

ABSTRACT
High mobility group box 1 (HMGB1) is a non-histone DNA-binding protein that is secreted into the extracellular milieu in response to inflammatory stimuli. The secreted HMGB1 mediates various inflammatory diseases, including periodontitis; however, the underlying mechanisms of HMGB1-induced periodontal inflammation are not completely understood. Here, we examined whether anti-HMGB1 neutralizing antibody inhibits periodontal progression and investigated the molecular pathology of HMGB1 in vitro and in vivo. In vitro analysis indicated that HMGB1, granulocyte-macrophage colony-stimulating factor (GM-CSF), and interleukin-1β (IL-1β) were secreted in response to tumor necrosis factor-α (TNF-α) stimuli in human gingival epithelial cells (HGECs) and human monocytic leukemia cells (THP-1) treated with phorbol myristate acetate. Increased levels of GM-CSF and IL-1β were observed in the conditioned media from TNF-α-stimulated HGECs and THP-1 in vitro. Simultaneous stimulation with TNF-α and anti-HMGB1 antibody significantly decreased TNF-α-induced inflammatory cytokine secretion. Experimental periodontitis was induced in mice using Porphyromonas gingivalis-soaked ligatures. The extracellular translocation was confirmed in gingival epithelia in the periodontitis model mice by immunofluorescence analysis. Systemic administration of anti-HMGB1 neutralizing antibody significantly inhibited translocation of HMGB1. The anti-HMGB1 antibody inhibited periodontal inflammation, expression of IL-1β and C-X-C motif chemokine ligand 1 (CXCL1), migration of neutrophils, and bone resorption, shown by bioluminescence imaging of myeloperoxidase activity, quantitative reverse transcription-PCR (RT-PCR), and micro-computed tomography analysis. These findings indicate that HMGB1 is secreted in response to inflammatory stimuli caused by periodontal infection, which is crucial for the initiation of periodontitis, and the anti-HMGB1 antibody attenuates the secretion of a series of inflammatory cytokines, consequently suppressing the progression of periodontitis.
KEYWORDS: periodontal disease, cytokine, infectious disease, molecular imaging, bone resorption
INTRODUCTION
High mobility group box 1 (HMGB1) is an essential non-histone DNA-binding nuclear protein in eukaryotes. HMGB1 is composed of three domains: two homologous DNA-binding domains, i.e., the A and B boxes, and a negatively charged C-terminal tail (1). HMGB1 contributes to organizing DNA structure and modulates the transcription of various genes, such as those encoding steroid hormone receptors, p53, and nuclear factor-κB (NF-κB), through chromatin remodeling (2). Since HMGB1-deficient mice die within 24 h of birth, this protein is thought to be crucial for survival (3). HMGB1 has been reported to function as a late inflammatory mediator in sepsis (4). HMGB1 is actively secreted by various cells (activated macrophages/monocytes, epithelial cells, and fibroblasts with various histogenic backgrounds) (5). It has been demonstrated that exposure to inflammatory stimuli such as lipopolysaccharides (LPS), tumor necrosis factor-α (TNF-α), or trauma causes the acetylation of lysine residues of HMGB1. Acetylation of HMGB1 results in its cytoplasmic accumulation and release from the cytoplasm to the extracellular compartment (6). Moreover, HMGB1 binds to several receptors, such as Toll-like receptor-2 (TLR-2) and TLR-4 (TLR-2/4) and receptor for advanced glycation end product (RAGE) at the B box domains, and induces a series of inflammatory responses (6). Thus, HMGB1 is now considered a crucial molecule, triggering multiple inflammatory signaling pathways.
HMGB1 has been reported to have a role as an inflammatory mediator not only in acute inflammation such as sepsis (7), traumatic brain injury (8, 9), and brain ischemia (10, 11, 12) but also in chronic and immunologic inflammation such as rheumatoid arthritis (13) and arteriosclerosis (14). Administration of anti-HMGB1 antibody (Ab) has resulted in marked alleviation in few of these diseases. Therefore, blocking of HMGB1 has been examined as a potential therapeutic target for these intractable diseases (8, 12, 15, 16).
Periodontitis, an osteoclastic and chronic inflammatory disease, is caused by immunoreaction to bacterial infection (17). In the initial steps of periodontal disease, gingival epithelial cells defend against bacterial infection and protect tissues by the secretion of various proteins such as antibacterial peptides and cytokines. Secreted interleukin-8 (IL-8), granulocyte macrophage colony-stimulating factor (GM-CSF), and monocyte chemotactic protein (MCP-1) from gingival tissues induce the migration of immune cells into the inflammation sites (18, 19). These immune cells in turn produce various cytokines such as IL-1β, IL-6, and TNF-α and defend against bacterial infection. However, an exaggerated response by the immune system can cause destruction of tissue and bone resorption (17). Recently, several reports have pointed out that HMGB1 is released in the gingival crevicular fluid from periodontitis patients and that TNF-α or butyric acid increases HMGB1 secretion in gingival epithelial cells in vitro (20, 21). LPS and IL-1β increased HMGB1 secretion in periodontal ligament fibroblasts in vitro, and the number of HMGB1-positive cells was increased in a rat periodontitis model (22). Taking the data together, it has been considered that HMGB1 may be associated with the progression of periodontitis. However, the details of the mechanism by which the released HMGB1 could influence the periodontal inflammatory cascade have yet to be elucidated. In this study, we hypothesized that secreted HMGB1 would be a crucial regulator that could initiate and prolong periodontal disease by regulating inflammatory cytokine production, which might be inhibited by administration of anti-HMGB1 neutralizing antibody. To reveal the role of HMGB1 during periodontal inflammation, we investigated the effects of the anti-HMGB1 neutralizing antibody in gingival epithelial cells and macrophages in vitro and in experimental periodontitis in mice.
RESULTS
HMGB1 secretion from THP-1 and HGECs by TNF-α stimuli.
To examine whether human monocytic leukemia cells (THP-1) and human gingival epithelial cells (HGECs) produce HMGB1 after inflammatory stimulation, we measured the amount of HMGB1 sequentially by enzyme-linked immunosorbent assay (ELISA). We used TNF-α as a stimulus for early inflammation and HMGB1 release in vitro because HMGB1 is a late mediator; it acts after bacterial infection or following stimulation with early cytokines such as TNF-α (25, 26). It is the major inflammatory cytokine involved in periodontitis and plays a key role in periodontal tissue breakdown (27, 28). The amount of HMGB1 increased as the experiment proceeded and showed a significant increase after 12 h in THP-1 and 24 h in HGECs. THP-1 released more HMGB1 than HGECs did (Fig. 1A).
FIG 1.

ELISA data showing the secretion of HMGB1 in HGECs and THP-1 stimulated by TNF-α and the effects of anti-HMGB1 antibody on inflammatory cytokines. (A) The supernatants of 10 ng/ml TNF-α-stimulated HGECs and THP-1 were analyzed for secreted HMGB1 using ELISA. Each experiment was performed three times. *, P < 0.05; **, P < 0.01 (one-way ANOVA and Dunnett's test). (B and C) The supernatants of 10 ng/ml TNF-α-stimulated HGECs and THP-1 with or without 50 μg/ml anti-HMGB1 antibody were analyzed by ELISA for levels of released IL-1β in THP-1 (B) and GM-CSF in HGECs (C). Each experiment was performed three times. One-way ANOVA and Tukey-Kramer tests were performed. *, P < 0.05; **, P < 0.01.
Anti-HMGB1 antibody inhibited production of inflammatory cytokines by TNF-α stimuli.
To examine the effects of anti-HMGB1 antibody on TNF-α-mediated inflammatory cytokine profiles, cytokine array analysis was performed using HGECs and THP-1 that were either left untreated or treated with anti-HMGB1 (see Fig. S1 in the supplemental material). On the basis of the array data and of previous studies that have reported cytokine-mediated mechanisms of periodontal inflammation and bone resorption (17, 19), secretion of IL-1β and GM-CSF was further examined by ELISA (Fig. 1B and C). In THP-1, the amount of IL-1β was also significantly increased by TNF-α stimuli. The release of IL-1β into media was decreased by anti-HMGB1 antibody treatment (Fig. 1B). In HGECs, the amount of GM-CSF was significantly increased by TNF-α stimuli and decreased by administration of anti-HMGB1 antibody (Fig. 1C).
Anti-HMGB1 antibody inhibited translocation of HMGB1 in vivo.
To examine the change in HMGB1 localization around the periodontal tissue in a murine periodontitis model, we performed immunofluorescence analysis on day 7. We confirmed the presence of control IgG antibody and anti-HMGB1 antibody in mouse serum (Fig. S2). The results indicated that HMGB1 was localized in the nuclei of gingival epithelia of the shams (Fig. 2E and F) but that the immunoreactivity of nuclear HMGB1 was markedly attenuated in the mice administered control IgG (Fig. 2I and J). The administration of anti-HMGB1 antibody retained the localization of the nuclear HMGB1 in a dose-dependent manner (Fig. 2M, N, Q, and R), indicating that 25 μg of anti-HMGB1 antibody inhibited the translocation of HMGB1 from the nucleus to the cytoplasm. No signal was observed with the control IgG antibody (Fig. S3).
FIG 2.

Immunofluorescence localization of HMGB1 in periodontitis mice. (A) Histological image from a healthy mouse (sham) at day 7 (low magnification). Bar, 500 μm. (B) Enlargement of the section of the image indicated in panel A. D, dentin; GE, gingival epithelium; CT, connective tissue; A, alveolar bone. Bar, 200 μm. We performed each staining at least three times. Images of the gingival junctional epithelium for HE and immunofluorescence of HMGB1 (green) and DAPI (blue) in sham and periodontitis mice are shown as follows: sham, panels C to F; control IgG administration group, panels G to J; anti-HMGB1 antibody administration group, panels K to N (10 μg/mice) and O to R (25 μg/mice). Merged images (F, J, N, and R) indicating colocalization are also shown. Bars, 100 μm (low magnification) (C to O) and 50 μm (high magnification) (D to R).
Anti-HMGB1 antibody inhibited inflammation in periodontal tissue.
Next, molecular imaging analysis was performed to measure myeloperoxidase (MPO) activity to examine the anti-inflammatory effects of the anti-HMGB1 antibody. The average level of total flux was 38.5% higher in the mice examined at 7 days than in the mice examined at 21 days. The average level of maximum radiance was also 82.7% higher in the mice examined at 7 days than in the mice examined at 21 days. Both total flux and maximum radiance in the mice examined at 7 days were significantly decreased by administration of anti-HMGB1 antibody (10 and 25 μg/ml) in a dose-dependent manner (total flux, 45.1% and 29.2%; maximum radiance, 45.7% and 28.2% [for control IgG in the mice examined at 7 days]). The level of total flux in the mice examined at 21 days was not significantly decreased by administration of 10 μg anti-HMGB1 antibody (51.8% of control IgG in the mice examined at 21 days) but was decreased by administration of 25 μg anti-HMGB1 antibody (45.0% of control IgG in the mice examined at 21 days) (Fig. 3I). The maximum radiance in the mice examined at 21 days was significantly decreased in a dose-dependent manner by administration of anti-HMGB1 antibody (57.9% and 49.0% of control IgG in the mice examined at 21 days) (Fig. 3J).
FIG 3.

Molecular imaging analysis examining the effects of anti-HMGB1 antibody on periodontal inflammation. The maxillae were extracted at day 7 (A to D) and day 21 (E to H) after ligature placement. Images of the signal intensity of MPO activity around periodontal tissue are shown as follows: sham, panels A and E; control IgG administration group, panels B and F; anti-HMGB1 antibody administration group, panels C and G (10 μg/mice) and D and H (25 μg/mice). (I and J) Results of comparisons of the levels of signal intensity with respect to total flux (I) and maximum radiance (J) are shown. We performed each experiment four times (A and C to F) or five times (B). One-way ANOVA and Tukey-Kramer tests were performed. *, P < 0.05; **, P < 0.01.
Anti-HMGB1 antibody inhibited neutrophil recruitment in periodontal tissue.
Immunohistochemistry was performed to examine the neutrophil recruitment in periodontal tissue, because MPO is highly activated during neutrophil phagocytosis. In sham samples, neutrophils localized in junctional epithelial cells but did not do so as much in connective tissue (Fig. 4B). In periodontitis tissue, abundant neutrophils localized in connective tissue in control samples and in samples from mice administered monoclonal antibody (MAb) at 10 μg (Fig. 4D, F, and I). Neutrophil recruitment was significantly inhibited in samples from mice administered MAb at 25 μg compared to control IgG-administered samples (Fig. 4H and I).
FIG 4.
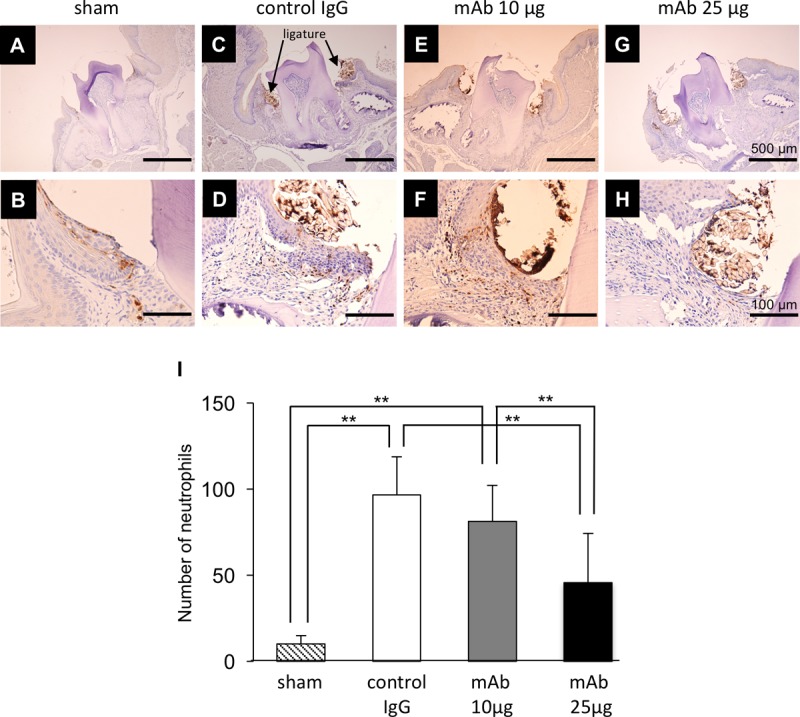
Immunostaining localization of neutrophils in periodontitis mice. Data represent immunostaining localization of neutrophils in sham and periodontitis mice after ligature placement at day 7. Typical images of gingival junctional epithelium are shown as follows: sham, panels A and B; control IgG administration group, panels C and D; anti-HMGB1 antibody administration group, panels E and F (10 μg/mice) and G and H (25 μg/mice). Bars, 500 μm (low magnification) (A, C, E, and G) and 100 μm (high magnification) (B, D, F, and H). (I) Numbers of neutrophils that migrated in gingival junctional epithelium within 200 μm2 are indicated. We performed each experiment three times. One-way ANOVA and Tukey-Kramer tests were performed. **, P < 0.01.
Anti-HMGB1 antibody inhibited alveolar bone resorption.
To examine whether the anti-HMGB1 antibody actually inhibits bone resorption, we examined bone volume after 7 days and 14 days by micro-computed tomography analysis. The computed tomography images taken at day 21 indicated that the bone volumes of treated mice were decreased compared to those of the shams (Fig. 5A to H). Quantitation of the images demonstrated that there was no obvious change of bone volume between all groups at day 7. At day 21, the bone volume of mice with control IgG was significantly decreased compared to that of shams. However, bone resorption was significantly inhibited by administration of anti-HMGB1 antibody in a dose-dependent manner (Fig. 5J).
FIG 5.
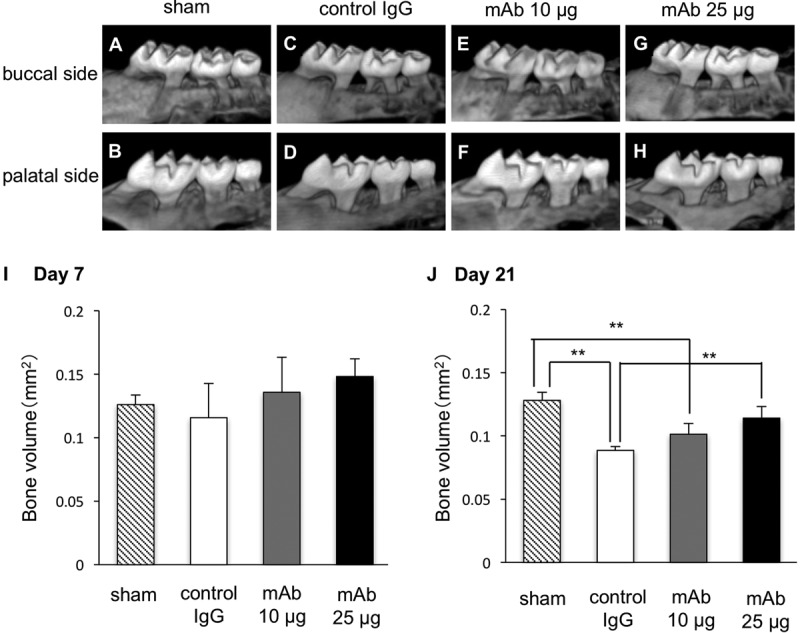
Computed tomography scan analysis examining the effects of anti-HMGB1 antibody on bone resorption. We captured images focusing on the buccal sides (A, C, E, and G) and palatal sides (B, D, F, and H) of periodontal tissue extracted at day 21 after ligature placement. (I and J) Bone volume ratios (test side/control side) were analyzed at day 7 (I) and day 21 (J). We performed each experiment three times (A, B, E, and F) or four times (C, D, G, and H). One-way ANOVA and Tukey-Kramer tests were performed. *, P < 0.05; **, P < 0.01.
Gene expression changes resulting from anti-HMGB1 antibody treatment in periodontal tissue.
We examined the changes in gene expression around the periodontal tissue on day 21 to confirm the in vitro results described above (Fig. 1). The gene expression level of GM-CSF was significantly decreased in samples from mice administered control IgG and was increased in samples from mice administered MAb at 25 μg compared to sham treatment results (Fig. 6A). The gene expression level of IL-1β was significantly increased in samples from mice administered control IgG compared to sham treatment. It was decreased in samples from mice administered MAb at 25 μg compared to control samples, but the difference was not significant (Fig. 6B). The gene expression level of CXCL-1 was increased in samples from mice administered control IgG and decreased in samples from mice administered MAb at 25 μg compared to sham treatment, but the difference was not significant (Fig. 6C).
FIG 6.

Quantitative RT-PCR showing the effects of anti-HMGB1 antibody on inflammatory cytokines. The mRNA expression levels seen with periodontal mice at day 7 were analyzed for GM-CSF (A), IL-1β (B), and CXCL1 (C) by quantitative RT-PCR. The abundance of each gene was determined relative to that of GAPDH mRNA, and the results are shown as fold induction data. We performed each experiment three times. One-way ANOVA and Dunnett's tests were performed. *, P < 0.05; **, P < 0.01.
DISCUSSION
Inflammatory cytokines and bacterial components such as LPS cause HMGB1 to be translocated from the nucleus to the cytoplasm, followed by extracellular secretion, which is crucial for the progression of inflammation (29). The present study demonstrated that the HGECs were capable of secreting HMGB1 as a result of TNF-α stimulation in vitro, and translocation of HMGB1 as a result of stimulation with Porphyromonas gingivalis-soaked ligatures was observed in murine gingival epithelial cells. Although the secretion of HMGB1 from HGECs was significantly lower than that of HMGB1 from THP-1, HGECs appear to play critical roles during periodontal inflammation as the first defense against bacterial invasion and as the initiator of essential immune responses by secreting HMGB1 and GM-CSF.
The murine periodontitis tissue exhibited strong MPO activity with no detectable bone resorption at day 7 (data not shown), while moderate MPO activity with obvious bone resorption was observed at day 21. The alteration of MPO activity is in accordance with the normal innate immune response. In early inflammation (equivalent to day 7), MPO is highly activated during neutrophil phagocytosis for killing invaded bacteria. Actually, recruitment of neutrophils in the periodontal tissue was associated with MPO activity. As inflammation proceeds (equivalent to day 21), the transition from neutrophils to monocytes shifts inflammation from acute to chronic and differentiation of macrophages and activation of osteoclasts are promoted (30). In this study, anti-HMGB1 antibody significantly inhibited early and late MPO activation. Moreover, the anti-HMGB1 antibody inhibited the translocation of HMGB1 from the nucleus to cytoplasm or extracellular space on day 7 in vivo. A previous study (27) and our in vitro data (Fig. 1; see also Fig. S4 in the supplemental material) indicated that the inflammatory stimulus caused HMGB1 transportation from the nucleus to the cytoplasm or extracellular space and led to HMGB1 secretion in these cells. Although we did not measure the level of HMGB1 secreted from gingival epithelial cells and macrophages in vivo, it is suggested that HMGB1 might be secreted into the extracellular space due to periodontitis, because the absence of HMGB1 in the nucleus and anti-HMGB1 antibody administration inhibited the transportation and the secretion of HMGB1. It was also suggested that the released HMGB1 might play crucial roles in the initiation of inflammation through the recruitment and activation of neutrophils and in prolonging inflammation, thus resulting in bone resorption.
The antibody array and ELISA were conducted to examine the biological significance of the released HMGB1. In HGECs, GM-CSF levels were remarkably increased by TNF-α stimuli and decreased by anti-HMGB1 antibody administration. GM-CSF can induce the growth and differentiation of granulocyte and monocyte progenitors (18), which may contribute to immunostimulation in inflamed periodontal tissue. In THP-1, IL-1β levels were also increased by TNF-α stimuli and decreased by anti-HMGB1 antibody treatment. IL-1β is considered an osteoclastogenic cytokine (31, 32). It induces differentiation and activation for mature osteoclasts (33). Anti-HMGB1 antibody significantly inhibited the secretion of the inflammatory cytokines that we examined, although the level was not sufficient for complete inhibition. Therefore, even in the presence of a higher concentration of anti-HMGB1 antibody, alveolar bone resorption was observed at only low levels. Alternative signaling pathways may exist that partially maintain an active state of the inflammatory cytokines in addition to GM-CSF and IL-1β. The additional molecular mechanisms underlying HMGB1-induced periodontal inflammation should be examined in future studies. To confirm the in vitro results, we examined gene expression changes caused by anti-HMGB1 administration in periodontal tissue in vivo. The reason that the results of the in vitro and in vivo GM-CSF experiments were not similar is that the extracted maxilla contained various tissues such as epithelial cells, connective tissue, immune cells, and bone. The levels of expression of IL-1β in the in vitro and in vivo assays were similar but not significant. We should have chosen other experiments such as in situ hybridization or laser microdissection to extract precise tissue, because periodontal tissue is complex and contains various cell types. The chemokine CXCL1, also named GRO-alpha, is a strong chemoattractant for neutrophils and was previously reported to be induced by LPS in oral fibroblasts (34). In our study, neutrophils localized in gingival fibroblasts and were inhibited by high MPO activity in periodontitis tissue and by administration of the anti-HMGB1 antibody. Although the differences were not significant, the levels of CXCL1 gene expression observed in vivo support our hypothesis.
HMGB1 can bind to TLR-2/4 and RAGE as a cytokine mediator (6). On the basis of all observations in this study, we propose that the autocrine release of HMGB1 plays critical roles in regulating periodontal inflammation. In early inflammation, gingival epithelial cells release various cytokines and chemokines, and HMGB1 is then translocated from the nucleus to the cytoplasm by TNF-α stimuli. The released HMGB1 induces the translocation in an autocrine-related manner; the released HMGB1 also induces GM-CSF secretion from gingival epithelial cells, resulting in differentiation and activation of immune cells. As inflammation proceeds via the continuous secretion of HMGB1, macrophages release more cytokines, chemokines, and HMGB1. Moreover, the released IL-1β promotes osteoclastogenesis and bone resorption. Therefore, periodontal inflammation is initiated, exacerbated, and prolonged by the HMGB1 secretion cycle. The present study demonstrated that anti-HMGB1 antibody succeeded in preventing prolonged immunostimulation and the bone-resorbing activity of osteoclasts by inhibiting the foregoing cytokine release in periodontal tissue.
In conclusion, the results reported here indicate that anti-HMGB1 antibody inhibited not only inflammation but also bone resorption in a murine periodontitis model. Autocrine HMGB1-regulating secretion of inflammatory cytokines, such as IL-1β, GM-CSF, and CXCL1, was involved in the mechanism in the early and late phases of the inflammation. Therefore, HMGB1 is a crucial regulator of the initiation and prolongation of periodontitis. Understanding the detailed molecular mechanism of HMGB1 is required to control periodontal inflammation.
MATERIALS AND METHODS
Reagents.
Recombinant human TNF-α (rhTNF-α) was purchased from R&D Systems (Minneapolis, MN). Rat monoclonal antibody (MAb) against HMGB1 (antibody no. 10-22, subclass IgG2a; antibody no. 4-1, subclass IgG2a) and class-matched control IgG MAb (rat subclass IgG2a; anti-keyhole limpet hemocyanin) were used as described previously (11). Anti-HMGB1 antibody no. 4-1 recognizes the B box, and the no. 10-22 antibody recognizes the C-terminal tail of HMGB1.
Cell culture.
Pooled progenitor human gingival epithelium cells (HGECs) were maintained in CnT-Prime epithelial cell culture medium (obtained from CELLnTEC Advanced Cell Systems, Bern, Switzerland). Accutase cell detachment solution (CELLnTEC) was used for detachment of the cells. HGECs were used for the experiments performed between passages 5 and 10. A human monocytic leukemia cell line (THP-1) was obtained from ATCC (Manassas, VA) and maintained in RPMI 1640 medium supplemented with 1% l-glutamine, 1 mM sodium pyruvate, 10% fetal bovine serum, and 10 mM HEPES, all of which were purchased from Thermo Fisher Scientific, Waltham, MA, at 37°C in a humidified 5% CO2 atmosphere. THP-1 were differentiated into macrophage-like cells following exposure to 100 nM phorbol myristate acetate (PMA) (Sigma-Aldrich, St. Louis, MO) for 48 h.
ELISA.
The amounts of HMGB1 protein released into the supernatant of both HGECs and THP-1 were measured using human HMGB1 ELISA kit II (Shinotest, Tokyo, Japan). The supernatant was harvested up to 48 h after exposure to TNF-α stimuli. The amount of released IL-1β and GM-CSF proteins in the supernatant of both HGECs and THP-1 was measured using a human ELISA kit (R&D Systems). The supernatant was harvested 48 h after the addition of TNF-α and anti-HMGB1 antibody. The absorbance at 490 nm was measured using a microplate reader (GeminiXPS; Molecular Devices, Sunnyvale, CA).
Induction of periodontitis. (i) Bacteria.
Porphyromonas gingivalis strain W83 (P. gingivalis) was grown in an anaerobic chamber (Mitsubishi Gas Company, Inc., Tokyo, Japan) using modified GAM broth (Nissui Pharmaceutical Co., Hiroshima, Japan). After incubation at 37°C under anaerobic conditions, the bacteria were harvested and washed three times with sterile phosphate-buffered saline (PBS). P. gingivalis was resuspended in PBS at 7.5 × 107 CFU/ml.
(ii) Mice.
All experiments involving studies performed with animals were approved by the Animal Care and Use Committee, Okayama University. Ten-week-old specific-pathogen-free (SPF) female C57BL/6J mice were obtained from Charles River Laboratories Japan (Kanagawa, Japan). Experimental periodontitis was induced as described previously (22) with slight modifications. Briefly, a 6-0 silk thread was used to ligate around the cervical portion of the maxillary second molar without damaging gingiva. P. gingivalis bacteria were infiltrated to the silk thread every 3 days. We assigned mice to 4 groups: (i) sham (mice without any treatment); (ii) control IgG (experimental periodontitis with administration of control IgG) (10 μg/mice); (iii) 10 μg MAb (experimental periodontitis with administration of anti-HMGB1 antibody at 10 μg/mice); (iv) 25 μg MAb (experimental periodontitis with administration of anti-HMGB1 antibody at 25 μg/mice). The antibody was administered by peritoneal injection every 3 days. The analysis that followed was performed at day 7 and day 21 after ligature placement.
In vivo imaging.
To measure myeloperoxidase (MPO) activity, a XenoLight RediJect inflammation probe (PerkinElmer, Waltham, MA) was administered at 150 μl/mice, and mice were sacrificed immediately after injection. To eliminate errors in measurement due to positional effects of the specimen, the dissected maxillae were trimmed to the same size and thickness. After verifying that the wavelengths from specimens positioned on a plate and from the emission filters of the device were almost the same across all samples (23), luminescent images were taken using a charge-coupled-device (CCD) camera within 20 min of injection. Luminescence intensity was measured using IVIS Spectrum (PerkinElmer), and a circular region of interest (ROI) was defined as a region which exhibited more than 50% of maximum luminescence in the inflammatory site of each mouse. The total flux (measured in photons per second) and maximum radiance (measured in photons per second per square centimeter per steradian) in the ROI were quantified using Living Image Software V4.4 (PerkinElmer) according to the manufacturer's instructions.
Micro-computed tomography scanning.
After in vivo imaging, the alveolar bone was fixed with 4% paraformaldehyde (pH 7.4; Wako Pure Chemical Industries, Ltd., Osaka, Japan) for 24 h and scanned using a desktop non-computed tomography system (Sky scan1174; Bruker Corporation, Billerica, MA).
The computed tomography settings were as follows: voltage, 50 mV; current, 800 kA; slice thickness, 7.5 μm. The images were reconstructed using NRecon and CTVol software (Bruker). The buccal alveolar bone volume from the mesial contact point to the distal contact point of the second molar was calculated using CTAn software (Bruker) according to the manufacturer's instructions.
Histological and immunofluorescence staining.
Paraformaldehyde-fixed tissue samples were decalcified for 1 week with 10% EDTA disodium salt dehydrate (Sigma-Aldrich) and embedded in paraffin wax. Coronal serial sections (3 μm thick) were mounted in serial order on poly-l-lysine-coated slides (Sakura Finetek Japan, Tokyo, Japan) and stained with hematoxylin and eosin (HE). Immunofluorescence staining was performed following standard procedures. Briefly, slides were boiled in 1 mM EDTA–PBS (pH 8.0) for 3 min using a microwave for antigen unmasking, and immunofluorescence staining was performed with a primary antibody against HMGB1 (rabbit polyclonal IgG; Shinotest) (1:100). The secondary antibody was Alexa Fluor 488 goat anti-rabbit IgG (Thermo Fisher Scientific) (1:100). Vectashield mounting medium with DAPI (4′,6-diamidino-2-phenylindole; Vector Laboratories, Burlingame, CA) was used for nuclear staining. We used three mice for each experiment.
Immunohistochemistry.
An avidin-biotin complex (ABC) system (Vectastain Elite ABC rat kit; Vector Laboratories, Burlingame, CA, USA) was used for immunohistochemical analysis along with the primary antibodies for Ly-6G (rat polyclonal IgG; BD Pharmingen, NJ) (1:100). The numbers of Ly-6G positive cells within the range of 20 μm2 from the clavicular epithelium were counted in at least 3 sections from each mouse.
Quantitative RT-PCR.
Total RNA was collected from maxillary gingiva using a Lysing Matrix F kit (MP Biomedicals, CA) and an RNeasy Plus kit (Qiagen, Hilden, Germany), and the reverse transcription reaction was carried out using 1 μg mRNA and 10 μM deoxynucleoside triphosphate (dNTP) mix, 50 μM oligo(dT)12–18 primer, 5× First Standard buffer, 0.1 M dithiothreitol, and SuperScript III reverse transcriptase according to the manufacturer's protocols (all were purchased from Thermo Fisher Scientific).
Primers were designed using Primer3 online software (http://frodo.wi.mit.edu/) as follows: for IL-1β, 5′-GTCGCTCAGGGTCACAAGAA-3′ and 5′-CCACACGTTGACAGCTAGGT-3′; for GM-CSF, 5′-GGCCAAAAATGAMGGAAGCCC-3′ and 5′-TGTGCCACATCTCTTGGTCC-3′; for CXCL1, 5′-GGTGTCCCCAAGTAACGGAG-3′ and 5′-TTGTCAGAAGCCAGCGTTCA-3′; and for GAPDH (glyceraldehyde-3-phosphate dehydrogenase), 5′-GAGTCAACGGATTTGGTCGT-3′ and 5′-GACAAGCTTCCCGTTCTCAG-3′. Quantitative RT-PCR was performed using an ABI 7300 system (Applied Biosystems, Foster City, CA) under conditions of 95°C for 10 min followed by 40 cycles at 95°C for 15 s and 60°C for 1 min in 96-well plates in a final volume of 20 μl containing SYBR green PCR master mix (Applied Biosystems). The relative quantities of transcripts were calculated by the comparative threshold cycle (ΔΔCT) method (24) using GAPDH as the endogenous control. The relative mRNA expression ratio was calculated at each time point as a ratio of stimulated results/nonstimulated results.
Statistical analysis.
Data are presented as means ± standard deviations (SD) of the results from at least three independent experiments. Statistical analyses were performed using one-way analysis of variance (ANOVA). The Tukey-Kramer test was used to determine statistical significance when multiple non-pairwise comparisons were involved. The Dunnett's test was used when multiple pairwise comparisons were involved. Values of P of <0.05 were considered significant and were determined using JMP software (version 12.0; SAS, Cary, NC).
Supplementary Material
ACKNOWLEDGMENTS
We thank all staff in the Okayama Medical Innovation Center and Public Laboratory at the Okayama University Graduate School of Medicine, Dentistry and Pharmaceutical Sciences for supporting the study on molecular imaging. We also thank the Department of Oral Rehabilitation and Regenerative Medicine at Okayama University Graduate School of Medicine, Dentistry and Pharmaceutical Sciences for supporting the study on non-computed tomography scanning.
This work was supported by research funds from a JSPS KAKENHI Grant-in-Aid for Young Scientists (B) (JP 24792327), the Kobayashi Magobei Memorial Medical Foundation, and the Ryobi-Teien Foundation, by a Grant-in-Aid for the COE projects by MEXT, Japan, entitled “Center of excellence for molecular and gene targeting therapies with micro-dose molecular imaging modalities,” and by a Translational Research Network Program (no. H27 seeds B-8-1) from Japan AMED. We declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00111-18.
REFERENCES
- 1.Bianchi ME, Beltrame M. 2000. Upwardly mobile proteins. Workshop: the role of HMG proteins in chromatin structure, gene expression and neoplasia. EMBO Rep 1:109–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Müller S, Ronfani L, Bianchi ME. 2004. Regulated expression and subcellular localization of HMGB1, a chromatin protein with a cytokine function. J Intern Med 255:332–343. doi: 10.1111/j.1365-2796.2003.01296.x. [DOI] [PubMed] [Google Scholar]
- 3.Calogero S, Grassi F, Aguzzi A, Voigtländer T, Ferrier P, Ferrari S, Bianchi ME. 1999. The lack of chromosomal protein Hmg1 does not disrupt cell growth but causes lethal hypoglycaemia in newborn mice. Nat Genet 22:276–280. doi: 10.1038/10338. [DOI] [PubMed] [Google Scholar]
- 4.Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L, Manogue KR, Faist E, Abraham E, Andersson J, Andersson U, Molina PE, Abumrad NN, Sama A, Tracey KJ. 1999. HMG-1 as a late mediator of endotoxin lethality in mice. Science 285:248–251. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- 5.Yang H, Wang H, Czura CJ, Tracey KJ. 2005. The cytokine activity of HMGB1. J Leukoc Biol 78:1–8. doi: 10.1189/jlb.1104648. [DOI] [PubMed] [Google Scholar]
- 6.Andersson U, Antoine DJ, Tracey KJ. 2014. The functions of HMGB1 depend on molecular localization and post-translational modifications. J Intern Med 276:420–424. doi: 10.1111/joim.12309. [DOI] [PubMed] [Google Scholar]
- 7.Wang H, Yang H, Tracey KJ. 2004. Extracellular role of HMGB1 in inflammation and sepsis. J Intern Med 255:320–331. doi: 10.1111/j.1365-2796.2003.01302.x. [DOI] [PubMed] [Google Scholar]
- 8.Okuma Y, Liu K, Wake H, Zhang J, Maruo T, Date I, Yoshino T, Ohtsuka A, Otani N, Tomura S, Shima K, Yamamoto Y, Yamamoto H, Takahashi HK, Mori S, Nishibori M. 2012. Anti-high mobility group box-1 antibody therapy for traumatic brain injury. Ann Neurol 72:373–384. doi: 10.1002/ana.23602. [DOI] [PubMed] [Google Scholar]
- 9.Okuma Y, Liu K, Wake H, Liu R, Nishimura Y, Hui Z, Teshigawara K, Haruma J, Yamamoto Y, Yamamoto H, Date I, Takahashi HK, Mori S, Nishibori M. 2014. Glycyrrhizin inhibits traumatic brain injury by reducing HMGB1-RAGE interaction. Neuropharmacology 85:18–26. doi: 10.1016/j.neuropharm.2014.05.007. [DOI] [PubMed] [Google Scholar]
- 10.Kim JB, Sig Choi J, Yu YM, Nam K, Piao CS, Kim SW, Lee MH, Han PL, Park JS, Lee JK. 2006. HMGB1, a novel cytokine-like mediator linking acute neuronal death and delayed neuroinflammation in the postischemic brain. J Neurosci 26:6413–6421. doi: 10.1523/JNEUROSCI.3815-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu K, Mori S, Takahashi HK, Tomono Y, Wake H, Kanke T, Sato Y, Hiraga N, Adachi N, Yoshino T, Nishibori M. 2007. Anti-high mobility group box 1 monoclonal antibody ameliorates brain infarction induced by transient ischemia in rats. FASEB J 21:3904–3916. doi: 10.1096/fj.07-8770com. [DOI] [PubMed] [Google Scholar]
- 12.Zhang J, Takahashi HK, Liu K, Wake H, Liu R, Maruo T, Date I, Yoshino T, Ohtsuka A, Mori S, Nishibori M. 2011. Anti-high mobility group box-1 monoclonal antibody protects the blood-brain barrier from ischemia-induced disruption In rats. Stroke 42:1420–1428. doi: 10.1161/STROKEAHA.110.598334. [DOI] [PubMed] [Google Scholar]
- 13.Andersson U, Erlandsson-Harris H. 2004. HMGB1 is a potent trigger of arthritis. J Intern Med 255:344–350. doi: 10.1111/j.1365-2796.2003.01303.x. [DOI] [PubMed] [Google Scholar]
- 14.Kalinina N, Agrotis A, Antropova Y, DiVitto G, Kanellakis P, Kostolias G, Ilyinskaya O, Tararak E, Bobik A. 2004. Increased expression of the DNA-binding cytokine HMGB1 in human atherosclerotic lesions: role of activated macrophages and cytokines. Arterioscler Thromb Vasc Biol 24:2320–2325. doi: 10.1161/01.ATV.0000145573.36113.8a. [DOI] [PubMed] [Google Scholar]
- 15.Suda K, Takeuchi H, Ishizaka A, Kitagawa Y. 2010. High-mobility-group box chromosomal protein 1 as a new target for modulating stress response. Surg Today 40:592–601. doi: 10.1007/s00595-009-4232-1. [DOI] [PubMed] [Google Scholar]
- 16.Kanellakis P, Agrotis A, Kyaw TS, Koulis C, Ahrens I, Mori S, Takahashi HK, Liu K, Peter K, Nishibori M, Bobik A. 2011. High-mobility group box protein 1 neutralization reduces development of diet-induced atherosclerosis in apolipoprotein e-deficient mice. Arterioscler Thromb Vasc Biol 31:313–319. doi: 10.1161/ATVBAHA.110.218669. [DOI] [PubMed] [Google Scholar]
- 17.Cekici A, Kantarci A, Hasturk H, Van Dyke TE. 2014. Inflammatory and immune pathways in the pathogenesis of periodontal disease. Periodontol 2000 64:57–80. doi: 10.1111/prd.12002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.al-Shami A, Bourgoin SG, Naccache PH. 1997. Granulocyte-macrophage colony-stimulating factor-activated signaling pathways in human neutrophils. I. Tyrosine phosphorylation-dependent stimulation of phosphatidylinositol 3-kinase and inhibition by phorbol esters. Blood 89:1035–1044. [PubMed] [Google Scholar]
- 19.Research, Science, and Therapy Committee of The American Academy of Periodontology. 1999. The pathogenesis of periodontal diseases. J Periodontol 70:457–470. doi: 10.1902/jop.1999.70.4.457. [DOI] [PubMed] [Google Scholar]
- 20.Morimoto Y, Kawahara KI, Tancharoen S, Kikuchi K, Matsuyama T, Hashiguchi T, Izumi Y, Maruyama I. 2008. Tumor necrosis factor-α stimulates gingival epithelial cells to release high mobility-group box 1. J Periodont Res 43:76–83. doi: 10.1111/j.1600-0765.2007.00996.x. [DOI] [PubMed] [Google Scholar]
- 21.Ebe N, Hara-Yokoyama M, Iwasaki K, Iseki S, Okuhara S, Podyma-Inoue KA, Terasawa K, Watanabe A, Akizuki T, Watanabe H, Yanagishita M, Izumi Y. 2011. Pocket epithelium in the pathological setting for HMGB1 release. J Dent Res 90:235–240. doi: 10.1177/0022034510385688. [DOI] [PubMed] [Google Scholar]
- 22.Nogueira AV, de Souza JA, de Molon RS, Mariano Pereira EDS, de Aquino SG, Giannobile WV, Cirelli JA. 2014. HMGB1 localization during experimental periodontitis. Mediators Inflamm 2014:816320. doi: 10.1155/2014/816320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tseng JC, Vasquez K, Peterson JD. 2015. Optical imaging on the IVIS SpectrumCT system: general and technical considerations for 2D and 3D imaging. Technical note, pre-clinical in vivo imaging. PerkinElmer, Inc, Waltham, MA. [Google Scholar]
- 24.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 25.Willenbrock S, Braun O, Baumgart J, Lange S, Junghanss C, Heisterkamp A, Nolte I, Bullerdiek J, Murua Escobar H. 2012. TNF-α induced secretion of HMGB1 from non-immune canine mammary epithelial cells (MTH53A). Cytokine 57:210–220. doi: 10.1016/j.cyto.2011.11.011. [DOI] [PubMed] [Google Scholar]
- 26.Page RC. 1991. The role of inflammatory mediators in the pathogenesis of periodontal disease. J Periodont Res 26:230–242. doi: 10.1111/j.1600-0765.1991.tb01649.x. [DOI] [PubMed] [Google Scholar]
- 27.Andersson U, Tracey KJ. 2011. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol 29:139–162. doi: 10.1146/annurev-immunol-030409-101323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Abe T, Hajishengallis G. 2013. Optimization of the ligature-induced periodontitis model in mice. J Immunol Methods 394:49–54. doi: 10.1016/j.jim.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kang R, Chen R, Zhang Q, Hou W, Wu S, Cao L, Huang J, Yu Y, Fan XG, Yan Z, Sun X, Wang H, Wang Q, Tsung A, Billiar TR, Zeh HJ, Lotze MT, Tang D. 2014. HMGB1 in health and disease. Mol Aspects Med 40:1–116. doi: 10.1016/j.mam.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Knorr M, Münzel T, Wenzel P. 2014. Interplay of NK cells and monocytes in vascular inflammation and myocardial infarction. Front Physiol 5:295. doi: 10.3389/fphys.2014.00295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hofbauer LC, Lacey DL, Dunstan CR, Spelsberg TC, Riggs BL, Khosla S. 1999. Interleukin-1beta and tumor necrosis factor-alpha, but not interleukin-6, stimulate osteoprotegerin ligand gene expression in human osteoblastic cells. Bone 25:255–259. doi: 10.1016/S8756-3282(99)00162-3. [DOI] [PubMed] [Google Scholar]
- 32.Zupan J, Jeras M, Marc J. 2013. Osteoimmunology and the influence of pro-inflammatory cytokines on osteoclasts. Biochem Med (Zagreb) 23:43–63. doi: 10.11613/BM.2013.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jimi E, Nakamura I, Duong LT, Ikebe T, Takahashi N, Rodan GA, Suda T. 1999. Interleukin 1 induces multinucleation and bone-resorbing activity of osteoclasts in the absence of osteoblasts/stromal cells. Exp Cell Res 247:84–93. doi: 10.1006/excr.1998.4320. [DOI] [PubMed] [Google Scholar]
- 34.Jönsson D, Amisten S, Bratthall G, Holm A, Nilsson BO. 2009. LPS induces GROalpha chemokine production via NF-kappaB in oral fibroblasts. Inflamm Res 58:791–796. doi: 10.1007/s00011-009-0049-z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.