

ABSTRACT
Retapamulin, a semisynthetic pleuromutilin derivative, is exclusively used for the topical short-term medication of impetigo and staphylococcal infections. In the present study, we report that retapamulin is adequately and rapidly metabolized in vitro via various metabolic pathways, such as hydroxylation, including mono-, di-, and trihydroxylation, and demethylation. Like tiamulin and valnemulin, the major metabolic routes of retapamulin were hydroxylation at the 2β and 8α positions of the mutilin moiety. Moreover, in vivo metabolism concurred with the results of the in vitro assays. Additionally, we observed significant interspecies differences in the metabolism of retapamulin. Until now, modifying the side chain was the mainstream method for new drug discovery of the pleuromutilins. This approach, however, could not resolve the low bioavailability and short efficacy of the drugs. Considering the rapid metabolism of the pleuromutilins mediated by cytochrome P450 enzymes, we propose that blocking the active metabolic site (C-2 and C-8 motif) or administering the drug in combination with cytochrome P450 enzyme inhibitors is a promising pathway in the development of novel pleuromutilin drugs with slow metabolism and long efficacy.
KEYWORDS: drug discovery and development, high-resolution mass spectrometry, metabolism, retapamulin
INTRODUCTION
Pleuromutilin, first discovered in 1951, exhibits modest in vitro activity against Gram-positive pathogens and mycoplasma (1). Mycoplasma infections have raised great concerns due to their high morbidity, mortality, and multidrug resistance (MDR) because of the bacterium's unique physical character, such as the absence of a cell wall (2, 3). Additionally, Staphylococcus aureus and Streptococcus pyogenes are key pathogens causing skin and skin structure infections (4, 5) and have also been reported to have acquired MDR. The emergence and prevalence of MDR have led to the reduction or even the loss of antibiotic efficacy, which definitely poses a serious threat to animal and even human health (6, 7). Antibiotics with novel modes of action are thus needed to address the growing problem of MDR.
Pleuromutilins have no target-specific cross-resistance to other antibacterial agents, which makes them appealing for human clinical development. Pleuromutilins possess a mirror image-like stereochemistry and have a unique mode of action (8, 9). They exert activity by binding to domain V of the 23S rRNA prokaryotic ribosome, whereas the peptidyltransferase cavity and the complex mode of action are responsible for the lack of cross-resistance (9, 10). The chemical structures of the pleuromutilins are illustrated in Fig. S1 in the supplemental material.
The antibacterial activity of pleuromutilins has been demonstrated to be markedly enhanced if the glycolic acid side chain at the C-14 position is replaced by other acyl residues (i.e., basic thioether side chains) (11, 12). Consequently, a large number of pleuromutilin derivatives exhibiting considerable antimicrobial activity have been synthesized and studied. For instance, tiamulin (TIA) and valnemulin (VLM) were successfully developed as therapeutic agents to treat enteric diseases in pigs and enzootic pneumonia in pigs and poultry (13–15). Moreover, VLM has also been used to treat immunocompromised patients with resistant mycoplasma infections (16). However, azamulin, another pleuromutilin derivative, displayed poor oral absorption due to its poor solubility in water and short half-life due to rapid metabolism and subsequent excretion (17). These poor pharmacokinetic (PK) characteristics are the bane of the pleuromutilin class, and efforts to overcome these demerits have been largely fruitless. Retapamulin (RTM) became the first approved pleuromutilin derivative for human use since 2007 (18). However, RTM is limited to topical treatment of impetigo and secondarily infected traumatic lesions.
Structure-activity relationship (SAR) studies indicated that the presence of a thioether group at the C-22 position and modifications at the C-14 side chain of pleuromutilin could alter the antibacterial activity of pleuromutilins (19, 20). However, a paramount requirement for novel drug development is the identification of principal routes of elimination and metabolism and the mechanism of drug-drug interactions, which is expensive, demanding, and laborious. This hurdle could be a reason contributing to the very slow development of pleuromutilin drugs and the approval of RTM for human use. RTM is not even available in any systemic dosage form. To date, only a few derivatives (such as valnemulin and tiamulin) have been successfully translated into clinical practice.
Acquiring metabolic information is critical in the process of drug discovery. According to the European Medicines Agency (EMA), pleuromutilin incubated with hepatic cytochrome P450 (CYP) enzymes is prone to be extensively hydroxylated at the 1β, 2β, and 8α positions (21). Recently, our research team has conducted many studies on the pharmacokinetics (22) and in vitro and in vivo metabolism of VLM and TIA (23, 24). However, until now, metabolic information about RTM has remained scarce. Therefore, the aim of present study was to comprehensively unravel the metabolic routes of RTM, providing indispensable information for new drug development of the pleuromutilins.
RESULTS
Fragmentation behavior of RTM.
RTM was found to ionize more efficiently in the positive mode than in the negative mode, generating a pseudomolecular precursor ion at m/z 518.3303. The first step in the collision-induced dissociation (CID) of RTM is the cleavage of the bond at the C-8 position, resulting in the generation of m/z 216.1054 (side chain) and m/z 303.2326 (mutilin moiety) product ions, wherein m/z 216.1054 is the most dominant product ion (Fig. 1). Further fragmentation of m/z 216.1054 resulted in the loss of a mercaptoacetic acid (C2H4O2S) moiety, resulting in the product ion m/z 124.1123, and subsequently generated a stable product ion, m/z 81.0704.
FIG 1.

MS/MS spectra of retapamulin along with its metabolites and their proposed fragment ion patterns.
On the other hand, the loss of H2O from the mutilin moiety (m/z 303.2326) led to the generation of the m/z 285.2210 product ion, which subsequently yielded m/z 245.1908 after the loss of the side chain at the C-12 position and, eventually, m/z 163.1123 after the neutral loss of a C6H10 moiety.
Metabolic profiles of RTM in rat, swine, and human liver microsomes.
In vitro metabolism assays of RTM using rat, swine, and human liver microsomes revealed various metabolic routes of RTM, yielding approximately 16 different metabolites (metabolites M1 to M16). Information regarding the retention time, accurate m/z value, and main fragment ions of each metabolite is described in Table S3 in the supplemental material. The metabolic routes and the annotation of these metabolites using tandem mass spectrometry (MS/MS) are described in succeeding sections.
Monohydroxylated or oxidative metabolites M1 to M6.
Six metabolites were found to have an m/z value of 534.325 at different retention times. These metabolites were considered monohydroxylated derivatives of RTM due to the mass difference associated with the addition of a hydroxyl (-OH) moiety within a 2-ppm mass error of the calculated exact mass. Metabolites M1 to M5 yielded similar product ions after CID MS/MS, but their retention times differed, as influenced by the location of the hydroxylation reaction in the parent molecule. For M1 to M5, CID produced product ions m/z 216, m/z 124, and m/z 81, which are similar to those of RTM, inferring that hydroxylation did not occur at the side chain of the molecule. In addition, characteristic ions m/z 301 and m/z 319, which are equivalent to m/z 285 and m/z 303 each with the addition of 16 Da, respectively, further demonstrated that hydroxylation or oxidation occurred in the mutilin moiety. Unfortunately, the exact point of hydroxylation within the mutilin structure could not be soundly elucidated merely on the basis of the MS/MS spectra. According to a report delivered by EMA, the 1β, 2β, and 8α positions of the mutilin structure are prone to hydroxylation. Moreover, Lykkeberg et al. described that 2β-OH-TIA and 8α-OH-TIA were the main hydroxylated metabolites of tiamulin, another pleuromutilin derivative (25), which was consistent with the finding of EMA regarding valnemulin, which is also a pleuromutilin derivative (21). On the basis of the results of these previous studies on structurally closely related compounds, the two major metabolites obtained from this study were regarded as 2β- and 8α-OH-RTM. Eventually, M1 and M2 were identified as 2β-OH-RTM and 8α-OH-RTM, respectively, after comparison to a standard of 8α-OH-mutilin. M3 to M5 were regarded as the products monohydroxylated on the mutilin moiety.
M6, on the contrary, yielded an MS/MS spectrum different from that of M1 to M5, such that the product ion m/z 232 instead of m/z 301 was produced. This characteristic product ion could come from the addition of 16 Da to m/z 216, which represented the side chain. Hence, M6 was regarded as a hydroxyl derivative of RTM on the side chain moiety, in contrast to the mutilin moiety for M1 to M5. On the basis of the chemical structure of RTM, the S-motif is the most likely point of hydroxylation.
Dihydroxylated (M7 to M12) and trihydroxylated (M13) metabolites.
The exact mass of M7 to M12 was measured to be m/z 550.31969, which represented the addition of 2 hydroxyl moieties to RTM, suggesting that these metabolites are dihydroxylated metabolites. As explained above for the monohydroxylated derivatives, the characteristic product ion m/z 216.10532 in M7 to M10 indicated that the side chain of RTM was maintained unchanged and the double hydroxylation occurred on the mutilin moiety. Based on previous reports (21), we propose M8 to be hydroxylated at the C-2 and C-8 positions of the mutilin structure. On the other hand, the characteristic fragment ion m/z 232.10005 in M11 and M12 implied that one hydroxylation is at the S-motif of the side chain. Furthermore, M13 had an m/z value of 566.31515 with a characteristic product ion of m/z 216.10532, which suggested that the side chain remained unchanged and all the triple hydroxylations occurred at the mutilin moiety.
N-Demethyl-RTM and N-demethyl-hydroxylated RTM.
Using high-resolution MS, the elemental composition of M14 was predicted to be C29H46O4NS+. On the basis of the exact mass, M14 lost a mass equivalent to that of a CH2 moiety, which indicated that M14 was potentially a demethyl-RTM. CID of M14 resulted in its characteristic product ion of m/z 202.08963, which was 14 Da less than m/z 216.1058, implying that the demethylation may have occurred at the N motif of the side chain. M15 and M16 shared the same predicted elemental component of C29H46O5NS and exact mass, which was 16 Da higher than that of M14. This suggested that these two metabolites could be the hydroxylated or oxidative products of M14, and hence, it is tentatively identified as an N-demethyl-RTM. The characteristic product ion of m/z 319.2273 was 16 Da larger than m/z 303.2324, which implied that the hydroxylation or the oxidative reaction occurred at the mutilin moiety. Hence, M15 and M16 were tentatively identified as N-demethyl-hydroxylated RTM derivatives.
Metabolic pathways and interspecies metabolic differences of RTM.
On the basis of the results of the in vitro metabolism assays using human, rat, and swine liver microsomes, the main metabolic routes of RTM were (mono-, di-, and tri-) hydroxylation at both the side chain and mutilin moiety and S-oxidation and N-demethylation in the side chain. Hydroxylation at the mutilin moiety was tentatively assigned to the C-2 and C-8 positions. Our proposed metabolic routes for RTM are depicted in Fig. 2.
FIG 2.

Proposed metabolic pathways of retapamulin in vitro and in vivo.
As shown in Fig. 3, it is clear that interspecies variation occurs in terms of RTM metabolism. For instance, M3 was found only in rat and swine liver microsomes, whereas M13 was produced solely by human liver microsomes. Contrary to swine and rat liver microsomes, merely a minority of the parent drug remained after incubation in human liver microsomes, which may explain the rapid metabolism of RTM. It is therefore essential to consider these differences when translating results from animal metabolism experiments into clinical studies.
FIG 3.

Recovery of retapamulin and generation of its metabolites after incubation with rat, swine, and human liver microsomes (RLMs, SLMs, and HLMs, respectively).
Nonetheless, it can be concluded that RTM is metabolized by rat, human, and swine liver microsomes into a variety of metabolites. For all liver microsomes tested, 2β-OH-RTM (M1) and 8α-OH-RTM (M2) were the major metabolites produced. Furthermore, the metabolites produced by rat liver microsomes resembled the metabolites produced by human liver microsomes more closely than those produced by swine liver microsomes.
Comparative metabolic characteristics of the pleuromutilins.
Extracted ion chromatograms (EICs) of RTM and its metabolites after incubation with human liver microsomes are shown in Fig. S2. The in vitro metabolism of the investigated pleuromutilins (TIA, VLM, and RTM) in human liver microsomes revealed that the major metabolites of the pleuromutilins were metabolites hydroxylated at the C-2 and C-8 positions of the mutilin moiety, which accounted for over 50% of the total metabolites produced (Fig. 4).
FIG 4.

Relative amounts of 2β- and 8α-hydroxylated metabolites and other metabolites of tiamulin, valnemulin, and retapamulin in human liver microsomes.
In vivo metabolism in rats.
Considering the similar metabolic pattern in liver microsomes of rats and human, the in vivo metabolism of RTM in rats was conducted to reflect the metabolic information in human, although not exactly as discussed above. The results from the in vivo experiments concurred with the in vitro results, such that RTM was rapidly metabolized in the body. Consequently, a total of 6 metabolites were identified, including 2 monohydroxylated metabolites (M1 and M2) and 4 dihydroxylated metabolites (M7, M8, M9, and M10). Similarly, the major metabolites of RTM were also monohydroxylated products at the 2β and 8α positions, which was consistent with the in vitro results. The other metabolites, like N-demethyl-RTM, were not detected in vivo, which could be due to their presence at trace levels. Furthermore, this difference also reflects the oral bioavailability of RTM, which is also affected by the metabolism of RTM by the intestines.
Metabolic phenotype of the pleuromutilins.
Recombinant human cytochrome enzymes (CYP enzymes 1A2, 2B6, 2C8, 2C9, 2C19, 2D6, and 3A4) were incubated with TIA, VLM, and RTM individually to determine the main CYP responsible for the metabolism of pleuromutilins. The results suggested that CYP3A4 played a vital role in the in vitro metabolism of the drugs and caused elimination of the parent compounds (Fig. 5). However, when the pleuromutilins were incubated with other cytochrome enzymes, such as CYP1A2 and CYP2B6, the levels of pleuromutilins remained unchanged (100%), indicating that CYP3A4 is the principal enzyme responsible for the metabolism of TIA, VLM, and RTM.
FIG 5.
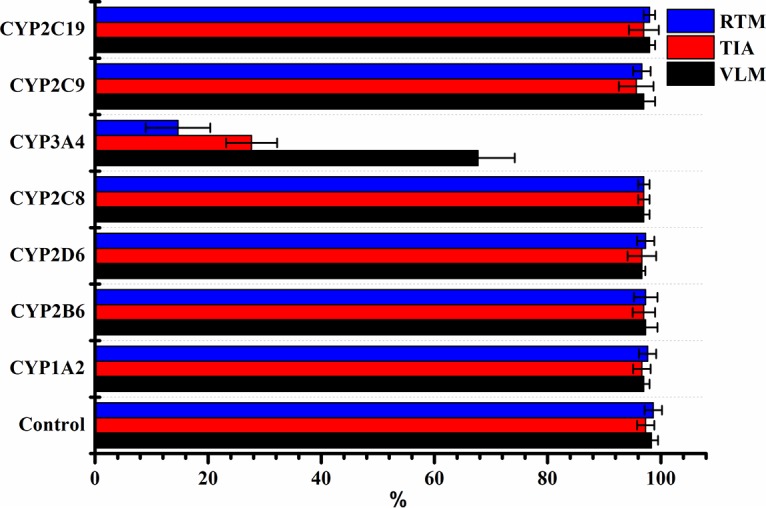
Relative amounts of tiamulin, valnemulin, and retapamulin remaining after incubation with human CYP enzymes (1A2, 2B6, 2D6, 2C8, 3A4, 2C9, or 2C19).
DISCUSSION
Recently, the emergence and rapid spread of methicillin-resistant Staphylococcus aureus (MRSA) isolates have fueled the need for the discovery and development of new antibiotics with novel modes of action, such as pleuromutilin antibiotics (26). However, a number of challenges, some of which are outlined later in this paper, have led to a significantly delayed entry of these drugs into successful clinical application, although like almost all new classes of antibiotics, pleuromutilins have made their way into veterinary medicine. However, very limited progress to make use of this underexploited antibiotic class for treatment in humans has been made.
It has previously been reported that modification of the pleuromutilin side chain at C-14 could be the most promising way to obtain a considerable enhancement in antibacterial activity (12). For instance, molecular modifications at the C-14 glycolic acid chain of pleuromutilin led to the discovery of two pleuromutilin derivatives, TIA and VAL. Additionally, pleuromutilin analogues which contain a purine ring have been reported to have excellent antibacterial activity. SAR studies showed that the presence of a thioether group at the C-22 position of pleuromutilin also enhances its antibacterial activity (19).
For the novel drug discovery and development of the pleuromutilin class, both academic and industrial research groups have focused on the modifications at the molecule's side chain (27), which is often optimized to obtain maximum in vitro activity, based on empirical and semiempirical SAR studies (28, 29). In vitro activity, however, could not be sufficiently extrapolated to explain in vivo biological activity. This could be attributed to some undesirable pharmacokinetic properties that reduce the in vivo activity and bioavailability, whereas the formation of reactive metabolites may contribute to increased toxicity. Thus, it is highly important to investigate the pharmacokinetics and metabolism of candidate drugs, as their metabolic fate could be considered prerequisites to in vivo efficacy. Moreover, as metabolism studies of new drug candidates are initially conducted in vitro and in animal models before clinical trials, it is important to compare the metabolism of animals and humans in order to obtain information on the appropriate selection of animal species for toxicity and in vivo metabolism studies (28).
High-resolution mass spectrometry has been widely used to aid with drug discovery and development (30–33). Using this technique, our research team has previously illustrated the pharmacokinetics (22) and metabolic profiles of VLM (24) and TIA (23). In the current study, the comparative metabolism of RTM in animal and human liver microsomes was investigated. Our results indicated that the pleuromutilins, including VLM, TIA, and RTM, were prone to be metabolized at the 2β and 8α motifs of the mutilin moiety. Furthermore, RTM is likely dihydroxylated and trihydroxylated, which may explain the extensive metabolism and poor bioavailability of RTM after oral administration.
Drugs with high metabolic clearance are usually subjected to extensive first-pass metabolism, resulting in low bioavailability. Hence, to improve the bioavailability of the drug, it could be useful to determine the main metabolizing enzymes responsible for the hydroxylation of RTM. Drug-metabolizing enzymes, including cytochrome P450s, perform numerous oxidative reactions with the carbon, oxygen, nitrogen, and sulfur atoms of substrates (34). Our results showed that the pleuromutilins were adequately metabolized by CYP3A4. We therefore hypothesize that an alternative approach to the development of new pleuromutilin derivatives is to block the active metabolic moiety within the molecule or to administer the drug in combination with a CYP3A4 inhibitor.
The concept of soft drugs was first put forward in 1980 (35). These so-called soft drugs evade oxidative metabolism and use hydrolytic enzymes to achieve predictable and controllable drug metabolism (36). Based on this principle, Bodor designed soft quaternary-type drugs containing three structural components: an acidic group, an aldehyde, and a tertiary amine (37). As for the highly lipophilic and poorly soluble compounds, like the pleuromutilins, a basic amine could be incorporated into the active metabolic site to increase the solubility and block the rapid metabolism. Several human immunodeficiency virus (HIV) protease inhibitors successfully applied the concept of sealing the active metabolic site by the use of, for instance, different substitutions at the N atom of the C-2 position. Consequently, one of the compounds, indinavir sulfate, is well absorbed after oral dosing and was approved for use for the treatment of AIDS (38). Thus, this concept for new drug discovery, blocking the active metabolic site, could be used for novel drug discovery of the pleuromutilins.
In conclusion, due to the surging drug resistance and the unique mode of action, semisynthesized pleuromutilin derivatives of the pleuromutilins with no cross-resistance have attracted considerable attention. The metabolic results obtained in vivo and in vitro suggested that the pleuromutilins, including TIA, VLM, and RTM, are rapidly metabolized into hydroxylated products at the C-2 and C-8 positions. The current pathway for the development of pleuromutilin antibiotic derivatives often focuses on the modifications at the side chain, which could improve the bioactivity only to some extent but not improve metabolic stability and bioavailability. Therefore, in this paper, we propose a promising approach to develop safe and efficacious drugs derived from pleuromutilins: to seal active metabolic sites (the C-2 and C-8 motifs) or to administer the drug in combination with a CYP3A4 inhibitor.
MATERIALS AND METHODS
Materials and chemicals.
Retapamulin was purchased from Ouhe Technology Co. Ltd. (Beijing, China). Pooled human, rat, and porcine liver microsomes were bought from the Research Institute for Liver Diseases Co. Ltd. (Shanghai, China) and stored at −80°C until use. NADPH was purchased from Sigma-Aldrich (Germany). Acetonitrile and formic acid were all of liquid chromatography (LC)-MS grade and were purchased from Fisher Chemical Co. (USA). 8α-OH-Mutilin was purchased from Argus Chemicals S.r.l. (Italy). Distilled water was obtained using a Milli-Q system (Millipore, USA). The other reagents were of analytical grade.
Animals.
Six Wistar rats (age, 6 weeks; weight, 200 to 220 g; 3 males and 3 females) were purchased from Vital River Laboratory Animal Technology Co. Ltd. (Beijing, China). The animals were acclimatized for 1 week under standardized conditions. All the animal procedures were approved by the Ethical Committee of China Agricultural University (Beijing, China) (2016-SYXK-0038).
Preparation of in vivo samples.
Rats (3 male rats and 3 female rats) were subjected to a 12-h fast before the in vivo metabolism study. The animals were individually housed in metabolic cages and administered RTM by oral gavage at a dosage of 15 mg/kg of body weight. Urine and fecal samples were gathered before RTM administration and 12 to 24 h postadministration. The samples were frozen at −20°C until further analysis. Extraction was performed by adding 10 ml ethyl acetate into 2.0 g of feces or 2.0 ml of urine, followed by vortexing for 5 min and centrifugation at 5,000 × g for 10 min at 4°C. The supernatant was obtained and evaporated to dryness with a gentle nitrogen flow at 45°C. The residue was then finally redissolved in 1.0 ml of 15% acetonitrile in aqueous solution and filtered through a 0.22-μm-pore-size micropore cellulose membrane filter before analysis using liquid chromatography–high-resolution mass spectrometry (LC-HRMS).
In vitro metabolism.
Recombinant human cytochrome enzymes (CYP enzymes 1A2, 2B6, 2C8, 2D6, 2C9, 2C19, and 3A4) were incubated with TIA, VLM, and RTM to investigate the roles of these enzymes in the metabolism of pleuromutilins. To initiate the in vitro metabolism, the reaction consisted of phosphate buffer (100 mM, pH 7.4), CYP enzymes (30 pmol), and NADPH (2 mM) along with 3 μM each TIA, VLM, and RTM. After incubation for 1 h at 37°C, 100 μl cold acetonitrile was added to terminate the reaction. Then the mixtures were vortexed and centrifuged at 12,000 × g for 5 min. The supernatant was gathered and evaporated to dryness under a gentle nitrogen stream. Finally, the residue was reconstituted with the initial mobile phase. The in vitro metabolism assays were performed in triplicate, and an additional sample without NADPH served as the negative control.
Instrument conditions.
In current study, the metabolic fate of the pleuromutilins, especially RTM, was investigated using a Thermo LC system equipped with a binary pump, an autosampler, and thermostatic column modules coupled to a Quadrupole-Exactive Plus (UHPLC-Q-Exactive Plus; Thermo Fisher Scientific, Bremen, Germany) high-resolution mass spectrometer consisting of a heated electrospray ionization source (HESI-II) in the positive mode. Chromatographic separation was achieved using a Hypersil Gold C18 column (100 mm by 2.1 mm; particle size, 1.9 μm; Thermo Fisher Scientific, Waltham, MA). The mobile phase consisted of solvent A (0.1% formic acid) and solvent B (acetonitrile containing 0.1% formic acid) with a flow rate of 0.3 ml min−1. The optimum gradient elution program was as follows: from 0 to 2.0 min, 15% to 35% solvent B; from 2.0 to 3.5 min, 35% to 50% solvent B; from 3.5 to 4.0 min, 50% to 65% solvent B; from 4.0 to 6.0 min, 65% to 80% solvent B; from 6.0 to 8.0 min, 80% to 100% solvent B; from 8.0 to 9.0 min, 100% to 100% solvent B; from 9.0 to 9.5 min, 100% to 15% solvent B; and from 9.5 to 10.0 min, 15% solvent B. The column temperature was maintained at 30°C, and the injection volume was 2 μl.
Typical parameters for mass spectrometry detection were set as follows: the spray voltage and capillary temperature were set at 3.5 kV and 320°C, respectively, and the sheath gas and auxiliary gas flows were set at 45 and 10 arbitrary units, respectively. For the full MS mode, the mass range was set between m/z 300 and 800, while the major parameters were set as follows: resolution, 70,000 full width at half maximum (FWHM); automatic gain control (AGC) target, 3 × 106; maximum injection time (IT), 100 ms. For the data-dependent MS2 mode, the major parameters were set as follows: resolution, 17,500; AGC target, 1 × 105; maximum IT, 100 ms. Mass accuracy was maintained within 2 ppm to ensure the reliability of the identifications.
Data analysis.
Acquired data (full scan and all ion fragmentation) were initially processed using Compound Discoverer (version 2.0) software, which could conveniently screen the possible metabolites in comparison with control samples. The mass window was set within 0.05 Da for expected and unexpected metabolites. Furthermore, false-positive identifications were eliminated on the basis of an accurate mass and the elemental composition of both precursor ions and all fragment ions.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (no. 31702296 and no. 31672599) and the Evaluation of Scientific Establishment of Maximum Residual Limits in Animal Products Foundation of China (no. GJFP201600706). Yanshen Li was supported by the National Natural Science Foundation of China (grant no. 31402246). Gerard Bryan Gonzales is a postdoctoral fellow of the Research Foundation—Flanders (FWO).
We declare no conflict of interest.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.02388-17.
REFERENCES
- 1.Ross JE, Jones RN. 2005. Quality control guidelines for susceptibility testing of retapamulin (SB-275833) by reference and standardized methods. J Clin Microbiol 43:6212–6213. doi: 10.1128/JCM.43.12.6212-6213.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wood PR, Kampschmidt JC, Dube PH, Cagle MP, Chaparro P, Ketchum NS, Kannan TR, Singh H, Peters JI, Baseman JB, Brooks EG. 2017. Mycoplasma pneumoniae and health outcomes in children with asthma. Ann Allergy Asthma Immunol 119:146–152.e2. doi: 10.1016/j.anai.2017.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee E, Cho HJ, Hong SJ, Lee J, Sung H, Yu J. 2017. Prevalence and clinical manifestations of macrolide resistant Mycoplasma pneumoniae pneumonia in Korean children. Korean J Pediatr 60:151–157. doi: 10.3345/kjp.2017.60.5.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bowler PG, Duerden BI, Armstrong DG. 2001. Wound microbiology and associated approaches to wound management. Clin Microbiol Rev 14:244–269. doi: 10.1128/CMR.14.2.244-269.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Robson MC, Heggers JP. 1981. Burn wound microbiology. Am J Clin Pathol 76:246–247. doi: 10.1093/ajcp/76.2.246. [DOI] [PubMed] [Google Scholar]
- 6.Nasiri MJ, Haeili M, Ghazi M, Goudarzi H, Pormohammad A, Imani Fooladi AA, Feizabadi MM. 2017. New insights in to the intrinsic and acquired drug resistance mechanisms in mycobacteria. Front Microbiol 8:681. doi: 10.3389/fmicb.2017.00681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lipinska N, Romaniuk A, Paszel-Jaworska A, Toton E, Kopczynski P, Rubis B. 2017. Telomerase and drug resistance in cancer. Cell Mol Life Sci 74:4121–4132. doi: 10.1007/s00018-017-2573-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hogenauer G. 1975. The mode of action of pleuromutilin derivatives. Location and properties of the pleuromutilin binding site on Escherichia coli ribosomes. Eur J Biochem 52:93–98. [DOI] [PubMed] [Google Scholar]
- 9.Hodgin LA, Hogenauer G. 1974. The mode of action of pleuromutilin derivatives. Effect on cell-free polypeptide synthesis. Eur J Biochem 47:527–533. [DOI] [PubMed] [Google Scholar]
- 10.Poulsen SM, Karlsson M, Johansson LB, Vester B. 2001. The pleuromutilin drugs tiamulin and valnemulin bind to the RNA at the peptidyl transferase centre on the ribosome. Mol Microbiol 41:1091–1099. doi: 10.1046/j.1365-2958.2001.02595.x. [DOI] [PubMed] [Google Scholar]
- 11.Hirokawa Y, Kinoshita H, Tanaka T, Nakamura T, Fujimoto K, Kashimoto S, Kojima T, Kato S. 2009. Pleuromutilin derivatives having a purine ring. Part 3: synthesis and antibacterial activity of novel compounds possessing a piperazine ring spacer. Bioorg Med Chem Lett 19:175–179. doi: 10.1016/j.bmcl.2008.10.127. [DOI] [PubMed] [Google Scholar]
- 12.Hirokawa Y, Kinoshita H, Tanaka T, Nakamura T, Fujimoto K, Kashimoto S, Kojima T, Kato S. 2009. Pleuromutilin derivatives having a purine ring. Part 2: influence of the central spacer on the antibacterial activity against Gram-positive pathogens. Bioorg Med Chem Lett 19:170–174. doi: 10.1016/j.bmcl.2008.10.123. [DOI] [PubMed] [Google Scholar]
- 13.Huang Q, Li J, Xia L, Xia X, Duan P, Shen J, Ding S. 2010. Residue depletion of valnemulin in swine tissues after oral administration. Anal Chim Acta 664:62–67. doi: 10.1016/j.aca.2010.01.058. [DOI] [PubMed] [Google Scholar]
- 14.Burch DG. 2008. Tiamulin activity against Brachyspira hyodysenteriae. Vet Rec 163:760. [PubMed] [Google Scholar]
- 15.Hannan PC, Windsor HM, Ripley PH. 1997. In vitro susceptibilities of recent field isolates of Mycoplasma hyopneumoniae and Mycoplasma hyosynoviae to valnemulin (Econor), tiamulin and enrofloxacin and the in vitro development of resistance to certain antimicrobial agents in Mycoplasma hyopneumoniae. Res Vet Sci 63:157–160. doi: 10.1016/S0034-5288(97)90010-2. [DOI] [PubMed] [Google Scholar]
- 16.Heilmann C, Jensen L, Jensen JS, Lundstrom K, Windsor D, Windsor H, Webster D. 2001. Treatment of resistant mycoplasma infection in immunocompromised patients with a new pleuromutilin antibiotic. J Infect 43:234–238. doi: 10.1053/jinf.2001.0910. [DOI] [PubMed] [Google Scholar]
- 17.Springer DM, Sorenson ME, Huang S, Connolly TP, Bronson JJ, Matson JA, Hanson RL, Brzozowski DB, LaPorte TL, Patel RN. 2003. Synthesis and activity of a C-8 keto pleuromutilin derivative. Bioorg Med Chem Lett 13:1751–1753. doi: 10.1016/S0960-894X(03)00297-X. [DOI] [PubMed] [Google Scholar]
- 18.Jacobs MR. 2007. Retapamulin: a semisynthetic pleuromutilin compound for topical treatment of skin infections in adults and children. Future Microbiol 2:591–600. doi: 10.2217/17460913.2.6.591. [DOI] [PubMed] [Google Scholar]
- 19.Hirokawa Y, Kinoshita H, Tanaka T, Nakata K, Kitadai N, Fujimoto K, Kashimoto S, Kojima T, Kato S. 2008. Water-soluble pleuromutilin derivative with excellent in vitro and in vivo antibacterial activity against gram-positive pathogens. J Med Chem 51:1991–1994. doi: 10.1021/jm8000136. [DOI] [PubMed] [Google Scholar]
- 20.Hirokawa Y, Kinoshita H, Tanaka T, Nakamura T, Fujimoto K, Kashimoto S, Kojima T, Kato S. 2008. Pleuromutilin derivatives having a purine ring. Part 1: new compounds with promising antibacterial activity against resistant Gram-positive pathogens. Bioorg Med Chem Lett 18:3556–3561. doi: 10.1016/j.bmcl.2008.05.011. [DOI] [PubMed] [Google Scholar]
- 21.European Agency for the Evaluation of Medicinal Products. 1998. Committee for Veterinary Medicinal Products. Valnemulin. Summary report, 339–398 European Agency for the Evaluation of Medicinal Products, London, United Kingdom: http://www.ema.europa.eu/docs/en_GB/document_library/Maximum_Residue_Limits_-_Report/2009/11/WC500015381.pdf. [Google Scholar]
- 22.Sun F, Fan R, Wang J, Xiong L, Shen J, Zhang S, Cao X. 2017. Pharmacokinetics of valnemulin after intravenous, intramuscular, and oral administration in layer chickens. J Vet Pharmacol Ther 40:415–418. doi: 10.1111/jvp.12389. [DOI] [PubMed] [Google Scholar]
- 23.Sun FF, Yang SP, Zhang HY, Zhou JH, Li Y, Zhang JZ, Jin Y, Wang ZH, Li YS, Shen JZ, Zhang SX, Cao XY. 2017. Comprehensive analysis of tiamulin metabolites in various species of farm animals using ultra-high-performance liquid chromatography coupled to quadrupole/time-of-flight. J Agric Food Chem 65:199–207. doi: 10.1021/acs.jafc.6b04377. [DOI] [PubMed] [Google Scholar]
- 24.Yang SP, Shi WM, Hu DF, Zhang SX, Zhang HY, Wang ZH, Cheng LL, Sun FF, Shen JZ, Cao XY. 2014. In vitro and in vivo metabolite profiling of valnemulin using ultraperformance liquid chromatography-quadrupole/time-of-flight hybrid mass spectrometry. J Agric Food Chem 62:9201–9210. doi: 10.1021/jf5012402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lykkeberg AK, Cornett C, Halling-Sorensen B, Hansen SH. 2006. Isolation and structural elucidation of tiamulin metabolites formed in liver microsomes of pigs. J Pharm Biomed Anal 42:223–231. doi: 10.1016/j.jpba.2006.03.023. [DOI] [PubMed] [Google Scholar]
- 26.Saravolatz LD, Pawlak J, Saravolatz SN, Johnson LB. 2013. In vitro activity of retapamulin against Staphylococcus aureus resistant to various antimicrobial agents. Antimicrob Agents Chemother 57:4547–4550. doi: 10.1128/AAC.00282-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hon KL, Lee VWY. 2017. Challenges for drug discovery and development in China. Expert Opin Drug Discov 12:105–113. doi: 10.1080/17460441.2017.1257115. [DOI] [PubMed] [Google Scholar]
- 28.Differding E. 2017. The drug discovery and development industry in India—two decades of proprietary small-molecule R&D. ChemMedChem 12:786–818. doi: 10.1002/cmdc.201700043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bajorath J, Overington J, Jenkins JL, Walters P. 2016. Drug discovery and development in the era of big data. Future Med Chem 8:1807–1813. doi: 10.4155/fmc-2014-0081. [DOI] [PubMed] [Google Scholar]
- 30.Lin W, Flarakos J, Du Y, Hu WY, He HD, Mangold J, Tanaka SK, Villano S. 2017. Pharmacokinetics, distribution, metabolism, and excretion of omadacycline following a single intravenous or oral dose of 14C-omadacycline in rats. Antimicrob Agents Chemother 61:e01784-16. doi: 10.1128/AAC.01784-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vale N, Gouveia MJ, Rinaldi G, Brindley PJ, Gartner F, Correia da Costa JM. 2017. Praziquantel for schistosomiasis: single-drug metabolism revisited, mode of action, and resistance. Antimicrob Agents Chemother 61:e02582-16. doi: 10.1128/AAC.02582-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wipf JRK, Riley MC, Kania SA, Bemis DA, Andreis S, Schwendener S, Perreten V. 2017. New macrolide-lincosamide-streptogramin B resistance gene erm(48) on the novel plasmid pJW2311 in Staphylococcus xylosus. Antimicrob Agents Chemother 61:e00066-17. doi: 10.1128/AAC.00066-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Potter BM, Xie LH, Vuong C, Zhang J, Zhang P, Duan D, Luong TL, Bandara Herath HM, Dhammika Nanayakkara NP, Tekwani BL, Walker LA, Nolan CK, Sciotti RJ, Zottig VE, Smith PL, Paris RM, Read LT, Li Q, Pybus BS, Sousa JC, Reichard GA, Marcsisin SR. 2015. Differential CYP 2D6 metabolism alters primaquine pharmacokinetics. Antimicrob Agents Chemother 59:2380–2387. doi: 10.1128/AAC.00015-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shalan H, Kato M, Cheruzel L. 2017. Keeping the spotlight on cytochrome P450. Biochim Biophys Acta 1866:80–87. doi: 10.1016/j.bbapap.2017.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rassokha TM, Kabachna AV, Pertsev IM. 1980. Current status of the industrial manufacture of soft drugs by factories of the Soiuzlikzasoby All-Union Manufacturing Association. Farm Zh 1980:71 (In Ukrainian.) [PubMed] [Google Scholar]
- 36.Stanczak A, Ferra A. 2006. Prodrugs and soft drugs. Pharmacol Rep 58:599–613. [PubMed] [Google Scholar]
- 37.Bodor N. 1994. Designing safer ophthalmic drugs by soft drug approaches. J Ocul Pharmacol 10:3–15. doi: 10.1089/jop.1994.10.3. [DOI] [PubMed] [Google Scholar]
- 38.Schouten JT. 1996. FDA approves 2 new protease inhibitors: ritonavir (Norvir) and Crixivan (indinavir sulfate). Food and Drug Administration. STEP Perspect 8:7–8. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.