of classical autophagy during C. burnetii infection and suggest a noncanonical bacterium-directed mode of p62 recruitment. A recent report by Newton et al. suggests that T4SS effectors may direct p62 to the PV (13). This group showed that a Cig2-deficient mutant is unable to form a PV that decorates with p62 or the cargo receptor NDP52. These intriguing results suggest that C. burnetii purposely recruits p62 to the PV using specialized effectors.
siRNA-based silencing of p62 indicates that the protein is not absolutely required for efficient infection and intracellular growth. Therefore, we predict that p62 regulates signaling events that impact the pathogen. The Nrf2-Keap1 pathway is an intriguing target due to its cytoprotective, antioxidant nature and role in other bacterial infections (34). C. burnetii potently inhibits host cell death (11, 35–39) and antagonizes bactericidal activities, such as reactive oxygen species production (40). Thus, we predict that the pathogen uses Nrf2 signaling to ensure survival of the host cell and establish a hospitable replication niche. Nrf2 levels are maintained in C. burnetii-infected cells and the protein translocates to the nucleus, a prerequisite for transcriptional activity. Nrf2 activation is regulated in a noncanonical manner by p62. Phosphorylated p62 (S349) binds Keap1, sequestering the protein from Nrf2 and allowing Nrf2 translocation to the nucleus (26). Indeed, the levels of p62 phosphorylated at S349 increased substantially throughout C. burnetii infection. Nuclear Nrf2 binds antioxidant response elements (ARE) that transcriptionally regulate numerous genes; however, ARE-regulated genes altered during C. burnetii infection have not been defined and are the subject of ongoing studies.
The Nrf2-Keap1 pathway has been studied primarily in the cancer arena. Few reports address the role of Nrf2 and downstream pathways in bacterial pathogenesis; however, a recent study highlights the potential role of Nrf2 during the host response to Streptococcus pneumoniae infection (41). Additionally, Salmonella is recognized by p62 in the cytosol of eukaryotic cells, and this event triggers Nrf2 activation (31). Nrf2 is predominantly expressed in alveolar epithelial cells and macrophages in the lung (42), making the protein an attractive target for C. burnetii due to the pulmonary nature of the pathogen and its preference for replication in hAMs. Additionally, C. burnetii reportedly antagonizes NADPH oxidase activity (40), and Nrf2 is likely involved in this response. Moreover, virulent C. burnetii avoids activation of the macrophage proinflammatory response that is tightly regulated by macrophages (43). Interestingly, p62 regulates the inflammatory response to closely related L. pneumophila (32). Thus, we are currently pursuing a potential role for p62 in regulation of the antioxidant and inflammatory response to C. burnetii. Moreover, the role of Nrf2 in C. burnetii parasitism of macrophages represents a novel area of research in the field.
Together, the current results point to novel events regulated by p62 that are subverted by C. burnetii. The Nrf2-Keap1 pathway is a critical cellular process involved in many human diseases but is understudied in the host response to bacterial pathogens. Understanding the contribution of this pathway and additional p62-dependent events to C. burnetii pathogenesis will advance knowledge of eukaryotic pathways required for infection and an effective host cellular response.
MATERIALS AND METHODS
Eukaryotic and bacterial cell culture.
Primary human alveolar macrophages (hAMs) were acquired postmortem by bronchoalveolar lavage from human donor lungs (National Disease Research Interchange) and processed as previously described (27). Cells were maintained in Dulbecco's modified Eagle medium–F-12 medium (DMEM–F-12) containing 10% fetal bovine serum (FBS), penicillin (50 U/ml), streptomycin sulfate (50 μg/ml), gentamicin sulfate (50 μg/ml), and amphotericin B (0.25 μg/ml) at 37°C with 5% CO2. Prior to infection, cells were washed with medium lacking antibiotics/antimycotics and cultured for 24 h to ensure the absence of contamination. Earle's balanced salt solution (EBSS) was used for nutrient starvation assays.
THP-1 cells (TIB-202; American Type Culture Collection [ATCC]) and HeLa cells (CCL-2; ATCC) were cultured in RPMI 1640 medium with 10% FBS at 37°C with 5% CO2. THP-1 cells were differentiated into macrophage-like cells prior to infection by treatment with phorbol 12-myristate 13-acetate (PMA; 200 nM) overnight. The medium containing PMA was then removed, and cells were cultured in normal medium at least 4 h prior to infection.
C. burnetii Nine Mile I (NMI; RSA493) and Nine Mile II (NMII; RSA439) were propagated in acidified citrate cysteine medium (ACCM) for 7 days at 37°C with 5% CO2 and 2.5% O2. The bacteria were washed three times and stored in sucrose phosphate buffer at −80°C prior to infection. Eukaryotic cell infections were performed at a multiplicity of infection of ∼10 by adding bacteria to the cells overnight and then removing extracellular bacteria by washing. All experiments with NMI were performed in the biosafety level 3 facility at the University of Arkansas for Medical Sciences, which is approved by the Centers for Disease Control and Prevention.
Transfection.
p62-specific or nontargeting SMARTpool siRNA was resuspended according to the manufacturer's instructions (Thermo). THP-1 cells were transfected with siRNA, rested, and then cultured in medium containing PMA for differentiation. GFP-p62 and domain mutants (GFP-p62-ΔUBA and GFP-p62-ΔLIR) were the generous gift of Terje Johansen (University of Tromso) and John H. Brumell (University of Toronto) and were previously described (28). Plasmids encoding these proteins were introduced into HeLa or THP-1 cells by Effectene transfection reagent-mediated transfection or nucleofection (Lonza), respectively, at 36 hpi with C. burnetii. At 18 h posttransfection, cells were fixed and processed for microscopy using antibodies directed against CD63 (PV membrane) and C. burnetii. For growth curve analysis, transfected cells were cultured in phenol red-free medium in black-wall 96-well plates and assessed by fluorescence at each time point using a Synergy H1 reader and accompanying software (BioTek). HeLa cells were transfected with plasmids encoding GFP-Nrf2 (Addgene) using Effectene, as described above.
Immunoblotting and cellular fractionalization analysis.
THP-1 cells or hAMs were harvested in lysis buffer comprised of 50 mM Tris, 5 mM EDTA, and 1% sodium dodecyl sulfate (SDS) with protease and phosphatase inhibitor cocktails (Sigma). Protein quantities were determined using a DC protein assay (Bio-Rad). Total protein (10 μg) from each sample was separated by 10% or 4 to 15% SDS-polyacrylamide gel electrophoresis. After transfer to a polyvinylidene difluoride (PVDF) membrane (Bio-Rad), the membranes were blocked in Tris-buffered saline with Tween 20 (TBS-T; 150 mM NaCl, 100 mM Tris-HCl [pH 7.6], 0.1% Tween 20) containing 5% nonfat milk at room temperature for 1 h. Antibodies directed against p62 (Sigma), phosphorylated p62 (S349), histone H3 (Cell Signaling), or Nrf2 (Abcam) were used at a 1:1,000 dilution in TBS-T with 5% nonfat milk or 5% bovine serum albumin. The membranes were washed three times with TBS-T. Secondary antibodies conjugated to horseradish peroxidase (HRP; Cell Signaling) were diluted in TBS-T with 5% nonfat milk and incubated with the membranes for 1 h at room temperature. After washing three times with TBS-T, the membranes were incubated with a chemiluminescent HRP substrate (Advansta WesternBright) and visualized using a Bio-Rad ChemiDoc imaging system or exposure to film. Equal protein loading was determined using a monoclonal antibody directed against β-tubulin (clone SAP.4G5; Sigma).
Cellular fractionations were performed using THP-1 cells (2 × 106) that were uninfected or infected for 72 h. The nuclear fraction of cells was isolated using a fractionation kit according to the manufacturer's instructions (BioVision). Samples were then processed as described above for immunoblot analysis to detect Nrf2 or histone H3 (positive nuclear control).
Immunofluorescence microscopy.
Eukaryotic cells were cultured on 12-mm glass coverslips in 24-well plates. The coverslips were washed three times with cold phosphate-buffered saline (PBS) and fixed with 100% ice-cold methanol for 3 min or 4% paraformaldehyde (PFA) for 15 min. After washing three times with cold PBS, the coverslips were blocked in PBS containing 0.5% BSA for 1 h at room temperature. PFA-fixed samples were blocked with PBS containing 0.5% BSA and 0.3% Triton X-100 for membrane permeabilization. After blocking, coverslips were incubated with primary antibodies directed against p62 (Cell Signaling), CD63 (BD Biosciences), or Nrf2 (Cell Signaling) for 1 h at room temperature. The coverslips were washed three times with cold PBS before addition of secondary antibodies conjugated to Alexa Fluor 488 or 594 (Invitrogen) for 1 h at room temperature. The coverslips were washed three times in PBS and then incubated with 6-diamidino-2-phenylindole (DAPI; Invitrogen) to detect DNA. The coverslips were mounted to slides using Mowiol mounting medium and allowed to cure overnight. Samples were visualized and captured using a Ti-Eclipse confocal microscope or a Ti-U microscope with a ×60 objective (Nikon), and analysis, including intensity profile generation, was performed with NIS-Elements software (Nikon). For quantification of GFP-Nrf2, NIS-Elements was used to score at least 500 cells per condition. Mean fluorescence intensities (MFIs) were determined for the GFP signal in nuclei from uninfected and NMII-infected cells. A 1.5-fold increase in the MFI over basal uninfected levels (∼1,000 MFI) was considered a positive result for nuclear Nrf2.
ACKNOWLEDGMENTS
We thank Terje Johansen and John H. Brumell for constructing and providing the GFP-p62 constructs.
This research was supported by funding to D.E.V. from the NIH (R01AI087669 and R21AI127931), the Arkansas Biosciences Institute, and the Center for Microbial Pathogenesis and Host Inflammatory Responses (NIH P20GM103625). R.C.K. was supported by the NIH (P01HL114471).
REFERENCES
- 1.Oyston PC, Davies C. 2011. Q fever: the neglected biothreat agent. J Med Microbiol 60:9–21. doi: 10.1099/jmm.0.024778-0. [DOI] [PubMed] [Google Scholar]
- 2.Maurin M, Raoult D. 1999. Q fever. Clin Microbiol Rev 12:518–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mazokopakis EE, Karefilakis CM, Starakis IK. 2010. Q fever endocarditis. Infect Disord Drug Targets 10:27–31. doi: 10.2174/187152610790410918. [DOI] [PubMed] [Google Scholar]
- 4.Anderson A, Bijlmer H, Fournier PE, Graves S, Hartzell J, Kersh GJ, Limonard G, Marrie TJ, Massung RF, McQuiston JH, Nicholson WL, Paddock CD, Sexton DJ. 2013. Diagnosis and management of Q fever—United States, 2013: recommendations from CDC and the Q Fever Working Group. MMWR Recommend Rep 62:1–30. [PubMed] [Google Scholar]
- 5.Voth DE, Heinzen RA. 2007. Lounging in a lysosome: the intracellular lifestyle of Coxiella burnetii. Cell Microbiol 9:829–840. doi: 10.1111/j.1462-5822.2007.00901.x. [DOI] [PubMed] [Google Scholar]
- 6.Moos A, Hackstadt T. 1987. Comparative virulence of intra- and interstrain lipopolysaccharide variants of Coxiella burnetii in the guinea pig model. Infect Immun 55:1144–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Narasaki CT, Toman R. 2012. Lipopolysaccharide of Coxiella burnetii. Adv Exp Med Biol 984:65–90. doi: 10.1007/978-94-007-4315-1_4. [DOI] [PubMed] [Google Scholar]
- 8.Shannon JG, Howe D, Heinzen RA. 2005. Virulent Coxiella burnetii does not activate human dendritic cells: role of lipopolysaccharide as a shielding molecule. Proc Natl Acad Sci U S A 102:8722–8727. doi: 10.1073/pnas.0501863102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beare PA, Gilk SD, Larson CL, Hill J, Stead CM, Omsland A, Cockrell DC, Howe D, Voth DE, Heinzen RA. 2011. Dot/Icm type IVB secretion system requirements for Coxiella burnetii growth in human macrophages. mBio 2:e00175-11. doi: 10.1128/mBio.00175-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Winchell CG, Graham JG, Kurten RC, Voth DE. 2014. Coxiella burnetii type IV secretion-dependent recruitment of macrophage autophagosomes. Infect Immun 82:2229–2238. doi: 10.1128/IAI.01236-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klingenbeck L, Eckart RA, Berens C, Luhrmann A. 2013. The Coxiella burnetii type IV secretion system substrate CaeB inhibits intrinsic apoptosis at the mitochondrial level. Cell Microbiol 15:675–687. doi: 10.1111/cmi.12066. [DOI] [PubMed] [Google Scholar]
- 12.Luhrmann A, Nogueira CV, Carey KL, Roy CR. 2010. Inhibition of pathogen-induced apoptosis by a Coxiella burnetii type IV effector protein. Proc Natl Acad Sci U S A 107:18997–19001. doi: 10.1073/pnas.1004380107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Newton HJ, Kohler LJ, McDonough JA, Temoche-Diaz M, Crabill E, Hartland EL, Roy CR. 2014. A screen of Coxiella burnetii mutants reveals important roles for Dot/Icm effectors and host autophagy in vacuole biogenesis. PLoS Pathog 10:e1004286. doi: 10.1371/journal.ppat.1004286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levine B, Kroemer G. 2008. Autophagy in the pathogenesis of disease. Cell 132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klionsky DJ. 2007. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol 8:931–937. doi: 10.1038/nrm2245. [DOI] [PubMed] [Google Scholar]
- 16.Lamark T, Kirkin V, Dikic I, Johansen T. 2009. NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle 8:1986–1990. doi: 10.4161/cc.8.13.8892. [DOI] [PubMed] [Google Scholar]
- 17.Mizushima N, Yoshimori T. 2007. How to interpret LC3 immunoblotting. Autophagy 3:542–545. doi: 10.4161/auto.4600. [DOI] [PubMed] [Google Scholar]
- 18.Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, Bamber BA, Bassham DC, Bergamini E, Bi X, Biard-Piechaczyk M, Blum JS, Bredesen DE, Brodsky JL, Brumell JH, Brunk UT, Bursch W, Camougrand N, Cebollero E, Cecconi F, Chen Y, Chin LS, Choi A, Chu CT, Chung J, Clarke PG, Clark RS, Clarke SG, Clavé C, Cleveland JL, Codogno P, Colombo MI, Coto-Montes A, Cregg JM, Cuervo AM, Debnath J, Demarchi F, Dennis PB, Dennis PA, Deretic V, Devenish RJ, Di Sano F, Dice JF, Difiglia M, Dinesh-Kumar S, Distelhorst CW, Djavaheri-Mergny M, et al. . 2008. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 4:151–175. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, Adhihetty PJ, Adler SG, Agam G, Agarwal R, Aghi MK, Agnello M, Agostinis P, Aguilar PV, Aguirre-Ghiso J, Airoldi EM, Ait-Si-Ali S, Akematsu T, Akporiaye ET, Al-Rubeai M, Albaiceta GM, Albanese C, Albani D, Albert ML, Aldudo J, Algül H, Alirezaei M, Alloza I, Almasan A, Almonte-Beceril M, Alnemri ES, Alonso C, Altan-Bonnet N, Altieri DC, Alvarez S, Alvarez-Erviti L, Alves S, Amadoro G, Amano A, Amantini C, Ambrosio S, Amelio I, Amer AO, Amessou M, Amon A, An Z, et al. . 2016. Guidelines for the use and interpretation of assays for monitoring autophagy. (3rd edition). Autophagy 12:1–222. doi: 10.1080/15548627.2015.1100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Katsuragi Y, Ichimura Y, Komatsu M. 2015. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J 282:4672–4678. doi: 10.1111/febs.13540. [DOI] [PubMed] [Google Scholar]
- 21.Rathinam VA, Vanaja SK, Fitzgerald KA. 2012. Regulation of inflammasome signaling. Nat Immunol 13:333–342. doi: 10.1038/ni.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shi CS, Shenderov K, Huang NN, Kabat J, Abu-Asab M, Fitzgerald KA, Sher A, Kehrl JH. 2012. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol 13:255–263. doi: 10.1038/ni.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, Kim M, Nishito Y, Iemura S, Natsume T, Ueno T, Kominami E, Motohashi H, Tanaka K, Yamamoto M. 2010. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol 12:213–223. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- 24.Ichimura Y, Komatsu M. 2010. Selective degradation of p62 by autophagy. Semin Immunopathol 32:431–436. doi: 10.1007/s00281-010-0220-1. [DOI] [PubMed] [Google Scholar]
- 25.Jiang T, Harder B, Rojo de la Vega M, Wong PK, Chapman E, Zhang DD. 2015. p62 links autophagy and Nrf2 signaling. Free Radic Biol Med 88:199–204. doi: 10.1016/j.freeradbiomed.2015.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ichimura Y, Waguri S, Sou YS, Kageyama S, Hasegawa J, Ishimura R, Saito T, Yang Y, Kouno T, Fukutomi T, Hoshii T, Hirao A, Takagi K, Mizushima T, Motohashi H, Lee MS, Yoshimori T, Tanaka K, Yamamoto M, Komatsu M. 2013. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol Cell 51:618–631. doi: 10.1016/j.molcel.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 27.Graham JG, MacDonald LJ, Hussain SK, Sharma UM, Kurten RC, Voth DE. 2013. Virulent Coxiella burnetii pathotypes productively infect primary human alveolar macrophages. Cell Microbiol 15:1012–1025. doi: 10.1111/cmi.12096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zheng YT, Shahnazari S, Brech A, Lamark T, Johansen T, Brumell JH. 2009. The adaptor protein p62/SQSTM1 targets invading bacteria to the autophagy pathway. J Immunol 183:5909–5916. doi: 10.4049/jimmunol.0900441. [DOI] [PubMed] [Google Scholar]
- 29.Howe D, Shannon JG, Winfree S, Dorward DW, Heinzen RA. 2010. Coxiella burnetii phase I and II variants replicate with similar kinetics in degradative phagolysosome-like compartments of human macrophages. Infect Immun 78:3465–3474. doi: 10.1128/IAI.00406-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lau A, Wang XJ, Zhao F, Villeneuve NF, Wu T, Jiang T, Sun Z, White E, Zhang DD. 2010. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between Keap1 and p62. Mol Cell Biol 30:3275–3285. doi: 10.1128/MCB.00248-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ishimura R, Tanaka K, Komatsu M. 2014. Dissection of the role of p62/SQSTM1 in activation of Nrf2 during xenophagy. FEBS Lett 588:822–828. doi: 10.1016/j.febslet.2014.01.045. [DOI] [PubMed] [Google Scholar]
- 32.Ohtsuka S, Ishii Y, Matsuyama M, Ano S, Morishima Y, Yanagawa T, Warabi E, Hizawa N. 2014. SQSTM1/p62/A170 regulates the severity of Legionella pneumophila pneumonia by modulating inflammasome activity. Eur J Immunol 44:1084–1092. doi: 10.1002/eji.201344091. [DOI] [PubMed] [Google Scholar]
- 33.Mostowy S, Sancho-Shimizu V, Hamon MA, Simeone R, Brosch R, Johansen T, Cossart P. 2011. p62 and NDP52 proteins target intracytosolic Shigella and Listeria to different autophagy pathways. J Biol Chem 286:26987–26995. doi: 10.1074/jbc.M111.223610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Digaleh H, Kiaei M, Khodagholi F. 2013. Nrf2 and Nrf1 signaling and ER stress crosstalk: implication for proteasomal degradation and autophagy. Cell Mol Life Sci 70:4681–4694. doi: 10.1007/s00018-013-1409-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.MacDonald LJ, Graham JG, Kurten RC, Voth DE. 2014. Coxiella burnetii exploits host cAMP-dependent protein kinase signalling to promote macrophage survival. Cell Microbiol 16:146–159. doi: 10.1111/cmi.12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vazquez CL, Colombo MI. 2010. Coxiella burnetii modulates Beclin 1 and Bcl-2, preventing host cell apoptosis to generate a persistent bacterial infection. Cell Death Differ 17:421–438. doi: 10.1038/cdd.2009.129. [DOI] [PubMed] [Google Scholar]
- 37.Voth DE, Heinzen RA. 2009. Sustained activation of Akt and Erk1/2 is required for Coxiella burnetii antiapoptotic activity. Infect Immun 77:205–213. doi: 10.1128/IAI.01124-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Luhrmann A, Roy CR. 2007. Coxiella burnetii inhibits activation of host cell apoptosis through a mechanism that involves preventing cytochrome c release from mitochondria. Infect Immun 75:5282–5289. doi: 10.1128/IAI.00863-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Voth DE, Howe D, Heinzen RA. 2007. Coxiella burnetii inhibits apoptosis in human THP-1 cells and monkey primary alveolar macrophages. Infect Immun 75:4263–4271. doi: 10.1128/IAI.00594-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hill J, Samuel JE. 2011. Coxiella burnetii acid phosphatase inhibits the release of reactive oxygen intermediates in polymorphonuclear leukocytes. Infect Immun 79:414–420. doi: 10.1128/IAI.01011-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gomez JC, Dang H, Martin JR, Doerschuk CM. 2016. Nrf2 modulates host defense during Streptococcus pneumoniae pneumonia in mice. J Immunol 197:2864–2879. doi: 10.4049/jimmunol.1600043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cho HY, Reddy SP, Kleeberger SR. 2006. Nrf2 defends the lung from oxidative stress. Antioxid Redox Signal 8:76–87. doi: 10.1089/ars.2006.8.76. [DOI] [PubMed] [Google Scholar]
- 43.Barker BR, Taxman DJ, Ting JP. 2011. Cross-regulation between the IL-1beta/IL-18 processing inflammasome and other inflammatory cytokines. Curr Opin Immunol 23:591–597. doi: 10.1016/j.coi.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]