

Abstract
We previously reported the discovery, validation, and structure-activity relationships of a series of piperidinyl ureas that potently inhibit the DCN1-UBE2M interaction. We demonstrated that compound 7 inhibits both the DCN1-UBE2M protein-protein interaction and DCN1-mediated cullin neddylation in biochemical assays and reduces levels of steady-state cullin neddylation in a squamous carcinoma cell line harboring DCN1 amplification. Although compound 7 exhibits good solubility and permeability, it is rapidly metabolized in microsomal models (CLint = 170 mL/min/kg). This work lays out the discovery of an orally bioavailable analog, NAcM-OPT (67). Compound 67 retains the favorable biochemical and cellular activity of compound 7 but is significantly more stable both in vitro and in vivo. Compound 67 is orally bioavailable, well tolerated in mice, and currently used to study the effects of acute pharmacologic inhibition of the DCN1-UBE2M interaction on the NEDD8/CUL pathway.
TOC image

Introduction
Ubiquitin-like protein (UBL) modification pathways have emerged as important targets for oncology drug discovery. Successful examples include clinical proteasome inhibitors (e.g., Bortezomib or Carfilzomib)2, E3 inhibitors3, and the NEDD8 E1 inhibitor (Pevonedistat)4–6. Here we present the discovery of an orally bioavailable small molecule inhibitor of the UBL NEDD8 co-E3 enzyme, defective in cullin neddylation 1 (DCN1). DCN1 is also known as DCUN1D1, DCNL1, or SCCRO (squamous cell carcinoma-related oncogene); we use “DCN1” hereafter. DCN1 is an oncogenic driver gene contained within a recurrent 3q26.3 chromosomal amplification that is common in squamous cell carcinomas (SCC) of the lung, head, and neck.7–13 DCN1 amplification is associated with malignant transformation, metastasis, and poorer patient survival rates.7–13
DCN1 is part of a hierarchical E1-E2-E3 enzymatic cascade that covalently attaches the UBL NEDD8 to target proteins14. The first step of this cascade is the ATP-dependent formation of an E1-NEDD8 thioester intermediate. Next, the NEDD8 is transferred from the E1 to a catalytic Cys on the E2. E3s then join the E2-NEDD8 complex and recruit the target protein. This complex consequently catalyzes the formation of an isopeptide bond connecting NEDD8’s C-terminus to the ɣ-amino group of a lysine side-chain on the target protein. DCN1 is a co-E3 that stimulates the neddylation reaction.15–16 Cullins, the best-characterized substrates of the NEDD8 pathway, are a family of ubiquitin E3 ligases that are activated by neddylation17–19. Cullins serve as the catalytic domain for approximately 300 human multi-protein CUL-RING E3 ligases (CRLs) that regulate ubiquitin (UB) conjugation.18, 20
The CRLs control diverse biological processes and their dysfunction is implicated in human diseases, including cancer.21–23 Thus, drug discovery efforts targeting the CRLs continues to grow.24–27 One approach to inhibiting CRL activity is to block cullin neddylation.28–31 Pevonedistat (MLN4924) is the most well characterized inhibitor of the NEDD8 pathway and is currently in phase II clinical trials for the treatment of cancer.6, 32–33 Pevonedistat is an adenosine sulfamate analogue that covalently inhibits the NEDD8 E1, thus inhibiting all neddylation.34 The success of Pevonedistat has stimulated discovery of other inhibitors of the neddylation pathway. Virtual screening has yielded both reversible and covalent small molecule inhibitors of the NEDD8 E1.35–38 Several rhodium(III) complexes also inhibit the NEDD8 E1, offering an alternative to small molecules.39–40 Recently, an inhibitor of the COP9 signalosome, responsible for de-neddylation of the CRLs, has been reported and also displays anti-tumor activity.41 We, and others, have recently reported the discovery of inhibitors of DCN1.42–44
While complete ablation of neddylation appears to be clinically efficacious,29 it is also toxic. In an effort to maintain efficacy and reduce toxicity, we sought more specific ways to interrogate the neddylation pathway.42 DCN1 plays a role in tumor formation, maintenance, and regulation of the neddylation pathway.7–17, 45–46 Therefore, we sought to develop DCN1 inhibitors that may offer advantages and/or alternative specificities compared to pan-neddylation or de-neddylation inhibitors.1, 42–43
We recently identified a class of piperidinyl ureas that inhibit the DCN1-UBE2M protein-protein interaction and thereby reduce steady-state neddylation of specific cullins.42 Subsequent iterative structure-enabled optimization of our high throughput screening hit (compound 2, Table 1, IC50 = 7.6 μM) yielded compound 7 (IC50 = 0.060 μM)1 that was 100-fold more potent than our initial hit.1, 42 Our optimization targeting five sub-pockets within the DCN1-UBE2M binding site is reported in the companion manuscript.1 The first three sub-pockets are the Ile, N-acetyl, and Leu, named for the UBE2M amino acid side chains that occupy these pockets. The last two sub-pockets are the urea and hinge pockets, named for the moieties from the chemotype that occupy them (Figure 1).42
Table 1. Rapid metabolism of early lead DCN1-UBE2M inhibitors.
Values for the half-life and intrinsic clearance were determined at a concentration of 0.8 μM using mouse microsomes and are represented as the means plus or minus standard deviation from three replicates. Compound preparation and potency was previously reported.1
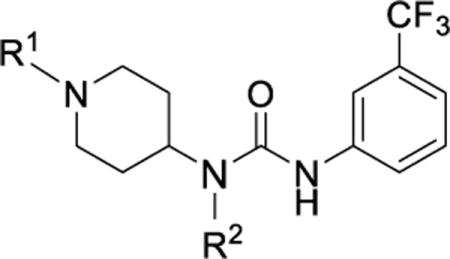
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
|
|
|
||||||||
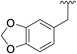
| No | R1 | R2 | t(1/2) (hr) | CLint (mL/min/kg) | No | R1 | R2 | t(1/2) (hr) | CLint (mL/min/kg) |
|
|
|
||||||||
| 1 |
|
|
< 0.1 | > 500 | 6 |
|
|
0.150 ± 0.019 | 361 ± 45.1 |
| 2 |
|
|
0.591 ± 0.0525 | 91.4 ± 8.12 | 7 |
|
|
0.327 ± 0.010 | 165 ± 25.9 |
| 3 |
|
|
0.137 ± 0.021 | 394 ± 55.2 | 8 |
|
|
0.245 ± 0.014 | 220 ± 12.8 |
| 4 |
|
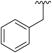
|
0.170 ± 0.016 | 318 ± 29.6 | 9 |
|
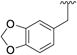
|
0.124 ± 0.006 | 434 ± 21.1 |
| 5 |
|
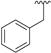
|
< 0.1 | > 500 | 10 |
|
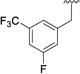
|
0.271 ± 0.014 | 199 ± 9.99 |
Figure 1.
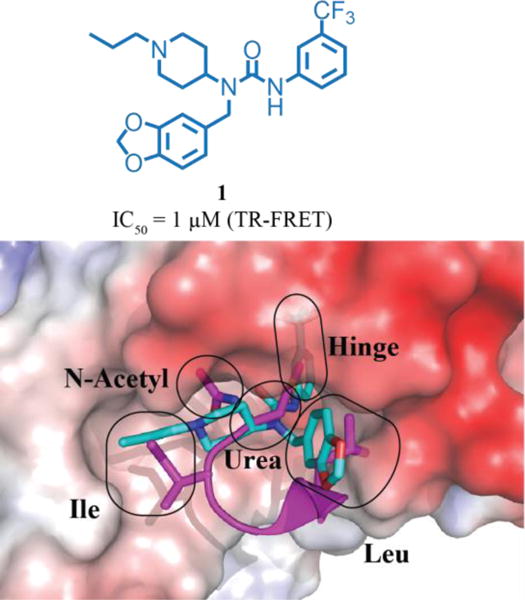
Top: Chemical structure of 1. Bottom: Overlay of 1(teal): DCN1 (electrostatic potential surface) (PDB: 6BG5)1 and UBE2MNAc (magenta): DCN1 (PDB: 3TDU) X-ray co-crystal structures, highlighting regions targeted for optimization1.
Examination of several X-ray co-crystal structures led us to identify four key drivers of potency. This finding is further exemplified by the 1:DCN1 X-ray co-structure shown in Figure 1 (PDB: 6BG5)1. The four potency drivers are: (1) the 4-aminopiperidinyl urea core that positions the hydrophobic alky and aryl ends of the molecule into the Ile and hinge pockets; (2) a key H-bond between the urea aryl N-H and the backbone amide of Gln114 in the urea pocket; (3) the induced hinge pocket, which affords tight steric packing of hydrophobic residues around the m-CF3 phenyl ring; and (4) the hydrophobic interactions of the methylbenzodioxole urea substituent within the Leu pocket (Figure 1).42 Compound 7 biochemically inhibits the DCN1-UBE2M protein-protein interaction and cullin neddylation.1 This compound also suppresses steady-state levels of cullin neddylation in HCC95 cells, a squamous carcinoma (SCC) cell line highly expressing DCN1.1 However, compound 7 is rapidly metabolized in microsomal and murine models. This report describes our continuing optimization efforts aimed at improving the clearance of compound 7. Our optimization strategy focused on 1) understanding the physical drivers for bioavailability (SPR); and 2) evaluating metabolic and toxic liabilities associated with the scaffold. This strategy has yielded a potent, orally bioavailable, and significantly more stable chemical probe, NAcM-OPT (67)42.
Results
Given the clear effect of compound 7 in cells1, we examined whether this compound possessed appropriate absorption, distribution, metabolism, excretion, and toxicology (ADME/T) characteristics to block DCN1 activity using in vivo models. Compound 7 was moderately soluble (57 ± 1.4 μM) and highly cell permeable (1400 ± 39 × 10−6 cm/s)1. However, compound 7 was unstable in mouse microsomes (CLint = 170 mL/min/kg, Table 1). We tested several structurally similar analogs and found that they were also rapidly metabolized (CLint > 150 mL/min/kg, Table 1). The most stable analog was the initial screening hit 2 (CLint = 90 mL/min/kg). Therefore, it seems as though the structural modifications made to improve potency also reduced metabolic stability. In particular, the benzyl urea substitution that was critical for potency and specificity due to its interactions within the Leu pocket1 (Figure 1) appears to increase rates of oxidative metabolism.
In order to ensure that the microsomal models accurately predict in vivo clearance and reveal any systemic toxicology exhibited by the series, we tested compound 7 in a single-dose intravenous (IV) pharmacokinetic (PK) study using three doses: 0.5, 1.0, and 1.5 mg/kg (Figure 2A). We did not observe any adverse reactions or compound-related side effects within 48 h after dosing (Supplementary Table 1). Compound 7 was rapidly cleared (t1/2 = 0.14 h, Clplasma = 42 L/h/kg) at all doses, suggesting that the microsomal models correctly predict clearance of compound 7 in vivo. Therefore, we sought to mitigate the scaffold’s inherent metabolic liabilities using microsomal models as a predictor of in vivo clearance.
Figure 2. Characterization of compound metabolism.
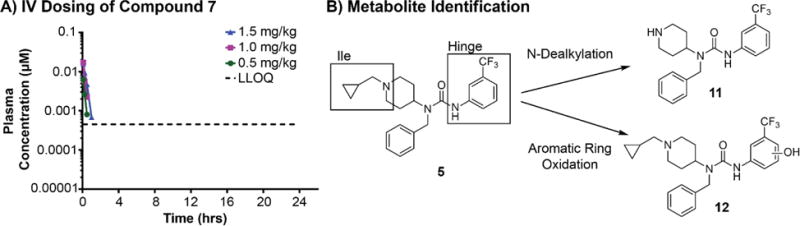
A) Compound 7 plasma concentrations over 24 h following IV administration 0.5, 1.0, and 1.5 mg/kg doses. Plasma concentrations from time points taken after 2 h were below the limit of quantitation and are thus excluded from the graph. B) Summary of metabolite identifications studies using compound 5. For details, see Supplementary Table 2.
We utilzed a recursive process of analog design, synthesis, testing, and design refinement to optimize the series. We assessed biochemical potency using our previously reported TR-FRET assay,42 which measures the binding of full-length DCN1 to a constrained peptide corresponding to N-terminally acetylated UBE2M15–16, 42. We used structurally directed modifications, as well as more empircal investigation of the moeity fitting into the induced fit hinge pocket to probe the steric and electronic requirements of binding47–51. We assessed the metabolic stability of all analogs exhibiting reasonable potency in mouse microsomes. Analogs that retained potency and posessed significantly improved microsomal stability were profiled in murine models to determine in vivo toxicity, clearance, and bioavailability.
To further guide the design of metabolically stable analogs, we conducted metabolite identification (Met-ID) studies using a close structural analog of compound 7, compound 5 (Figure 2B). This study identified N-dealkylation (11) of the piperidine ring in the Ile targeting substituent and oxidation of the aromatic ring of the urea (12), targeting the hinge region, as the primary metabolic transformations (Figure 2B; Supplementary Table 2). Time-course metabolite formation studies suggest that these modifications take place sequentially; oxidation of the aromatic ring occurs first, followed by N-dealkylation (Supplementary Table 2). Although we were unable to identify the specific oxidized carbon on the aromatic ring, we hypothesize that the carbons ortho- or para- to the amino group are the most likely the sites of oxidation. This hypothesis is based on the electron donating nature of the amino substituent and on previous reports52–53. Therefore, we targeted optimization of the Ile and hinge moieties. We used both a rational, structure-based, design and an empirical parallel chemistry approach to understand the key structural elements that drive the structure-activity relationship (SAR) and structure-property relationship (SPR) that define metabolic liabilities.
Following these findings, we sought to maintain potency and improve metabolic stability by leaving invariant the benzyl group (targeting the Leu pocket) of compound 7 and varying the alkyl side chain (targeting the Ile pocket) and aniline (targeting the hinge pocket) (Figure 1). We devised two synthetic routes that permitted late stage introduction of either substituent (Scheme 1; routes i, ii). Both routes were amenable to parallel synthesis.
Scheme 1. General synthetic routes for generating analogs.
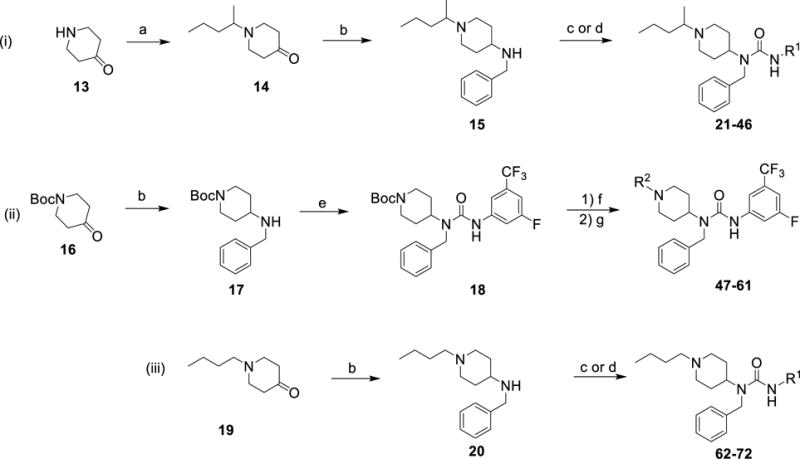
(a) potassium carbonate, 2-iodopentane, acetonitrile, 80 °C, 16 h, 30-48% (b) benzylamine, sodium triacetoxyborohydride, acetic acid, dichloromethane, rt, 16 h, 90-100%; (c) R1-isocyanate, N,N-diisopropylethylamine, dichloromethane, rt, 2 h, 40-80%; (d) R1-amine, 1,1′-carbonyldiimidazole, tetrahydrofuran, 16 h, 30-55%; (e) 3-Fluoro-5-(trifluoromethyl)phenyl isocyanate, N,N-diisopropylethylamine, dichloromethane, rt, 2 h, 80-99%; (f) trifluoroacetic acid, dichloromethane, 0 °C - rt, 16 h, 95%; (g) R2 (aldehyde or ketone), dibutyltin (IV) chloride, phenylsilane, tetrahydrofuran, rt, 16 h, 40-75%.
Synthesis of aniline analogs began by alkylation of 4-piperidone (13) with 2-iodopentane (Scheme 1; routes i)42 to generate ketone 14. Reductive amination of 14 with benzylamine furnished the secondary amine 1542. 15 was then either coupled directly with isocyanates or with anilines using cabonyldiimidazole (CDI) as a coupling agent (21 – 46, Tables 2 & 3)54–57. Given the wider availability of anilines, we used the CDI-mediated reaction for our parallel chemistry. For this reaction, it was critical to allow CDI to react with the aniline prior to addition of the secondary amine 15. The optimized procedure required no work-up and direct purification by HPLC chromatography afforded moderate yields (30-55%) of the desired products. The scalability and ease of this synthetic route allowed us to prepare roughly 140 analogs variant in the aniline substituent.
Table 2. Optimization of the phenyl urea occupying the hinge pocket.
IC50 values were generated using our TR-FRET binding assay and are represented as the mean of three replicates with errors reported as the 95% confidence interval.
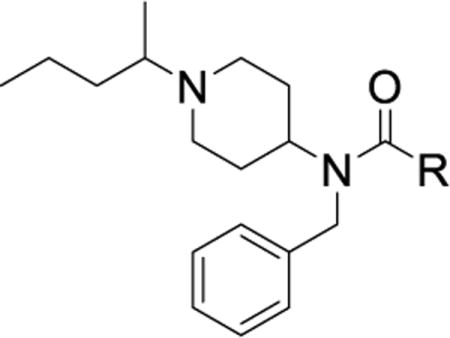
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
|
|
|
||||||||
| No | R4 | IC50 (μM) (95% Clf | t(1/2) (hr) | CLint’ (mL/min/kg) | No | R4 | IC50 (μM) (95% Cl)a | t(1/2) (hr) | CLint’ (mL/min/kg) |
|
|
|
||||||||
| 21 |
|
1.69 (0.640-4.46) |
≤0.10 | ≥ 500 | 26 |
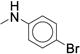
|
0.068 (0.049-0.095) |
0.386 ± 0.021 | 148 ± 7.63 |
| 22 |
|
0.304 (0.239-0.386) |
≥0.10 | ≥500 | 27 |
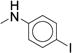
|
0.054 (0.044-0.066) |
0.498 ± 0.013 | 115 ± 2.78 |
| 23 |
|
0.353 (0.288-0.434) |
0.223 ± 0.021 | 256 ± 22.4 | 28 |
|
0.356 (0.241-0.527) |
0.160 ± 0.015 | 356 ± 31.9 |
| 24 |
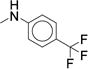
|
0.314 (0.229-0.432) |
0.455 ± 0.012 | 125 ± 3.17 | 29 |
|
0.654 (0.445-0.961) |
0.177 ± 0.003 | 323 ± 4.46 |
| 25 |
|
0.112 (0.082-0.152) |
0.368 ± 0.027 | 156 ± 10.7 | 30 |
|
1.72 (1.23-2.41) |
ND | ND |
95% confidence intervals were generated based on non-linear regression using the R drc package with the four-parameter log-logistic function (LL2.4)59. Values for the half-life and intrinsic clearance were determined at a concentration of 0.8 μM using mouse microsomes and are represented as the means plus or minus standard deviation from three replicates. ND = data not determined.
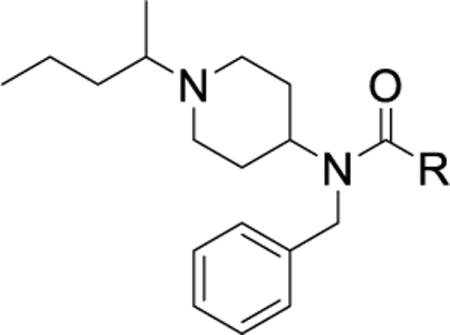
Table 3. Second round of optimization of the phenyl urea occupying the hinge pocket.
IC50 values were generated using our TR-FRET binding assay and are represented as the mean of three replicates with errors reported as the 95% confidence interval.
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
|
|
|
||||||||
| No | R4 | IC50 (μM) (95% Cl)a | t(1/2) (hr) | CLint’ (mL/min/kg) | No | R4 | IC50 (μM) (95% Cl)a | t(1/2) (hr) | CLint’ (mL/min/kg) |
|
|
|
||||||||
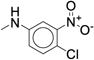
| 31 |
|
0.082 (0.060-0.112) |
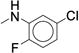
0.379 ± 0.027 | 151 ± 10.2 | 39 |
|
> 34 | 0.249 ± 0.015 | 230 ± 13.1 |
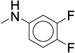
| 32 |
|
0.817 (0.666-1.00) |
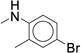
0.189 ± 0.013 | 302 ± 15.2 | 40 |
|
9.43 (6.16-14.5) |
ND | ND |
| 33 |
|
0.241 (0.212-0.273) |
0.150 ± 0.013 | 381 ± 31.9 | 41 |
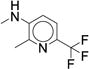
|
13.3 (11.7-15.1) |
> 4 | < 13.5 |
| 34 |
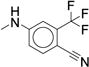
|
0.260 (0.202-0.333) |
0.932 ± 0.056 | 61.3 ± 3.49 | 42 |
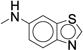
|
0.626 (0.338-1.16) |
0.239 ± 0.025 | 240 ± 24.0 |
| 35 |
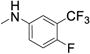
|
0.290 (0.240-0.350) |
0.393 ± 0.027 | 145 ± 9.51 | 43 |
|
0.077 (0.044-0.133) |
^ 0.10 | ≥500 |
| 36 |
|
0.058 (0.045-0.075) |
0.196 ± 0.016 | 291 ± 22.4 | 44 |
|
0.990 (0.718-1.37) |
< 0.10 | > 500 |
| 37 |
|
0.481 (0.390-0.594) |
<0.10 | >500 | 45 |
|
0.018 (0.014-0.025) |
<0.10 | > 500 |
| 38 |
|
7.44 (6.28-8.81) |
0.808 ± 0.062 | 66.9 ± 5.15 | 46 |
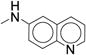
|
> 20 | ND | ND |
95% confidence intervals were generated based on non-linear regression using the R drc package with the four-parameter log-logistic function (LL2.4)59. Values for the half-life and intrinsic clearance were determined at a concentration of 0.8 μM using mouse microsomes and are represented as the means plus or minus standard deviation from three replicates. ND = data not determined.
Synthesis of analogs targeting variation of the alkyl sidechain started with reductive amination of 1-N-boc-4-piperidone (16) with benzylamine to give 17. A limited range of anilines with electron-withdrawing substitutions provided appropriate activity and the potential for increased stability. Thus, the secondary amine 17 was coupled with 3-fluoro-5-(trifluoromethyl)phenyl isocyanate to give 18. After BOC de-protection, Lewis acid-mediated reductive amination58 of 18 with either ketones or aldehydes, afforded the targeted analogs (47 – 61, Table 4) in moderate to good yields (40-75%). We used this route to prepare roughly 45 analogs variant in the alkyl side chain (Supplementary Dataset 1). Compounds prepared by parallel synthesis were characterized for purity (≥ 95% as characterized by UV/ELSD) and identity using UPLC-LRMS (Supplementary Dataset 1). Compounds that proved critical to our SAR analysis were further characterized using 1H NMR and HRMS (Supplementary Information).
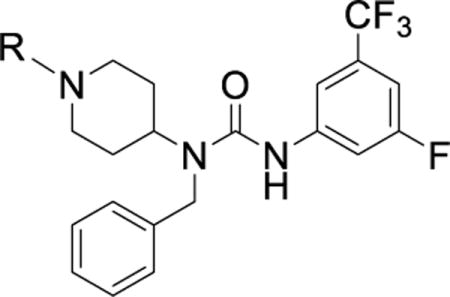
Table 4. Optimization of the alkyl piperidine.
IC50 values were generated using our TR-FRET binding assay and are represented as the mean of three replicates with errors reported as the 95% confidence interval.
| ||||||||
|---|---|---|---|---|---|---|---|---|
|
|
|
|
||||||
| No | R4 | IC50 (μM) (95% cna | No | R4 | IC50 (μM) (95% Cl)a | No | R4 | IC50 (μM) (95% Cl)a |
|
|
|
|
||||||
| 47 |
|
0.256 (0.095-0.685) |
52 |
|
>28 | 57 |
|
>25 |
| 48 |
|
1.79 (1.36-2.36) |
53 |
|
4.49 (3.86-5.21) |
58 |
|
4.27 (2.27-8.04) |
| 49 |
|
3.21 (2.12-4.85) |
54 |
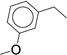
|
> 28 | 59 |
|
> 24 |
| 50 |
|
>24 | 55 |
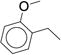
|
>25 | 60 |
|
> 22 |
| 51 |
|
>24 | 56 |
|
>28 | 61 |
|
> 21 |
95% confidence intervals were generated based on non-linear regression using the R drc package with the four-parameter log-logistic function (LL2.4)59.
Our studies revealed that the n-butyl alkyl sidechain was as potent but more metabolically stable than the 2-pentyl substitution (Table 5). Thus, we synthesized a final set of analogs starting from commercially available n-butyl piperidinone 19 (Scheme 1; route iii)42. This two-step procedure is readily scalable and has been used to prepare more than five grams of our top compound 67 with > 98% purity as characterized by UV/ELSD and NMR analysis42.
Table 5. Final optimization of n-butylpiperidines.
IC50 values were generated using our TR-FRET binding assay and are represented as the mean of three replicates with errors reported as the 95% confidence interval.
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
|
|
|
||||||||
| No | R4 | IC50 (μM) (95% Cl)a | t(1/2) (hr) | CLint’ (mL/min/kg) | No | R4 | IC50 (μM) (95% Cl)a | t(1/2) (hr) | CLint’ (mL/min/kg) |
|
|
|
||||||||
| 62 |
|
3.57 (2.87-4.43) |
<0.10 | >500 | 68 |
|
4.27 (2.95-6.16) |
<0.10 | >500 |
| 63 |
|
0.358 (0.248-0.515) |
≥0.10 | ≥500 | 69 |
|
0.583 (0.437-0.777) |
≥0.10 | ≥500 |
| 64 |
|
1.30 (0.947-1.78) |
0.178 ± 0.002 | 305 ± 4.02 | 70 |
|
0.612 (0.266-1.41) |
1.94 ± 0.189 | 27.9 ± 2.71 |
| 65 |
|
1.85 (1.48-2.31) |
ND | ND | 47 |
|
0.256 (0.095-0.685) |
2.43 ± 0.531 | 22.3 ± 4.88 |
| 66 |
|
4.25 (2.72-6.64) |
5.155 ± 0.015 | 349 ± 22.5 | 71 |
|
0.325 (0.214-0.496) |
0.286 ± 0.022 | 189 ± 14.5 |
| 67 |
|
0.045 (0.013-0.153) |
2.11 ± 0.336 | 25.6 ± 4.08 | 72 |
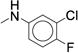
|
1.09 (0.925-1.28) |
1.40 ± 0.059 | 38.7 ± 1.63 |
95% confidence intervals were generated based on non-linear regression using the R drc package with the four-parameter log-logistic function (LL2.4)59. Values for the half-life and intrinsic clearance were determined at a concentration of 0.8 μM using mouse microsomes and are represented as the means plus or minus standard deviation from three replicates. ND = data not determined.
First, we explored the SAR/SPR of the aniline urea substituent targeting the hinge pocket. Our previous SAR studies and X-ray co-structures revealed that to permit deeper compound binding, the hinge pocket undergoes a structural rearrangement to produce a hydrophobic cavity composed of alkyl and aromatic residues (Ile86, Phe89, Val102, Ile105, Ala106, Phe109, Ala111, Phe117, Phe122, Phe164)1, 42. Throughout all of our investigations, an aromatic ring with a small hydrophobic substituent proved optimal for filling this sub-pocket. Compound 7 (IC50 = 0.060 μM), the most potent analog, utilizes a meta-trifluoromethylphenyl aniline that fits perfectly into the hinge sub-pocket1. Previous work suggests that although meta- and para-substitutions were tolerated, modestly increased steric bulk or ortho-substitution eliminate binding1. Thus, we used the parallel chemistry route to rapidly produce diverse analogs while paying particular attention to roughly isosteric para- and meta-substituted aniline rings (Table 2).
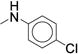
Initial exploration of mono-substituted phenyl ureas (Table 2, 21 – 30) revealed that only the para-bromo (26) and para-iodo (27) analogs were as potent as compound 7, with half maximal inhibitory concentrations (IC50’s) of roughly 0.060 μM. All of the mono-substituted phenyl ureas were rapidly cleared in the microsomal assay (CLint > 120 mL/min/kg). Halogen substituents were generally more potent than the corresponding alkyl substituents, suggesting that while hydrophobicity is necessary, the presence of an electron-withdrawing group, and potentially a hydrogen bond acceptor, is important for potency (25 – 27). This result suggested that we pursue the common strategy of decreasing the rate of metabolic oxidation of aromatic rings by decreasing their electron density. Indeed, such analogs were slightly more stable in microsomal models than compound 7 (CLint = 170 mL/min/kg vs. between 120 and 160 mL/min/kg, Table 2).
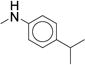
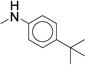
In concordance with previous studies,1 larger substitutions such as i-Pr (23, 29) and t-Bu (30) significantly reduced analog potency (Table 2, Supplementary Dataset 1). Similar to the para- series, the meta-substituted compounds’ potency appeared to improve with hydrophobic electron-withdrawing substituents. The meta-trifluoromethylphenyl (7, IC50 = 0.060 μM) was more potent than the meta-chlorophenyl (22, IC50 = 0.300 μM), a result that stands in stark contrast to the para-substituted analogs (p-CF3 (24) IC50 = 0.300 μM; p-Cl (25), IC50 = 0.100 μM). The meta-i-Pr analog (23, IC50 = 0.350 μM) was more potent than the para-i-Pr analog (29, IC50 = 0.650 μM). The meta-fluoro analog (21) was less potent (IC50 = 1.70 μM), likely due to its inability to fill the available pocket. Together, these results suggest that the meta-position affords slightly more room for increased steric bulk compared to the para-position. The results also highlight that subtle variations in size and electronic properties can significantly affect potency when there is induced fit in the binding site. The para-iodo analog (27, IC50 = 0.050 μM) was the only compound that maintained the potency of compound 7 and increased stability (CLint = 120 mL/min/kg). However, the observed increase in stability (CLint = 170 mL/min/kg vs. 120 mL/min/kg) was not sufficient to render the compound suitable for in vivo studies. We next investigated the di-substituted phenyl ureas with the intent to identify a substituent with improved potency and stability.
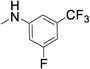
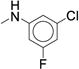
The targeted di-substituted phenyl ureas (Table 3) were designed to solidify the trends observed for the mono-substituted analogs. That is, the analogs should incorporate small hydrophobic and electron-withdrawing substituents at either the ortho- or para-position. These substitutions appear to electronically and sterically block oxidation of the aromatic ring without negatively affecting potency. We first tested meta- and para-di-substituted analogs (31 – 35). These analogs retained moderate potency with the meta-nitro, para-chloro analog (31) exhibiting an IC50 value below 0.100 μM. However, due to the potential toxicology associated with the nitro substituent60, this compound was deprioritized. These compounds were also subject to rapid metabolism. The meta-trifluoromethyl, para-nitrile derivative 34 (IC50 = 0.260 μM) was the only compound that was significantly more stable in the microsomal model than compound 7 (CLint = 60 mL/min/kg vs. 120 mL/min/kg). Among the phenyl ureas with bis-meta substitution patterns, only the meta-trifluoromethyl, meta-fluoro analog 36 was equipotent to compound 7 (IC50 = 0.060 μM). The corresponding meta-chloro, meta-fluoro analog 37 was significantly less potent, further emphasizing the tightly constrained nature of the SAR. All of the bis-meta analogs were rapidly metabolized.
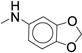
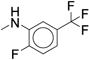
We next examined ortho-, meta-di-substituted phenyl ureas. We observed reduced potency even in the presence of small ortho-substituents (Table 3, 38 – 41), which is consistent with previous SAR data1. This series led to the emergence of the pyridine analog 41 - our first compound to exhibit good metabolic stability (CLint < 13 mL/min/kg). Unfortunately, this compound, like other pyridine-containing analogs, exhibited significantly reduced potency (IC50 = 13 μM, Supplementary Dataset 1)1, 42. Examination of fused bicyclic aryl ureas revealed a general trend that the hydrophobic carbocyclic compounds were more potent than their heterocyclic counterparts (42 – 46). The 2-napthyl analog (46) afforded our first significant gain in potency (IC50 = 0.020 μM). However, all of the fused bicyclic analogs were rapidly metabolized in the microsomal model (Table 3). This result illustrates a common phenomenon in medicinal chemistry: as one parameter is optimized, another in compromised. Based on these analogs, we hypothesized that reducing the electron density of the aniline ring would mitigate the oxidation of the aromatic ring, but the scope of the SAR would be narrow.
In parallel to our investigations of aniline analogs, we sought to address N-dealkylation of the alkyl-piperidine by exploring various substitutions occupying the Ile pocket. Initially, we fixed the aniline substituent as the meta-CF3, meta-F, based on inhibitory potency. Using this base, we screened a range of analogs replacing the 2-pentyl piperidine substitution that targets the Ile sub-pocket (Figure 1). Our previous studies closely explored alkyl substituents at this position and revealed that an n-butyl group was preferred and branched alkyl chains improved potency1. We have also previously prepared simple benzyl and aryl analogs. To expand our SAR, we explored several other alkyl, aryl, and heteroaryl substituents (Table 4, 47 – 61). Unfortunately, with the exception of the n-butyl analog 47 (IC50 = 0.260 μM), all of the substituents led to significantly reduced potency (Table 4, Supplementary Dataset 1). Thus, we selected only compound 47 for further evaluation in the microsomal model.
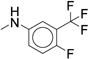
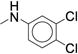
Microsomal modeling revealed that compound 47 was significantly more stable than compound 7 (CLint = 22 mL/min/kg vs. 170 mL/min/kg), with only a five-fold reduction in biochemical potency (IC50 = 0.260 μM vs. 0.060 μM). Comparison of 47 to the 2-pentyl derivative 36 revealed that replacing the 2-pentyl substitution with an n-butyl substitution markedly increased stability (CLint = 22 mL/min/kg vs. 290 mL/min/kg). Assuming this was a general trend, we examined other aryl ureas containing the n-butylpiperidine (Table 5). The di-substituted analogs were more stable than the corresponding mono-substituted aryl urea analogs. The di-substituted para-fluoro, meta-trifluoromethyl (70) or meta-fluoro, meta-trifluoromethyl (47) were more stable than the ortho-fluoro, meta-trifluoromethyl (69). This result supports our hypothesis that the site of oxidation is most likely the carbon para- to the amino substituent. Compound 67, which contains a meta-chloro, para-chloro phenyl urea afforded the best balance of potency and stability. Compound 67 (IC50 = 0.050 μM) retains the biochemical potency of our optimized hit compound 7 but is significantly more stable in microsomal models with a calculated intrinsic clearance of 25 mL/min/kg.
In order to determine whether we had altered the landscape of primary metabolites or simply decreased their rate of formation, we conducted metabolite identification studies with 67 (Supplementary Table 3). These studies revealed that replacing the meta-trifluoromethyl group of the aniline with a meta-, para-dichloro group effectively decreased the rate of microsomal oxidation. However, oxidation of the aniline ring remained a minor metabolic pathway. N-dealkylation was now the predominant metabolism event. Additionally, we observed oxidation of the benzyl urea substituent targeting the Leu pocket. These results indicate that further optimization may yield even more stable compounds. We believed we had successfully increased simulated oxidative stability without negatively affecting potency, and therefore sought to characterize the toxicology and pharmacokinetic characteristics of compound 67 in murine models.
We first conducted a low-dose IV PK experiment to understand the gross toxicity and clearance of compound 67 (Figure 3A). Similar to compound 7, 67 failed to induce any measureable adverse reactions during the 48 h monitoring period following IV administration (Supplementary Table 4, 5). The observed Cmax of compound 7 (Figure 2A) and 67 (Figure 3A) shows a non-linear increase from 0.5 to 1.5 mg/kg. Both compounds exhibit similar and large volumes of distribution (87.3 – 176 L/kg). The IV half-life of 67 is 3-4 hours, which is significantly longer than the half-life of compound 7 (t1/2 = 0.14-0.2 h). The increased metabolic stability of compound 67 correlates with the results from the microsomal model, which showed that this compound has a six-fold lower clearance in mouse microsomes (Supplementary Table 6). Due to the comparatively shorter half-life and lower Cmax, the AUClast of compound 7 is much smaller than that of compound 67. Therefore, compound 67 is the preferred early lead for preliminary in vivo studies. Based on these results, we determined that it was unethical to conduct an oral dosing study for compound 7. However, we did conduct an oral PK experiment with compound 67 (Figure 3B).
Figure 3. Preliminary pharmacokinetic profiling of compound 67.
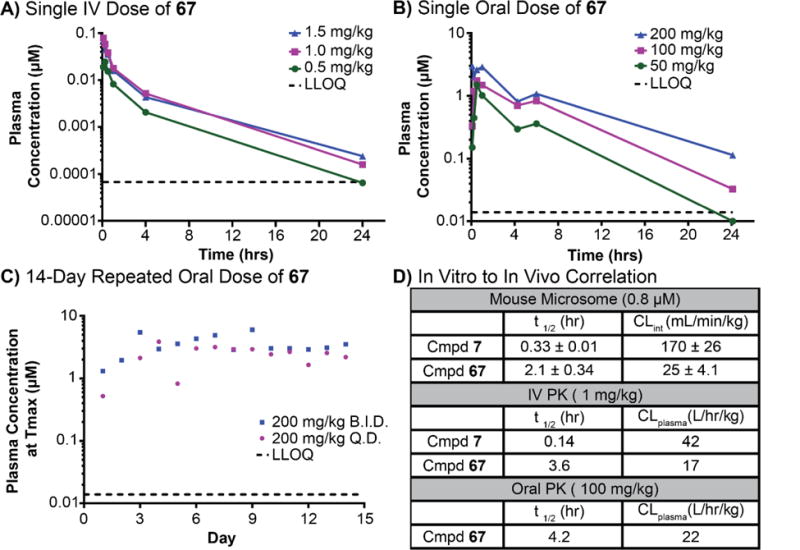
A) Time course of the drug plasma concentration over 24 h resulting from single dose intravenous administration of 67 at doses of 0.5, 1.0, and 1.5 mg/kg. B) Time course of the drug plasma concentration over 24 h resulting from single oral doses of 67 at 50, 100, and 200 mg/kg. C) Repeated dose-exposure studies to determine toxicological responses. Subjects were dosed orally with 200 mg/kg either once a day (QD) or twice a day (BID) and drug plasma concentration was measured 1 h after drug administration. D) Table comparing the in vitro and in vivo clearance data. Results support our hypothesis that the microsomal models predict in vivo clearance. Additional PK parameters are in Supplementary Table 6.
When dosed orally as a suspension in EtOH/PG/PEG/PBS (10/10/40/39; v/v/v/v; pH = 7.4) and 1% (w/v) 2-hydroxy-β-cyclodextrin (MW~1396 Da), compound 67 displayed a half-life of 3-5 h. This observation concurs with the IV half-life of 3-4 h and suggests that 1) the total exposure is driven by clearance and 2) levels of absorption are optimal. The observed Cmax and AUClast show a dose-dependent increase from 1.5 to 3 μM and 3.4 to 17.1 μM*h, respectively. The apparent volume of distribution and clearance are similar to those of IV. Based on these data, compound 67 has an oral bioavailability of 88% (calculated using IV dose at 1.5 mg/kg and PO dose at 50 mg/kg). This apparent bioavailability (Fin vivo) is close to what we predicted from in vitro clearance (Fin vitro = 78%), calculated by 1 - CL/Qh61. Compound 67 did not cause a significant adverse reaction at any dose tested during the 48 h monitoring period following oral administration (Supplementary Table 7, 8).
In order to enable efficacy modeling in mice, we investigated the toxicology and pharmacokinetics of compound 67 during repeat-dose experiments over a period of 14 days using either once (QD) or twice (BID) daily schedule with a fixed oral dose of 200 mg/kg. Throughout the 14-day dosing period, no adverse reactions or compound-related side effects were observed (Supplementary Table 9, 10). Dosed animals in both groups experienced minor weight loss (3-5%) during the first 3 days, but resumed normal growth within 5 days (Supplementary Figure 1). In order to determine if there were significant changes in metabolism and clearance during the study period, drug plasma concentrations were monitored one hour post administration after each oral dose (Figure 3C). C1h was 2.4 ± 0.93 μM (QD) and 3.3 ± 1.5 (BID), and was consistent with earlier data from our single oral dose study (C1h = 3 μM at 200 mg/kg). Therefore, compound 67 is well tolerated, possesses good oral bioavailability, and is suitable for use in animal efficacy models.
The details of the biochemical selectivity and cellular activity of compound 67 (NAcM-OPT), along with an irreversible inhibitor (NAcM-COV), and a structurally related negative control (NAcM-NEG), have recently been disclosed.42 Briefly, compound 67 maintains the high level of selectivity for inhibition of DCN1/2 over the highly homologous remaining human DCN isoforms.42 Compound 67 effectively disrupts the DCN1-UBE2M interaction and UBE2M interactome in cells.42 In HCC95, a squamous carcinoma cell line highly expressing DCN1, either treatment with compound 67 or knockdown of DCN1 with shRNA selectively reduces steady-state levels of neddylated CUL1 and CUL3 while CUL2, CUL4A, and CUL5 are relatively unaffected.42 Compound 67 inhibits steady-state levels of neddylated CUL1 and CUL3 in a dose-dependent manner (Figure 4A). Consistent with our previous studies,42 treatment of HCC95 cells with compound 67 does not elicit gross substrate stabilization (Figure 4B). However, at longer exposures, which oversaturate MLN4924 treated protein levels making direct comparison no longer possible, compounds 67 elicits a dose-dependent increase in the known CUL1/CUL3 substrates NRF2 and p27 (Figure 4B) but not other known CUL1/CUL3 substrates Cylin E or p21 (Supplementary Figure 2).
Figure 4.
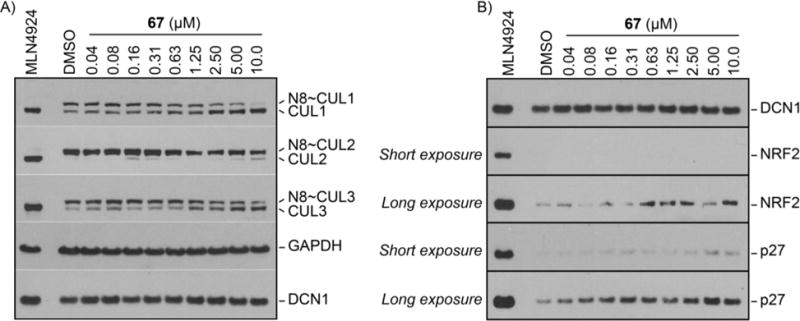
Western blot for inhibition of cellular neddylation and increased levels of substrates at 48 hours by DMSO, MLN4924 (positive control, 1 μM, single dose at T = 0), and compound 67 (indicated concentration, dosing at T = 0, and T = 24 hr). A) Dose-dependent inhibition of steady-state levels of CUL1 and CUL3 neddylation. B) Dose-dependent effects of 67 on steady-state levels of NRF2 and p27.
Discussion and Conclusions
In the present study, we employed both a rational, structure-based, design as well as an empirical chemistry approach to uncover the key structural elements critical for binding to DCN1 and driving compound metabolism. Our studies yielded compound 67, which is equipotent to earlier leads like compound 7 but is significantly more bioavailable. By developing short, scalable, synthetic routes, we rapidly prepared over 200 analogs (Supplementary Dataset 1) to select compounds with better PK profiles. A combinatory approach of microsomal modeling and in vivo PK allowed us to confidently conclude that the microsomal models correctly predict in vivo clearance. Metabolite identification studies focused our attention on the most rapidly metabolized moieties of the inhibitor series: the aniline and alkyl side chain of the piperidine. The structure-based design incorporated our previously described X-ray co-structures and SAR data1, 42 and allowed us to narrow our focus to the substituents that were most likely to yield potent molecules. In total, 38 of the newly synthesized analogs possessed potency values less than or equal to 360 nM in our TR-FRET binding assay (Supplementary Dataset 1). In parallel to the rational design approach, we employed an empirical approach to discover unexpectedly potent analogs, such as the bicyclic compound 45, which appears to bypass the stringent steric restrictions in the hinge sub-pocket, as suggested by our previous data. In the face of a known induced fit binding event, we believe it is imperative to keep testing the limits of the design model. The ease and scalability of the synthetic routes allowed us to produce ample material for both the PK and extended dosing studies. The synthetic routes will also allow us to produce sufficient quantities for future in vivo studies.
We observed two general SAR/SPR trends. First, the incorporation of small hydrophobic and electronegative substituents at either or both of the meta- and para-positions of the aniline ring electronically and sterically blocked oxidation of the aromatic ring without negatively affecting potency. Second, the SAR of the alkyl piperidine substituent was highly restricted only tolerating small hydrophobic substituents.1 Subsequent metabolite identification studies using the optimized compound 67 revealed that although incorporating the di-chlorophenyl ring reduced the oxidation of the aniline substituent. N-dealkylation of the 4-aminopiperidine and oxidation of the benzyl urea substituent targeting the Leu pocket were still prevalent. Future synthetic efforts will focus on these regions as well as on accessing the currently unoccupied N-acetyl pocket, which likely affords the best opportunity for potency gains.16
We are currently using compound 67 and others to dissect the effects of acute pharmacologic inhibition of the DCN1-UBE2M interaction on the NEDD8/CUL pathway in both cellular and in vivo models. The data suggest that inhibiting this NEDD8 E2-E3 interaction has significantly different biological consequences from inhibition of the upstream NEDD8 E1 by Pevonedistat or other E1 inhibitors. Pevonedistat acts by irreversibly inhibiting the E1 enzyme that initiates the NEDD8 tri-enzyme cascade, completely blocking all neddylation. Compound 67 selectively reduces the steady-state neddylation of specific cullins. The most striking phenotypic difference is that unlike E1 inhibition, compound 67 does not grossly stabilize canonical Cullin-RING ligase substrates.42 However, compound 67 does elicit a minor and selective increase in the levels of two known CRL substrates, NRF2 and p27. Compound 67 also inhibits the anchorage-independent growth of HCC95 cells, a phenotypic consequence associated with DCN1-amplification and hallmark of cancer in this squamous cell carcinoma line.42 Future experiments, outside the scope of this work, will focus on understanding the mechanisms driving the observed selectivity and phenotypic consequences associated with acute DCN inhibition in cells and murine models. These results suggest that selective regulation of the neddylation pathway still influences tumor progression, at least in cells harboring DCN1 amplification. However, we are clear to point out that selective inhibition of the DCN1-UBE2M interaction is not grossly toxic to the proliferation of monolayer cultures. Given the high levels of DCN1 amplification exhibited in many human cancers,7, 11, 13 selective inhibition of DCN1-mediated neddylation could provide a more targeted therapy relative to the complete ablation of neddylation afforded by Pevonedistat or other NEDD8 E1 inhibitors. Recent reports of a second class of DCN1 inhibitors demonstrates a similar spectrum of activity.43–44
In conclusion, the development of compound 67 permits a more selective and acute pharmacologic regulation of this highly complex and dynamic pathway that is already helping to discover previously unappreciated biological roles of DCN1-mediated neddylation,42, 62 and hopefully will provide further insight into tumor biology in areas with significant unmet clinical need.
Experimental Section
TR-FRET Assay1, 42
The TR-FRET assay was carried out in black 384-well microtiter plates at a final volume of 20 μL per well. To screen library compounds, the assay cocktail was prepared as a mixture of 50 nM Biotin-DCN1, 20 nM Ac-UBE2M12-AlexaFluor488, 2.5 nM Tb-Streptavidin (ThermoFisher) in assay buffer (25 mM HEPES, 100 mM NaCl, 0.1% Triton X-100, 0.5 mM DTT, pH 7.5). The assay cocktail was then incubated for 1 h at room temperature and distributed using a WellMate instrument (Matrix). Compounds to be screened were added to assay plates from DMSO stock solutions by pin transfer using 50SS pins (V&P Scientific). The assay mixture was incubated for 1 h at room temperature prior to measuring the TR-FRET signal with a PHERAstar FS plate reader (BMG Labtech) equipped with excitation modules at 337 nm and emissions at 490 and 520 nm. We set the integration start to 100 μs and the integration time to 200 μs. The number of flashes was set to 100. The ratio of 520:490 was used as TR-FRET signal in calculations. Assay endpoints were normalized from 0% (DMSO only) to 100% inhibition (unlabeled competitor peptide) for hit selection and curve fitting. All compounds were tested in triplicate or more.
Liver Microsomal Stability
We mixed 0.633 mL of mouse liver microsome (20 mg/mL, female CD9 mice, Fisher Scientific, #NC9567486) with 0.051 ml of 0.5 M EDTA solution and 19.316 ml potassium phosphate buffer (0.1 M, pH 7.4, 37°C) to produce 20 ml of liver microsome solution. One part 10 mM DMSO compound stock was mixed with 4 parts acetonitrile to make 2 mM diluted compound stock in DMSO and acetonitrile. We added 29.1 μL diluted compound stock to 2.3 mL liver microsomal solution and ran the mixture through a vortex to make final microsomal solution with 0.8 μM compound. We dispensed 180 μL of the mixed solutions into a 96-well storage plate (pION Inc., MA, #110323). Five time points (0-4 hr) were taken for each microsome assay. For the 0 h time point, 450 μL pre-cooled (4 °C) internal standard (5 μM caffeine in methanol) was added to the deep wells before the reaction started. For other time points, 1.25 ml of microsome assay solution A (Fisher Scientific, #NC9255727) was combined with 0.25 ml of solution B (Fisher Scientific, #NC9016235) in 3.5 ml of potassium phosphate buffer (0.1 M, pH 7.4). We added 45 μL of this A+B solution to each of the 96 wells in the storage or reaction plate. The reaction plate was then sealed and incubated at 37 °C and shaken at a speed of 60 rpm. At 0.5, 1, 2, and 4 h, we added 450 μL of pre-cooled internal standard (4 μg/ml caffeine in methanol) to the respective wells in the reaction plate. The liquid was then transferred to a quenched plate. The quenched plate was centrifuged (Eppendorf, model 5810R) at 4000 rpm for 20 minutes at 4 °C. We transferred 200 μL of supernatant to a 96-well plate and analyzed using UPLC-MS (Waters Inc.). Selected ion recording (SIR) was used to detect the compounds and internal standard. The log peak area ratio (compound peak area / internal standard peak area) was plotted against time and the slope was used to calculate the elimination rate constant [k = (−2.303) * slope]. The half-life (h) was calculated as t (1/2) = 0.693 / k. All compounds were tested in triplicate.
Characterization of key compounds and intermediates
All moisture-sensitive reactions were performed using syringe-septum techniques under an atmosphere of dry N2 unless otherwise noted. Prior to use, all glassware was dried in an oven at 120° C for at least 6 h or flame-dried under an atmosphere of dry nitrogen. Methylene chloride and acetonitrile were dried using an aluminum oxide column. Deuterated chloroform was stored over anhydrous potassium carbonate. Reactions were monitored by TLC analysis (pre-coated silica gel 60 F254 plates, 250 μm layer thickness) and visualized using an ultraviolet lamp (254 nm) or by staining with either Vaughn’s reagent (4.8 g of (NH4)6Mo7O24·4H2O and 0.2 g of Ce(SO4)2 in 100 mL of a 3.5 N H2SO4) or a potassium permanganate solution (1.5 g of KMnO4 and 1.5 g of K2CO3 in 100 mL of a 0.1% NaOH solution). Unless otherwise specified, commercially available reagents were used as received. Flash column chromatography was performed using a Biotage Isolera One and Biotage KP-SIL SNAP cartridges. Melting points were acquired on Buchi Melting Point B-545. All NMR data was collected at room temperature in CDCl3 or (CD3)2SO on a 400 or 500 MHz Bruker instrument. Chemical shifts (δ) are reported in parts per million (ppm) with internal CHCl3 (δ 7.26 ppm for 1H and 77.00 ppm for 13C), internal DMSO (δ 2.50 ppm for 1H and 39.52 ppm for 13C), or internal TMS (δ 0.0 ppm for 1H and 0.0 ppm for 13C) as the references. 1H NMR data are reported as follows: chemical shift; multiplicity (s = singlet, bs = broad singlet, d = doublet, t = triplet, q = quartet, p = pentet, sext = sextet, sep = septet, m= multiplet, dd = doublet of doublets, dt = doublet of triplets, td = triplet of doublets, qd = quartet of doublets); coupling constant(s) (J) in Hertz (Hz); and integration. Purity was assessed by LC/MS/UV/ELSD using a Waters Acquity UPLC-MS with the purity being assigned as the average determined by UV/ELSD. All compounds were confirmed to ≥ 95% purity. Compounds that proved critical to our SAR analysis were further characterized using 1H NMR and HRMS.
N-benzyl-1-(pentan-2-yl)piperidin-4-amine (15)
We added acetic acid (2.11 mL, 36.9 mmol, 1.2 equiv) to a stirred solution of 1442 (5.20 g, 30.7 mmol, 1 equiv) and benzylamine (3.36 ml, 30.7 mmol, 1 equiv) in CH2Cl2 (50 mL). The resulting cloudy mixture was stirred at room temperature for 1 h and sodium triacetoxyborohydride (8.46 g, 39.9 mmol, 1.3 equiv) was added portion-wise over 10 min. The resulting heterogeneous mixture was stirred at room temperature overnight (ca. 16 h), quenched with a saturated aqueous solution of 1M sodium hydroxide, extracted with EtOAc (×2), dried (MgSO4), filtered, and concentrated under reduced pressure to give 8.0 g (100% yield) of the desired product (15) as a pale yellow oil. 1H NMR (500 MHz, Chloroform-d) δ 7.36 – 7.28 (m, 4H), 7.26 – 7.17 (m, 1H), 3.81 (s, 2H), 2.81 – 2.76 (m, 2H), 2.59 (sext, J = 6.6 Hz, 1H), 2.51 – 2.46 (m, J = 1H), 2.32 (t, J = 11.2 Hz, 1H), 2.19 (t, J = 11.3 Hz, 1H), 1.98 – 1.83 (m, 2H), 1.70 – 1.22 (m, 6H), 0.96 (d, J = 6.5 Hz, 3H), 0.90 (t, J = 7.1 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 140.8, 128.4, 128.0, 126.8, 59.0, 54.4, 50.7, 48.5, 45.7, 35.7, 33.1, 32.9, 20.2, 14.3, 14.1. HRMS (ESI+) m/z calcd for C17H29N2 [M+H]+, 261.2331, found 261.2327.
tert-butyl 4-(benzylamino)piperidine-1-carboxylate (17)
We added acetic acid (2.76 mL, 48.2 mmol, 1.2 equiv) to a stirred solution of 1-Boc-4-piperidone (8.00 g, 40.2 mmol, 1 equiv) and benzylamine (4.39 mL, 40.2 mmol, 1 equiv) in dry CH2Cl2 (150 mL). The resulting cloudy mixture was stirred at room temperature for 1 h and sodium triacetoxyborohydride (11.4 g, 60.3 mmol, 1.5 equiv) was added portion-wise over 30 minutes. The resulting heterogeneous mixture was stirred at room temperature overnight (ca. 16 h), quenched with a saturated aqueous solution of NaHCO3, extracted with EtOAc (×2), dried (MgSO4), filtered, and concentrated under reduced pressure. The crude mixture was purified by chromatography on SiO2 (MeOH/CH2Cl2, 2% to 15%) to give 11.4 g (98% yield) of the desired product of a colorless oil. 1H NMR (500 MHz, Chloroform-d) δ 7.37 – 7.32 (m, 4H), 7.30 – 7.23 (m, 1H), 4.20 – 3.90 (m, 3H), 3.85 (s, 2H), 2.85 – 2.75 (m, 2H), 2.72 – 2.67 (m, 1H), 1.89 (d, J = 12.8 Hz, 2H), 1.47 (s, 9H), 1.40 – 1.32 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 154.7, 139.3, 128.5, 128.2, 127.2, 79.4, 54.1, 50.4, 42.6, 42.1, 31.9, 28.4. HRMS (ESI+) m/z calcd for C17H27N2O2 [M+H]+, 291.2073, found 291.2067.
tert-butyl 4-(1-benzyl-3-(3-fluoro-5-(trifluoromethyl)phenyl)ureido)piperidine-1-carboxylate (18)
To a stirred solution of 17 (2.90 g, 9.99 mmol, 1 equiv) in CH2Cl2 (50 mL), we added 1-isocyanato-3-(trifluoromethyl)benzene (1.44 mL, 9.99 mmol, 1.1 equiv). The reaction mixture was stirred at room temperature overnight (ca. 16 h), diluted with EtOAc, washed with a saturated aqueous solution of NaHCO3, brine, dried (MgSO4), filtered, and concentrated under reduced pressure to give a pale yellow liquid. The crude mixture was purified by chromatography on SiO2 (MeOH/CH2Cl2, 1% to 10%) to give 4.8 g (97% yield) of the desired product 18 as a white solid. 1H NMR (400 MHz, Chloroform-d) δ 7.49 – 7.40 (m, 2H), 7.39 – 7.21 (m, 4H), 7.11 (s, 1H), 6.90 (d, J = 8.1 Hz, 1H), 6.62 (s, 1H), 4.60 – 4.52 (m, 1H), 4.48 (s, 2H), 4.36 – 4.01 (m, 2H), 2.90 – 2.65 (m, 2H), 1.79 (d, J = 12.3 Hz, 2H), 1.66 – 1.50 (q, J = 12.3 Hz, 2H), 1.44 (s, 9H). 13C NMR (101 MHz, Chloroform-d) δ 162.6 (d, J = 246.4 Hz), 155.0, 154.5, 141.4 (d, J = 11.3 Hz), 132.2 (qd, J = 33.5, 9.4 Hz), 129.4, 128.2, 126.0, 123.1 (q, J = 273.5 Hz), 111.7, 109.9 (d, J = 26.2 Hz), 106.7 (d, J = 25.0 Hz), 79.8, 53.0, 46.2, 43.2, 30.0, 28.4. HRMS (ESI−) m/z calcd for C25H28F4N3O3 [M-H]−, 494.2067, found 494.2058.
General reductive amination procedure.58
To a stirred solution of 1-benzyl-3-(3-fluoro-5-(trifluoromethyl)phenyl)-1-(piperidin-4-yl)urea (0.040 g, 0.101 mmol, 1 equiv) in tetrahydrofuran (1 ml), we added the aldehyde or ketone (0.202 mmol, 2 equiv) and dibutyltin (IV) chloride (1.53 mg, 0.005 mmol, 0.05 equiv). After stirring the heterogeneous mixture for 5 minutes at room temperature, we added phenylsilane (0.025 ml, 0.202 mmol, 2 equiv). The resulting solution was stirred at room temperature overnight (ca. 16 h). For some more sterically encumbered ketones, we observed less than 20% conversion after overnight reaction. For these ketones, we added an extra 3-4 equiv of ketone and 2 equiv of phenylsilane and the reaction was stirred at room temperature for an additional 24 h. After the starting material was completely consumed, the crude mixture was diluted with DMSO, filtered through a 0.45 micron filter, and subjected to HPLC chromatography.
General isocyanate addition
To a stirred solution of 1-benzylpiperidin-4-amine (0.250 g, 1.314 mmol, 1.2 equiv) in CH2Cl2 (6 mL), we added 1-isocyanato-3-(trifluoromethyl)benzene (1.53 mL, 1.10 mmol, 1 equiv) and N,N-diisopropylethylamine (0.286 mL, 1.64 mmol, 1.5 equiv). The resulting mixture was stirred at room temperature overnight (ca. 16 h), concentrated under reduced pressure, and purified by chromatography on SiO2.
General CDI-mediated isocyanate addition
We added carbonyldiimidazole (0.026 g, 0.158 mmol, 1.3 equiv) to a stirred solution of aniline (0.146 mmol, 1.2 equiv) in tetrahydrofuran (1 ml). The mixture was stirred at room temperature for 3 h, followed by the addition of N-benzyl-1-butylpiperidin-4-amine (20) (0.030 g, 0.122 mmol, 1 equiv). The resulting solution was stirred at room temperature overnight (ca. 16 h). The crude mixture was diluted with DMSO, filtered through a 0.45 micron filter, and directly subjected to HPLC chromatography.
Reproduction of characterization from previously reported key compounds
1-benzyl-1-(1-(pentan-2-yl)piperidin-4-yl)-3-(3-(trifluoromethyl)phenyl)urea (7).42
Note: characterization was previously reported42 but is reproduced below for convenience: 1H NMR (500 MHz, Chloroform-d) δ 7.57 (s, 1H), 7.42 – 7.36 (m, 2H), 7.34-7.30 (m, 3H), 7.29 – 7.17 (m, 4H), 6.50 (s, 1H), 4.53 (s, 2H), 4.37 (tt, J = 12.1, 4.2 Hz, 1H), 2.84-2.80 (m, 2H), 2.57 (sext, J = 7.0 Hz, 1H), 2.42 (td, J = 11.6, 2.5 Hz, 1H), 2.27 (td, J = 11.6, 2.3 Hz, 1H), 1.83 – 1.76 (m, 2H), 1.69 (dqd, J = 24.1, 12.0, 3.8 Hz, 2H), 1.47 (dq, J = 12.5, 5.2 Hz, 1H), 1.41 – 1.13 (m, 3H), 0.95 (d, J = 6.6 Hz, 3H), 0.89 (t, J = 7.1 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 155.4, 139.6, 137.5, 130.8 (q, J = 31.5 Hz), 129.1, 129.0, 127.1, 126.0, 123.9 (q, J = 273.4 Hz, 1C), 122.6, 119.2 (q, J = 3.8 Hz), 116.3 (q, J = 3.8 Hz), 58.8, 53.2, 49.5, 46.1, 46.0, 35.7, 30.6, 30.4, 20.1, 14.1, 14.0. m/z (APCI-pos) M+1 = 448.2. HRMS (ESI+) m/z calcd for C25H33F3N3O [M+H]+, 448.2575 found 448.2575.
1-benzyl-1-(1-butylpiperidin-4-yl)-3-(3,4-dichlorophenyl)urea. NAcM-OPT (67).42
Note: characterization was previously reported42 but is reproduced below for convenience: m.p.: 118-120 °C. 1H NMR (500 MHz, Chloroform-d) δ 7.42 – 7.39 (m, 3H), 7.36 – 7.29 (m, 3H), 7.21 (d, J = 8.7 Hz, 1H), 6.91 (d, J = 8.7 Hz, 1H), 6.27 (s, 1H), 4.49 (s, 2H), 4.44 – 4.38 (m, 1H), 2.99 (d, J = 11.2 Hz, 2H), 2.31 (t, J = 7.8 Hz, 2H), 2.05 (t, J = 11.7 Hz, 2H), 1.85 – 1.65 (m, 4H), 1.44 (p, J = 7.5 Hz, 2H), 1.30 (sext, J = 7.4 Hz, 2H), 0.90 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, Chloroform-d) δ 155.3, 138.6, 137.4, 132.4, 130.1, 129.3, 128.0, 126.1, 125.9, 121.2, 118.8, 58.4, 53.1, 52.9, 46.1, 30.1, 29.4, 20.8, 14.0. HRMS (ESI+) m/z calcd for C23H30Cl2N3O [M+H]+ 434.1766, found 434.1761.
Supplementary Material
Acknowledgments
American Lebanese Syrian Associated Charities. We acknowledge the High Throughput Biosciences Center, Medicinal Chemistry Center, Compound Management, and High Throughput Analytical Chemistry Centers in Chemical Biology and Therapeutics; Hartwell Center, and Veterinary pathology cores of St. Jude Children’s Research Hospital for use of their personnel and facilities. This study was funded through B.A.S., HHMI, and NIH R37GM069530, P30CA021765; J.T.H., NIH F32GM113310; American Syrian Lebanese Associated Charities, and St Jude Children’s Research Hospital.
Abbreviations
- DCN1
defective in cullin neddylation 1
- UBE2M
NEDD8-conjugating enzyme Ubc12
- SCC
Squamous cell carcinoma
- CRL
cullin ring ligases
- NEDD8
Neural Precursor Cell Expressed, Developmentally Down-Regulated 8
- SAR
structure-activity relationship
- SPR
structure-property relationship
- TR-FRET
time-resolved fluorescence resonance energy transfer
- HTS
high-throughput screen
- t-Bu
tert-butyl
- NAE
NEDD8-activating enzyme
- RBX1
Ring-Box 1, E3 Ubiquitin Protein Ligase
- CUL1
cullin 1
- AF
AlexaFluor 488
- IV
intravenous
- PO
per os (by mouth)
- CLint
intrinsic clearence
- Cmax
maximum achieved concentration
- t(1/2)
half-life (time taken for the concentration of analyte to fall to half its original value)
- AUClast
area under the plasma concentration-time curve from time zero to time of last measurable concentration
Footnotes
Supporting Information:
Characterization details for the other synthetic compounds as well as additional information regarding the biological studies are described in Supporting Information.
Author Contributions:
All authors contributed to specific parts of the manuscript. J.T.H and R.K.G. assume responsibility for the manuscript in its entirety. All authors have given approval to the final version of the manuscript.
References
- 1.Hammill JT, Scott DC, Min J, Connelly M, Holbrook G, Zhu F, Matheny A, Yang L, Singh B, Schulman BA, Guy RK. Piperidinyl ureas chemically control defective in cullin neddylation 1 (DCN1)-mediated cullin neddylation. J Med Chem. 2018 doi: 10.1021/acs.jmedchem.7b01277. citation to be provided at time of publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang J, Wu P, Hu Y. Clinical and marketed proteasome inhibitors for cancer treatment. Curr Med Chem. 2013;20(20):2537–2551. doi: 10.2174/09298673113209990122. [DOI] [PubMed] [Google Scholar]
- 3.Lydeard JR, Harper JW. Inhibitors for E3 ubiquitin ligases. Nat Biotechnol. 2010;28(7):682–684. doi: 10.1038/nbt0710-682. [DOI] [PubMed] [Google Scholar]
- 4.Ciechanover A. Intracellular protein degradation: from a vague idea through the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting. Bioorg Med Chem. 2013;21(12):3400–3410. doi: 10.1016/j.bmc.2013.01.056. [DOI] [PubMed] [Google Scholar]
- 5.Bassermann F, Eichner R, Pagano M. The ubiquitin proteasome system - implications for cell cycle control and the targeted treatment of cancer. Biochim Biophys Acta. 2014;1843(1):150–162. doi: 10.1016/j.bbamcr.2013.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, Brownell JE, Burke KE, Cardin DP, Critchley S, Cullis CA, Doucette A, Garnsey JJ, Gaulin JL, Gershman RE, Lublinsky AR, McDonald A, Mizutani H, Narayanan U, Olhava EJ, Peluso S, Rezaei M, Sintchak MD, Talreja T, Thomas MP, Traore T, Vyskocil S, Weatherhead GS, Yu J, Zhang J, Dick LR, Claiborne CF, Rolfe M, Bolen JB, Langston SP. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458(7239):732–736. doi: 10.1038/nature07884. [DOI] [PubMed] [Google Scholar]
- 7.Sarkaria I, Oc P, Talbot SG, Reddy PG, Ngai I, Maghami E, Patel KN, Lee B, Yonekawa Y, Dudas M, Kaufman A, Ryan R, Ghossein R, Rao PH, Stoffel A, Ramanathan Y, Singh B. Squamous cell carcinoma related oncogene/DCUN1D1 is highly conserved and activated by amplification in squamous cell carcinomas. Cancer Res. 2006;66(19):9437–9444. doi: 10.1158/0008-5472.CAN-06-2074. [DOI] [PubMed] [Google Scholar]
- 8.Estilo CL, OC P, Ngai I, Patel SG, Reddy PG, Dao S, Shaha AR, Kraus DH, Boyle JO, Wong RJ, Pfister DG, Huryn JM, Zlotolow IM, Shah JP, Singh B. The role of novel oncogenes squamous cell carcinoma-related oncogene and phosphatidylinositol 3-kinase p110alpha in squamous cell carcinoma of the oral tongue. Clin Cancer Res. 2003;9(6):2300–2306. [PubMed] [Google Scholar]
- 9.Oc P, Sarkaria I, Talbot SG, Reddy P, Dao S, Ngai I, Shaha A, Kraus D, Shah J, Rusch V, Ramanathan Y, Singh B. SCCRO (DCUN1D1) induces extracellular matrix invasion by activating matrix metalloproteinase 2. Clin Cancer Res. 2008;14(21):6780–6789. doi: 10.1158/1078-0432.CCR-08-0719. [DOI] [PubMed] [Google Scholar]
- 10.Talbot SG, Oc P, Sarkaria IS, Ghossein R, Reddy P, Ngai I, Cordeiro CN, Wong RJ, Kris MG, Rusch VW, Singh B. Squamous cell carcinoma related oncogene regulates angiogenesis through vascular endothelial growth factor-A. Ann Surg Oncol. 2004;11(5):530–534. doi: 10.1245/ASO.2004.03.014. [DOI] [PubMed] [Google Scholar]
- 11.Huang G, Singh B. Coamplification and cooperation: toward identifying biologically relevant oncogenes. Clin Cancer Res. 2013;19(20):5549–5551. doi: 10.1158/1078-0432.CCR-13-2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang J, Qian J, Hoeksema MD, Zou Y, Espinosa AV, Rahman SM, Zhang B, Massion PP. Integrative genomics analysis identifies candidate drivers at 3q26-29 amplicon in squamous cell carcinoma of the lung. Clin Cancer Res. 2013;19(20):5580–5590. doi: 10.1158/1078-0432.CCR-13-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen Y, McGee J, Chen X, Doman TN, Gong X, Zhang Y, Hamm N, Ma X, Higgs RE, Bhagwat SV, Buchanan S, Peng SB, Staschke KA, Yadav V, Yue Y, Kouros-Mehr H. Identification of druggable cancer driver genes amplified across TCGA datasets. PloS one. 2014;9(5):e98293. doi: 10.1371/journal.pone.0098293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 15.Scott DC, Sviderskiy VO, Monda JK, Lydeard JR, Cho SE, Harper JW, Schulman BA. Structure of a RING E3 trapped in action reveals ligation mechanism for the ubiquitin-like protein NEDD8. Cell. 2014;157(7):1671–1684. doi: 10.1016/j.cell.2014.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scott DC, Monda JK, Bennett EJ, Harper JW, Schulman BA. N-terminal acetylation acts as an avidity enhancer within an interconnected multiprotein complex. Science. 2011;334(6056):674–678. doi: 10.1126/science.1209307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duda DM, Borg LA, Scott DC, Hunt HW, Hammel M, Schulman BA. Structural insights into NEDD8 activation of cullin-RING ligases: conformational control of conjugation. Cell. 2008;134(6):995–1006. doi: 10.1016/j.cell.2008.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Enchev RI, Schulman BA, Peter M. Protein neddylation: beyond cullin-RING ligases. Nat Rev Mol Cell Biol. 2015;16(1):30–44. doi: 10.1038/nrm3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lydeard JR, Schulman BA, Harper JW. Building and remodelling Cullin-RING E3 ubiquitin ligases. EMBO Rep. 2013;14(12):1050–1061. doi: 10.1038/embor.2013.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deshaies RJ, Joazeiro CA. RING domain E3 ubiquitin ligases. Annu Rev Biochem. 2009;78:399–434. doi: 10.1146/annurev.biochem.78.101807.093809. [DOI] [PubMed] [Google Scholar]
- 21.Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol. 2005;6(1):9–20. doi: 10.1038/nrm1547. [DOI] [PubMed] [Google Scholar]
- 22.Lu A, Pfeffer SR. A CULLINary ride across the secretory pathway: more than just secretion. Trends Cell Biol. 2014;24(7):389–399. doi: 10.1016/j.tcb.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cui D, Xiong X, Zhao Y. Cullin-RING ligases in regulation of autophagy. Cell Div. 2016;11:8–22. doi: 10.1186/s13008-016-0022-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bielskiene K, Bagdoniene L, Mozuraitiene J, Kazbariene B, Janulionis E. E3 ubiquitin ligases as drug targets and prognostic biomarkers in melanoma. Medicina (Kaunas) 2015;51(1):1–9. doi: 10.1016/j.medici.2015.01.007. [DOI] [PubMed] [Google Scholar]
- 25.Bulatov E, Ciulli A. Targeting Cullin-RING E3 ubiquitin ligases for drug discovery: structure, assembly and small-molecule modulation. Biochem J. 2015;467(3):365–386. doi: 10.1042/BJ20141450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang X, Dixit VM. Drugging the undruggables: exploring the ubiquitin system for drug development. Cell Res. 2016;26(4):484–498. doi: 10.1038/cr.2016.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weathington NM, Mallampalli RK. Emerging therapies targeting the ubiquitin proteasome system in cancer. J Clin Invest. 2014;124(1):6–12. doi: 10.1172/JCI71602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rulina AV, Mittler F, Obeid P, Gerbaud S, Guyon L, Sulpice E, Kermarrec F, Assard N, Dolega ME, Gidrol X, Balakirev MY. Distinct outcomes of CRL-Nedd8 pathway inhibition reveal cancer cell plasticity. Cell Death Dis. 2016;7(12):e2505. doi: 10.1038/cddis.2016.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abidi N, Xirodimas DP. Regulation of cancer-related pathways by protein NEDDylation and strategies for the use of NEDD8 inhibitors in the clinic. Endocr Relat Cancer. 2015;22(1):T55–70. doi: 10.1530/ERC-14-0315. [DOI] [PubMed] [Google Scholar]
- 30.Tanaka T, Nakatani T, Kamitani T. Inhibition of NEDD8-conjugation pathway by novel molecules: potential approaches to anticancer therapy. Mol Oncol. 2012;6(3):267–275. doi: 10.1016/j.molonc.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Soucy TA, Smith PG, Rolfe M. Targeting NEDD8-activated cullin-RING ligases for the treatment of cancer. Clin Cancer Res. 2009;15(12):3912–3916. doi: 10.1158/1078-0432.CCR-09-0343. [DOI] [PubMed] [Google Scholar]
- 32.Milhollen MA, Narayanan U, Soucy TA, Veiby PO, Smith PG, Amidon B. Inhibition of NEDD8-activating enzyme induces rereplication and apoptosis in human tumor cells consistent with deregulating CDT1 turnover. Cancer Res. 2011;71(8):3042–3051. doi: 10.1158/0008-5472.CAN-10-2122. [DOI] [PubMed] [Google Scholar]
- 33.Milhollen MA, Traore T, Adams-Duffy J, Thomas MP, Berger AJ, Dang L, Dick LR, Garnsey JJ, Koenig E, Langston SP, Manfredi M, Narayanan U, Rolfe M, Staudt LM, Soucy TA, Yu J, Zhang J, Bolen JB, Smith PG. MLN4924, a NEDD8-activating enzyme inhibitor, is active in diffuse large B-cell lymphoma models: rationale for treatment of NF-{kappa}B-dependent lymphoma. Blood. 2010;116(9):1515–1523. doi: 10.1182/blood-2010-03-272567. [DOI] [PubMed] [Google Scholar]
- 34.Brownell JE, Sintchak MD, Gavin JM, Liao H, Bruzzese FJ, Bump NJ, Soucy TA, Milhollen MA, Yang X, Burkhardt AL, Ma J, Loke HK, Lingaraj T, Wu D, Hamman KB, Spelman JJ, Cullis CA, Langston SP, Vyskocil S, Sells TB, Mallender WD, Visiers I, Li P, Claiborne CF, Rolfe M, Bolen JB, Dick LR. Substrate-assisted inhibition of ubiquitin-like protein-activating enzymes: the NEDD8 E1 inhibitor MLN4924 forms a NEDD8-AMP mimetic in situ. Mol Cell. 2010;37(1):102–111. doi: 10.1016/j.molcel.2009.12.024. [DOI] [PubMed] [Google Scholar]
- 35.Lu P, Liu X, Yuan X, He M, Wang Y, Zhang Q, Ouyang PK. Discovery of a novel NEDD8 activating enzyme inhibitor with piperidin-4-amine scaffold by structure-based virtual screening. ACS Chem Biol. 2016;11(7):1901–1907. doi: 10.1021/acschembio.6b00159. [DOI] [PubMed] [Google Scholar]
- 36.Leung CH, Chan DS, Yang H, Abagyan R, Lee SM, Zhu GY, Fong WF, Ma DL. A natural product-like inhibitor of NEDD8-activating enzyme. Chem Commun. 2011;47(9):2511–2513. doi: 10.1039/c0cc04927a. [DOI] [PubMed] [Google Scholar]
- 37.Zhong HJ, Ma VP, Cheng Z, Chan DS, He HZ, Leung KH, Ma DL, Leung CH. Discovery of a natural product inhibitor targeting protein neddylation by structure-based virtual screening. Biochimie. 2012;94(11):2457–2460. doi: 10.1016/j.biochi.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 38.Zhang S, Tan J, Lai Z, Li Y, Pang J, Xiao J, Huang Z, Zhang Y, Ji H, Lai Y. Effective virtual screening strategy toward covalent ligands: identification of novel NEDD8-activating enzyme inhibitors. J Chem Inf Model. 2014;54(6):1785–1797. doi: 10.1021/ci5002058. [DOI] [PubMed] [Google Scholar]
- 39.Zhong HJ, Wang W, Kang TS, Yan H, Yang Y, Xu L, Wang Y, Ma DL, Leung CH. A Rhodium(III) complex as an inhibitor of neural precursor cell expressed, developmentally down-regulated 8-activating enzyme with in vivo activity against inflammatory bowel disease. J Med Chem. 2017;60(1):497–503. doi: 10.1021/acs.jmedchem.6b00250. [DOI] [PubMed] [Google Scholar]
- 40.Zhong HJ, Yang H, Chan DS, Leung CH, Wang HM, Ma DL. A metal-based inhibitor of NEDD8-activating enzyme. PloS one. 2012;7(11):e49574. doi: 10.1371/journal.pone.0049574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schlierf A, Altmann E, Quancard J, Jefferson AB, Assenberg R, Renatus M, Jones M, Hassiepen U, Schaefer M, Kiffe M, Weiss A, Wiesmann C, Sedrani R, Eder J, Martoglio B. Targeted inhibition of the COP9 signalosome for treatment of cancer. Nat Commun. 2016;7:13166. doi: 10.1038/ncomms13166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scott DC, Hammill JT, Min J, Rhee DY, Connelly M, Sviderskiy VO, Bhasin D, Chen Y, Ong SS, Chai SC, Goktug AN, Huang G, Monda JK, Low J, Kim HS, Paulo JA, Cannon JR, Shelat AA, Chen T, Kelsall IR, Alpi AF, Pagala V, Wang X, Peng J, Singh B, Harper JW, Schulman BA, Guy RK. Blocking an N-terminal acetylation-dependent protein interaction inhibits an E3 ligase. Nat Chem Biol. 2017;13(8):850–857. doi: 10.1038/nchembio.2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou H, Lu J, Liu L, Bernard D, Yang CY, Fernandez-Salas E, Chinnaswamy K, Layton S, Stuckey J, Yu Q, Zhou W, Pan Z, Sun Y, Wang S. A potent small-molecule inhibitor of the DCN1-UBC12 interaction that selectively blocks cullin 3 neddylation. Nat Commun. 2017;8(1):1150. doi: 10.1038/s41467-017-01243-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou H, Zhou W, Zhou B, Liu L, Chern TR, Chinnaswamy K, Lu J, Bernard D, Yang CY, Li S, Wang M, Stuckey J, Sun Y, Wang S. High-affinity peptidomimetic inhibitors of the DCN1-UBC12 protein-protein interaction. J Med Chem. 2018;61(5):1934–1950. doi: 10.1021/acs.jmedchem.7b01455. [DOI] [PubMed] [Google Scholar]
- 45.Broderick SR, Golas BJ, Pham D, Towe CW, Talbot SG, Kaufman A, Bains S, Huryn LA, Yonekawa Y, Carlson D, Hambardzumyan D, Ramanathan Y, Singh B. SCCRO promotes glioma formation and malignant progression in mice. Neoplasia. 2010;12(6):476–484. doi: 10.1593/neo.10202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yoo J, Lee SH, Lym KI, Park SY, Yang SH, Yoo CY, Jung JH, Kang SJ, Kang CS. Immunohistochemical expression of DCUN1D1 in non-small cell lung carcinoma: its relation to brain metastasis. Cancer Res Treat. 2012;44(1):57–62. doi: 10.4143/crt.2012.44.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hansch C, Fujita T. p-σ-π Analysis. A Method for the correlation of biological activity and chemical structure. J Am Chem Soc. 1964;86(8):1616–1626. [Google Scholar]
- 48.Hansch C, Leo A, Taft RW. A survey of Hammett substituent constants and resonance and field parameters. Chem Rev. 1991;91(2):165–195. [Google Scholar]
- 49.Hammett LP. The effect of structure upon the reactions of organic compounds. benzene derivatives. J Am Chem Soc. 1937;59(1):96–103. [Google Scholar]
- 50.Hansch C, Maloney PP, Fujita T, Muir RM. Correlation of biological activity of phenoxyacetic acids with hammett substituent constants and partition coefficients. Nature. 1962;194(4824):178–180. [Google Scholar]
- 51.Cherkasov A, Muratov EN, Fourches D, Varnek A, Baskin II, Cronin M, Dearden J, Gramatica P, Martin YC, Todeschini R, Consonni V, Kuz’min VE, Cramer R, Benigni R, Yang C, Rathman J, Terfloth L, Gasteiger J, Richard A, Tropsha A. QSAR modeling: where have you been? where are you going to? J Med Chem. 2014;57(12):4977–5010. doi: 10.1021/jm4004285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sun H, Scott DO. Metabolism of 4-aminopiperidine drugs by cytochrome P450s: molecular and quantum mechanical insights into drug design. ACS Med Chem Lett. 2011;2(8):638–643. doi: 10.1021/ml200117z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rydberg P, Ryde U, Olsen L. Prediction of activation energies for aromatic oxidation by cytochrome P450. J Phys Chem A. 2008;112(50):13058–13065. doi: 10.1021/jp803854v. [DOI] [PubMed] [Google Scholar]
- 54.Carling RW, Moore KW, Moyes CR, Jones EA, Bonner K, Emms F, Marwood R, Patel S, Patel S, Fletcher AE, Beer M, Sohal B, Pike A, Leeson PD. 1-(3-Cyanobenzylpiperidin-4-yl)-5-methyl-4-phenyl-1, 3-dihydroimidazol-2-one: a selective high-affinity antagonist for the human dopamine D(4) receptor with excellent selectivity over ion channels. J Med Chem. 1999;42(14):2706–2715. doi: 10.1021/jm991029k. [DOI] [PubMed] [Google Scholar]
- 55.Archibald JL, Beardsley DR, Bisset GM, Fairbrother P, Jackson JL, Opalko A, Ward TJ. Antihypertensive ureidopiperidines. J Med Chem. 1980;23(8):857–861. doi: 10.1021/jm00182a009. [DOI] [PubMed] [Google Scholar]
- 56.McNulty J, Nair JJ, Singh M, Crankshaw DJ, Holloway AC. Potent and selective inhibition of human cytochrome P450 3A4 by seco-pancratistatin structural analogs. Bioorg Med Chem Lett. 2010;20(7):2335–2339. doi: 10.1016/j.bmcl.2010.01.157. [DOI] [PubMed] [Google Scholar]
- 57.Seifert K, Buttner A, Rigol S, Eilert N, Wandel E, Giannis A. Potent small molecule Hedgehog agonists induce VEGF expression in vitro. Bioorg Med Chem. 2012;20(21):6465–6481. doi: 10.1016/j.bmc.2012.08.026. [DOI] [PubMed] [Google Scholar]
- 58.Apodaca R, Xiao W. Direct reductive amination of aldehydes and ketones using phenylsilane: catalysis by dibutyltin dichloride. Org Lett. 2001;3(11):1745–1748. doi: 10.1021/ol015948s. [DOI] [PubMed] [Google Scholar]
- 59.Ritz C, Streibig JC. Bioassay Analysis Using R. J Stat Softw. 2005;1(5):1–22. [Google Scholar]
- 60.Purohit V, Basu AK. Mutagenicity of nitroaromatic compounds. Chem Res Toxicol. 2000;13(8):673–692. doi: 10.1021/tx000002x. [DOI] [PubMed] [Google Scholar]
- 61.Hallifax D, Houston JB. Evaluation of hepatic clearance prediction using in vitro data: emphasis on fraction unbound in plasma and drug ionisation using a database of 107 drugs. J Pharm Sci. 2012;101(8):2645–2652. doi: 10.1002/jps.23202. [DOI] [PubMed] [Google Scholar]
- 62.Huang G, Kaufman AJ, Xu K, Manova K, Singh B. Squamous cell carcinoma-related oncogene (SCCRO) neddylates Cul3 protein to selectively promote midbody localization and activity of Cul3KLHL21 protein complex during abscission. J Biol Chem. 2017;292(37):15254–15265. doi: 10.1074/jbc.M117.778530. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.