

ABSTRACT
APX001 is the prodrug of APX001A, which is a first-in-class small molecule with a unique mechanism of action that inhibits the fungal enzyme Gwt1 in the glycosylphosphatidylinositol (GPI) biosynthesis pathway. The goal of the present study was to determine which pharmacokinetic/pharmacodynamic (PK/PD) index and magnitude best correlated with efficacy in the murine disseminated candidiasis model for Candida albicans (n = 5), C. glabrata (n = 5), and C. auris (n = 4). MIC values ranged from 0.002 to 0.03 mg/liter for C. albicans, from 0.008 to 0.06 mg/liter for C. glabrata, and from 0.004 to 0.03 mg/liter for C. auris. Plasma APX001A pharmacokinetic measurements were performed in mice after oral administration of 4, 16, 64, and 256 mg/kg of body weight APX001. Single-dose pharmacokinetic studies exhibited maximum plasma concentration (Cmax) values of 0.46 to 15.6 mg/liter, area under the concentration-time curve (AUC) from time zero to infinity (AUC0–inf) values of 0.87 to 70.0 mg · h/liter, and half-lives of 1.40 to 2.75 h. A neutropenic murine disseminated candidiasis model was utilized for all treatment studies, and drug dosing was by the oral route. Dose fractionation was performed against C. albicans K1, with total doses ranging from 4 to 1,024 mg/kg/day of APX001 fractionated into regimens of dosing every 3, 6, 8, and 12 h for a 24-h treatment duration. Nonlinear regression analysis was used to determine which PK/PD index best correlated with efficacy on the basis of the reduction in the number of CFU/kidney at 24 h. The 24-h free-drug AUC/MIC ratio (fAUC0–24/MIC) was the PK/PD index that best correlated with efficacy (coefficient of determination [R2] = 0.88). Treatment studies with the remaining strains utilized regimens of 1 to 256 mg/kg of APX001 administered every 6 h for a 24-h duration with C. albicans and a 96-h study duration with C. glabrata and C. auris. The dose required to achieve 50% of the maximum effect (ED50) and stasis fAUC/MIC targets were as follows: for C. albicans, 3.67 ± 3.19 and 20.60 ± 6.50, respectively; for C. glabrata, 0.38 ± 0.21 and 1.31 ± 0.27, respectively; and for C. auris, 7.14 ± 4.54 and 14.67 ± 8.30, respectively. The present studies demonstrated in vitro and in vivo APX001A and APX001 potency, respectively, against C. albicans, C. glabrata, and C. auris. These results have potential relevance for clinical dose selection and evaluation of susceptibility breakpoints. The identification of a lower AUC/MIC ratio target for C. glabrata suggests that species-specific susceptibility breakpoints should be explored.
KEYWORDS: APX001, Candida, pharmacodynamics
INTRODUCTION
Invasive fungal infections have become increasingly common and remain associated with a high rate of morbidity and mortality, despite currently available antifungal therapies (1–4). Candida albicans is still the leading cause of candidemia, but other species of Candida (non-albicans Candida species) now comprise >50% of bloodstream infections in many parts of the world (5). Multidrug resistance among C. glabrata isolates (6) and the outbreak of infections caused by C. auris species further limit treatment options (7, 8). Therefore, antifungal drugs with novel mechanisms of action which lack cross-resistance to existing antifungals are desirable for the treatment of invasive fungal infections.
APX001, the methyl-phosphate prodrug of the active moiety APX001A, is a first-in-class broad-spectrum antifungal agent for the treatment of invasive fungal infections, including species resistant to existing antifungal drug classes (9–11). APX001A inhibits the fungal enzyme Gwt1 that is part of the glycosylphosphatidylinositol (GPI) biosynthesis pathway (12). Both APX001A and APX001 have been evaluated extensively in preclinical in vitro and in vivo studies and exhibit broad-spectrum activity against pathogenic yeasts (e.g., Candida spp.) and molds (e.g., Aspergillus spp.), including multidrug-resistant strains (e.g., Fusarium, Scedosporium, and fungi from the Mucorales order) (13–16).
The current studies included pharmacokinetic (PK)/pharmacodynamic (PD) evaluation of the APX001 efficacy against C. albicans, C. glabrata, and C. auris in the neutropenic mouse model of disseminated candidiasis to determine the PK/PD index and the magnitude predictive of efficacy to assist with further clinical development of optimal dosing strategies.
RESULTS
In vitro susceptibility testing.
The MIC values of APX001A and comparators (fluconazole and micafungin) are shown in Table 1. APX001A showed potent antifungal activity against these clinical strains, with similar MIC ranges for the three Candida species. The APX001A MIC values varied 15-fold for the tested organisms.
TABLE 1.
In vitro activities of APX001A and comparators
| Species | Isolate | MIC (mg/liter) |
||
|---|---|---|---|---|
| APX001A | Fluconazole | Micafungin | ||
| C. albicans | K1 | 0.008 | 0.5 | 0.016 |
| 98-210 | 0.03 | 16 | 0.016 | |
| 580 | 0.004 | 0.5 | 0.008 | |
| 2-76 | 0.002 | 0.25 | 0.008 | |
| 98-17 | 0.03 | 16 | 0.03 | |
| C. glabrata | 10956 | 0.016 | 2 | 0.25 |
| 5592 | 0.06 | 32 | 0.016 | |
| 35315 | 0.016 | 0.25 | 0.06 | |
| 513 | 0.03 | 4 | 0.016 | |
| 5376 | 0.008 | 2 | 0.008 | |
| C. auris | B11104 | 0.03 | >256 | 0.25 |
| B11221 | 0.008 | 128 | 1 | |
| B11219 | 0.004 | >256 | 4 | |
| B11804 (C54007) | 0.008 | 2 | 0.5 | |
Drug pharmacokinetics.
Single-dose pharmacokinetics of APX001A are shown in Fig. 1. After an oral dose of APX001, the APX001A exposure increased in a dose-dependent manner across the dose range. The maximum plasma concentration (Cmax) values ranged from 0.455 to 15.6 mg/liter. The area under the concentration-time curve (AUC) from time zero to infinity (AUC0–inf) values ranged from 0.868 to 70.0 mg · h/liter and were linear across the 4- to 256-mg dosing range (coefficient of determination [R2] = 0.97). The elimination half-life (t1/2) ranged from 1.40 to 2.75 h.
FIG 1.

Single-dose plasma pharmacokinetics of APX001A. Four different doses of the prodrug APX001 that varied by 4-fold concentrations on a milligram-per-kilogram basis were administered to mice by the oral route. Groups of three mice were sampled for each time point. Each symbol represents the mean ± SD for three animals. Shown in the key is the maximum plasma concentration (Cmax), the area under the concentration curve from time zero to infinity (AUC), and the elimination half-life (t1/2).
PK/PD index determination.
At the start of therapy, mice had 4.10 ± 0.16 log10 CFU/kidney of C. albicans K1, and the organism grew 3.71 ± 0.26 log10 CFU/kidney after 24 h in untreated control mice. Escalating doses of APX001 resulted in concentration-dependent killing. The highest doses studied reduced the organism burden from 0.75 ± 0.09 to 1.84 ± 0.24 log10 CFU/kidney compared to the numbers at the start of therapy. The dose-response relationship for the four dosing intervals against C. albicans K1 is shown in Fig. 2. The dose-response curves were relatively similar among the four dosing intervals.
FIG 2.
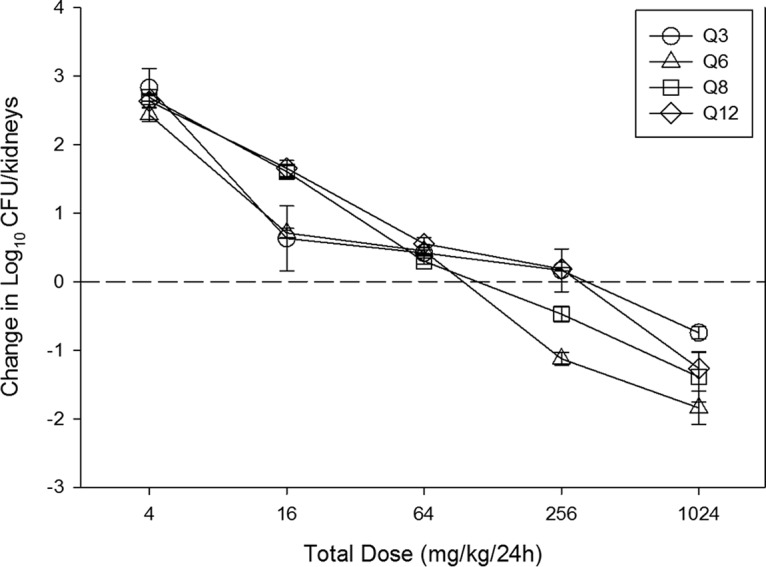
Relationship between the APX001 dosing interval and efficacy against C. albicans K1 in a murine neutropenic disseminated candidiasis model. Each symbol represents the mean ± SD for three animals. The error bars represent the standard deviations. The dashed horizontal line represents net stasis over the treatment period. Points above the line represent net growth, and points below the line represent net killing (cidal activity).
The relationships between the microbiological effect and each of the pharmacodynamic indices, 24-h free-drug AUC/MIC ratio (fAUC0–24/MIC), free-drug Cmax/MIC ratio (fCmax/MIC), and the percentage of the dosing interval during which plasma free-drug levels exceeded the MIC for each of the dosage regimens studied (TMIC), against C. albicans K1 are shown in Fig. 3. The strongest relationship was seen when the results were correlated with the fAUC0–24/MIC ratio, with an R2 value of 88%. Regression with fCmax/MIC (R2 = 82%) and TMIC (R2 = 7.5%) resulted in less strong relationships. Consideration of bound or unbound drug levels did not appreciably impact the relationships between efficacy and PK/PD indices.
FIG 3.

Pharmacodynamic regression of the in vivo dose fractionation study with APX001 against C. albicans K1. Each symbol represents the mean and standard deviation for three mice. The dose data are expressed as fAUC0–24/MIC, fCmax/MIC, and the percentage of the dosing interval during which plasma free-drug levels exceeded the MIC for each of the dosage regimens studied (TMIC). R2 is the coefficient of determination. Also shown for each PD index is the maximum effect (Emax), the PD index value associated with 50% of the maximum effect (ED50), and the slope of the relationship, or the Hill coefficient (N). The line drawn through the data points is the best-fit line based upon the sigmoid Emax formula.
Treatment efficacy and pharmacodynamic magnitude determination.
At the start of therapy, mice had 4.04 ± 0.3 log10 CFU/kidney, and the burden increased 3.30 ± 0.4 log10 CFU/kidney in the untreated controls at the end of therapy. The initial burden and growth in the controls for each organism are listed in Table 2. The in vivo dose-response curves for each Candida species are shown in Fig. 4A1, B1, and C1. Dose-dependent activity was observed with all Candida species groups. The maximal reduction in the C. albicans burden in APX001-treated mice compared to the burden in the untreated controls ranged from 0.99 to −1.54 log10 CFU/kidney, and static efficacy was reached in three of five C. albicans isolates. For C. glabrata, the maximal reduction was −0.72 to −1.62 log10 CFU/kidney, stasis was observed in all five strains, and 1-log kill below stasis was reached in four of five strains. For C. auris, the maximal reduction was 0.21 to −1.02 log10 CFU/kidney, with static efficacy being achieved in three of four strains. Potency was more pronounced against C. glabrata on the basis of the dose-response curves. The relationship between the PK/PD index fAUC0–24/MIC and treatment effect is shown in Fig. 4A2, B2, and C2. The coefficients of determination (R2) were strong, ranging from 0.67 to 0.90.
TABLE 2.
Static doses and associated AUC/MIC values in the neutropenic disseminated candidiasis modela
| Species | Isolate or parameter | Log10 no. of CFU/kidneys |
ED50 |
Stasis |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Start | Growth | Maximum kill | 24-h dose (mg/kg) | 24-h tAUC/MIC | 24-h fAUC/MIC | 24-h dose (mg/kg) | 24-h tAUC/MIC | 24-h fAUC/MIC | ||
| C. albicans | K1 | 4.16 | 3.40 | −1.37 | 87.3 | 3,413 | 6.83 | 265.5 | 12,952 | 25.91 |
| 98-210 | 3.36 | 4.05 | 0.99 | 43.1 | 371 | 0.742 | NA | |||
| 580 | 4.24 | 3.09 | −0.93 | 51.3 | 3,361 | 6.72 | 86.0 | 6,678 | 13.35 | |
| 2-76 | 3.54 | 3.78 | −1.54 | 17.7 | 1,999 | 4.00 | 76.5 | 11,270 | 22.54 | |
| 98-17 | 4.39 | 2.90 | 0.11 | 5.71 | 41.9 | 0.084 | NA | |||
| Mean | 3.94 | 3.44 | −0.55 | 41.0 | 1,837 | 3.67 | 142.5 | 10,300 | 20.60 | |
| Median | 4.16 | 3.40 | −0.93 | 43.1 | 1,999 | 4.00 | 86.0 | 11,270 | 22.54 | |
| SD | 0.46 | 0.48 | 1.07 | 31.8 | 1,597 | 3.19 | 106.7 | 3,248 | 6.50 | |
| C. glabrata | 10956 | 3.85 | 3.64 | −1.22 | 4.7 | 65 | 0.13 | 28.1 | 432.8 | 0.866 |
| 5592 | 3.91 | 3.13 | −0.72 | 29.9 | 124 | 0.247 | 125.2 | 734.2 | 1.47 | |
| 35315 | 4.07 | 3.15 | −1.41 | 20.9 | 307 | 0.61 | 41.9 | 675.5 | 1.35 | |
| 513 | 3.86 | 3.56 | −1.15 | 22.5 | 178 | 0.36 | 69.4 | 647.2 | 1.29 | |
| 5376 | 4.17 | 3.15 | −1.62 | 10.32 | 284.1 | 0.568 | 26.0 | 792.3 | 1.59 | |
| Mean | 3.97 | 3.33 | −1.22 | 17.7 | 192 | 0.38 | 58.1 | 656.4 | 1.31 | |
| Median | 3.91 | 3.15 | −1.22 | 20.9 | 178 | 0.36 | 41.9 | 675.5 | 1.35 | |
| SD | 0.14 | 0.25 | 0.34 | 10.1 | 103 | 0.21 | 41.3 | 136.9 | 0.27 | |
| C. auris | B11104 | 4.18 | 3.31 | 0.21 | 24.8 | 682.0 | 1.36 | NA | ||
| B11221 | 4.32 | 3.24 | −0.94 | 47.5 | 3,095.1 | 6.19 | 100.2 | 4,213.0 | 8.25 | |
| B11219 | 4.31 | 2.36 | −0.99 | 106.4 | 4,469.2 | 8.94 | 78.6 | 5,864.2 | 11.73 | |
| B11804 | 4.18 | 3.46 | −1.02 | 671.3 | 6,034.5 | 12.07 | 243.3 | 12,022 | 24.04 | |
| Mean | 4.25 | 3.09 | −0.69 | 212.5 | 3,570.2 | 7.14 | 140.7 | 7,336.4 | 14.67 | |
| Median | 4.25 | 3.28 | −0.97 | 77.0 | 3,782.2 | 7.56 | 100.2 | 5,864.2 | 11.73 | |
| SD | 0.08 | 0.50 | 0.60 | 307.8 | 2,269.2 | 4.54 | 89.5 | 4,150.2 | 8.30 | |
NA, not achieved; tAUC, total drug AUC; fAUC, free (non-protein-bound) drug AUC.
FIG 4.

In vivo dose effect of APX001 against select C. albicans (A1 and A2), C. glabrata (B1 and B2), and C. auris (C1 and C2) strains in the mouse disseminated candidiasis model. Each symbol represents the mean and standard deviation for three mice. Five total drug dose levels were fractionated into a regimen of administration every 6 h. The burden of organisms was measured at the start and the end of therapy. The study period was 24 h for C. albicans and 96 h for C. glabrata and C. auris. The horizontal dashed line at 0 represents the burden of organisms in the kidneys of the mice at the start of therapy. Data points below the line represent killing, and points above the line represent growth.
Calculation of the doses necessary to achieve the 50% of the maximum effect (ED50) and a static effect against multiple organisms are shown in Table 2. The ED50 and stasis fAUC/MIC0–24 targets for each organism group were as follows: for C. albicans, 3.67 ± 3.19 and 20.60 ± 6.50, respectively; for C. glabrata, 0.38 ± 0.21 and 1.31 ± 0.27, respectively; and for C. auris, 7.14 ± 4.54 and 14.67 ± 8.30, respectively.
DISCUSSION
Candida spp. are the most frequent organisms encountered in invasive fungal infections among hospitalized patients (3). Immunocompromised or immunosuppressed patients, including solid-organ or hematopoietic stem cell transplant recipients and individuals who are on immunosuppressive drug regimens, are especially at risk for invasive fungal infection, which is associated with a high rate of morbidity and mortality (5). Although C. albicans remains the most common pathogen, the species distribution of Candida spp. has changed considerably over the past decade, with a substantial emergence of non-albicans Candida spp. being seen in different health care settings worldwide (6). More recently, multidrug-resistant C. auris has emerged as an important health care-associated fungal pathogen associated with high rates of clinical treatment failure (7, 8). However, current available antifungal drugs have limitations in terms of spectrum of activity, toxicity, pharmacokinetic variability, and drug interactions. These drug limitations, along with the steady emergence of strains with resistance, prompted an expanded search for new antifungal agents with novel mechanisms of action.
APX001 is a first-in-class, intravenous (i.v.) and oral (p.o.) broad-spectrum antifungal agent in clinical development for the treatment of invasive fungal infections (9, 12). The active moiety APX001A inhibits the inositol acyltransferase in fungal GPI biosynthesis but not the human homolog, Pig-W (12). The goal of the present study was to determine the PK/PD index for this new antifungal and also the amount of drug relative to the MIC or the magnitude of the predictive pharmacodynamic index required for the treatment efficacy of APX001. In addition, we wanted to determine whether the pharmacodynamic target was similar among Candida species.
Our studies demonstrated APX001 to be active against C. albicans, C. glabrata, and C. auris, including against isolates resistant to fluconazole and micafungin. This potent activity has been observed in previous in vitro studies, with MIC values ranging from ≤0.008 to 0.016 mg/liter for C. albicans and from ≤0.008 to 0.06 mg/liter for C. glabrata. APX001A has also shown equally potent activities against fluconazole-resistant (14, 16) and caspofungin-resistant (16) Candida strains.
In vivo pharmacodynamic evaluation of antimicrobial activity requires the integration of the aforementioned in vitro potency (MIC), drug pharmacokinetics, and antimicrobial activity over time (17). A previous in vivo pharmacokinetic study with APX001 demonstrated a short half-life of 2.2 h in mice, but only at the 1-mg/kg of body weight dosage (13). Our study expanded the dose range (4 to 256 mg/kg), and a similar half-life (1.40 to 2.75 h) was observed with a linear PK profile (R2 = 0.97) across the dose range. Concentration-dependent activity has been shown in both the previous in vivo study (13) and our current study. The PK/PD indexes associated with the efficacy of antibiotics characterized by this pattern of activity are AUC/MIC and Cmax/MIC. The present study demonstrated a stronger correlation with the AUC/MIC index.
In the pharmacodynamic analyses, we considered both the protein-bound and the unbound drug concentrations, since the majority of antibacterial and antifungal pharmacodynamic experiments have shown that free-drug concentrations are more relevant. We considered the fAUC0–24/MIC to be the PK/PD index most closely correlated with treatment efficacy, based on the results of the dose fractionation experiments performed. As a first-in-class antifungal, the nonclinical PK/PD target endpoint for APX001 that correlates with the clinical response is unknown. Previous studies in this model for the triazole class have shown the relevance of the ED50 endpoint for forecasting outcomes in patients (18–21). Conversely, the net stasis endpoint in the neutropenic murine disseminated candidiasis model was predictive of the clinical outcome for the echinocandin class (22). In current study, we reported both ED50 and stasis endpoints. The fAUC0–24/MIC targets required for ED50 and stasis for C. albicans were 3.67 ± 3.19 and 20.60 ± 6.50, respectively. The targets for C. auris were relatively similar at 7.14 ± 4.54 and 14.65 ± 8.79, respectively. However, the targets for C. glabrata were significantly lower at 0.38 ± 0.21 and 1.24 ± 0.26, respectively. A similar species-specific PK/PD target difference has been observed for the echinocandins (23, 24). The stasis targets for APX001A in the current study were also comparable to those for micafungin and anidulafungin, which were near 20 (23, 25).
In summary, the present studies demonstrate that APX001 has concentration-dependent in vivo efficacy against C. albicans, C. glabrata, and C. auris. The fAUC0–24/MIC ratios were very highly associated with in vivo APX001 activity. These PD characteristics support a dosing strategy that maximizes the AUC. The identification of a lower AUC/MIC ratio target for C. glabrata than for other Candida species suggests that species-specific susceptibility breakpoints should be explored. The clinical relevance of these PK/PD targets should be considered in future clinical development of this promising antifungal.
MATERIALS AND METHODS
Organisms, media, and antibiotic.
Fourteen clinical Candida isolates were used for the in vivo treatment studies, including five C. albicans, five C. glabrata, and four C. auris isolates (Table 1), all of which belong to biosafety level 2. The organisms were chosen on the basis of their similar fitness in the animal model, as defined by the amount of growth in control animals over 24 h for C. albicans or 96 h for C. glabrata and C. auris (Table 2). We also attempted to choose strains with various susceptibilities to the study drug as well as other antifungal drug classes. The organisms were grown, subcultured, and quantified on Sabouraud's dextrose agar (SDA) plates. APX001A for in vitro studies and APX001 for in vivo studies were supplied by the study sponsor (Amplyx Pharmaceuticals, Inc., San Diego, CA). APX001A was prepared on the day of use by dissolving it with 700 mM HCl and subsequent dilution in RPMI to the required concentrations. APX001 was prepared in 0.21 M NaOH at a concentration of 100 mg/ml and then diluted with sterile 5% glucose solution to the required concentrations.
In vitro susceptibility testing.
The MICs of APX001A for the various isolates were determined using a broth microdilution method in accordance with the guidelines presented in Clinical and Laboratory Standards Institute (CLSI) document M27-A3 (26). All MIC assays were performed in duplicate on three separate occasions. The MIC values of APX001A were defined as the lowest concentration at which a prominent decrease in growth turbidity (that is, a 50% reduction in growth determined spectrophotometrically) relative to the turbidity of the compound-free control at 600 nm was observed. The median MIC from replicate assays is reported and was utilized in PK/PD analyses.
Murine disseminated candidiasis model.
Six-week-old, specific-pathogen-free, female ICR/Swiss mice weighing 23 to 27 g were used for all studies (Harlan Sprague-Dawley, Indianapolis, IN). The animals for the present studies were maintained in accordance with the criteria of the Association for Assessment and Accreditation of Laboratory Animal Care. All animal studies were approved by the Animal Research Committee of the William S. Middleton Memorial Veterans Hospital.
Mice were rendered neutropenic (neutrophils, <100/mm3) by injecting them with cyclophosphamide (Mead Johnson Pharmaceuticals, Evansville, IN) subcutaneously 4 days (150 mg/kg) and 1 day (100 mg/kg) before infection and 2 days after infection (100 mg/kg). Previous studies have shown that this regimen produces neutropenia (neutrophils, <100/mm3) in this model for the 96-h study period (27). Organisms were subcultured on SDA 24 h prior to infection. The inoculum was prepared by placing three to five colonies into 5 ml of sterile pyrogen-free 0.9% saline that had been warmed to 35°C. The final inoculum was adjusted to a 0.6 transmittance at 530 nm. The fungal counts of the inoculum determined by viable counts of C. albicans, C. glabrata, and C. auris on SDA were 6.29 ± 0.03, 6.15 ± 0.10, and 6.30 ± 0.07 log10 CFU/ml, respectively.
Disseminated infection with the Candida organisms was achieved by injection of 0.1 ml of inoculum via the lateral tail vein 2 h prior to the start of drug therapy. We utilized a treatment period of 24 h for C. albicans and 96 h for C. glabrata and C. auris to allow comparison of the effects of other antifungals with those of APX001 against these pathogens in this model. At the end of the study period, the animals were sacrificed by CO2 asphyxiation. After sacrifice, the kidneys of each mouse were removed and placed in sterile 0.9% saline at 4°C. The homogenate was then serially diluted 1:10, and aliquots were plated on SDA for viable fungal colony counts after incubation for 24 h at 35°C. The lower limit of detection was 100 CFU/ml. Results were expressed as the mean number of CFU per kidney for three mice. No-treatment and zero-hour controls were included in all experiments.
Pharmacokinetics.
Single-dose plasma pharmacokinetics of APX001A were performed in mice administered a single oral dose (0.2 ml/dose) of APX001 at dose levels of 4, 16, 64, and 256 mg/kg. Groups of three mice were sampled at each time point (seven time points, consisting of 0.25, 0.5, 1, 2, 4, 8, and 12 h) and dose level. Samples were then centrifuged for 5 min at 4,000 rpm, and plasma was removed and frozen at −20°C until assay. Plasma concentrations of APX001A (the active moiety) were determined using liquid chromatography-tandem mass spectrometry (LC-MS/MS) by the sponsor. The lower limit of detection of the LC-MS/MS assay was 10 ng/ml. Pharmacokinetic measures, including elimination half-life (t1/2), area under the concentration-time curve (AUC), and maximum plasma concentration (Cmax), were calculated using a noncompartmental model. t1/2 was determined by linear least-squares regression. AUC was calculated from the mean concentrations using the trapezoidal rule. Pharmacokinetic estimates for dose levels that were not directly measured were calculated using linear interpolation for dose levels between those with measured kinetics (e.g., between 4 and 256 mg/kg) and linear extrapolation for dose levels above or below the highest and lowest dose levels with kinetic measurements. Protein binding of 99.8% was used for estimation of free-drug concentrations, based on a report from the sponsor.
PK/PD index determination.
A dose fractionation study design was undertaken to determine the PK/PD index (AUC/MIC, Cmax/MIC, or time above the MIC) that was predictive of efficacy for APX001. Fourfold increasing doses (range, 4 to 1,024 mg/kg) of APX001 were fractionated into dosing regimens in which the drug was administered every 3 h (q3h), every 6 h (q6h), every 8 h (q8h), and every 12 h (q12h). Mice were infected with isolate C. albicans K1 as described above and administered APX001 by oral gavage. After 24 h the mice were euthanized and the CFU count in the kidneys was determined. To determine which PK/PD index was most closely linked with efficacy, the number of yeast cells in the kidneys at the end of 24 h of therapy was correlated with (i) the 24-h free-drug AUC/MIC ratio (fAUC/MIC), (ii) the free-drug Cmax/MIC ratio (fCmax/MIC), and (iii) the percentage of the dosing interval during which plasma free-drug levels exceeded the MIC for each of the dosage regimens studied (TMIC). The correlation between efficacy and each of the three PK/PD indices was determined by nonlinear regression based upon the Hill equation: E = (Emax × DN)/(ED50N + DN), where E is the effector, in this case, the log10 change in the number of CFU per kidney between treated mice and untreated controls after the 24-h period of study; Emax is the maximum effect; D is the 24-h total dose; ED50 is the dose required to achieve 50% of the Emax; and N is the slope of the dose-effect curve. The values for the pharmacodynamic parameters Emax, ED50, and N were calculated using nonlinear least-squares regression. The coefficient of determination (R2) was used to estimate the variance that might be due to regression with each of the PK/PD indices.
PK/PD index magnitude studies.
Dose-response experiments using the disseminated candidiasis model were performed for five C. albicans, five C. glabrata, and four C. auris isolates as described above. The dose range consisted of 4-fold increases (range, 1 to 256 mg/kg/6 h) in drug concentration with administration by the oral route. The treatment duration was 24 h for C. albicans and 96 h for C. glabrata and C. auris. The dose-response relationships were quantified, and the relationship between the PK/PD index fAUC/MIC and treatment efficacy was determined using the sigmoid Emax model. These PK/PD relationships were examined utilizing the plasma total and free-drug concentrations from pharmacokinetic studies. The coefficient of determination (R2) from this model was used to assess the strength of this relationship. The doses required to produce 50% of the maximal effect (ED50) and a net static effect (static dose) compared to the number of CFU at the start of therapy for multiple pathogens in the disseminated candidiasis model were determined utilizing the plasma total and free-drug concentrations and the following equation: log10 D = {log10[E/(Emax − E)]/N} + log ED50, where E is the control growth for dose (D) and E is the control. The associated 24-h total and free-drug AUC/MIC targets were calculated.
REFERENCES
- 1.Antinori S, Milazzo L, Sollima S, Galli M, Corbellino M. 2016. Candidemia and invasive candidiasis in adults: a narrative review. Eur J Intern Med 34:21–28. doi: 10.1016/j.ejim.2016.06.029. [DOI] [PubMed] [Google Scholar]
- 2.Rodloff C, Koch D, Schaumann R. 2011. Epidemiology and antifungal resistance in invasive candidiasis. Eur J Med Res 16:187–195. doi: 10.1186/2047-783X-16-4-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pfaller MA, Diekema DJ. 2007. Epidemiology of invasive candidiasis: a persistent public health problem. Clin Microbiol Rev 20:133–163. doi: 10.1128/CMR.00029-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Horn DL, Neofytos D, Anaissie EJ, Fishman JA, Steinbach WJ, Olyaei AJ, Marr KA, Pfaller MA, Chang CH, Webster KM. 2009. Epidemiology and outcomes of candidemia in 2019 patients: data from the prospective antifungal therapy alliance registry. Clin Infect Dis 48:1695–1703. doi: 10.1086/599039. [DOI] [PubMed] [Google Scholar]
- 5.Kullberg BJ, Arendrup MC. 2015. Invasive candidiasis. N Engl J Med 373:1445–1456. doi: 10.1056/NEJMra1315399. [DOI] [PubMed] [Google Scholar]
- 6.Pfaller MA, Diekema DJ, Gibbs DL, Newell VA, Ellis D, Tullio V, Rodloff A, Fu W, Ling TA, Global Antifungal Surveillance Group. 2010. Results from the ARTEMIS DISK Global Antifungal Surveillance Study, 1997 to 2007: a 10.5-year analysis of susceptibilities of Candida species to fluconazole and voriconazole as determined by CLSI standardized disk diffusion. J Clin Microbiol 48:1366–1377. doi: 10.1128/JCM.02117-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chowdhary A, Voss A, Meis JF. 2016. Multidrug-resistant Candida auris: ‘new kid on the block’ in hospital-associated infections? J Hosp Infect 94:209–212. doi: 10.1016/j.jhin.2016.08.004. [DOI] [PubMed] [Google Scholar]
- 8.Clancy CJ, Nguyen MH. 2017. Emergence of Candida auris: an international call to arms. Clin Infect Dis 64:141–143. doi: 10.1093/cid/ciw696. [DOI] [PubMed] [Google Scholar]
- 9.Gonzalez-Lara MF, Sifuentes-Osornio J, Ostrosky-Zeichner L. 2017. Drugs in clinical development for fungal infections. Drugs 77:1505–1518. doi: 10.1007/s40265-017-0805-2. [DOI] [PubMed] [Google Scholar]
- 10.Osherov N, Kontoyiannis DP. 2017. The anti-Aspergillus drug pipeline: is the glass half full or empty? Med Mycol 55:118–124. doi: 10.1093/mmy/myw060. [DOI] [PubMed] [Google Scholar]
- 11.Osherov N. 2012. The top three areas of basic research on Aspergillus fumigatus in 2011. Ann N Y Acad Sci 1273:74–77. doi: 10.1111/j.1749-6632.2012.06798.x. [DOI] [PubMed] [Google Scholar]
- 12.Watanabe NA, Miyazaki M, Horii T, Sagane K, Tsukahara K, Hata K. 2012. E1210, a new broad-spectrum antifungal, suppresses Candida albicans hyphal growth through inhibition of glycosylphosphatidylinositol biosynthesis. Antimicrob Agents Chemother 56:960–971. doi: 10.1128/AAC.00731-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hata K, Horii T, Miyazaki M, Watanabe NA, Okubo M, Sonoda J, Nakamoto K, Tanaka K, Shirotori S, Murai N, Inoue S, Matsukura M, Abe S, Yoshimatsu K, Asada M. 2011. Efficacy of oral E1210, a new broad-spectrum antifungal with a novel mechanism of action, in murine models of candidiasis, aspergillosis, and fusariosis. Antimicrob Agents Chemother 55:4543–4551. doi: 10.1128/AAC.00366-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miyazaki M, Horii T, Hata K, Watanabe NA, Nakamoto K, Tanaka K, Shirotori S, Murai N, Inoue S, Matsukura M, Abe S, Yoshimatsu K, Asada M. 2011. In vitro activity of E1210, a novel antifungal, against clinically important yeasts and molds. Antimicrob Agents Chemother 55:4652–4658. doi: 10.1128/AAC.00291-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pfaller MA, Duncanson F, Messer SA, Moet GJ, Jones RN, Castanheira M. 2011. In vitro activity of a novel broad-spectrum antifungal, E1210, tested against Aspergillus spp. determined by CLSI and EUCAST broth microdilution methods. Antimicrob Agents Chemother 55:5155–5158. doi: 10.1128/AAC.00570-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pfaller MA, Hata K, Jones RN, Messer SA, Moet GJ, Castanheira M. 2011. In vitro activity of a novel broad-spectrum antifungal, E1210, tested against Candida spp. as determined by CLSI broth microdilution method. Diagn Microbiol Infect Dis 71:167–170. doi: 10.1016/j.diagmicrobio.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 17.Craig WA. 1998. Pharmacokinetic/pharmacodynamic parameters: rationale for antibacterial dosing of mice and men. Clin Infect Dis 26:1–10. doi: 10.1086/516284. [DOI] [PubMed] [Google Scholar]
- 18.Andes D, van Ogtrop M. 1999. Characterization and quantitation of the pharmacodynamics of fluconazole in a neutropenic murine disseminated candidiasis infection model. Antimicrob Agents Chemother 43:2116–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Andes D, Marchillo K, Stamstad T, Conklin R. 2003. In vivo pharmacodynamics of a new triazole, ravuconazole, in a murine candidiasis model. Antimicrob Agents Chemother 47:1193–1199. doi: 10.1128/AAC.47.4.1193-1199.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Andes D, Marchillo K, Stamstad T, Conklin R. 2003. In vivo pharmacokinetics and pharmacodynamics of a new triazole, voriconazole, in a murine candidiasis model. Antimicrob Agents Chemother 47:3165–3169. doi: 10.1128/AAC.47.10.3165-3169.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Andes D, Marchillo K, Conklin R, Krishna G, Ezzet F, Cacciapuoti A, Loebenberg D. 2004. Pharmacodynamics of a new triazole, posaconazole, in a murine model of disseminated candidiasis. Antimicrob Agents Chemother 48:137–142. doi: 10.1128/AAC.48.1.137-142.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Andes D, Ambrose PG, Hammel JP, Van Wart SA, Iyer V, Reynolds DK, Buell DN, Kovanda LL, Bhavnani SM. 2011. Use of pharmacokinetic-pharmacodynamic analyses to optimize therapy with the systemic antifungal micafungin for invasive candidiasis or candidemia. Antimicrob Agents Chemother 55:2113–2121. doi: 10.1128/AAC.01430-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Andes DR, Diekema DJ, Pfaller MA, Marchillo K, Bohrmueller J. 2008. In vivo pharmacodynamic target investigation for micafungin against Candida albicans and C. glabrata in a neutropenic murine candidiasis model. Antimicrob Agents Chemother 52:3497–3503. doi: 10.1128/AAC.00478-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Andes D, Diekema DJ, Pfaller MA, Bohrmuller J, Marchillo K, Lepak A. 2010. In vivo comparison of the pharmacodynamic targets for echinocandin drugs against Candida species. Antimicrob Agents Chemother 54:2497–2506. doi: 10.1128/AAC.01584-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Andes D, Diekema DJ, Pfaller MA, Prince RA, Marchillo K, Ashbeck J, Hou J. 2008. In vivo pharmacodynamic characterization of anidulafungin in a neutropenic murine candidiasis model. Antimicrob Agents Chemother 52:539–550. doi: 10.1128/AAC.01061-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clinical and Laboratory Standards Institute. 2008. Reference method for broth dilution antifungal susceptibility testing of yeasts, 3rd ed Approved standard M27-A3 Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 27.Andes D. 2005. Use of an animal model of disseminated candidiasis in the evaluation of antifungal therapy. Methods Mol Med 118:111–128. [DOI] [PubMed] [Google Scholar]