

Abstract
Alcohol consumption during pregnancy places the fetus at risk for permanent physical, cognitive, and behavioral impairments, collectively termed fetal alcohol spectrum disorders (FASD). However, prenatal alcohol exposure (PAE) outcomes vary widely, and growing evidence suggests that maternal nutrition is a modifying factor. Certain nutrients, such as iron, may modulate FASD outcomes. Untreated gestational iron deficiency (ID) causes persistent neurodevelopmental deficits in the offspring that affect many of the same domains damaged by PAE. Although chronic alcohol consumption enhances iron uptake and elevates liver iron stores in adult alcoholics, alcohol-abusing premenopausal women often have low iron reserves due to menstruation, childbirth, and poor diet. Recent investigations show that low iron reserves in during pregnancy are strongly associated with a worsening of several hallmark features in FASD including reduced growth and impaired associative learning. This review discusses recent clinical and animal model findings that maternal ID worsens fetal outcomes in response to PAE. It also discusses underlying mechanisms by which PAE disrupts maternal and fetal iron homeostasis. We suggest that alcohol-exposed, ID pregnancies contribute to the severe end of the FASD spectrum.
Keywords: Fetal alcohol spectrum disorders, gestational iron deficiency, hepcidin, neurodevelopment, maternal/fetal nutrition
Maternal nutrition and FASD
Prenatal alcohol exposure (PAE) negatively affects embryonic and fetal development. Hallmark PAE characteristics include specific facial dysmorphology, growth restriction, restricted brain growth, and permanent neurobehavioral impairments (Hoyme et al. 2016; Kabel et al. 2016). The impact of PAE upon the offspring is variable, and patterns and quantity of alcohol intake do not entirely explain the differential outcomes. This suggests that additional factors may modify FASD risk; these include maternal parity and age, maternal weight, maternal and fetal genetics relating to alcohol metabolism, and socioeconomic status (Burd et al. 2012; Kelly et al. 2012; May and Gossage 2011; Warren and Li 2005).
Growing evidence suggests that another modifying factor is maternal nutrition. Population-based studies propose an association between poor maternal nutrition and increased FASD risk (May et al. 2014; May et al. 2016). Research predominantly in animal models has identified individual nutrients that may interact with PAE, such as zinc, choline, iron, copper, selenium, methionine, folate, and essential fatty acids (Keen et al. 2010; Young et al. 2014; Zeisel 2013). Alcohol abuse may exacerbate nutrient deficiencies because the alcohol calories displace nutrient-dense foods and because alcohol may impair nutrient absorption and utilization, in part by disrupting liver and gut function (Bishehsari et al. 2017), but also by increasing intestinal bleeding and nutrient losses in stool. Alcohol-exposed pregnancies may also have a lower likelihood of prenatal vitamin use (Amos-Kroohs et al. 2016; Carlson et al. 2017).
Randomized controlled population studies are currently underway to assess whether micronutrient supplementation improves outcomes in alcohol-exposed pregnancies (Coles et al. 2015; Kable et al. 2015; Wozniak et al. 2015). Prenatal multivitamin and mineral plus choline supplementation improved aspects of infant cognition, including basic learning and memory of visual stimuli (Coles et al. 2015; Kable et al. 2015). Postnatal choline supplementation improved memory in young children diagnosed with FASD (Wozniak et al. 2015). A deeper understanding of how alcohol alters macronutrient and micronutrient requirements during pregnancy will improve our ability to mitigate alcohol’s perinatal damage.
Iron is the most common nutritional deficiency in child-bearing women and is crucial for healthy neurodevelopment. Here, we discuss current evidence that supports an important role for maternal iron status as a modulator of FASD risk.
Perinatal iron deficiency
Iron is an essential cofactor for proteins that mediate oxidation-reduction reactions, DNA replication and repair, iron regulation, cell cycle control, and mitochondrial electron transfer and energy generation (Zhang 2014). It is also essential for heme synthesis, which enables oxygen transport and utilization in the body (Evstatiev and Gasche 2012). Iron is obtained from the diet; the current U.S. and Canadian recommended daily allowance (RDA) is 8mg/day for adult males and 18mg/day for adult premenopausal females (Panel on Micronutrients et al. 2001). Diet affects iron status, with vegetarian and nutrient-poor diets posing a greater risk of iron deficiency (ID); this is partially because non-heme iron (in plant-based foods) has lower bioavailability than does heme iron (in animal-based foods) (Beard et al. 1996). Once iron is absorbed, losses are minimal except through bleeding. Thus, iron overload and its ability to damage tissue pose a problem for those who absorb excessive iron, as in hemochromatosis or adult alcoholics.
ID is the most common single micronutrient deficiency in the world (Alwan et al. 2015; Milman 2006) and is a significant public health problem for most countries (World Health Organization 2015). ID occurs when iron stores are low but the turnover of hematological profiles is not yet affected (Camaschella 2015). If untreated, ID may lead to iron-deficient anemia (IDA), wherein iron stores decline to levels inadequate to support ongoing red blood cell formation, causing weakness, exhaustion, and difficulty concentrating (Camaschella 2015). Many factors increase the risk for ID and IDA including blood loss, menstruation, intestinal parasites, frequent blood donations, chronic infection, and intestinal conditions that impair iron absorption, such as celiac disease. ID predominantly affects women of childbearing age, due to the iron losses from menstruation and childbirth, and affects children, due to their rapid growth. The prevalence of ID and IDA is higher in pregnancy, and IDA affects 18-38% of pregnant women worldwide (Lopez et al. 2016; World Health Organization 2015). The worldwide prevalence of ID is twice that of IDA (Camaschella 2015), and studies in Western nations report ID in as many as 60% of pregnant women (Arija et al. 2013; da Costa et al. 2016; Harvey et al. 2016; Ribot et al. 2012).
Because of the key role for iron in cell proliferation, growth places high demands upon iron requirements. This partially explains why pregnant women and children have the greatest vulnerability to IDA (World Health Organization 2015). ID and IDA are also common in pregnancy because iron intake is often insufficient to meet gestational needs (Livock et al. 2016; May et al. 2014; May et al. 2016; Nair and Iyengar 2009; Sukchan et al. 2010; Zenter et al. 2008). A healthy pregnancy requires about 1240mg of iron (RDA 27 mg/day) to support maternal and fetal red blood cell production, provide iron for growing fetal and placental tissues, and accommodate blood loss at birth (Milman 2006). However, many women enter pregnancy with insufficient iron stores to support their pregnancy, and even women with adequate iron stores before pregnancy may become ID by parturition (Allen 2000). External factors also increase maternal risk for gestational ID and IDA, including poor healthcare access, increased parity, short inter-pregnancy interval, vegetarianism, limited access to high quality foods, adolescence, and low socioeconomic status (Callander and Schofield 2016; Milman 2006). Although iron supplements alleviate ID, compliance is low because iron’s pro-oxidant activity irritates the gut lining and produces gastrointestinal disturbances such as nausea and constipation.
Infants are also susceptible to ID and IDA. Iron levels in human milk are insufficient to support infant growth beyond approximately four months of age. Thus, prenatal placental iron transfer is essential for healthy postnatal development. The newborn acquires about 80% of her iron endowment during the third trimester (Baker et al. 2010). Maternal characteristics including smoking, hypertension, psychological distress, and diabetes limit placental iron transfer and reduce the fetal iron endowment (Armony-Sivan et al. 2013; Baker et al. 2010; Georgieff 2008). Prematurity is also a risk factor for infant ID even in iron-sufficient mothers, because the birth occurs prior to deposition of the aforementioned iron endowment. After birth, infant factors including male gender, greater postnatal growth rate, lower SES, and lower hemoglobin indicate greater risk for ID and IDA (Lozoff et al. 2006b).
Consequences of gestational ID
Gestational ID negatively impacts both mother and child. In humans, maternal anemia and ID increase the risk for preeclampsia, prematurity, and small-for-gestational-age babies (Alwan et al. 2015; Ribot et al. 2012; Scholl 2005). Maternal ID also reduces postnatal growth. In the infant, low fetal iron status results in poorer reflexes, indicating neurologic abnormality (Armony-Sivan et al. 2004). ID infants also have poor auditory recognition memory at 2 months of age (Geng et al. 2015). Furthermore, low cord serum ferritin levels are associated with worsened language skills and fine-motor skills at age 5 (Tamura et al. 2002). Multiple studies link perinatal ID with decreased cognitive capacity, altered socioemotional functioning, reduced motor ability, and diminished overall brain functioning (Georgieff 2011), and many of these outcomes persist even after normalization of iron levels (Algarín et al. 2003, Congdon et al. 2012; Lozoff et al. 2006a), suggesting that ID interferes with healthy brain development.
Animal studies have identified several mechanisms through which ID impairs neurodevelopment. Many of the enzymes involved in neurotransmitter metabolism are iron-dependent, and their impairment could alter developmental processes that are monoamine-dependent (-Lozoff and Georgieff 2006). Studies confirm alterations in neurotransmitter production that depend on the brain region involved and the extent and timing of ID (Beard et al. 2006; Georgieff 2008; Ward et al. 2007). For example, gestational ID elevates extracellular dopamine and norepinephrine levels and reduces dopamine receptors and monoamine transporters (Lozoff and Georgieff 2006). In the glutamatergic system, ID increases glutamate:glutamine ratios in the striatum and decreases GABA shunt enzyme activity, suggesting a suppression of glutamatergic neurotransmission (Kim and Wessling-Resnick 2014; Ward et al. 2007). Gestational ID decreases myelination, which may contribute to the decreased reflexes and slower neural processing seen in these infants (Georgieff 2007). Perinatal ID also alters neuronal fatty acid profiles, reduces energy production in the hippocampus and frontal cortex, and delays neuronal maturation, outcomes that may contribute to these functional deficits (Georgieff 2008; Georgieff 2011).
Many of the neurobehavioral abnormalities observed in children who experienced perinatal ID are similar to those in children with PAE. We hypothesized that iron and alcohol might interact mechanistically to influence PAE outcomes. An understanding of this interaction requires a discussion of normal iron metabolism (as reviewed in Evstatiev and Gasche 2012; Martins et al. 2017; and Winter et al. 2014).
Overview of normal iron metabolism
Dietary iron (Fe) is primarily absorbed in the duodenum and upper jejunum. The duodenal membrane-associated cytochrome b ferroreductase (DcytB) and intestinal divalent metal transporter-1 (DMT1) mediate the reduction and import of non-heme iron into enterocytes (Figure 1, upper panel). Heme iron is imported via heme carrier protein 1 (HCP-1). At the mucosal contraluminal surface, ferroportin (FPN) exports dietary iron into the serum, where enterocyte-bound hephaestin oxidizes ferrous iron (Fe2+) to its ferric form (Fe3+). Ferric iron binds its plasma transporter, transferrin (Tf), and travels to target tissues, where Fe-Tf binds transferrin receptor 1 (TfRc1) on cell membranes. The Fe-Tf-TfRc1 complex is internalized by clathrin-mediated endocytosis, and ferric iron is released in the vesicle’s low pH. The metalloreductase STEAP3 reduces Fe3+ to Fe2+, and DMT1 exports the iron into the cytosol, where it is bound to proteins and employed in various metabolic and mitochondrial reactions, or is stored within ferritin. Tf and TfRc1 are recycled back to the cell surface, where TfRc1 is reused and Tf enters the extracellular space. This process differs in immature red blood cells where iron is incorporated into hemoglobin, accounting for nearly two-thirds of body iron.
Figure 1. Iron metabolism in the human body.
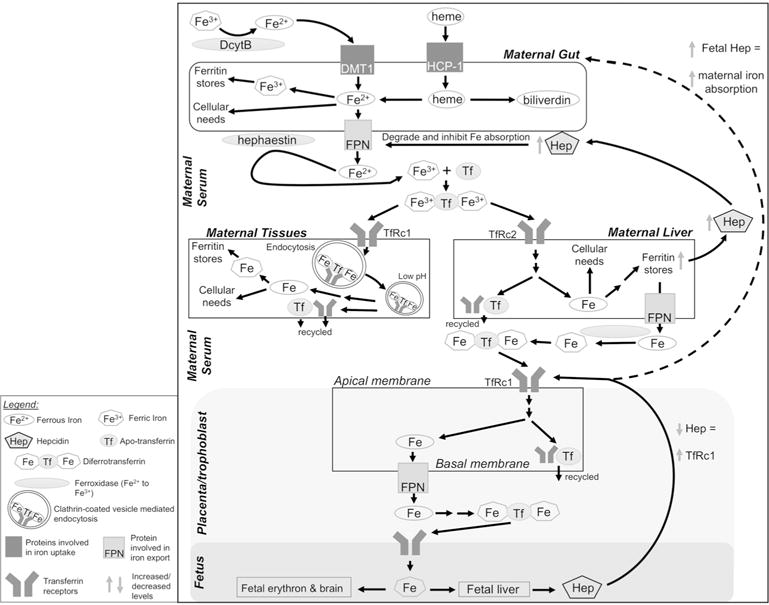
Upper panel: Dietary non-heme iron (Fe3+) is reduced to Fe2+ via duodenal cytochrome b (DcytB) and imported into enterocytes by divalent metal ion transporter-1 (DMT1). Heme is imported via heme-carrier protein-1 (HCP-1). In the enterocyte, Fe2+ may be used as a protein cofactor, oxidized to Fe3+ for storage in ferritin, or exported from the cell by ferroportin (FPN). Enterocyte-bound hephaestin oxidizes Fe2+, and two Fe3+ bind apotransferrin (Tf) to form diferrotransferrin in the serum. Middle panel: Target tissues, including liver, present transferrin receptors (TfRc1 or TfRc2) and acquire diferrotransferrin via endocytosis of clarithin-coated vesicles. Decreased vesicular pH releases Fe3+, which is reduced to Fe2+ via metalloreductase STEAP3 and transported out of the vesicle by DMT1 (not shown). Intracellular Fe2+ is used as a protein cofactor or stored in ferritin. Apo- transferrin and TfRc are recycled to the cell surface. Increased liver iron upregulates hepatic hepcidin (Hep) production. Hep causes FPN degradation to decrease iron absorption. Lower panel: During pregnancy, TfRc1 on the apical membrane of placenta/trophoblast enables fetal iron transfer. Diferrotransferrin is internalized as above and the iron is stored, utilized, or transported across the basal membrane via FPN. Iron enters fetal circulation for utilization and distribution to target tissues. Low fetal Fe leads to low hepcidin which signals increased placental TfRc1; fetal hepcidin may also increase iron absorption in maternal enterocytes, but the underlying mechanism is unknown (indicated by dashed arrow). DcytB, duodenal cytochrome b; DMT1, divalent metal ion transporter-1; Fe, iron; FPN, ferroportin; HCP-1, heme-carrier protein-1; Hep, hepcidin; Tf, transferrin; TfRc1/2, transferrin receptor-1 or -2.
Free iron is a potent oxidant and catalyzes Fenton reactions in tissues and blood. To reduce that risk and enhance iron’s limited solubility (10−18 M as Fe3+) (Martins et al. 2017), iron is normally protein-bound. Serum and tissue iron levels that exceed protein binding is known as iron overload. Iron overload propagates liver damage (as liver is the principal iron storage site), causes widespread inflammation, and potentially promotes early death (Fleming and Ponka 2012). Since iron is lost only during menstruation or trauma, several systems tightly control iron absorption and transport to avoid iron excess (Figure 1, middle panel). A major regulator is the hepatic peptide hepcidin. In response to high dietary iron, hepcidin is induced by several signals including the hemojuvelin/BMP/SMAD pathway. Hepcidin travels to the enterocyte where it binds contraluminal FPN, promoting the latter’s degradation and blocking further dietary iron absorption; the sequestered iron is lost when enterocytes are shed. Hepcidin also interacts with macrophage FPN to enhance their iron sequestration and limit iron availability for erythropoiesis. Inflammatory cytokines such as IL-6 and IL1β induce hepcidin through the Jak/STAT pathway and contribute to the anemia of inflammation (Wang and Babitt 2016). Iron status is also regulated post-transcriptionally via iron regulatory protein-1 (IRP1) and IRP2, which bind iron-responsive elements (IRE) in the 5′ and 3′ untranslated regions (UTR) of iron-related protein mRNAs and control their translation in response to cellular needs. For example, under ID, IRP binding to the IRE in the 5′ UTR of ferritin suppresses ribosome binding and protein translation to reduce cellular ferritin content.
Similar processes regulate iron metabolism and transport during pregnancy (Cetin et al. 2011; Fuchs and Ellinger 2004; McArdle et al. 2014). Fetal iron demand enhances apical placental TfRc1 on trophoblasts to increase their binding and internalization of maternal-derived Tf-Fe (Figure 1, lower panel). At the basal placenta surface, FPN exports the iron into fetal serum where Tf delivers it to fetal tissues. Iron priority appears to be fetus > maternal hematocrit > maternal iron stores (Gambling et al. 2011), but details on the regulation of this maternal-fetal communication remain incompletely described. Animal studies suggest the fetal liver may influence maternal gut absorption, plasma concentration, maternal liver storage, and placental transfer (Gambling et al. 2011). A key player is placental TfRc1. TfRc1 levels increase as maternal iron absorption decreases and positively correlate with fetal iron accumulation (Bradley et al. 2004; Gambling et al. 2011).
Although the placenta adapts to maternal ID, stressors such as alcohol alter placental function and might impair iron transport (Cetin et al. 2011). The timing of ID is also important and affects different fetal processes. Iron transferred in the first two-thirds of pregnancy predominantly serves to promote fetal growth and development, whereas the large amounts of iron transferred in the third trimester enrich fetal iron stores for postnatal growth needs (Gambling et al. 2004, 2009).
Iron and alcohol in adults
That alcohol modifies iron utilization was first suggested by associations between high alcohol consumption and hepatic iron overload (Powell 1966), and subsequent demonstrations of elevated serum ferritin, serum free iron, and transferrin-iron saturation (Chapman et al. 1982; Friedman et al. 1988; Gordeuk et al. 2008; Ioannou et al. 2004; Milman and Kirchoff 1996; Suzuki et al. 2002). Elevated hepatic iron may contribute to alcoholic liver cirrhosis (Harrison-Findik 2009). Within a few weeks of drinking cessation, elevated serum iron markers decline (Bell et al. 1994).
One link between alcohol consumption and iron overload in adults is hepcidin. Normally, upregulated under conditions of iron overload (Nemeth et al. 2004) (see Figure 1), chronic alcohol suppresses liver hepcidin expression, thus limiting ferroportin degradation and increasing dietary absorption despite elevated hepatic iron stores (Bridle et al. 2006; Harrison-Findik 2009; Harrison-Findik et al. 2007; Ohtake et al. 2007). How chronic alcohol elevates hepcidin synthesis is not entirely clear. Alcohol may modulate hepcidin through oxidative stress and inflammatory cytokines such as IL-6. Studies suggest that cytokines inhibit the DNA-binding ability of transcription factor CCAAT/enhancer-binding protein alpha (C/EBP-α), which then cannot bind the C/EBP-α binding sites within the hepcidin promoter to initiate hepcidin transcription (Harrison-Findik 2009; Harrison-Findik et al. 2006; Harrison-Findik et al. 2009). However, more recent studies implicate liver hypoxia as a driving force in alcohol-induced hepcidin suppression (Anderson et al. 2012; Heritage et al. 2009). Ethanol stabilizes hepatocyte hypoxia-inducible factor alpha (HIFα), leading to proteasomal degradation of C/EBP-α and blocking hepcidin induction in response to high iron (Anderson et al. 2012). Regardless of the exact mechanism, alcohol limits hepcidin production and thereby maintains duodenal iron absorption in the face of iron overload. Hepcidin suppression is apparent within days of alcohol exposure (Harrison-Findik 2009).
Hepcidin suppression in response to alcohol consumption in adults contributes to the alcohol-induced tissue damage, particularly in the liver (Harrison-Findik 2009). Chronic alcohol abusers have higher serum levels of free iron than do former alcohol abusers, and free iron is higher in those with liver cirrhosis than in non-cirrhotics (De Feo et al. 2001). Excess circulating and stored iron caused by alcohol consumption may be a critical step in alcoholic liver damage (Pietrangelo 1998; Zhu et al. 2012).
Iron and alcohol in pregnancy: Population studies
In 1973, a case study noted that some alcoholic women became anemic during pregnancy (Jones et al. 1973). Streissguth et al. (1983) found that pregnant women who drank heavily were more iron depleted than women with lower levels of alcohol consumption, although this was a small cohort and none of the women had progressed to IDA. Carter et al. (2007) were the first to directly associate PAE with infant IDA in a prospective study in a Western Cape Province population in South Africa with high rates of drinking. Recruited mothers either drank heavily during pregnancy or abstained or drank lightly. At 6 and 12 months of age, infants who were prenatally exposed to binge drinking (more than 4 drinks on occasion) were almost 4 times more likely to be diagnosed with IDA than infants who were not prenatally exposed. A second study found that children prenatally exposed to alcohol were 6.5 times more likely to be ID than control children at 5 years of age (Carter et al. 2012). Infants with IDA and PAE were significantly smaller than PAE infants who were not IDA, and alcohol decreased infant length at 6.5 and 12 months only in the presence of IDA (Carter et al. 2007). IDA during infancy enhanced the negative impact of PAE on child growth and resulted in overall lower weight-for-age and height-for-age Z-scores. In contrast, low food security, which was used as a measure of dietary inadequacy, was unrelated to growth outcomes (Carter et al. 2012). This suggested that the reduced growth that partially characterizes FASD is unrelated to food insecurity per se but rather to a reduced availability of select nutrients such as iron.
This strong association between maternal ID and poor growth outcomes in FASD is unlikely to be unique to this particular South African cohort, given the high incidence of marginal iron status among child-bearing age women across all socioeconomic groups and the prevalence of gestational drinking across populations. An understanding of the iron-PAE relationship is likely to yield novel insights into the mitigation of FASD.
Iron and alcohol in pregnancy: Animal models
Animal studies provide direct evidence that maternal iron status modifies gestational outcomes in the alcohol-exposed pregnancy. Rufer et al. (2012) created a rat model wherein mothers received an iron sufficient (IS) (100 ppm) or iron deficient (ID) (20 ppm) diet; the ID mothers exhibited low liver iron stores but were not anemic, modeling the common condition in pregnancy. Alcohol exposure (0, 3.5, 5 g/kg) temporally targeted the fetal brain growth spurt, which is especially vulnerable to alcohol during the human third trimester and in rat postnatal days 4 through 9 (West and Goodlett 1990). In PAE offspring, maternal ID reduced postnatal growth and, in males, interacted with ID to further impede growth, consistent with the clinical findings of Carter et al. (2007, 2012) and suggesting a role for iron status in PAE’s growth inhibition. Maternal ID and PAE synergized to impair associative learning in the offspring, not only in a simple cerebellum-dependent task (delay eyeblink conditioning), but also in associative tasks involving other brain regions including contextual fear conditioning (amygdala and hippocampus), auditory-cued conditioning (amygdala), and trace eyeblink conditioning (cerebellum and hippocampus) (Huebner et al. 2015; Rufer et al. 2012). The iron-alcohol interaction was selective, as other behaviors including open field behavior, gait, balance, and muscle strength were unaffected by the ID-alcohol interaction (Rufer et al. 2012).
Analysis of brain morphology revealed a partial explanation, as rats from alcohol-consuming ID mothers had reduced cellularity in the hippocampal (CAI) and cerebellar (interpositus nucleus) regions that mediate eyeblink associative learning; cellularity tightly correlated with alcohol exposure and iron status (Huebner et al. 2015). The ID-alcohol combination also increased neuronal apoptosis, and ID-alcohol interacted to significantly reduce cerebellar myelin content (Rufer et al. 2012) to levels beyond the impact of ID or alcohol alone (Georgieff 2011; Wilhelm and Guizzetti 2016). Because the learning and cellular deficiencies in PAE rats are lessened in the IS offspring compared to ID offspring, this work suggests that maternal iron adequacy can partially ameliorate alcohol’s neurodevelopmental damage (Huebner et al. 2015).
It is unclear how iron and alcohol interact to alter fetal brain development. It is not simply due to regions having differential iron content, because regions typically having lower iron content (such as the hippocampus) are as vulnerable to ID as are regions having higher iron content (such as the cerebellum) (Connor and Benkovic 1992; Georgieff et al. 2015; Hare et al. 2012). One likely explanation is that both ID and alcohol impair white matter formation in the brain. Perinatal alcohol reduces white matter content, delays myelination, causes disorganized white matter tracts, decreases oligodendrocyte differentiation, and increases cell death (Wilhelm and Guizzetti 2016; Wozniak et al. 2009). Iron is in high supply in the white matter, with oligodendrocytes and glial cells regulating iron delivery to neurons and storing iron when it is not needed (Connor and Benkovic 1992; Gerber and Connor 1989). Iron is also essential for energy generation in the mitochondria, and ID reduces the amount of iron available for metabolic reactions, specifically in the iron-sulfur clusters of the electron transport chain; this could lower ATP generation and create a “metabolic brown-out” in developing tissues (Georgieff 2011).
A novel mechanistic insight from Miller et al. (1995) suggested that PAE directly alters fetal iron metabolism. In the study, pregnant rats were fed a nutritionally adequate liquid diet with a high alcohol dose (up to 6.7% v/v of liquid diet) that contributed 37% of total daily calories from gestational day 11 through 20. Although the alcohol exposure ended at birth, PAE reduced brain iron concentrations in the adolescent and adult offspring (Miller et al. 1995). Brain expression of iron regulatory proteins (ferritin and transferrin) did not accurately reflect iron status, and PAE altered their normal developmental pattern, such that the iron regulatory proteins responded too slowly to changing iron levels. Iron mobility within the cerebral cortex, as measured by transferrin saturation, was greatly reduced in PAE adult rats, suggesting that alcohol interferes with brain iron distribution. PAE appears to disrupt the normal temporal relationships between iron and major iron regulatory proteins well after alcohol exposure ends.
Additional mechanistic insights emerged from Huebner et al. (2016), who fed pregnant rats with IS (100 ppm) or ID (4-5 ppm) diets and provided them with either 5.0 g ethanol/kg body weight or isocaloric maltodextrin by daily gavage from gestational day 13.5 through 19.5. Although both IS and ID dams had normal hematological values, their PAE fetuses were anemic with significantly reduced red cell counts, hematocrit, and hemoglobin. This is a striking outcome, given that fetal erythropoiesis is largely hepatic and that PAE fetal livers had significantly elevated non-heme and total iron content. This suggests that PAE renders liver iron unavailable for erythropoiesis. PAE also limits iron availability to the developing brain, and total brain iron in the IS-PAE fetuses was reduced to levels seen in ID-alone. Thus, PAE creates fetal anemia and brain iron deficiency, even though the mother is iron-adequate. The altered fetal iron distribution is explained by the three-fold elevation in both fetal and maternal hepcidin in response to PAE that was independent of iron status. This is in marked contrast to non-pregnant adults, in whom alcohol consumption decreases hepcidin expression (Ohtake et al. 2007). As a negative regulator of iron import, elevated hepcidin would limit iron availability and, consistent with this, the PAE brain fails to upregulate its Tf, TfRc, and ferritin content, even though brain iron is reduced, suggesting that alcohol disrupts the signals that normally govern cellular responses to altered iron status.
Altered iron metabolism also occurs in a moderate PAE paradigm (blood alcohol concentration of 120 mg/dl) that used an otherwise nutritionally adequate sheep model (Sozo et al. 2013). In that study, third trimester alcohol exposure (0.75 g/kg maternal body weight) reduced fetal liver iron content and hepcidin production and increased placental ferroportin expression. Although this study found opposing results from the above rat studies, this could reflect the lower alcohol dose, the model’s herbivore metabolism, or other unknown variables.
How might alcohol stimulate hepcidin production and disconnect it from iron status? Insights emerge from the anemia of chronic inflammation, in which cytokines including IL6 and IL-1β signal through the JAK/STAT pathway to increase hepcidin transcription through the STAT3 response element within the hepcidin promoter; signaling through the BMP/SMAD pathway may also play a role (Wang and Babitt 2016). Elevated hepcidin signals the duodenum to reduce its iron import and signals the liver and macrophages to store circulating iron, thus limiting iron’s availability for processes such as erythropoiesis and brain development. Alcohol is proinflammatory, especially at binge or chronic intakes, and it stimulates cytokine production (Bishehsari et al. 2017). In preliminary studies, we find that PAE increases both maternal and fetal cytokine production in liver and fetal brain, including IL6, IL1β, TNFα, and IFNγ (Huebner et al. submitted). Sustained elevations in hepcidin could also limit placental iron transport and promote liver iron sequestration at the expense of the expanding fetal erythron and developing brain. Current knowledge of the interaction between alcohol and iron during pregnancy is summarized in Figure 2, and studies are underway to evaluate this further.
Figure 2. Altered maternal and fetal iron metabolism in iron limiting states with or without alcohol exposure.
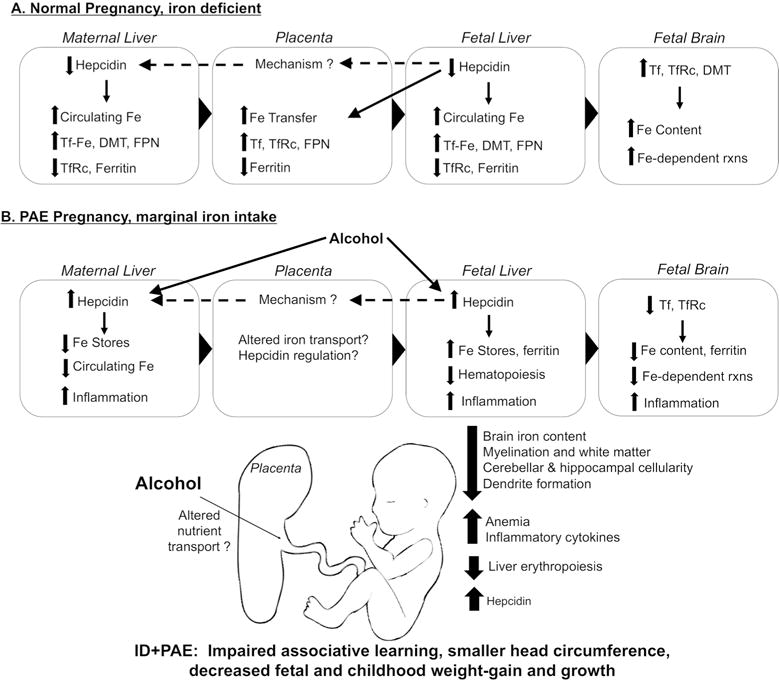
Modifications in maternal and fetal iron uptake, utilization, and distribution in (A) an otherwise-normal pregnancy with iron deficiency, and (B) prenatal alcohol exposure (PAE) pregnancy with marginal iron intake. (A) In the iron-deficient (ID) pregnancy, maternal hepcidin is downregulated to enhance her dietary iron absorption and mobilize her liver iron, thus elevating circulating iron available for placental transport. Simultaneously, fetal hepcidin decreases to meet fetal iron demands and bring more iron across the placenta via Tf, TfR, and FPN. Under low hepcidin, fetal liver mobilizes iron stores to increase circulating iron, and fetal brain upregulates Tf, TfRc, and DMT to import that iron. Fetal hepcidin may affect maternal hepcidin levels through unclear mechanisms. (B) In prenatal alcohol exposure (PAE), alcohol increases maternal hepcidin regardless of her actual iron status. Fetal liver hepcidin also increases, promoting iron storage in fetal liver and preventing iron mobilization for other functions such as hematopoiesis. Thus the fetus become anemic. Fetal brain fails to upregulate Tf and TfRc to increase its iron uptake, and brain iron content remains inadequate, contributing to deficits in neuronal development. Overall, PAE-induced changes in iron metabolism harm fetal brain development, impair learning, cause fetal anemia, and decrease fetal and postnatal growth.
Conclusion
The interaction between iron status and PAE is a significant public health issue for both well-nourished and malnourished populations. Despite years of public education, women still drink during pregnancy, with 10.2% of pregnant women in the U.S. reporting alcohol use and 3.1% reporting binge drinking in the past 30 days (Tan et al. 2015). Global rates of gestational drinking are similar. Rates of drinking in regions of Canada may be higher, especially in Aboriginal populations (Popova et al. 2017). Gestational ID and low iron stores remain common in women across the socioeconomic spectrum, and it may not be coincidental that populations with exceptional rates of FASD have high rates of ID and poor nutrient intakes (Camaschella 2015; May et al. 2016). ID without anemia adversely affects brain development (Georgieff 2008), yet women are screened and treated for IDA and not necessarily for low iron stores with normal hematology. Measures such as serum ferritin, ferritin/transferrin receptor ratio, and zinc protoporphyrin/heme ratio are more sensitive indicators of iron status than are hematologic values, and the latter two measures are less influenced by inflammation, which can occur in some pregnancies and obscure ID. Identification and treatment of marginal iron status is important since, in our rat model, PAE outcomes are worsened when the mother is ID but not anemic, and even in iron sufficiency, PAE reduces fetal iron distribution and utilization (Huebner et al. 2015, 2016; Ruber et al. 2012). However, iron adequacy clearly mitigated some consequences of ID-PAE. In light of this, certain simple interventions may improve fetal outcomes in alcohol-exposed pregnancies. These include aggressive diagnosis and treatment of gestational ID, iron supplementation of mother and/or infant, and delayed cord clamping at delivery, regardless of whether the mother consumes alcohol during pregnancy. A prospective study in the Ukraine recently showed that maternal multivitamin supplementation including iron improves cognitive scores in 6-month-old infants prenatally exposed to alcohol (Coles et al. 2015). Studies in non-alcohol-exposed pregnancies suggest that postnatal iron supplementation further improves cognitive and psychomotor performance (Beard et al. 2007; Hermoso et al. 2011), suggesting a similar potential to improve alcohol-exposed pregnancies. Maternal supplementation should be limited to actual insufficiencies, since excess iron consumption can cause iron overload and is linked to gestational diabetes, hypertension, and metabolic syndrome (Casanueva and Viteri 2003). Accurate screening and diagnosis of ID is essential so that mothers and infants who truly need iron can receive it.
In conclusion, PAE adversely affects neurodevelopment, and maternal iron status has a significant influence such that ID magnifies alcohol’s fetal damage. Animal models demonstrate that even when the mother is iron-adequate, PAE alters normal iron distribution, creating fetal anemia and fetal brain iron deficiency, while fetal liver iron stores increase. The commensurate increases in maternal-fetal hepcidin and cytokines may partially underlie this dysregulated iron homeostasis. Additional work is needed to further characterize the iron-alcohol interaction and thereby optimize supplementation and intervention. The timing of alcohol exposure and iron intervention likely matters, given the existence of critical points for fetal iron transfer (Gambling et al. 2009). A detailed understanding of iron-alcohol interactions in developing brain will clarify mechanisms underlying the cognitive defects of FASD (Huebner et al. 2015; Rufer et al. 2012). While explanatory mechanisms for the connection between iron metabolism and alcohol during pregnancy are still being explored, current knowledge supports the use of interventions including iron assessment and supplementation to improve fetal outcomes in both alcohol-exposed and unexposed pregnancies.
Acknowledgments
Supported by award #R01 AA22999 from the National Institutes of Health to S.M.S.
References
- Algarín C, Peirano P, Garrido M, Pizarro F, Lozoff B. Iron deficiency anemia in infancy: long-lasting effects on auditory and visual system functioning. Pediatr Res. 2003;53(2):217–223. doi: 10.1203/01.PDR.0000047657.23156.55. [DOI] [PubMed] [Google Scholar]
- Allen LH. Anemia and iron deficiency: effects on pregnancy outcome. Am J Clin Nutr. 2000;71(5):1280S–1284S. doi: 10.1093/ajcn/71.5.1280s. [DOI] [PubMed] [Google Scholar]
- Alwan NA, Cade JE, McArdle HJ, Greenwood DC, Hayes HE, Simpson NAB. Maternal iron status in early pregnancy and birth outcomes: insights from the Baby’s Vascular health and Iron in Pregnancy study. Br J Nutr. 2015;113(12):1985–1992. doi: 10.1017/S0007114515001166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amos-Kroohs RM, Fink BA, Smith CJ, Chin L, Van Calcar SC, Wozniak JR, Smith SM. Abnormal eating behaviors are common in children with fetal alcohol spectrum disorders. J Pediatr. 2016;169:194–200. doi: 10.1016/j.jpeds.2015.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson ER, Taylor M, Xue X, Martin A, Moons DS, Omary MB, Shah YM. The hypoxia-inducible factor-C/EBPα axis controls ethanol-mediated hepcidin repression. Mol Cell Biol. 2012;32(19):4068–4077. doi: 10.1128/MCB.00723-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arija V, Ribot B, Aranda N. Prevalence of iron deficiency states and risk of haemoconcentration during pregnancy according to initial iron stores and iron supplementation. Public Health Nutr. 2013;16(8):1371–1378. doi: 10.1017/S1368980013000608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armony-Sivan R, Eidelman AI, Lanir A, Sredni D, Yehuda S. Iron status and neurobehavioral development of premature infants. J Perinatol. 2004;24:757–762. doi: 10.1038/sj.jp.7211178. [DOI] [PubMed] [Google Scholar]
- Armony-Sivan R, Aviner S, Cojocaru L, Fytlovitch S, Ben-Alon D, Eliassy A, Babkoff H, Lozzof B, Anteby E. Prenatal maternal stress predicts cord-blood ferritin concentration. J Perinat Med. 2013;41(3):259–265. doi: 10.1515/jpm-2012-0125. [DOI] [PubMed] [Google Scholar]
- Baker RD, Greer FR, THE COMMITTEE ON NUTRITION Clinical report-diagnosis and prevention of iron deficiency and iron-deficiency anemia in infants and young children (0-3 years of age) Pediatrics. 2010;126(5):1040–1050. doi: 10.1542/peds.2010-2576. [DOI] [PubMed] [Google Scholar]
- Beard JL, Dawson H, Pinero DJ. Iron metabolism: a comprehensive review. Nutr Rev. 1996;54(10):295–317. doi: 10.1111/j.1753-4887.1996.tb03794.x. [DOI] [PubMed] [Google Scholar]
- Beard JL, Felt B, Schallert T, Burhans M, Connor JR, Georgieff MK. Moderate iron deficiency in infancy: biology and behavior in young rats. Behav Brain Res. 2006;170(2):224–232. doi: 10.1016/j.bbr.2006.02.024. [DOI] [PubMed] [Google Scholar]
- Beard JL, Unger EL, Bianco LE, Paul T, Rundle SE, Jones BC. Early postnatal iron repletion overcomes lasting effects of gestational iron deficiency in rats. J Nutr. 2007;137(5):1176–1182. doi: 10.1093/jn/137.5.1176. [DOI] [PubMed] [Google Scholar]
- Bell H, Skinningsrud A, Raknerud N, Try K. Serum ferritin and transferrin saturation in patients with chronic alcoholic and non-alcoholic liver diseases. J Intern Med. 1994;236(3):315–322. doi: 10.1111/j.1365-2796.1994.tb00802.x. [DOI] [PubMed] [Google Scholar]
- Bishehsari F, Magno E, Swanson G, Desai V, Voigt RM, Forsyth CB, Keshavarzian A. Alcohol and gut-derived inflammation. Alcohol Res. 2017;38(2):e1–9. [PMC free article] [PubMed] [Google Scholar]
- Bradley J, Leibold EA, Harris ZL, Wobken JD, Clarke S, Zumbrennen KB, Eisenstein RS, Georgieff MK. Influence of gestational age and fetal iron status on IRP activity and iron transporter protein expression in third-trimester human placenta. Am J Physiol Regul Integr Comp Physiol. 2004;287(4):R894–901. doi: 10.1152/ajpregu.00525.2003. [DOI] [PubMed] [Google Scholar]
- Bridle KR, Cheung TK, Murphy TL, Walters MM, Anderson GJ, Crawford DHG, Fletcher LM. Hepcidin is down-regulated in alcoholic liver injury: implications for the pathogenesis of alcoholic liver disease. Alcohol Clin Exp Res. 2006;30(1):106–112. doi: 10.1111/j.1530-0277.2006.00002.x. [DOI] [PubMed] [Google Scholar]
- Burd L, Blair J, Dropps K. Prenatal alcohol exposure, blood alcohol concentrations and alcohol elimination rates for the mother, fetus and newborn. J Perinatol. 2012;32:652–659. doi: 10.1038/jp.2012.57. [DOI] [PubMed] [Google Scholar]
- Callander EJ, Schofield DJ. Is there a mismatch between who gets iron supplementation and who needs it? A cross-sectional study of iron supplements, iron deficiency anaemia and socio-economic status in Australia. Br J Nutr. 2016;115(4):703–708. doi: 10.1017/S0007114515004912. [DOI] [PubMed] [Google Scholar]
- Camaschella C. Iron-deficiency anemia. N Engl J Med. 2015;372(19):1832–1843. doi: 10.1056/NEJMra1401038. [DOI] [PubMed] [Google Scholar]
- Carlson CR, Uriu-Adams JY, Chambers CD, Yetushok L, Zymak-Zakutnya N, Chan PH, Schafer JJ, Wertelecki W, the CIFASD. Keen CL. Vitamin D deficiency in pregnant Ukrainian women: effects of alcohol consumption on vitamin D status. J Am Coll Nutr. 2017;36(1):44–56. doi: 10.1080/07315724.2016.1174091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter RC, Jacobson SW, Molteno CD, Jacobson JL. Fetal alcohol exposure, iron-deficiency anemia, and infant growth. Pediatrics. 2007;120(3):559–567. doi: 10.1542/peds.2007-0151. [DOI] [PubMed] [Google Scholar]
- Carter RC, Jacobson JL, Molteno CD, Jiang H, Meintjes EM, Jacobson SW, Duggan C. Effects of heavy prenatal alcohol exposure and iron deficiency anemia on child growth and body composition through age 9 years. Alcohol Clin Exp Res. 2012;36(11):1973–1982. doi: 10.1111/j.1530-0277.2012.01810.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casanueva E, Viteri FE. Iron and oxidative stress in pregnancy. J Nutr. 2003;133(5 Suppl 2):1700S–1708S. doi: 10.1093/jn/133.5.1700S. [DOI] [PubMed] [Google Scholar]
- Cetin I, Berti C, Mandò C, Parisi F. Placental iron transport and maternal absorption. Ann Nutr Metab. 2011;59(1):55–58. doi: 10.1159/000332133. [DOI] [PubMed] [Google Scholar]
- Chapman RW, Morgan MY, Laulicht M, Hoffbrand AV, Sherlock S. Hepatic iron stores and markers of iron overload in alcoholics and patients with idiopathic hemochromatosis. Dig Dis Sci. 1982;27(10):909–916. doi: 10.1007/BF01316575. [DOI] [PubMed] [Google Scholar]
- Coles CD, Kable JA, Keen CL, Jones KL, Wertelecki W, Granovska IV, Pashtepa AO, Chambers CD, the CIFASD Dose and timing of prenatal alcohol exposure and maternal nutritional supplements: developmental effects on 6-month-old infants. Matern Child Health J. 2015;19(12):2605–2614. doi: 10.1007/s10995-015-1779-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Congdon EL, Westerlund A, Algarin CR, Peirano PD, Gregas M, Lozoff B, Nelson CA. Iron deficiency in infancy is associated with altered neural correlates of recognition memory at 10 years. J Pediatr. 2012;160(6):1027–1033. doi: 10.1016/j.jpeds.2011.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor JR, Benkovic SA. Iron regulation in the brain: histochemical, biochemical, and molecular considerations. Ann Neurol. 1992;32:S51–S61. doi: 10.1002/ana.410320710. [DOI] [PubMed] [Google Scholar]
- da Costa AG, Vargas S, Clode N, Graca LM. Prevalence and risk factors for iron deficiency anemia and iron depletion during pregnancy: a prospective study. Acta Med Port. 2016;29(9):514–518. doi: 10.20344/amp.6808. [DOI] [PubMed] [Google Scholar]
- De Feo TM, Fargion S, Duca L, Cesana BM, Boncinelli L, Lozza P, Cappellini MD, Fiorelli G. Non-transferrin-bound iron in alcohol abusers. Alcohol Clin Exp Res. 2001;25(10):1494–1499. doi: 10.1097/00000374-200110000-00013. [DOI] [PubMed] [Google Scholar]
- Evstatiev R, Gasche C. Iron sensing and signalling. Gut. 2012;61:933–952. doi: 10.1136/gut.2010.214312. [DOI] [PubMed] [Google Scholar]
- Fleming RE, Ponka P. Iron overload in human disease. N Engl J Med. 2012;366(4):348–359. doi: 10.1056/NEJMra1004967. [DOI] [PubMed] [Google Scholar]
- Friedman IM, Kraemer HC, Mendoza FS, Hammer LD. Elevated serum iron concentration in adolescent alcohol users. Am J Dis Child. 1988;142(2):156–159. doi: 10.1001/archpedi.1988.02150020058027. [DOI] [PubMed] [Google Scholar]
- Fuchs R, Ellinger I. Endocytic and transcytotic processes in villous syncytiotrophoblast: role in nutrient transport to the human fetus. Traffic. 2004;5(10):725–738. doi: 10.1111/j.1600-0854.2004.00221.x. [DOI] [PubMed] [Google Scholar]
- Gambling L, Andersen HS, Czopek A, Wojciak R, Krejpcio Z, McArdle HJ. Effect of timing of iron supplementation on maternal and neonatal growth and iron status of iron-deficient pregnant rats. J Physiol (Lond) 2004;561(1):195–203. doi: 10.1113/jphysiol.2004.068825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambling L, Czopek A, Andersen HS, Holtrop G, Srai SKS, Krejpcio Z, McArdle HJ. Fetal iron status regulates maternal iron metabolism during pregnancy in the rat. Am J Physiol Regul Integr Comp Physiol. 2009;296(4):R1063–R1070. doi: 10.1152/ajpregu.90793.2008. [DOI] [PubMed] [Google Scholar]
- Gambling L, Lang C, McArdle HJ. Fetal regulation of iron transport during pregnancy. Am J Clin Nutr. 2011;94(6):1903S–1907S. doi: 10.3945/ajcn.110.000885.Am. [DOI] [PubMed] [Google Scholar]
- Geng F, Mai X, Zhan J, Xu L, Zhao Z, Georgieff M, Shao J, Lozoff B. Impact of fetal-neonatal iron deficiency on recognition memory at 2 months of age. J Pediatr. 2015;167(6):1226–1232. doi: 10.1016/j.jpeds.2015.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgieff MK. Nutrition and the developing brain: nutrient priorities and measurement. Am J Clin Nutr. 2007;85(2):614S–620S. doi: 10.1093/ajcn/85.2.614S. [DOI] [PubMed] [Google Scholar]
- Georgieff MK. The role of iron in neurodevelopment: fetal iron deficiency and the developing hippocampus. Biochem Soc Trans. 2008;36(Pt 6):1267–1271. doi: 10.1042/BST0361267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgieff MK. Long-term brain and behavioral consequences of early iron deficiency. Nutr Rev. 2011;69(S1):S43–S48. doi: 10.1111/j.1753-4887.2011.00432.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgieff MK, Brunette KE, Tran PV. Early life nutrition and neural plasticity. Dev Psychopathol. 2015;27(2):411–423. doi: 10.1017/S0954579415000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber MR, Connor JR. Do oligodendrocytes mediate iron regulation in the human brain? Ann Neurol. 1989;26(1):95–98. doi: 10.1002/ana.410260115. [DOI] [PubMed] [Google Scholar]
- Gordeuk VR, Diaz SF, Onojobi GO, Kasvosve I, Debebe Z, Edossa A, Pantin JM, Xiong S, Nekhai S, Nouraie M, Tsukamoto H, Taylor RE. Ferroportin q248h, dietary iron, and serum ferritin in community African-Americans with low to high alcohol consumption. Alcohol Clin Exp Res. 2008;32(11):1947–1953. doi: 10.1111/j.1530-0277.2008.00782.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hare DJ, Lee JK, Beavis AD, van Gramberg A, George J, Adlard PA, Finkelstein DI, Doble PA. Three-dimensional atlas of iron, copper, and zinc in the mouse cerebrum and brainstem. Anal Chem. 2012;84(9):3990–3997. doi: 10.1021/ac300374x. [DOI] [PubMed] [Google Scholar]
- Harrison-Findik DD. Is the iron regulatory hormone hepcidin a risk factor for alcoholic liver disease? World J Gastroenterol. 2009;15(10):1186–1193. doi: 10.3748/wjg.15.1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison-Findik DD, Schafer D, Klein E, Timchenko NA, Kulaksiz H, Clemens D, Fein E, Andriopoulos B, Pantopoulos K, Gollan J. Alcohol metabolism- mediated oxidative stress down-regulates hepcidin transcription and leads to increased duodenal iron transporter expression. J Biol Chem. 2006;281(32):22974–22982. doi: 10.1074/jbc.M602098200. [DOI] [PubMed] [Google Scholar]
- Harrison-Findik DD, Klein E, Crist C, Evans J, Timchenko N, Gollan J. Iron-mediated regulation of liver hepcidin expression in rats and mice is abolished by alcohol. Hepatology. 2007;46(6):1979–1985. doi: 10.1002/hep.21895. [DOI] [PubMed] [Google Scholar]
- Harrison-Findik DD, Klein E, Evans J, Gollan J. Regulation of liver hepcidin expression by alcohol in vivo does not involve Kupffer cell activation or TNF-α signaling. Am J Physiol Gastrointest Liver Physiol. 2009;296(1):G112–118. doi: 10.1152/ajpgi.90550.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey T, Zkik A, Auges M, Clavel T. Assessment of iron deficiency and anemia in pregnant women: an observational French study. Womens Health (Lond) 2016;12(1):95–102. doi: 10.2217/whe.15.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heritage ML, Murphy TL, Bridle KR, Anderson GJ, Crawford DHG, Fletcher LM. Hepcidin regulation in wild-type and Hfe knockout mice in response to alcohol consumption: evidence for an alcohol-induced hypoxic response. Alcohol Clin Exp Res. 2009;33(8):1391–1400. doi: 10.1111/j.1530-0277.2009.00969.x. [DOI] [PubMed] [Google Scholar]
- Hermoso M, Vucic V, Vollhardt C, Arsic A, Roman-Viñas B, Iglesia-Altaba I, Gurinovic M, Koletzko B. The effect of iron on cognitive development and function in infants, children and adolescents: a systematic review. Ann Nutr Metab. 2011;59(2–4):154–165. doi: 10.1159/000334490. [DOI] [PubMed] [Google Scholar]
- Hoyme HE, Kalberg WO, Elliott AJ, Blankenship J, Buckley D, Marais AS, Manning MA, Robinson LK, Adam MP, Abdul-Rahman O, Jewett T, Coles CD, Chambers C, Jones KL, Adnams CM, Shah PE, Riley EP, Charness ME, Warren KR, May PA. Updated clinical guidelines for diagnosing fetal alcohol spectrum disorders. Pediatrics. 2016;138(2):e20154256–e20154256. doi: 10.1542/peds.2015-4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huebner SM, Tran TD, Rufer ES, Crump PM, Smith SM. Maternal iron deficiency worsens the associative learning deficits and hippocampal and cerebellar losses in a rat model of fetal alcohol spectrum disorders. Alcohol Clin Exp Res. 2015;39(11):2097–2107. doi: 10.1111/acer.12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huebner SM, Blohowiak SE, Kling PJ, Smith SM. Prenatal alcohol exposure alters fetal iron distribution and elevates hepatic hepcidin in a rat model of fetal alcohol spectrum disorders. J Nutr. 2016;146(6):1180–1188. doi: 10.3945/jn.115.227983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioannou GN, Dominitz JA, Weiss NS, Heagerty PJ, Kowdley KV. The effect of alcohol consumption on the prevalence of iron overload, iron deficiency, and iron deficiency anemia. Gastroenterology. 2004;126(5):1293–1301. doi: 10.1053/j.gastro.2004.01.020. [DOI] [PubMed] [Google Scholar]
- Jones KL, Smith DW, Ulleland CN, Streissguth AP. Pattern of malformation in offspring of chronic alcoholic mothers. The Lancet. 1973;1(7815):1267–1271. doi: 10.1016/s0140-6736(73)91291-9. [DOI] [PubMed] [Google Scholar]
- Kable JA, Coles CD, Keen CL, Uriu-Adams JY, Jones KL, Yevtushok L, Kulikovsky Y, Wertelecki W, Pedersen TL, Chambers CD, the CIFASD The impact of micronutrient supplementation in alcohol-exposed pregnancies on information processing skills in Ukrainian infants. Alcohol. 2015;49(7):647–656. doi: 10.1016/j.alcohol.2015.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kable JA, O’Connor MJ, Olson HC, Paley B, Mattson SN, Anderson SM, Riley EP. Neurobehavioral disorder associated with prenatal alcohol exposure (ND-PAE): proposed DSM-5 diagnosis. Child Psychiatry Hum Dev. 2016;47(2):335–346. doi: 10.1007/s10578-015-0566-7. [DOI] [PubMed] [Google Scholar]
- Keen CL, Uriu-Adams JY, Skalny A, Grabeklis A, Grabeklis S, Green K, Yevtushok L, Wertelecki WW, Chambers CD. The plausibility of maternal nutritional status being a contributing factor to the risk for fetal alcohol spectrum disorders: the potential influence of zinc status as an example. BioFactors. 2010;36(2):125–135. doi: 10.1002/biof.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly YJ, Sacker A, Gray R, Kelly J, Wolke D, Head J, Quigley MA. Light drinking during pregnancy: still no increased risk for socioemotional difficulties or cognitive deficits at 5 years of age? J Epidemiol Community Health. 2012;66(1):41–48. doi: 10.1136/jech.2009.103002. [DOI] [PubMed] [Google Scholar]
- Kim J, Wessling-Resnick M. Iron and mechanisms of emotional behavior. J Nutr Biochem. 2014;25(11):1101–1107. doi: 10.1016/j.jnutbio.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livock M, Anderson PJ, Lewis S, Bowden S, Muggli E, Halliday J. Maternal micronutrient consumption periconceptionally and during pregnancy: a prospective cohort study. Public Health Nutr. 2016;20(2):294–304. doi: 10.1017/S1368980016002019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez A, Cacoub P, Macdougall IC, Peyrin-Biroulet L. Iron deficiency anaemia. The Lancet. 2016;387(10021):907–916. doi: 10.1016/S0140-6736(15)60865-0. [DOI] [PubMed] [Google Scholar]
- Lozoff B, Georgieff MK. Iron deficiency and brain development. Semin Pediatr Neurol. 2006;13(3):158–165. doi: 10.1016/j.spen.2006.08.004. [DOI] [PubMed] [Google Scholar]
- Lozoff B, Jimenez E, Smith JB. Double burden of iron deficiency in infancy and low socioeconomic status. JAMA Pediatr. 2006a;160(11):1108–1113. doi: 10.1001/archpedi.160.11.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozoff B, Kaciroti N, Walter T. Iron deficiency in infancy: applying a physiologic framework for prediction. Am J Clin Nutr. 2006b;84(6):1412–1421. doi: 10.1093/ajcn/84.6.1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins AC, Almeida JI, Lima IS, Kapitão AS, Gozzelino R. Iron metabolism and the inflammatory response. IUBMB Life. 2017;69(6):442–450. doi: 10.1002/iub.1635. [DOI] [PubMed] [Google Scholar]
- May PA, Gossage JP. Maternal risk factors for fetal alcohol spectrum disorders. Alc Res Health. 2011;34(1):15–26. [PMC free article] [PubMed] [Google Scholar]
- May PA, Hamrick KJ, Corbin KD, Hasken JM, Marais AS, Brooke LE, Blankenship J, Hoyme HE, Gossage JP. Dietary intake, nutrition, and fetal alcohol spectrum disorders in the Western Cape Province of South Africa. Reprod Toxicol. 2014;46:31–39. doi: 10.1016/j.reprotox.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May PA, Hamrick KJ, Corbin KD, Hasken JM, Marais AS, Blankenship J, Hoyme HE, Gossage JP. Maternal nutritional status as a contributing factor for the risk of fetal alcohol spectrum disorders. Reprod Toxicol. 2016;59:101–108. doi: 10.1016/j.reprotox.2015.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McArdle HJ, Gambling L, Kennedy C. Iron deficiency during pregnancy: the consequences for placental function and fetal outcome. Proc Nutr Soc. 2014;73(1):9–15. doi: 10.1017/S0029665113003637. [DOI] [PubMed] [Google Scholar]
- Miller MW, Roskams AJI, Connor JR. Iron regulation in the developing rat brain: effect of in utero ethanol exposure. J Neurochem. 1995;65(1):373–380. doi: 10.1046/j.1471-4159.1995.65010373.x. [DOI] [PubMed] [Google Scholar]
- Milman N. Iron and pregnancy- a delicate balance. Ann Hematol. 2006;85:559–565. doi: 10.1007/s00277-006-0108-2. [DOI] [PubMed] [Google Scholar]
- Milman N, Kirchhoff M. Relationship between serum ferritin, alcohol intake, and social status in 2235 Danish men and women. Ann Hematol. 1996;72(3):145–151. doi: 10.1007/s002770050153. [DOI] [PubMed] [Google Scholar]
- Nair KM, Iyengar V. Iron content, bioavailability & factors affecting iron status of Indians. Indian J Med Res. 2009;130(5):634–645. [PubMed] [Google Scholar]
- Nemeth E, Rivera S, Gabayan V, Keller C, Taudorf S, Pedersen BK, Ganz T. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest. 2004;113(9):1271–1276. doi: 10.1172/JCI20945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtake T, Saito H, Hosoki Y, Inoue M, Miyoshi S, Suzuki Y, Fujimoto Y, Kohgo Y. Hepcidin is down-regulated in alcohol loading. Alcohol Clin Exp Res. 2007;31(S1):2S–8S. doi: 10.1111/j.1530-0277.2006.00279.x. [DOI] [PubMed] [Google Scholar]
- Panel on Micronutrients, Subcommittees on Upper Reference Levels of Nutrients and of Interpretation and Use of Dietary Reference Intakes, and Standing Committee on the Scientific Evaluation of Dietary Reference Intakes. Iron Dietary Reference Intakes for Vitamin A, Vitamin K, Arsenic, Boron, Chromium, Copper, Iodine, Iron, Manganese, Molybdenum, Nickel, Silicon, Vanadium, and Zinc. National Academies Press; 2001. [DOI] [PubMed] [Google Scholar]
- Pietrangelo A. Iron, oxidative stress and liver fibrogenesis. J Hepatol. 1998;28:8–13. doi: 10.1016/S0168-8278(98)80368-1. [DOI] [PubMed] [Google Scholar]
- Popova S, Lange S, Probst C, Parunashvili N, Rehm J. Prevalence of alcohol consumption during pregnancy and fetal alcohol spectrum disorders among the general and Aboriginal populations in Canada and the United States. Eur J Med Genet. 2017;60(1):32–48. doi: 10.1016/j.ejmg.2016.09.010. [DOI] [PubMed] [Google Scholar]
- Powell LW. Normal human iron storage and its relation to ethanol consumption. Australasian Annals of Medicine. 1966;15(2):110–115. doi: 10.1111/imj.1966.15.2.110. [DOI] [PubMed] [Google Scholar]
- Ribot B, Aranda N, Viteri F, Hernández-Martínez C, Canals J, Arija V. Depleted iron stores without anaemia early in pregnancy carries increased risk of lower birthweight even when supplemented daily with moderate iron. Hum Reprod. 2012;27(5):1260–1266. doi: 10.1093/humrep/des026. [DOI] [PubMed] [Google Scholar]
- Rufer ES, Tran TD, Attridge MM, Andrzejewski ME, Flentke GR, Smith SM. Adequacy of maternal iron status protects against behavioral, neuroanatomical, and growth deficits in fetal alcohol spectrum disorders. PLoS ONE. 2012;7(10):1–12. doi: 10.1371/journal.pone.0047499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sozo F, Dick AM, Bensley JG, Kenna K, Brien JF, Harding R, De Matteo R. Alcohol exposure during late ovine gestation alters fetal liver iron homeostasis without apparent dysmorphology. Am J Physiol Regul Integr Comp Physiol. 2013;304(12):R1121–R1129. doi: 10.1152/ajpregu.00479.2012. [DOI] [PubMed] [Google Scholar]
- Streissguth AP, Barr HM, Labbe RF, Smith JR, Darby BL, Smith NJ, Martin DC, Doan RN. Alcohol use and iron status in pregnant women. Alcohol Clin Exp Res. 1983;7(2):227–230. doi: 10.1111/j.1530-0277.1983.tb05447.x. [DOI] [PubMed] [Google Scholar]
- Sukchan P, Liabsuetrakul T, Chongsuvivatwong V, Songwathana P, Sornsrivichai V, Kuning M. Inadequacy of nutrients intake among pregnant women in the Deep South of Thailand. BMC Public Health. 2010;10:1–8. doi: 10.1186/1471-2458-10-572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y, Saito H, Suzuki M, Hosoki Y, Sakurai S, Fujimoto Y, Kohgo Y. Up-regulation of transferrin receptor expression in hepatocytes by habitual alcohol drinking is implicated in hepatic iron overload in alcoholic liver disease. Alcohol Clin Exp Res. 2002;26(8 Suppl):26S–31S. doi: 10.1097/01.ALC.0000026830.27338.23. [DOI] [PubMed] [Google Scholar]
- Tamura T, Goldenberg RL, Hou J, Johnston KE, Cliver SP, Ramey SL, Nelson KG. Cord serum ferritin concentrations and mental and psychomotor development of children at five years of age. J Pediatr. 2002;140(2):165–170. doi: 10.1067/mpd.2002.120688. [DOI] [PubMed] [Google Scholar]
- Tan CH, Denny CH, Cheal NE, Sniezek JE, Kanny D. Alcohol use and binge drinking among women of childbearing age- United States, 2011-2013. MMWR Morb Mortal Wkly Rep. 2015;64(37):1042–1046. doi: 10.15585/mmwr.mm6437a3. [DOI] [PubMed] [Google Scholar]
- Scholl TA. Iron status during pregnancy: setting the stage for mother and infant. Am J Clin Nutr. 2005;81(suppl):1218S–1222S. doi: 10.1093/ajcn/81.5.1218. [DOI] [PubMed] [Google Scholar]
- Wang CY, Babitt JL. Hepcidin regulation in the anemia of inflammation. Curr Opin Hematol. 2016;23(3):189–197. doi: 10.1097/MOH.0000000000000236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward KL, Tkac I, Jing Y, Felt B, Beard J, Connor J, Schallert T, Georgieff MK, Rao R. Gestational and lactational iron deficiency alters the developing striatal metabolome and associated behaviors in young rats. Jour Nutr. 2007;137(4):1043–1049. doi: 10.1093/jn/137.4.1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren KR, Li TK. Genetic polymorphisms: impact on the risk of fetal alcohol spectrum disorders. Birth Defects Res. 2005;73(4):195–203. doi: 10.1002/bdra.20125. [DOI] [PubMed] [Google Scholar]
- West JR, Goodlett CR. Teratogenic effects of alcohol on brain development. Ann Med. 1990;22(5):319–325. doi: 10.3109/07853899009147914. [DOI] [PubMed] [Google Scholar]
- Wilhelm CJ, Guizzetti M. Fetal alcohol spectrum disorders: an overview from the glia perspective. Frontiers in Integrative Neuroscience. 2016;9:1–16. doi: 10.3389/fnint.2015.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter WE, Bazydlo LAL, Harris NS. The molecular biology of human iron metabolism. Lab Med. 2014;45(2):92–102. doi: 10.1309/LMF28S2GIMXNWHMM. [DOI] [PubMed] [Google Scholar]
- World Health Organization. The global prevalence of anaemia in 2011 WHO Report. 2015. [Google Scholar]
- Wozniak JR, Muetzel RL, Mueller BA, McGee CL, Freerks MA, Ward EE, Nelson ML, Chang PN, Lim KO. Microstructural corpus callosum anomalies in children with prenatal alcohol exposure: an extension of previous diffusion tensor imaging findings. Alcohol Clin Exp Res. 2009;33(10):1825–1835. doi: 10.1111/j.1530-0277.2009.01021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak JR, Fuglestad AJ, Eckerle JK, Fink BA, Hoecker HL, Boys CJ, Radke JP, Kroupina MG, Miller NC, Brearly AM, Zeisel SH, Georgieff MK. Choline supplementation in children with fetal alcohol spectrum disorders: a randomized, double-blind, placebo-controlled trial. Am J Clin Nutr. 2015;102(5):1113–1125. doi: 10.3945/ajcn.114.099168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JK, Giesbrecth HE, Eskin MN, Aliani M, Suh M. Nutrition implications for fetal alcohol spectrum disorder. Adv Nutr. 2014;5(7):675–692. doi: 10.3945/an.113.004846.A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisel SH. Nutrition in pregnancy: the argument for including a source of choline. Int J Womens Health. 2013;5:193–199. doi: 10.2147/IJWH.S36610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zentner LEA, Rondó PHC, Duran MC, Oliveira JM. Relationships of blood lead to calcium, iron, and vitamin C intakes in Brazilian pregnant women. Clin Nutr. 2008;27(1):100–104. doi: 10.1016/j.clnu.2007.10.005. [DOI] [PubMed] [Google Scholar]
- Zhang C. Essential functions of iron-requiring proteins in DNA replication, repair and cell cycle control. Protein Cell. 2014;5(10):750–760. doi: 10.1007/s13238-014-0083-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu R, Wang Y, Zhang L, Guo Q. Oxidative stress and liver disease. Hepatology Research. 2012;42(8):741–749. doi: 10.1111/j.1872-034X.2012.00996.x. [DOI] [PubMed] [Google Scholar]