


Abstract
In-depth and reproducible protein measurement in many biological samples is often critical for pharmaceutical/biomedical proteomics but remains challenging. MS1-based quantification using quadrupole/ultrahigh-field Orbitrap (Q/UHF-Orbitrap) holds great promise, but the critically important experimental approaches enabling reliable large-cohort analysis have long been overlooked. Here we described an IonStar experimental strategy achieving excellent quantitative quality of MS1 quantification. Key features include: (i) an optimized, surfactant-aided sample preparation approach provides highly efficient (>75% recovery) and reproducible (<15% CV) peptide recovery across large cell/tissue cohorts; (ii) a long column with modest gradient length (2.5 h) yields the optimal balance of depth/throughput on a Q/UHF-Orbitrap; (iii) a large-ID trap not only enables highly reproducible gradient delivery as for the first time observed via real-time conductivity monitoring, but also increases quantitative loading capacity by >8-fold and quantified >25% more proteins; (iv) an optimized HCD-OT markedly outperforms HCD-IT when analyzing large cohorts with high loading amounts; (v) selective removal of hydrophobic/hydrophilic matrix components using a novel selective trapping/delivery approach enables reproducible, robust LC–MS analysis of >100 biological samples in a single set, eliminating batch effect; (vi) MS1 acquired at higher resolution (fwhm = 120 k) provides enhanced S/N and quantitative accuracy/precision for low-abundance species. We examined this pipeline by analyzing a 5 group, 20 samples biological benchmark sample set, and quantified 6273 unique proteins (≥2 peptides/protein) under stringent cutoffs without fractionation, 6234 (>99.4%) without missing data in any of the 20 samples. The strategy achieved high quantitative accuracy (3–6% media error), low intragroup variation (6–9% media intragroup CV) and low false-positive biomarker discovery rates (3–8%) across the five groups, with quantified protein abundances spanning >6.5 orders of magnitude. Finally, this strategy is straightforward, robust, and broadly applicable in pharmaceutical/biomedical investigations.
Keywords: quantitative proteomics, LC–MS, label-free, MS1-based, proteomics, large cohorts, missing data, false-positive biomarker discovery rate, Orbitrap, ion current
Graphical abstract
INTRODUCTION
For quantitative proteomic research in biomedical and pharmaceutical fields, it is often critical to employ a relatively large number of biological replicates (e.g., patients, animals or various cell conditions) to enhance quantitative reliability and statistical power, and to minimize the false-positive biomarker discovery arising from the typical large interindividual variability.1,2 Label-free approaches are often preferred over labeling methods owing to the theoretically unlimited replicate capacity, more flexible sample preparation options and lower cost.3–7 However, it is challenging to achieve reliable quantification of larger biological cohorts (e.g., ≥20) using label-free strategies.8 First, highly robust, reproducible and well-controlled sample preparation, LC separation and MS analysis across large cohorts is critical but difficult to achieve with most existing label-free analysis strategies.4,9,10 Second, missing data remains a prominent issue for both labeling and label-free approaches.11,12 While missing data may arise from biological factors, currently the vast majority of missing data in quantitative proteomics are due to technical reasons.13,14 Conceivably, poor efficiency and reproducibility in sample preparation and LC–MS analysis would significantly contribute to missing data.15 Another primary source is the quantitative strategy; for instance, most MS2-based quantification methods (e.g., spectral counting, MS2 total ion current) show high missing data arising from poor reproducibility of data-dependent acquisition (DDA).11 To enable more reproducible proteomic measurements, MS2-based “data-independent acquisition (DIA)” methods were developed.11,16 Though representing a ground-breaking advance, these methods often carry limitations associated with the high-interference spectra17 and limited depth.18
Another label-free quantitative strategy is based on MS1 precursor chromatographic peak areas (i.e., ion current-based approach).14 Although DDA is often used to assign peptide ID to quantitative features (i.e., MS1 ion current peak areas of the same peptide across all samples), the quantitative features themselves are acquired in a strictly non-data-dependent manner. Therefore, MS1 strategy has a great potential to alleviate the missing data problem that plagues DDA-based proteomics quantification.15
Optimal experimental strategies are essential to fully exploit these potentials; however this issue has long been overlooked and thereby not yet been extensively investigated. For example, it remains challenging for most methods to analyze large cohorts due to suboptimal experimental efficiency and reproducibly across many biological samples;8,9 moreover, the reported missing-value-rates of popular MS1-based quantitative methods were substantially >10% in >10 technical replicates.11,19 Previously we reported a reproducible ion current-based workflow with much lower missing data, but with quite limited depth of proteomics analysis (e.g., quantification of <2000 proteins in human cell lysates).20 Furthermore, three additional issues have not been addressed: first, we observed using a trap substantially promoted run-to-run reproducibility for LC separation, but the underlying mechanisms were not well-characterized; second, it is possible to perform a selective trapping/delivery strategy to improve reproducibility, sensitivity and robustness, but this potential has not been exploited;15 third, it was reported the large-ID trap enables loading of higher amount of peptide digests,21 but it is not clear as to the effects of larger loading mass on quantitative sensitivity of peptides of different polarities.
Finally, the use of quadrupole/ultrahigh-field Orbitrap (Q/UHF-Orbitrap) provides substantially higher MS resolution, scan speed and sensitivity than the lower-field instruments,22 which holds great promises for in-depth and reproducible MS1-based proteomics analysis. However, so far the vast majority of developmental efforts on this platform is centered on achieving more peptide identifications in a few of samples.23,24 Despite of the tremendous advances in this regard,23,24 the existing strategies are not likely suitable for MS1 quantification in larger cohorts. Moreover, as the key parameters of MS1-based strategy are heavily dependent on the performance and the characteristics of the MS analyzer,14 an extensive investigation of the experimental approaches in order to take advantage of this new platform in MS1 quantification, is highly desirable but have not been adequately addressed thus far. To name a few examples: (i) previously we reported a 7-h gradient on a long column is optimal on a lower-field Orbitrap;15,25 however, the characteristics of Q/UHF-Orbitrap may shift the optima, e.g., higher-resolution MS1 markedly enhance the quantification and identification selectivity and faster MS scans may shorten the duty cycle thus reducing the need of long-gradient separation; (ii) recent works showed HCD-IT provided the highest ID number among several fragmentation/scan options on a tribrid UHF-Orbitrap instrument;23 whether this holds true for a large cohort and larger loading amounts remains to be examined; (iii) previously up to 60 k resolution for MS1;7,15,25 conceivably using higher MS1 resolution, as made practical by the new high-field-Orbitrap, could enhance quantitative selectivity and decrease noise. This potential has not been evaluated yet.
In this study, we investigated comprehensively the key experimental parameters including these for sample preparation, LC separation and MS analysis on the MS1 quantification of larger cohort, and developed a new “IonStar” experimental pipeline enabling consistent, sensitive and reliable data acquisition for many biological replicates, and thus contributing to in-depth proteomic quantification with high accuracy, precision, reproducibility, and low missing data and low false-positives. The performance in quantification of the changes of lower-abundance proteins was evaluated by analyzing a 20 samples, 5 groups sample set.
EXPERIMENTAL PROCEDURES
Sample Sets Used in This Study
Human Cell Samples for Development of Sample Preparation, Separation and MS Identification/Quantification
To benchmark and optimize the methods, a series of protein lysates from human MIA PaCa-2 cell line were employed. For evaluation of the sample preparation efficiency and reproducibility, we prepared 48 human MIA PaCa-2 cell samples that were treated by different regimen and collected at different time points. For development and optimization of LC and MS methods, a pooled sample is used. Additional details are in Supplementary Methods.
Spike-In Sample Set Benchmarking Quantitative Performance
In order for an extensive evaluation of the quantitative performances of the developed strategy, we prepared a set of spike-in samples by spiking small, variable levels of DH5α E. coli digest (representing altered proteins) into a large, constant level of human cell Panc-1 cell digest, as shown in Supplementary Figure S5. Protein extraction, digestion, and LC–MS analysis were performed using optimized procedures specified below. There are five groups each with a different level of spiked-in E. coli digest: 1 fold (labeled A), 1.5 folds (B), 2 folds (C), 2.5 folds (D) and 3 folds (E), with four replicates per group (N = 4) and totally 20 samples. Samples were analyzed by LC–MS in an alternating sequence in order to avoid bias. Group A was used as control group for ratio calculation. p-value of each comparison pair was calculated by two-sample Student’s t test.
Protein Extraction
Briefly, samples were placed in an ice-cold lysis buffer (containing 50 mM Tris-FA, 150 mM NaCl, 0.5% sodium deoxycholate and 2% SDS, 1.5% IGEPAL CA-630, pH 8.0) including Complete protease inhibitor cocktail tablet and PhosSTOP phosphatase inhibitor cocktail tablet (Roche Applied Science, Indianapolis, IN). The mixture was homogenized with a Polytron homogenizer (Kinematica AG, Switzerland) by repeating the homogenization (at 15 000 rpm, 5–10 s) and cooling (about 20 s) cycles for 5–10 times. Sonication was then performed using a probe. Each sonication cycle takes 20s, whereas each sample subjects to 3–5 cycles until the solution became pellucid. Then the mixture was centrifuged at 20 000g under 4 °C for 1 h. The supernatant was carefully transferred, and the protein concentration for each sample was determined by bicinchoninic acid assay (BCA) kit (Pierce Biotechnology, Inc., Rockford, IL) before storage under −80 °C.
Surfactant-Aided On-Pellet Digestion
Portions of 100 µg total proteins from each sample were transferred and then reduced by addition of 5 mM Dithiothreitol (DTT) for 30 min, followed by alkylation by addition of 20 mM iodoacetamide (IAA) and incubated in darkness for 30 min. The reduction and alkylation of proteins were both conducted under 37 °C with rigorous oscillation in an Eppendorf Thermomixer (Eppendorf, Hauppauge, NY) at 200 rpm. The proteins were precipitated by stepwise addition of 6 volumes of chilled acetone with incessant vortexing, and were incubated under −20 °C for >4 h. After centrifugation at 20 000g under 4 °C for 30 min, the supernatants were discarded and the pellets containing precipitated proteins were washed with 500 µL of chilled acetone/water mixture (85/15, v/v %) and left to partially air-dry. For the on-pellet digestion, trypsin at an enzyme/substrate ratio of 1:20 (w/w), dissolved in 100 µL of Tris buffer (50 mM, pH 8.5), was added to the precipitated protein pellets, and the mixture was incubated under 37 °C for 6 h with constant vortexing in an Eppendorf Thermomixer. Digestion was terminated by addition of 1% formic acid.
Nano LC/UHF-Orbitrap LUMOS MS Analysis
The optimized LC–MS conditions are shown here, while the process of optimization is in the following sections. The nano-RPLC (reverse-phase liquid chromatography) system is consisted of a Dionex Ultimate 3000 nano-LC system and an Ultimate 3000 gradient micro-LC system with a WPS-3000 autosampler. The flow set up is shown in Supplementary Figure S2. A GE Healthcare Life Sciences (Princeton, NJ) zero-dead-volume conductivity sensor (taken from a MDLC system and data collected using Unicorn package) was connected between trap and column for debugging and real time flow monitoring. Mobile phase A and B were 0.1% formic acid in 2% acetonitrile and 0.1% formic acid in 88% acetonitrile, respectively. Four µL of samples containing 4 µg peptides were loaded onto a reversed-phase trap (300 µm ID × 5 mm), with 1% mobile phase B at a flow rate of 10 µL/min, and the trap was washed for 3 min. A series of nano flow gradients (flow rate at 250 nL/min) was used to back-flush the trapped samples onto the nano-LC column (75-µm ID × 100 cm, packed with 3 µm Peptidemap C18) for separation, and the optimized gradient profile was as following: 4–13% B over 15 min; 13% to 28% B over 110 min; 28% to 44% B over 5 min; 44% to 60% B over 5 min; 60% to 97% B in 1 min, and finally isocratic at 97% B for 17 min. The trap was switched offline at 45 min to prevent hydrophobic matrix components from entering the column. The nano-LC column was heated at 52 °C to improve both chromatographic resolution and reproducibility. An Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific, San Jose, CA) was used for MS analysis. For detailed MS parameters, see Supplementary Methods.
Protein Identification and Quantification
We employed a stringent set of criteria for protein identification, including low peptide and protein FDR (<1%) and ≥2 peptides per protein. An ion current-based data processing method (IonStar processing pipleline) was employed for MS1 quantification in large cohorts. More details are in Supplementary Methods.
RESULTS AND DISCUSSION
1. Development and Optimization of the IonStar Experimental Procedures for MS1 Quantification Based on an UHF-Orbitrap Platform
Here we extensively developed and optimized the IonStar experimental procedures for MS1 quantification, including (i) a new sample preparation protocol enabling reproducible and robust extraction, cleanup and digestion of biological samples and providing high and consistent protein and peptide recovery; (ii) nanoflow LC related developments (e.g., investigations of gradient delivery stabilization and enhanced quantitative loading capacity, and a selective trapping/delivery strategy) to achieve sensitive, in-depth and robust analysis of many samples with high run-to-run reproducibility; and (iii) optimal MS detection approaches for selective and sensitive procurement of high-resolution MS1 ion currents as well as extensive peptide identification; as the Q/UHF-Orbitrap affords dramatically improved sensitivity, cycle-time and rapid high-resolution measurement over lower-field-Oribitrap and thus substantially changes the detectable proteomics landscape,23,26 we developed new optimal MS approaches for MS1 ion current-based quantification using this platform. Taken together, these elements would provide a solid foundation for accurate, reproducible and in-depth ion current-based quantification in larger cohorts.
1.1. Surfactant-Aided Precipitation/On-Pellet-Digestion for Efficient and Reproducible Sample Preparation
To achieve reliable proteomic profiling of larger biological cohorts, it is critically important to develop a robust sample preparation procedure for efficient and reproducible proteins extraction, cleanup and digestion. Previously we reported an overnight on-pellet-digestion method for reproducible sample preparation;27 recently, our lab has described a surfactant-aided-precipitation/on-pellet-digestion(SOD) strategy for targeted LC/SRM-MS analysis of protein drugs, which affords rapid digestion of a target mAb in plasma and tissues.28 Nonetheless, this method has not been investigated in proteomics studies. Here we modified this protocol (e.g., investigation of detergent composition, proteolytic conditions, etc.) to attain optimal sample treatment for global proteomics quantification. Briefly, cell or tissue samples are thoroughly lysed using polytron homogenizer and sonication in a strong buffer containing an optimized cocktail of ionic and nonionic detergents (i.e., IGEPAL CA-630, SDS, sodium deoxycholate, details in Experimental Procedures). This procedure was found to achieve efficient disruption of cellular structures, as well as exhaustive and consistent protein extraction including membrane proteins. A following organic solvent precipitation step removes detergents and nonprotein components. The use of buffered detergent-cocktail prior to precipitation effectively removed detrimental matrix components such as phospholipids and fatty acids, as we showed previously,28 and the thorough protein denaturation by the detergents and precipitation permits robust and effective proteolytic cleavage. Although it was previously found an efficient on-pellet-digestion of antibody drug was achieved in only 30 min at 37 °C,28 for proteomics, we observed it takes 2 h to obtain the maximal protein identification numbers using a nano-LC coupled to a lower-field-Oritrap, while 6 h for a Q/UHF-Orbitrap (data not shown), likely due to the longer time required to accomplish digestion of low-abundance, hydrophobic proteins that were not detectable with lower-field-Orbitrap. The optimized SOD procedure provides ~20–40% more peptide identifications than popular methods such as in-solution digestion, in-gel digestion, FASP, and precipitation/on-pellet-digestion we described previously.15 An example for treating large-cohorts of biological samples is shown in Figure 1. The optimized SOD method achieved high and reproducible protein and peptide recoveries, robustly across the 48 biological samples (with >75% recovery and <15% CV, sample details are in Supplementary Methods). This level of efficiency and reproducibility constitutes a solid foundation for reliable large-cohort analysis.
Figure 1.

Efficient and reproducible sample processing strategy across many biological samples. In this example, 48 pancreactic cancer cell samples were collected at 16 different treatment/time groups (N = 3/group). Detailed information on this set is in Experimental Procedures and Supplementary Methods. (A) Protein yields by group using the detergent-cocktail extraction procedure with polytron/sonication; (B) peptide recovery in each group using the surfactant-aided precipitation/on-pellet digestion (SOD) procedure developed for proteomics in this study.
1.2. Chromatographic and Trapping Strategies for Extensive, Highly Reproducible and Sensitive Analysis Across Large Cohorts
Extensive, reproducible and robust chromatographic separation across all biological samples is another critical requirement for reliable label-free quantification. Previously we reported a trap-column system featuring 7-h gradient separation, zero-dead-volume (ZDV)-Tee liquid junction and 2-µm particles.15 Nonetheless, the effects of configurations on separation reproducibility was not well understood or optimized at the time. In this study, we extensively investigated the mechanisms underlying separation reproducibility and devised a new, optimal chromatographic procedure. In the new design, a large-ID trap was placed on a ZDV valve, and bidirectional flows for loading and analysis were employed for reliable peak compression while preventing hydrophobic and hydrophilic components from entering the nano-LC–MS. An online nanoscale conductivity sensor was used to evaluate gradient delivery and for real-time debugging. The schematics is in Supplementary Figure S1. Moreover, it was discovered the use of 3-µm rather than 2-µm particles carries markedly better robustness for analysis of large cohorts, with a slight trade-off of peak capacity that was later found not critical. This design was found to be very resistant to contamination and individual variations in biological matrices (as is often the case in most clinical/preclinical studies), and provides rugged quantitative analysis of >100 complex biological samples in a single set (cf. Figure 2E).
Figure 2.

The use of large-ID trap enables reproducible and robust LC–MS analysis of large biological cohort. Conductivity profiles of three consecutive 2-h runs (A) without and (B) with a 300 µm-ID trap, as monitored by an online conductivity sensor placed in front of the column. By providing homogeneous mobile phase mixing and dampened pump noise, the large-ID trap achieves high run-to-run reproducibility in gradient delivery to the downstream nano column. The use of large-ID trap with selective trapping/delivery strategy also prevents hydrophilic and hydrophobic matrix components from compromising the nano-LC–MS system, and thus enabling robust analysis of many biological samples. For example, (C) shows a conductivity spike after injection of a cell digest, indicative of intensive hydrophilic matrix components (e.g., salts and polar organic molecules) entering the LC–MS system without using the large-ID trap; conversely, in (D), using a large-ID trap eliminated such spike. In (C) and (D) high-flow binary pumps with online mixer and flow splitting were used to eliminate the effects of mixing. (E) shows it is feasible to switch the trap off the column at 45 min during a 180 min gradient to remove hydrophobic components without losing peptide ID for a human cell sample; (F) shows selective delivery at 45 min resulted in significantly improved robustness than less-selective methods, <15% signal decrease for analysis of 100 biological samples.
1.2.1. Effects of Column and Gradient Lengths on Depth of Identification
Since most MS1-based methods rely on MS2 fragmentation information to assign peptide ID to quantified features, an in-depth MS2 identification is important. Previously we reported a 100 cm-long column with a 7-h gradient provided optimal identification when a low-field LTQ/Orbitrap (without quad isolation) was employed.10,15,29 Nonetheless, the much higher sensitivity, depth and cycle speed by the hybrid Q/UHF-Orbitrap30 likely shifts the optima of chromatographic conditions. Here we optimized the column length and gradient time by balancing the considerations of throughput and depth of proteomic analysis. Four column lengths (25–100 cm) and four gradient times (40 min to 5 h) were investigated using human cell digests (N = 3 replicates per condition). Representative results are shown in Supplementary Figure S2. With the exception of the 40 min gradient, it was found protein ID increases significantly (p < 0.01) with the length of column, and thus the 100 cm column was determined optimal. Although a 5-h gradient yields the highest protein ID, the increase of ID from 2.5-h to 5-h gradient is much less drastic than those from 1.5 to 2.5-h gradient. This observation is quite different from the observations on a lower-field Orbtirap, where longer gradients provided substantial benefits over shorter gradients.15 We speculate this is because the much faster scan rate, parallelizable MS1/MS2 processes, and higher sensitivity by the Quadrupole-UHF Orbitrap markedly decreases the need of achieving very high peak capacity. Considering the importance of a reasonable throughput for analysis of large cohorts, the 2.5-h gradient was chosen.
1.2.2. A Large-ID Trap and a Selective Trapping/Delivery Strategy Enabled Robust and Reproducible Separation of Large Cohorts
Previously we observed the use of a trap upstream of the nanocolumn may help to improve the reproducibility of nano-LC separation,15 but the underlying mechanism has yet not been fully investigated, nor was this approach further developed or optimized.
Here we hypothesized the use of a large-ID trap may contribute to high LC–MS analytical reproducibility and robustness in two ways: first, the trap could enable reproducible gradient delivery to the downstream column by providing homogeneous mobile phase mixing; second, a selective peptide trapping/delivery strategy can prevent detrimental hydrophobic and hydrophilic matrix components from entering the nano-LC–MS system, and thus effectively improving the reproducibility and robustness for both chromatographic separation and MS1 signal detection.
To examine the first point, we connected a ZDV conductivity sensor to the inlet of the column to monitor the consistency of gradient delivery in a real-time manner (Supplementary Figure S2). Figure 2A and B shows conductivity profiles for three consecutive runs without and with trap implemented, respectively. When the large-ID trap was not used in the setup, the fluctuating conductivity profiles (Figure 2A) reflect irreproducible gradient delivery to the column, most likely due to incomplete gradient mixing and pump noises (prevalent problems for typical nano-LC systems7,15), which compromises LC–MS reproducibility by causing not only variations of peptide retention times owing to peptide’s strong dependency on the retention factor from the mobile phase composition,31 but also fluctuation in ionization arising from the micro-pulsation of the liquid phase compressibility and conductivity. Conversely, shown in Figure 2B, the use of an upstream large-ID trap enabled homogeneous mixing and dampened pump noise, as indicated by the smooth and reproducible conductivity profiles. As it is impractical to make a nanoscale mobile phase mixer, using a trap to enable homogeneous gradient mixing at nanoflow is a straightforward solution, which also provides benefits of selective sample clean up and enhanced analytical sensitivity (discussed below). Traps with a length of 5 mm and ID of 75–500 µm were evaluated and a 300 µm ID trap (i.e., 4:1 ID ratio for trap: column) was chosen based on balanced considerations on mixing efficiency, delay volume and loading capacity.
To examine the second point, we developed a novel selective trapping/delivery approach with the large-ID trap. The purpose is efficient recovery of peptides while preventing hydrophilic and hydrophobic matrix/buffer components from entering the nano-LC–MS system, which would otherwise severely compromise analytical reproducibility especially when analyzing a large number of biological samples. Injection without a trap introduces salts and polar compounds into the LC–MS, as evident by the >15 min spike of conductivity (Figure 2C), whereas this effect was eliminated by using the large-ID trap using a low-organic buffer (Figure 2D). More importantly, accumulation of components much more hydrophobic than typical peptides such as lipids and fatty acids on the column could rapidly deteriorate chromatographic and ionization performances.32 To remove these compounds, we experimentally identified the optimal trap-to-column delivery time, i.e. when the peptides are completely delivered from the trap to the column while more hydrophobic nonprotein components remain on the trap. Right at this time point, we switch the trap offline and employed high-organic mobile phase to flush the hydrophobic compounds to waste. We investigated delivery time from 15 to 120 min, and typical results of peptide ID vs delivery time is shown in Figure 2E. Significant loss of peptide ID were not observed until the delivery time was shortened to 30 min, suggesting 45 min is sufficient to deliver peptides from trap to column; this notion is supported by peptide intensity data as well (not shown). Interestingly, delivery time of 45 and 65 min resulted in more peptide identification (p < 0.01) than 120 min delivery (i.e., representing a nonselective delivery). This phenomenon was repetitively observed in different experimental settings. Further investigation suggested the decreased matrix effect by shorter delivery time most likely contributes to the increased ID. Consequently, 45 min delivery is determined optimal. This selective delivery approach substantially improves the robustness and reproducibility of LC–MS analysis of large cohorts. As shown in Figure 2F, with 45 min delivery, only <15% signal drop for analysis of >100 biological samples was observed without appreciable loss of chromatographic resolution (data not shown), indicating excellent capacity to analyze large biological cohorts. Such minor drop in intensity can be readily corrected by a normalization step. By comparison, using 120 min delivery only <20 samples can be reliably analyzed (assuming signal drop >20% being unacceptable). Such high level of LC–MS reproducibility constitutes another solid foundation for robust large-cohort analysis.
1.2.3. Increased MS1 Quantitative Sensitivity by High-Capacity Loading on the Large-ID Trap
The sensitivity of nano-LC is often limited by the low loading capacity for biological samples.33 Here we speculated the large-ID trap in conjunction with a relatively shallow gradient provides significantly improved loading capacity and thus the quantitative sensitivity for MS1-based quantification. To examine this hypothesis, we tested the quantitative performance of this system by injecting different amounts of a pooled human cell digest. We define that a loading amount exceeds the quantitative loading capacity of the system if one of the following is violated: (i) < 20% peptide peak broadening compared to a low, 0.25-µg loading; and (ii) S/N of peptides increases linearly with the loading amounts. As evident in Figure 3A and B, hydrophilic peptides (i.e., peptides eluting in the first 15 min of the elution window) are more susceptible to overcapacity most likely due to their lower affinity to the C18 sorbents; therefore these peptides determine the quantitative loading capacity. Without using a large-ID trap, compromised quantitative linearity was observed at 1 µg loading, and a rational quantitative loading capacity determined as ~0.5 µg (Figure 3A). Conversely, when the large-ID trap was employed, the S/N of hydrophilic peptides is almost linear for loading of up to 6 µg peptides, without appreciable peak broadening (Figure 3A). To ensure the robustness of the procedure, an optimal loading amount of 4 µg peptides is adopted in the current system. The high loading capacity markedly improves the sensitivity of ion current measurement, as exemplified in Figure 3B. Owing to the improved sensitivity, the 4-µg loading led to >25% more protein ID and >26.7% peptide ID than 0.5-µg loading without a trap, as shown in Supplementary Figure S3.
Figure 3.

Enhanced depth and quantitative sensitivity by high-capacity loading on the large-ID trap. A human cell digest sample was used (N = 3/condition). Hydrophilic, midpolarity, and hydrophobic peptides refer to groups of 10 randomly selected peptides respectively eluting within the first 5 min, center 5 min and the last 5 min of the entire peptide elution window. (A) Mean normalized MS1 intensity in each group as a function of total peptide loading amounts without trap and with the large-ID trap. Optimal loading amounts (i.e., upper limit of the quantitative loading, without causing >20% peak broadening or intensity does not increase linearly with the loading amount) are 0.5 µg when without trap, whereas 4 µg with trap. The loading capacity appears to be determined by hydrophilic peptides and an optimal loading amount of 4 µg was chosen for this system. (B) Exemplifies the linear increase of intensity of peptide ion-currents with higher loading amounts, resulting in improved sensitivity.
1.3. Optimal MS2 Identification Approach under High Loading Amount and Larger Sample Numbers
For ion current-based quantification, peptide/protein identification is important to assign peptide ID of the quantified ion current peaks. Although it was reported that HCD-IT (i.e., HCD fragment scan performed in a dual-cell ion-trap) achieved the best protein ID number among various choices on a hybrid Q/UHF-Orbitrap using 1-µg loading and without using a trap,23 the high-capacity loading achieved in this study and the use of lager cohorts may shift the dynamics of MS2 data acquisition. Consequently, we investigated the performance of various fragmentation and detection strategies in this study.
In line with previous observations,23 we found that MS2 fragmentation using quadrupole isolation and HCD generated slightly more identifications than in-trap-CID (data not shown), which is probably because HCD tends to offer more comprehensive backbone fragmentations and reduced window effects. We then compared the two analyzers for MS2 scan (i.e., dual-cell ion trap, IT vs Orbitrap, OT), while the MS1 scans were performed with OT in both cases. Despite that IT provides much higher scan rate and better absolute sensitivity (i.e., more detectable MS2 fragments) than OT, its resolution and mass accuracy are substantially lower. According to our preliminary data, though HCD-IT generated 30% more MS2 spectra than HCD-OT in analysis of the same samples, the percentage of successful PSM achieved by HCD-IT was ~20% comparing to ~50% achieved by HCD-OT when the same protein/peptide FDR were applied, which is again in agreement with previous observation.34 Then we evaluated the performance of HCD-OT vs HCD-IT at different loading amounts (i.e., 1, 2, and 4 µg of digested peptides). Interestingly, it was observed that HCD-IT identified more proteins than HCD-OT (N = 3) when loading amount was 2 µg or lower (in line with previous reports), most likely because the higher sensitivity of IT helps to identify more lower-abundance proteins at lower loading amount. Nonetheless HCD-OT outperforms HCD-IT at 4-µg loading when utilizing the large-ID trap (p < 0.01, N = 3/condition, Figure 4A), likely reflecting the fact the high resolution/accuracy of OT enables more confident identification and lower FDR,34,35 which appears to be a determining factor at higher loading amounts. We further discovered the advantage of high-confidence identification by HCD-OT is much more pronounced when analyzing larger number of samples, which is typically the case in pharmaceutical and biomedical investigations. HCD-OT was found to identify >15% more ID cumulatively than HCD-IT at 20 runs and 4-µg loading (Figure 4B). Moreover, while the gain in protein number along with increased number of runs nearly reached a plateau at 20 runs for HCD-IT, the ID by HCD-OT continues to trend upward. We speculate this is because false-positive identification by HCD-OT increases at a much slower pace along with the increase of runs than HCD-IT, owing to the high confidence of HCD-OT identification. Consequently, the proteomics analysis conducted in this study employs loading of 4 µg samples and HCD-OT for MS2 analysis to achieve in-depth identification and quantification.
Figure 4.

Comparison of identification results acquired by HCD-OT vs HCD-IT (i.e., HCD fragments scanned by either Orbitrap or dual-cell Ion-Trap) at different loading amounts and numbers of samples. (A) Protein identification at different loading amounts with N = 3/condition, indicating superior performance of HCD-OT (p < 0.05) at high loading amount. (B) The advantage of confident identification by HCD-OT becomes much more prominent when larger numbers of human cell samples are analyzed (cumulative ID number, which is important for MS1 quantification). As a result, HCD-OT was chosen to assign ID to ion-current quantitative features for large biological cohorts.
1.4. High-Resolution MS1 for Accurate, Sensitive and Selective Quantification
For MS1 quantification, acquisition of peptide ion currents at higher resolutions likely enhances selectivity by lowering chemical noises arising from matrix components with close m/z, which in turn improves quantitative sensitivity, especially for lower-abundance proteins.36 A previous study showed that S/N increased proportionally to the square root of resolution increase on an Orbitrap Fusion.37 Here we examined whether high resolution MS1 with rapid scan cycle by UHF-Orbitrap could improve the quantitative performance for ion current-based quantification in complex biological samples. Significantly increased S/N and lower chemical noise for low-abundance peptides were observed at 120 k vs 60 k and 30 k, as exemplified in Figure 5A. In terms of quantification of a spiked-in sample set, using 120 k resolution afforded markedly better accuracy and precision for protein quantification than 60 k and 30 k (Figure 5B). Finally, higher resolution also enables more accurate mass measurement, as reflected by the much narrower distribution of mass differences than that of lower resolution (Supplementary Figure S4).
Figure 5.

High-resolution MS1 measurement with narrow-width extraction improves selectivity, sensitivity and quantitative accuracy and precision. (A) Examples for MS1 ion currents of low-abundance peptides under resolutions of 30 k, 60 k and 120 k, acquired in consecutive scan events and decreasing extraction window widths corresponding to the resolutions in the same LC–MS analysis of a human cell digest. (B) Comparison of quantitative accuracy and precision under different MS1 resolutions for quantification of human proteins (true ratio = 3.0 between the two groups and N = 3/group) that were spiked in E. coli. lysate. Details of the sample set is in Supplemental Methods.
Though quantitative performances increased marginally beyond 120 k resolution (data not shown), 120 k was determined optimal for an UHF-Orbitrap, to maintain a reasonable throughput.
2. Extensive Evaluation of IonStar for Proteomics Quantification
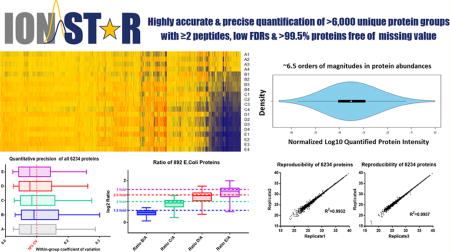
We comprehensively evaluated the quantitative performances of the developed IonStar Experimental Strategy with a multigroup benchmark sample set. The design is shown in Supplementary Figure S5. Briefly, we spiked small (<6% of total protein mass), various portions of E. coli lysate (i.e., true-positive proteins, 1, 1.5, 2, 2.5, and 3 folds across the five groups, N = 4/group) into a much larger portion, constant amount of human cell lysate (i.e., true-negative proteins), and then prepared and analyzed with the developed experimental workflow, without any prefractionation. After removing shared peptides between the two species, a total of 6273 proteins were quantified with stringent criteria including low peptide/protein FDR for identification, strict quality control for feature generation and ≥2 peptides/protein. Among these, 6234 (99.4%) proteins were quantified in all 20 samples without missing value. This low level of missing value and excellent depth in proteomics analysis compared favorably to previously reported MS2-DIA methods,16,17 which is contributed by the reproducible, in-depth and robust experimental strategy developed here and the comprehensive feature generation by an ion current-based data processing method.15,36 Protein quantification results were provided in Supplementary Table S1.
Heatmap visualization of all the protein ratios is shown in Figure 6A. Hierarchy clustering results in a clear-cut between E. coli proteins and human proteins. Owing to the high sensitivity of the technique, quantified proteins span a wide protein abundance value range of 6.5 orders of magnitude, as shown Figure 6B.
Figure 6.

The developed IonStar analytical strategy achieved accurate, reproducible and in-depth quantification. A benchmark sample set (20 samples, five groups) is analyzed. Using an ion current-based data processing method, 6234 (out of 6273 in total) proteins were quantified without missing data. (A) Heatmap visualization of normalized log2 ratios. Hierarchy clustering shows a clear classification of E. coli proteins (in black lines) from human proteins. (B) Violin plot distribution of normalized log10 abundances of the quantified proteins. (C) Reproducibility evaluation of protein quantitative values between two LC–MS analyses of the same sample. Proteins with missing values were removed. (D) Precision evaluation of all protein quantitative values in the each group (N = 4/group). (E) Quantitative accuracy evaluation for all quantified E. coli proteins. Dashed lines denote theoretical ratios. (F) False-positives in discovery of significantly different proteins. False Altered proteins Discovery Rates (FADR, defined as FP/FP+TP) are shown for each comparison.
For relative quantification, reproducibility of protein measurement profoundly affects both quantitative accuracy and precision. Therefore, we evaluated reproducibility of the method by correlating the quantitative values between two LC–MS analyses of the same sample. As shown in Figure 6C, high correlation (R2 > 0.99) were observed for the quantification of >6000 proteins. This high level of analytical reproducibility resulted in excellent quantitative precision among proteins, with median intragroup CV of 6–9% for all quantified proteins across the five groups (Figure 6D). The accuracies for quantification of E. coli proteins (i.e., the true positives) are summarized in Figure 6E. Median errors of E. coli protein ratios (i.e., relative deviation from the theoretical values) of all 4 comparison pairs were lower than 6%, suggesting superb quantitative accuracy despite of the wide dynamic range of E. coli proteins.
Finally, we evaluated the performance of IonStar in correctly discovering significantly different proteins, one of the ultimate goals for quantitative proteomics. False-positives represent a prominent problem that leads to incorrect biological clues and waste of resources in downstream analysis and validation.38 In the benchmark data set, false altered proteins discovery rate (FADR, defined in a previous publication10) can be easily calculated because all E. coli proteins are true-positives and all human proteins are true-negatives. By applying a ratio cutoff determined by an Experimental Null method we described10 and p-value cutoff of <0.05, 2.96–7.94% FADR were achieved across the 4 comparison pairs, indicating reliable discovery (Figure 6F). In group pairs where ratio differences were 2–3 fold, >80% of true positives were correctly discovered with low numbers of false-positives; even for the pair with a quite subtle change (50%), >60% sensitivity was achieved. Considering all the true positives were spiked in at low-abundances and spanning a very wide dynamic range, the strategy demonstrated excellent ability in discovering altered proteins.
An application of the IonStar proteomics experimental strategy in characterization of temporary proteomic responses to combinational chemotherapy is shown in Supplementary Figure S6. Gemcitabine is the standard-of-care chemotherapeutics for treating pancreatic cancer (PaCA), however is subjected to high occurrences of drug resistance largely because of induced epithelial-mesenchymal transition and eventually tumor metastasis.39,40 Therefore, combining drugs capable of inhibiting epithelial-mesenchymal transition appears to be a viable solution to Gem resistance,41 and one potential candidate is fibroblast growth factor receptor (FGFR) inhibitors.42,43 We have discovered that Gemcitabine combining BGJ398, an FGFR inhibitor,44 significantly suppressed cell growth/mobility and triggered sustained cell cycle arrest in Gem-resistant human PaCA cell lines (i.e., MIA-RG8). To further investigation underlying mechanisms, we applied the IonStar proteomics strategy to comprehensively characterize the temporal drug-responsive proteome changes. In total, >6000 unique protein groups were quantified without any fractionation and with at least 2 peptides and low FDRs, ~99.5% free of missing values in all the 39 biological samples. These reproducibly quantified proteins likely accounting for almost 50% of expressed human cellular proteome,45 demonstrating excellent depth of quantification. By applying a ratio threshold of 1.4 fold change and ANOVA p-value threshold of 0.05, 1302 proteins were determined as altered proteins. These proteins are closely related to key biological process such as cell cycle, apoptosis, cell migration and adhesion. A number of interesting biological insights were observed (details not elaborated in this technical paper) and the biological validation is ongoing.
CONCLUSION
For reliable and informative pharmaceutical and biomedical investigations, the ability to reproducibly and accurately quantify proteins across many biological replicates is critically important. MS1 ion current-based method carries great potential, because of the theoretically unlimited capacity for biological replicates and that MS1 ion currents are acquired in a nondata-independent manner. Nonetheless, this potential has not been fully exploited by current experimental methods. For instance, existing sample preparation and LC–MS approaches for MS1-based quantification may lack the necessary robustness and reproducibility to handle large biological cohorts; moreover, the new Q/UHF-Orbitrap platforms hold great promise for substantial improvement of the depth and selectivity for MS1 quantification, but the remarkably enhanced scan speed and sensitivity by this technique substantially shift the optimal experimental conditions for MS1 ion current measurement and feature identification. Thus, far the sample preparation, LC separation and MS detection approaches, which has not been adequately developed for MS1 quantification.
Here we addressed this need by extensive development and evaluation of an experimental pipeline taking full advantage of MS1-based quantification and permitting in-depth, high-quality quantification of larger cohorts. These include a new surfactant-aided sample preparation, strategies for reproducible and robust separation of large cohorts, in-depth MS identification, as well as selective and sensitive MS1 quantification. Furthermore, we disered using a large-ID trap provided three important benefits: (i) substantially enhanced reproducibility of gradient delivery and dampened pump noise as for the first time observed via an in-line conductivity sensor; (ii) the trap increased the quantitative loading capacity by >8-fold and thus improving sensitivity and quantified >25% more proteins; and (iii) the selective trapping/delivery strategy prevented hydrophobic and hydrophilic matrix components from entering the LC–MS system, enabling reproducible and robust separation of >100 biological samples consecutively, compared to less than 20 samples without a selective delivery strategy. Finally, many key optimal parameters on this platform are markedly different from these previously developed based on lower-field Orbitrap platform, to name a few: first, more extensive digestion is needed due to the enhanced depth of analysis by Q/UHF-Orbitrap; second, a long column with a modest gradient length (as opposed to a long gradient) is found optimal owing to the higher scan speed and sensitivity; third, because of the faster duty cycle and low-false-positive feature, HCD-OT markedly outperforms HCD-IT in protein identification when large cohorts and higher loading amount (enabled by a large-ID trap) are employed; fourth, the rapid high-resolution MS1 scan permitted practical higher resolution (fwhm = 120 k) ion current acquisition, achieving enhanced S/N and selectivity for low-abundance species.
We extensively evaluated this optimized strategy for proteomic quantification. The strategy showed efficient and reproducible peptide recoveries across 48 biological samples. With a spiked-in benchmark sample set, we were able to quantify 6273 proteins under stringent cutoffs and ≥2 peptides per protein, with only 0.6% proteins with missing data across the 20 samples unique proteins. The strategy achieved good quantitative performance, including high accuracy (2.7–6.1% median error across the five groups), low intragroup-variation (~6–9% median CV) and low false-positive biomarker discovery rate (2.96–7.94%), with quantified protein abundances spanning 6.5 orders of magnitude. Finally, we demonstrated a successful application in investigation of novel combinatory chemotherapies.
To our knowledge, this was the first time a label-free protein measurement achieves such depth and low missing data in a larger cohort.
Taken together, the experimental strategy described here provides a solid foundation for accurate, reproducible and in-depth MS1 ion current-based quantification. The method may serve as a promising alternative to MS2-DIA approaches for reproducible and reliable proteomic measurement in large cohorts.
Supplementary Material
Acknowledgments
This work is supported, in part, by NIH grants HD071594 (JQ), GM121174 (JQ), AI125746(JQ), NS094181(JQ), HL103411 (JQ), NS096104(JQ), by a Center of Protein Therapeutics Industrial Award (JQ) and by an NIH CTSA award UL1TR001412 (JQ). We also thank Qingxiang Lin and Dr. Straubinger for providing cell treatment samples for the application.
Footnotes
ASSOCIATED CONTENT
The authors declare no competing financial interest.
All the raw files of the benchmark data set will be available on ProteomeXchange Consortium via the PRIDE partner repository with accession number PXD005590.
References
- 1.Rifai N, Gillette MA, Carr SA. Protein biomarker discovery and validation: the long and uncertain path to clinical utility. Nat. Biotechnol. 2006;24:971–983. doi: 10.1038/nbt1235. [DOI] [PubMed] [Google Scholar]
- 2.Mallick P, Kuster B. Proteomics: a pragmatic perspective. Nat. Biotechnol. 2010;28:695–709. doi: 10.1038/nbt.1658. [DOI] [PubMed] [Google Scholar]
- 3.Schilling B, Rardin MJ, MacLean BX, Zawadzka AM, Frewen BE, Cusack MP, Sorensen DJ, Bereman MS, Jing EX, Wu CC, Verdin E, Kahn CR, MacCoss MJ, Gibson BW. Platform-independent and Label-free Quantitation of Proteomic Data Using MS1 Extracted Ion Chromatograms in Skyline APPLICATION TO PROTEIN ACETYLATION AND PHOSPHORYLATION. Mol. Cell. Proteomics. 2012;11:202–214. doi: 10.1074/mcp.M112.017707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, Mann M. Accurate Proteome-wide Label-free Quantification by Delayed Normalization and Maximal Peptide Ratio Extraction, Termed MaxLFQ. Mol. Cell. Proteomics. 2014;13:2513–2526. doi: 10.1074/mcp.M113.031591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Higgs RE, Knierman MD, Gelfanova V, Butler JP, Hale JE. Label-free LC–MS method for the identification of biomarkers. Methods Mol. Biol. 2008;428:209–230. doi: 10.1007/978-1-59745-117-8_12. [DOI] [PubMed] [Google Scholar]
- 6.Merl J, Ueffing M, Hauck SM, von Toerne C. Direct comparison of MS-based label-free and SILAC quantitative proteome profiling strategies in primary retinal Muller cells. Proteomics. 2012;12:1902–1911. doi: 10.1002/pmic.201100549. [DOI] [PubMed] [Google Scholar]
- 7.Qu J, Lesse AJ, Brauer AL, Cao J, Gill SR, Murphy TF. Proteomic expression profiling of Haemophilus influenzae grown in pooled human sputum from adults with chronic obstructive pulmonary disease reveal antioxidant and stress responses. BMC Microbiol. 2010;10:162. doi: 10.1186/1471-2180-10-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gillet LC, Navarro P, Tate S, Rost H, Selevsek N, Reiter L, Bonner R, Aebersold R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol. Cell. Proteomics. 2012;11 doi: 10.1074/mcp.O111.016717. O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nahnsen S, Bielow C, Reinert K, Kohlbacher O. Tools for label-free peptide quantification. Mol. Cell. Proteomics. 2013;12:549–556. doi: 10.1074/mcp.R112.025163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shen X, Hu Q, Li J, Wang J, Qu J. Experimental Null Method to Guide the Development of Technical Procedures and to Control False-Positive Discovery in Quantitative Proteomics. J. Proteome Res. 2015;14:4147–4157. doi: 10.1021/acs.jproteome.5b00200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bruderer R, Bernhardt OM, Gandhi T, Miladinovic SM, Cheng LY, Messner S, Ehrenberger T, Zanotelli V, Butscheid Y, Escher C, Vitek O, Rinner O, Reiter L. Extending the limits of quantitative proteome profiling with data-independent acquisition and application to acetaminophen-treated three-dimensional liver micro-tissues. Mol. Cell. Proteomics. 2015;14:1400–1410. doi: 10.1074/mcp.M114.044305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang B, Kall L, Zubarev RA. DeMix-Q: Quantification-Centered Data Processing Workflow. Mol. Cell. Proteomics. 2016;15:1467–1478. doi: 10.1074/mcp.O115.055475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Webb-Robertson BJ, Wiberg HK, Matzke MM, Brown JN, Wang J, McDermott JE, Smith RD, Rodland KD, Metz TO, Pounds JG, Waters KM. Review, evaluation, and discussion of the challenges of missing value imputation for mass spectrometry-based label-free global proteomics. J. Proteome Res. 2015;14:1993–2001. doi: 10.1021/pr501138h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tu C, Sheng Q, Li J, Shen X, Zhang M, Shyr Y, Qu J. ICan: an optimized ion-current-based quantification procedure with enhanced quantitative accuracy and sensitivity in biomarker discovery. J. Proteome Res. 2014;13:5888–5897. doi: 10.1021/pr5008224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nouri-Nigjeh E, Sukumaran S, Tu C, Li J, Shen X, Duan X, DuBois DC, Almon RR, Jusko WJ, Qu J. Highly multiplexed and reproducible ion-current-based strategy for large-scale quantitative proteomics and the application to protein expression dynamics induced by methylprednisolone in 60 rats. Anal. Chem. 2014;86:8149–8157. doi: 10.1021/ac501380s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Selevsek N, Chang CY, Gillet LC, Navarro P, Bernhardt OM, Reiter L, Cheng LY, Vitek O, Aebersold R. Reproducible and Consistent Quantification of the Saccharomyces cerevisiae Proteome by SWATH-mass spectrometry. Mol. Cell. Proteomics. 2015;14:739–749. doi: 10.1074/mcp.M113.035550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rost HL, Rosenberger G, Navarro P, Gillet L, Miladinovic SM, Schubert OT, Wolski W, Collins BC, Malmstrom J, Malmstrom L, Aebersold R. OpenSWATH enables automated, targeted analysis of data-independent acquisition MS data. Nat. Biotechnol. 2014;32:219–223. doi: 10.1038/nbt.2841. [DOI] [PubMed] [Google Scholar]
- 18.Guo TN, Kouvonen P, Koh CC, Gillet LC, Wolski WE, Rost HL, Rosenberger G, Collins BC, Blum LC, Gillessen S, Joerger M, Jochum W, Aebersold R. Rapid mass spectrometric conversion of tissue biopsy samples into permanent quantitative digital proteome maps. Nat. Med. 2015;21:407. doi: 10.1038/nm.3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen YY, Chambers MC, Li M, Ham AJ, Turner JL, Zhang B, Tabb DL. IDPQuantify: combining precursor intensity with spectral counts for protein and peptide quantification. J. Proteome Res. 2013;12:4111–4121. doi: 10.1021/pr400438q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tu C, Bu Y, Vujcic M, Shen S, Li J, Qu M, Hangauer D, Clements JL, Qu J. Ion Current-Based Proteomic Profiling for Understanding the Inhibitory Effect of Tumor Necrosis Factor Alpha on Myogenic Differentiation. J. Proteome Res. 2016;15:3147–3157. doi: 10.1021/acs.jproteome.6b00321. [DOI] [PubMed] [Google Scholar]
- 21.Fisk JC, Li J, Wang H, Aletta JM, Qu J, Read LK. Proteomic analysis reveals diverse classes of arginine methylproteins in mitochondria of trypanosomes. Mol. Cell. Proteomics. 2013;12:302–311. doi: 10.1074/mcp.M112.022533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Makarov A, Denisov E, Lange O. Performance evaluation of a high-field Orbitrap mass analyzer. J. Am. Soc. Mass Spectrom. 2009;20:1391–1396. doi: 10.1016/j.jasms.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 23.Hebert AS, Richards AL, Bailey DJ, Ulbrich A, Coughlin EE, Westphall MS, Coon JJ. The one hour yeast proteome. Mol. Cell. Proteomics. 2014;13:339–347. doi: 10.1074/mcp.M113.034769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ting YS, Egertson JD, Payne SH, Kim S, MacLean B, Kall L, Aebersold R, Smith RD, Noble WS, MacCoss MJ. Peptide-Centric Proteome Analysis: An Alternative Strategy for the Analysis of Tandem Mass Spectrometry Data. Mol. Cell. Proteomics. 2015;14:2301–2307. doi: 10.1074/mcp.O114.047035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tu C, Li J, Jiang X, Sheflin LG, Pfeffer BA, Behringer M, Fliesler SJ, Qu J. Ion-current-based proteomic profiling of the retina in a rat model of Smith-Lemli-Opitz syndrome. Mol. Cell. Proteomics. 2013;12:3583–3598. doi: 10.1074/mcp.M113.027847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Senko MW, Remes PM, Canterbury JD, Mathur R, Song Q, Eliuk SM, Mullen C, Earley L, Hardman M, Blethrow JD, Bui H, Specht A, Lange O, Denisov E, Makarov A, Horning S, Zabrouskov V. Novel parallelized quadrupole/linear ion trap/Orbitrap tribrid mass spectrometer improving proteome coverage and peptide identification rates. Anal. Chem. 2013;85:11710–11714. doi: 10.1021/ac403115c. [DOI] [PubMed] [Google Scholar]
- 27.Duan X, Young R, Straubinger RM, Page B, Cao J, Wang H, Yu H, Canty JM, Qu J. A straightforward and highly efficient precipitation/on-pellet digestion procedure coupled with a long gradient nano-LC separation and Orbitrap mass spectrometry for label-free expression profiling of the swine heart mitochondrial proteome. J. Proteome Res. 2009;8:2838–2850. doi: 10.1021/pr900001t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.An B, Zhang M, Johnson RW, Qu J. Surfactant-aided precipitation/on-pellet-digestion (SOD) procedure provides robust and rapid sample preparation for reproducible, accurate and sensitive LC–MS quantification of therapeutic protein in plasma and tissues. Anal. Chem. 2015;87:4023–4029. doi: 10.1021/acs.analchem.5b00350. [DOI] [PubMed] [Google Scholar]
- 29.Tu C, Mammen MJ, Li J, Shen X, Jiang X, Hu Q, Wang J, Sethi S, Qu J. Large-scale, ion-current-based proteomics investigation of bronchoalveolar lavage fluid in chronic obstructive pulmonary disease patients. J. Proteome Res. 2014;13:627–639. doi: 10.1021/pr4007602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scheltema RA, Hauschild JP, Lange O, Hornburg D, Denisov E, Damoc E, Kuehn A, Makarov A, Mann M. The Q Exactive HF, a Benchtop mass spectrometer with a pre-filter, high-performance quadrupole and an ultra-high-field Orbitrap analyzer. Mol. Cell. Proteomics. 2014;13:3698–3708. doi: 10.1074/mcp.M114.043489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dolan JW. Mobile phase proportioning problems: a case study. LC-GC. 1989;7:18–24. [Google Scholar]
- 32.Lott K, Li J, Fisk JC, Wang H, Aletta JM, Qu J, Read LK. Global proteomic analysis in trypanosomes reveals unique proteins and conserved cellular processes impacted by arginine methylation. J. Proteomics. 2013;91:210–225. doi: 10.1016/j.jprot.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qu J, Straubinger RM. Improved sensitivity for quantification of proteins using triply charged cleavable isotope-coded affinity tag peptides. Rapid Commun. Mass Spectrom. 2005;19:2857–2864. doi: 10.1002/rcm.2138. [DOI] [PubMed] [Google Scholar]
- 34.Frese CK, Altelaar AFM, Hennrich ML, Nolting D, Zeller M, Griep-Raming J, Heck AJR, Mohammed S. Improved Peptide Identification by Targeted Fragmentation Using CID, HCD and ETD on an LTQ-Orbitrap Velos. J. Proteome Res. 2011;10:2377–2388. doi: 10.1021/pr1011729. [DOI] [PubMed] [Google Scholar]
- 35.Michalski A, Damoc E, Hauschild JP, Lange O, Wieghaus A, Makarov A, Nagaraj N, Cox J, Mann M, Horning S. Mass spectrometry-based proteomics using Q Exactive, a high-performance benchtop quadrupole Orbitrap mass spectrometer. Mol. Cell. Proteomics. 2011;10 doi: 10.1074/mcp.M111.011015. M111.011015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tu C, Li J, Sheng Q, Zhang M, Qu J. Systematic assessment of survey scan and MS2-based abundance strategies for label-free quantitative proteomics using high-resolution MS data. J. Proteome Res. 2014;13:2069–2079. doi: 10.1021/pr401206m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kelstrup CD, Jersie-Christensen RR, Batth TS, Arrey TN, Kuehn A, Kellmann M, Olsen JV. Rapid and deep proteomes by faster sequencing on a benchtop quadrupole ultra-high-field Orbitrap mass spectrometer. J. Proteome Res. 2014;13:6187–6195. doi: 10.1021/pr500985w. [DOI] [PubMed] [Google Scholar]
- 38.Margolin AA, Ong SE, Schenone M, Gould R, Schreiber SL, Carr SA, Golub TR. Empirical Bayes analysis of quantitative proteomics experiments. PLoS One. 2009;4:e7454. doi: 10.1371/journal.pone.0007454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jia Y, Xie J. Promising molecular mechanisms responsible for gemcitabine resistance in cancer. Genes & Diseases. 2015;2:299–306. doi: 10.1016/j.gendis.2015.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang R, Cheng L, Xia J, Wang Z, Wu Q, Wang Z. Gemcitabine resistance is associated with epithelial-mesenchymal transition and induction of HIF-1alpha in pancreatic cancer cells. Curr. Cancer Drug Targets. 2014;14:407–417. doi: 10.2174/1568009614666140226114015. [DOI] [PubMed] [Google Scholar]
- 41.Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, Wu C-C, LeBleu VS, Kalluri R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. 2015;527:525–530. doi: 10.1038/nature16064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ahmad I, Iwata T, Leung HY. Mechanisms of FGFR-mediated carcinogenesis. Biochim. Biophys. Acta, Mol. Cell Res. 2012;1823:850–860. doi: 10.1016/j.bbamcr.2012.01.004. [DOI] [PubMed] [Google Scholar]
- 43.Tomlinson DC, Baxter EW, Loadman PM, Hull MA, Knowles MA. FGFR1-Induced Epithelial to Mesenchymal Transition through MAPK/PLCγ/COX-2-Mediated Mechanisms. PLoS One. 2012;7:e38972. doi: 10.1371/journal.pone.0038972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guagnano V, Furet P, Spanka C, Bordas V, Le Douget M, Stamm C, Brueggen J, Jensen MR, Schnell C, Schmid H, Wartmann M, Berghausen J, Drueckes P, Zimmerlin A, Bussiere D, Murray J, Graus Porta D. Discovery of 3-(2,6-Dichloro-3,5-dimethoxy-phenyl)-1-{6-[4-(4-ethyl-piperazin-1-yl)-phenylamino]-pyrimidin-4-yl}-1-methyl-urea (NVP-BGJ398), A Potent and Selective Inhibitor of the Fibroblast Growth Factor Receptor Family of Receptor Tyrosine Kinase. J. Med. Chem. 2011;54:7066–7083. doi: 10.1021/jm2006222. [DOI] [PubMed] [Google Scholar]
- 45.Kim MS, Pinto SM, Getnet D, Nirujogi RS, Manda SS, Chaerkady R, Madugundu AK, Kelkar DS, Isserlin R, Jain S, Thomas JK, Muthusamy B, Leal-Rojas P, Kumar P, Sahasrabuddhe NA, Balakrishnan L, Advani J, George B, Renuse S, Selvan LD, et al. A draft map of the human proteome. Nature. 2014;509:575–581. doi: 10.1038/nature13302. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.