

Background
Clobazam is an antiepileptic drug (AED) which has been approved by the FDA for use in the treatment of Lennox-Gastaut Syndrome in patients 2 years and older. Outside of the U.S., clobazam is also used as an adjunctive therapy for refractory epilepsy and to treat other epileptic syndromes and anxiety [1-3]. It is a 1,5-benzodiazepine, distinguished by the presence of a nitrogen atom at positions 1 and 5 of the benzodiazepine ring rather than at positions 1 and 4 as seen in the 1,4-benzodiazepines (e.g. diazepam) [4]. At the time of writing, clobazam is the only 1,5-benzodiazepine in clinical use [4]. The main metabolite of clobazam, norclobazam, is also clinically active and exerts an antiepileptic effect [5, 6].
Epileptic seizures are thought to be caused by excessive excitatory action potentials in neurons [7]. Clobazam and norclobazam exert their effects by binding to postsynaptic GABAA receptors in the brain, ultimately causing hyperpolarization of the neuron to create an inhibitory signal [8]. This hyperpolarization increases the action potential threshold, thereby reducing the frequency of action potentials and the likelihood of seizures [7]. This is discussed in greater detail in the Pharmacodynamics section below.
Clobazam is generally considered to cause fewer side-effects and cutaneous reactions, such as Stevens-Johnson Syndrome (SJS) or toxic epidermal necrolysis (TEN), at a lower rate than other AEDs, such as the 1,4-benzodiazepine carbamazepine [1, 9, 10]. Cases of SJS/TEN have been reported in patients taking clobazam, often in combination with the 1,4-benzodiazepine lamotrigine and valproic acid [11-13]. As lamotrigine is associated with a high risk of SJS/TEN [14] and clobazam and lamotrigine are not thought to interact with each other [15], it seems likely that lamotrigine is inducing SJS/TEN in these cases.
The FDA has identified 21 cases of SJS/TEN from both the US and abroad, many of these in children. As a result, they have released a warning about the potential for clobazam to cause SJS/TEN at https://www.fda.gov/drugs/drugsafety/ucm377204.htm. However, it should be noted that, in 19 of these cases, the patient was taking one or more concomitant drugs which are also associated with SJS/TEN [16], many of which are themselves considered to be high risk for SJS/TEN [14]. Indeed, a recent case-control study of 480 cases of SJS/TEN found no causal association between clobazam and SJS/TEN [17].
Pharmacokinetics
As norclobazam is the active metabolite of clobazam, many studies have compared the pharmacokinetics of the two molecules. A study of the single-dose pharmacokinetics of norclobazam in both healthy volunteers and epilepsy patients found that administration of 30mg norclobazam resulted in a roughly 50% increase in the AUC for norclobazam than administration of 30mg clobazam. The smaller norclobazam AUC following clobazam administration is to be expected, and correlates to the fraction of clobazam that is metabolized to norclobazam (discussed in greater detail in the next section).
Norclobazam is considerably more persistent in the body than clobazam, having a half-life of around twice that of clobazam (71-82 hours and 36-42 hours, respectively) and, at therapeutic doses, a serum concentration 3-5 times higher than that of clobazam [18]. It has been found that norclobazam has a shorter half-life when administered directly than when it is formed in the body from clobazam metabolism [19]. It has been suggested that this may be due to the presence of residual clobazam in the body which is still available for metabolism into norclobazam following clobazam administration.
The different half-lives of clobazam and norclobazam are reflected in the time needed for both drugs to reach steady state concentrations. In multi-dose studies, clobazam reaches a steady-state concentration within one week of beginning treatment, while a steady-state concentration of norclobazam can take up to 3 weeks to be established [18, 20]. Steady-state concentrations of norclobazam have been found to be higher in females than in males [19].
Studies of patients who became seizure free after using clobazam also found that, despite receiving a low dose of clobazam and having undetectable serum concentrations of clobazam, the patients had high serum concentrations of norclobazam [21]. These observations strongly suggest that the actions of norclobazam account for much of the clinical activity of clobazam.
Absorption, distribution, metabolism and excretion
Clobazam is available in tablet or oral suspension form [22] and is rapidly absorbed from the gut, with a Tmax of 0.5-3 hours [23-25], with Tmax of norclobazam following at 3-56 hours [25, 26]. The rate of absorption of clobazam is not affected by a patient’s age or sex [24]. Once absorbed, clobazam distributes rapidly throughout the body [22]. The volume of distribution increases with age and is consistently higher in females than in males, regardless of age [24].
Taking clobazam with food has been found to cause a slight reduction in the Cmax of clobazam as well as a delayed Tmax, however there was no distinguishable effect on AUC or the elimination half-life [27] and current prescribing information states that clobazam can be taken with or without food [22]. However, a case study of a patient beginning a ketogenic diet while taking clobazam showed a 42% decrease in serum concentrations of clobazam [28]. The authors hypothesize that the ketogenic diet may increase cytochrome P450 enzyme activity, leading to increased metabolism of the drug [28].
Upon entry into the blood, both clobazam and norclobazam are highly protein-bound (90% and 89% respectively). The majority of this binding is to plasma albumin with some binding to α1-acid-glycoprotein also observed [29]. Only unbound clobazam and norclobazam molecules are able to cross the blood-brain barrier [29] and the fraction of non-bound clobazam in the blood increases with age, partially due to a decreasing concentration of plasma albumin [24]. Given that clobazam and norclobazam are lipophilic, have low molecular weights and are only able to form 2-3 hydrogen bonds, it seems likely that they are both able to diffuse freely across the blood-brain barrier without the involvement of a specific transporter [30].
Clobazam is extensively metabolized in the liver by cytochrome P450 enzymes, with only 3% of the original dose being excreted unchanged [22, 31, 32]. Around 70% of clobazam molecules are demethylated at nitrogen-1 to form norclobazam [6, 33]; a reaction primarily catalyzed by CYP3A4, with CYP2C19 and CYP2B6 playing a minor role [32]. The relative contributions of CYP3A4 and CYP2C19 to clobazam metabolism can be demonstrated in enzyme inhibition studies. The AUC of clobazam undergoes a 54% increase when CYP3A4 is inhibited, while inhibition of CYP2C19 increases the AUC of clobazam by 27% [15]. The overall rate of norclobazam formation decreases with age in males, but not females [24].
The remaining 27% of clobazam forms eight other metabolites [6], all of which are considered to be inactive. The most commonly detected of these inactive metabolites is 4′hydroxyclobazam, which is produced by CYP2C18 and CYP2C19 [32]. Both norclobazam and 4′hydroxyclobazam are further metabolized to the inactive metabolite 4′-hydroxy-N-desmethylclobazam [6]. Norclobazam is predominantly metabolized by CYP2C19 but can also by hydroxylated by CYP2C18 [32]. The demethylation of 4′-hydroxyclobazam is carried out by CYP2C9, CYP2B6 and CYP3A4 [32]. Clobazam and norclobazam’s status as CYP2C19 substrates is unusual when compared with the 1,4-benzodiazepines, as only diazepam is metabolized by the same enzyme (see the PharmGKB benzodiazepine pharmacokinetics pathway at https://www.pharmgkb.org/pathway/PA165111375).
Numerous studies investigating drug-drug interactions with clobazam have shown that induction of CYP3A4 and/or inhibition of CYP2C19, for example by omeprazole (a CYP2C19 inhibitor) or etravirine (a CYP3A4 inducer and CYP2C19 inhibitor), can increase plasma concentrations of norclobazam [33-38]. Similarly, induction of CYP2C19 increases norclobazam elimination from the body [36]. There have been indications that clobazam is itself a CYP3A4 inducer and could therefore self-induce its own metabolism [20, 39], but these have yet to be fully confirmed. Clobazam is known to inhibit CYP2D6, affecting the pharmacokinetics of drugs such as dextromethorphan, which undergoes an 95% increase in its AUC in the presence of clobazam [15].
Clobazam and its metabolites are excreted in the urine and feces [6], with the rate of elimination of clobazam increasing with age [24]. There is some evidence that hepatic impairment can affect the pharmacokinetics of clobazam metabolism, most notably the elimination of norclobazam [40, 41]. This is reflected in the prescribing information, which recommends that patients with hepatic impairment are given reduced doses of clobazam [22].
Pharmacodynamics
Clobazam and norclobazam act on postsynaptic GABAA receptors. These receptors are pentameric and are composed of a combination of α, β, γ, δ, ε or ρ subunits [4]. The inclusion of α and β subunits is required for functional GABAA receptors [42], while γ subunits are necessary for benzodiazepine binding [43]. Benzodiazepines bind allosterically to GABAA receptors [44] at the benzodiazepine binding site, located between γ and α subunits [45]. For more information on benzodiazepine pharmacodynamics, see the PharmGKB benzodiazepine pharmacodynamics pathway at https://www.pharmgkb.org/pathway/PA165111376.
Unlike the 1,4-benzodiazepines, which are full agonists of the GABAA receptor, clobazam is considered to be a selective agonist and does not interact with GABAA receptors containing γ1 or α6 subunits [46, 47]. Although clobazam does binds to receptors containing the γ2 subunit, like the 1,4-benzodiazepines, it is less dependent on the presence of the F77 residue in the γ2 subunit for binding [48].
There is conflicting information about clobazam and norclobazam’s relative binding affinities to the α1 and α2 subunits of GABAA receptors. Some studies have found an increased affinity for α2 over α1 subunits [4, 49, 50], while Hammer et al. saw no difference in clobazam binding affinities for α1 and α2 subunits and only a marginal increase in norclobazam’s binding affinity for α2 subunits compared to α1 [51].
There is no direct evidence of how the binding of clobazam or norclobazam to a GABAA receptor increases GABA binding and/or an increased influx of chloride ions. Work with the 1,4-benzodiazepine diazepam indicates that binding of a GABAA agonist in the BZD binding site increases the likelihood that the receptor will enter the preactivated state after GABA has bound, making it more likely that the receptor will activate and initiate chloride ion flow across the membrane [52]
Clobazam has been found to act on GABAA receptors involved in phasic inhibition (i.e. those involved in temporary inhibition of excitatory action potentials), but does not interact with receptors involved in tonic (long-term) inhibition [53]. Although it is more persistent in the body than its parent drug and appears likely to confer much of the clinical effect of clobazam, in vitro studies suggest that norclobazam has a lower activity at GABAA receptors than clobazam [54].
Although the majority of research focuses on the interactions between clobazam or norclobazam and GABAA receptors, there are other proteins which could be affected by the drug. A study of clobazam treatment in rats has suggested that the clinical effects of clobazam and norclobazam may extend to transporter proteins. Clobazam treatment increased expression of the GABA transporter GAT3 and the glutamate transporter GLT-1 in the hippocampus [55].
Clobazam is considered less likely to cause some severe side effects in patients, such as somnolence and physical dependency, than the 1,4-benzodiazepines [56], possibly because clobazam is a selective agonist [49, 51]. Mouse neurons treated with clobazam show a reduced upregulation of L-type high-voltage calcium channels compared to 1,4-benzodiazepines. Upregulation of these calcium channels is thought to be involved in the establishment of physical dependence and addiction [57].
Pharmacogenomics
There is a considerable amount of evidence in the scientific literature that CYP2C19 polymorphisms affect clobazam pharmacokinetics. Over 30 different CYP2C19 alleles have been identified so far (https://www.pharmvar.org/gene/CYP2C19), many of which affect the enzyme’s activity. The most frequently found alleles are *1, associated with normal levels of enzyme activity, *2 and *3, which encode enzymes with a decreased level of activity, and *17, which is linked to increased levels of enzymatic activity [58, 59].
It is possible to assign one of five different CYP2C19 metabolizer phenotypes to an individual based on the combination of CYP2C19 alleles that they carry, as compared to the ‘normal’ activity levels conferred by the *1 allele [60]. These are described in Table 1, with examples of common genotypes. It should be noted that the CYP2C19 alleles exhibit a gene dose effect [61, 62], where an individual with a *1/*2 diplotype (i.e. one functional and one non-functional allele) will have a different phenotype compared to individuals who are homozygous for either the *1 or *2 allele.
Table 1. CYP2C19 metabolizer phenotypes.
Overview of the different CYP2C19 metabolizer phenotypes with example diplotypes. The abbreviation for each phenotype is given in parentheses.
| CYP2C19 phenotype | Enzyme activity level compared to Normal Metabolizer | Example diplotypes |
|---|---|---|
| Ultrarapid metabolizer (UM) | Greatly increased | *17/*17 |
| Rapid metabolizer (RM) | Increased | *1/*17 |
| Normal metabolizer (NM) | - | *1/*1 |
| Intermediate metabolizer (IM) | Decreased | *1/*2, *1/*3 |
| Poor metabolizer (PM) | Greatly decreased or absent | *2/*2, *2/*3, *3/*3 |
Because CYP2C19 is the main enzyme involved in the metabolism of norclobazam to 4′-hydroxy-N-desmethylclobazam, norclobazam persists in CYP2C19 poor metabolizers (i.e. patients carrying two alleles conferring decreased or no CYP2C19 activity) for longer than in other CYP2C19 phenotypes. Patients with a CYP2C19 poor metabolizer (PM) phenotype can be distinguished by a high norclobazam C/D ratio and can have serum concentrations of norclobazam up to 7-fold higher than those seen in other CYP2C19 phenotypes [22, 63].
Clearance of clobazam is also decreased in patients carrying one or more non-functional CYP2C19 alleles, although the associated increase in clobazam concentrations is marginal compared to the increase in norclobazam concentrations. As an example, patients with the CYP2C19 PM phenotype experience a 30-50% increase in clobazam serum concentrations compared to those with two functional alleles (e.g. *1/*1), but a 7-fold increase in norclobazam concentrations [62, 63]. Clobazam C/D ratios are minimally affected by a patient’s CYP2C19 phenotype and development of tolerance to clobazam was unaffected by a patient’s CYP2C19 phenotype [62, 63].
A case study of two patients with high C/D ratios of norclobazam found that one patient was homozygous for CYP2C19*2, while the other patient carried one CYP2C19*2 allele. As rare and private CYP2C19 mutations were not screened for in this study, it is possible that this second patient carried a second non-functional allele which would confer a PM phenotype [64].
As norclobazam is the active metabolite of clobazam, CYP2C19 IMs and PMs can experience a greater clinical response (i.e. reduction of or freedom from seizures) to clobazam treatment than other phenotypes and start to see treatment efficacy at a lower dose. In a study of 110 epilepsy patients, Seo et al. found that 23.8% of IMs and 21.7% of PMs became seizure-free following clobazam treatment, compared to 8.1% of NMs (OR = 6.12 for IMs and 7.83 for PMs) [62]. Similarly, Hashi et al. treated 50 epilepsy patients with a low dose (2.5mg/day) of clobazam and observed that almost all PMs had a reduction in seizure frequency, while IMs and NMs had no change in seizure frequency (p <0.01) [59]. It is assumed that patients who carry one or more CYP2C19*17 alleles, and therefore have a rapid metabolizer or ultrarapid metabolizer phenotype, would have decreased exposure to clobazam compared to NMs, however, no research has been published to confirm this.
While the CYP2C19 PM phenotype can improve treatment outcomes, patients with this phenotype are potentially at greater risk of clobazam toxicity, symptoms of which can include severe somnolence, CNS depression and ataxia, as a result of clobazam treatment [22, 62, 65, 66]. It has been suggested that therapeutic drug monitoring (TDM) of the norclobazam C/D ratio in patients could be an effective way to identify CYCP2C19 PMs when no genetic information is available [65, 67], thus alerting the clinician to the potential for clobazam toxicity. Further to this, the prescribing information for clobazam recommends dosage adjustment for CYP2C19 PMs, starting at a lower dose of 5mg/day and slowly increasing the dose as necessary (see https://www.pharmgkb.org/chemical/PA10888/label for further details) [22].
There are indications that polymorphisms in other genes could also impact on clobazam pharmacokinetics. Saruwatari et al. found that the P450 oxidoreductase POR*28 TT genotype was associated with a 30% reduction in the serum concentrations of both clobazam and norclobazam as well as a 44% increase in the oral clearance rate of clobazam [61]. The mechanism behind these effects is as yet unclear.
Conclusion
Genetic variation in CYP2C19 can affect norclobazam metabolism, impacting both treatment outcome and experience of side effects in patients. Poor metabolizers have higher serum concentrations of norclobazam than intermediate and normal metabolizers and should be monitored to ensure that they do not suffer from norclobazam toxicity.
The majority of pharmacogenetic studies involving clobazam have focused on poor metabolizers or on populations where poor metabolizer alleles are prevalent (e.g. Japanese) [58] and there appears to be no work investigating the effects of a CYP2C19 ultrarapid or rapid metabolizer phenotype on clobazam and norclobazam pharmacokinetics. Given that the *17 allele, which increases CYP2C19 activity, is more frequent in European, African and admixed American populations than the Asian populations which have been studied to date, there is a need to ensure that the impact of all CYP2C19 phenotypes on norclobazam metabolism is investigated.
There may be other genes which affect clobazam pharmacokinetics, such as POR, and further investigation is merited. As norclobazam is an active metabolite, it is vital for future studies to measure both clobazam and norclobazam pharmacokinetics in subjects in order to fully understand the effects of genetic variation on clobazam treatment in patients.
Figure 1.
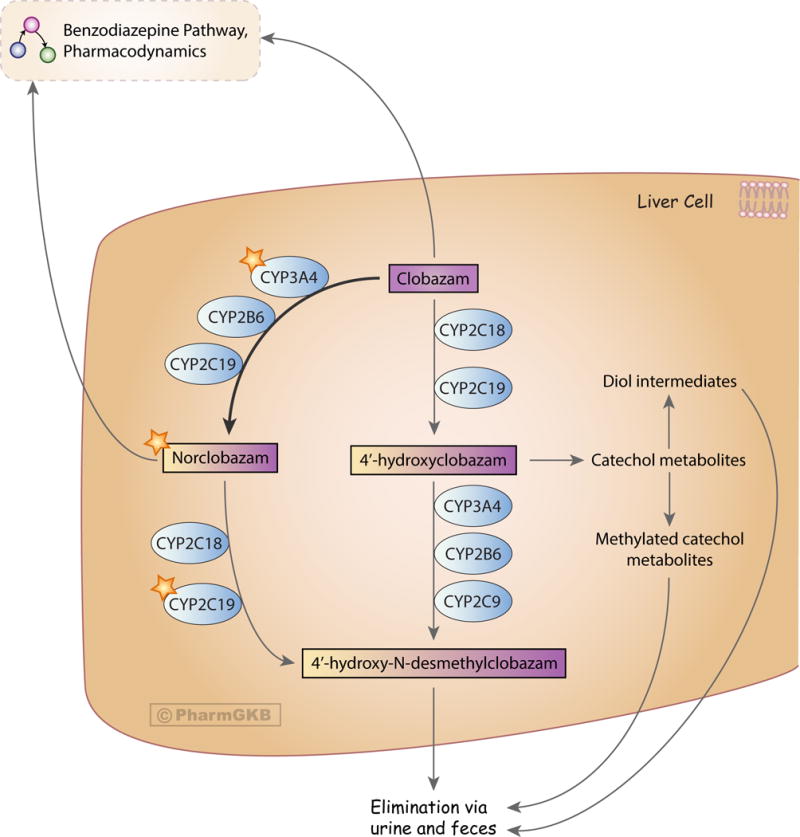
Stylized diagram showing clobazam metabolism in the liver. A fully clickable version of this figure can be found at https://www.pharmgkb.org/pathway/PA166170042.
Acknowledgments
The authors thank Caroline Thorn and Michelle Whirl-Carrillo for critical reading of the manuscript. This work is supported by the NIH/NIGMS grant GM61374.
Footnotes
Conflict of interest: RBA is a stockholder in Personalis Inc. and 23andMe, and a paid advisor for Karius
References
- 1.Wheless JW, Phelps SJ. Clobazam: a newly approved but well-established drug for the treatment of intractable epilepsy syndromes. J Child Neurol. 2013;28(2):219–29. doi: 10.1177/0883073812463609. [DOI] [PubMed] [Google Scholar]
- 2.Allen JW, et al. Clobazam as adjunctive treatment in refractory epilepsy. Br Med J (Clin Res Ed) 1983;286(6373):1246–7. doi: 10.1136/bmj.286.6373.1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gastaut H, Low MD. Antiepileptic properties of clobazam, a 1-5 benzodiazepine, in man. Epilepsia. 1979;20(4):437–46. doi: 10.1111/j.1528-1157.1979.tb04825.x. [DOI] [PubMed] [Google Scholar]
- 4.Sankar R. GABA(A) receptor physiology and its relationship to the mechanism of action of the 1,5-benzodiazepine clobazam. CNS Drugs. 2012;26(3):229–44. doi: 10.2165/11599020-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 5.Haigh JR, et al. N-desmethylclobazam: a possible alternative to clobazam in the treatment of refractory epilepsy? Br J Clin Pharmacol. 1987;23(2):213–8. doi: 10.1111/j.1365-2125.1987.tb03032.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Volz M, et al. Kinetics and metabolism of clobazam in animals and man. Br J Clin Pharmacol. 1979;7(Suppl 1):41s–50s. doi: 10.1111/j.1365-2125.1979.tb04664.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stafstrom CE, Carmant L. Seizures and epilepsy: an overview for neuroscientists. Cold Spring Harb Perspect Med. 2015;5(6) doi: 10.1101/cshperspect.a022426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nakamura F, et al. Effects of clobazam and its active metabolite on GABA-activated currents in rat cerebral neurons in culture. Epilepsia. 1996;37(8):728–35. doi: 10.1111/j.1528-1157.1996.tb00643.x. [DOI] [PubMed] [Google Scholar]
- 9.Arif H, et al. Comparison and predictors of rash associated with 15 antiepileptic drugs. Neurology. 2007;68(20):1701–9. doi: 10.1212/01.wnl.0000261917.83337.db. [DOI] [PubMed] [Google Scholar]
- 10.Blaszczyk B, Lason W, Czuczwar SJ. Antiepileptic drugs and adverse skin reactions: An update. Pharmacol Rep. 2015;67(3):426–34. doi: 10.1016/j.pharep.2014.11.009. [DOI] [PubMed] [Google Scholar]
- 11.Dang CD, Beets-Shay L, Kahn EC. Toxic epidermal necrolysis triggered by clobazam: a case report in a 13-year-old girl. Pediatr Dermatol. 2015;32(3):e102–3. doi: 10.1111/pde.12527. [DOI] [PubMed] [Google Scholar]
- 12.Yapici AK, et al. Stevens-Johnson Syndrome triggered by a combination of clobazam, lamotrigine and valproic acid in a 7-year-old child. Ann Burns Fire Disasters. 2014;27(3):121–5. [PMC free article] [PubMed] [Google Scholar]
- 13.Ertam I, Sezgin AO, Unal I. A case with Stevens Johnson syndrome triggered by combination of clobazam, lamotrigine, and valproic acid treatment. Int J Dermatol. 2009;48(1):98–9. doi: 10.1111/j.1365-4632.2009.03865.x. [DOI] [PubMed] [Google Scholar]
- 14.Mockenhaupt M, et al. Stevens-Johnson syndrome and toxic epidermal necrolysis: assessment of medication risks with emphasis on recently marketed drugs. The EuroSCAR-study. J Invest Dermatol. 2008;128(1):35–44. doi: 10.1038/sj.jid.5701033. [DOI] [PubMed] [Google Scholar]
- 15.Walzer M, et al. Pharmacokinetic drug interactions between clobazam and drugs metabolized by cytochrome P450 isoenzymes. Pharmacotherapy. 2012;32(4):340–53. doi: 10.1002/j.1875-9114.2012.01028.x. [DOI] [PubMed] [Google Scholar]
- 16.Food and Drug Administration. FDA Drug Safety Communication: FDA warns of serious skin reactions with the anti-seizure drug Onfi (clobazam) and has approved label changes. 2013 08/30/2017]; Available from: https://www.fda.gov/Drugs/DrugSafety/ucm377204.htm–tabs-2.
- 17.Frey N, et al. The risk of Stevens-Johnson syndrome and toxic epidermal necrolysis in new users of antiepileptic drugs. Epilepsia. 2017;58(12):2178–2185. doi: 10.1111/epi.13925. [DOI] [PubMed] [Google Scholar]
- 18.Rupp W, et al. Pharmacokinetics of single and multiple doses of clobazam in humans. Br J Clin Pharmacol. 1979;7(Suppl 1):51s–57s. doi: 10.1111/j.1365-2125.1979.tb04665.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pullar T, et al. Pharmacokinetics of N-desmethylclobazam in healthy volunteers and patients with epilepsy. Br J Clin Pharmacol. 1987;24(6):793–7. doi: 10.1111/j.1365-2125.1987.tb03247.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Leon J, Spina E, Diaz FJ. Clobazam therapeutic drug monitoring: a comprehensive review of the literature with proposals to improve future studies. Ther Drug Monit. 2013;35(1):30–47. doi: 10.1097/FTD.0b013e31827ada88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kinoshita M, et al. Efficacy of low-dose, add-on therapy of clobazam (CLB) is produced by its major metabolite, N-desmethyl-CLB. J Neurol Sci. 2007;263(1–2):44–8. doi: 10.1016/j.jns.2007.05.025. [DOI] [PubMed] [Google Scholar]
- 22.Lundbeck US. ONFI Prescribing Information. 2016 [Google Scholar]
- 23.Tedeschi G, Riva R, Baruzzi A. Clobazam plasma concentrations: pharmacokinetic study in healthy volunteers and data in epileptic patients. Br J Clin Pharmacol. 1981;11(6):619–22. doi: 10.1111/j.1365-2125.1981.tb01180.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Greenblatt DJ, et al. Clobazam kinetics in the elderly. Br J Clin Pharmacol. 1981;12(5):631–6. doi: 10.1111/j.1365-2125.1981.tb01281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jawad S, Richens A, Oxley J. Single dose pharmacokinetic study of clobazam in normal volunteers and epileptic patients. Br J Clin Pharmacol. 1984;18(6):873–7. doi: 10.1111/j.1365-2125.1984.tb02558.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rouini M, et al. Simultaneous determination of clobazam and its major metabolite in human plasma by a rapid HPLC method. J Chromatogr B Analyt Technol Biomed Life Sci. 2005;823(2):167–71. doi: 10.1016/j.jchromb.2005.06.031. [DOI] [PubMed] [Google Scholar]
- 27.Divoll M, et al. Clobazam kinetics: intrasubject variability and effect of food on adsorption. J Clin Pharmacol. 1982;22(1):69–73. doi: 10.1002/j.1552-4604.1982.tb05711.x. [DOI] [PubMed] [Google Scholar]
- 28.Kverneland M, et al. Modified Atkins diet may reduce serum concentrations of antiepileptic drugs. Acta Neurol Scand. 2015;131(3):187–90. doi: 10.1111/ane.12330. [DOI] [PubMed] [Google Scholar]
- 29.Patsalos PN, et al. Serum protein binding of 25 antiepileptic drugs in a routine clinical setting: A comparison of free non-protein-bound concentrations. Epilepsia. 2017;58(7):1234–1243. doi: 10.1111/epi.13802. [DOI] [PubMed] [Google Scholar]
- 30.Pardridge WM. Drug transport across the blood-brain barrier. J Cereb Blood Flow Metab. 2012;32(11):1959–72. doi: 10.1038/jcbfm.2012.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Iwasaki T, et al. Correlating blood and urinary concentrations of clobazam doses in Japanese children and adolescents with intractable epilepsy. Biomed Chromatogr. 2017;31(4) doi: 10.1002/bmc.3848. [DOI] [PubMed] [Google Scholar]
- 32.Giraud C, et al. In vitro characterization of clobazam metabolism by recombinant cytochrome P450 enzymes: importance of CYP2C19. Drug Metab Dispos. 2004;32(11):1279–86. [PubMed] [Google Scholar]
- 33.Levy RH, et al. Analysis of parent drug-metabolite relationship in the presence of an inducer. Application to the carbamazepine-clobazam interaction in normal man. Drug Metab Dispos. 1983;11(4):286–92. [PubMed] [Google Scholar]
- 34.Naccarato M, et al. A case of a potential drug interaction between clobazam and etravirine-based antiretroviral therapy. Antivir Ther. 2012;17(3):589–92. doi: 10.3851/IMP1953. [DOI] [PubMed] [Google Scholar]
- 35.Geffrey AL, et al. Drug-drug interaction between clobazam and cannabidiol in children with refractory epilepsy. Epilepsia. 2015;56(8):1246–51. doi: 10.1111/epi.13060. [DOI] [PubMed] [Google Scholar]
- 36.Tolbert D, et al. Drug-metabolism mechanism: Knowledge-based population pharmacokinetic approach for characterizing clobazam drug-drug interactions. J Clin Pharmacol. 2016;56(3):365–74. doi: 10.1002/jcph.603. [DOI] [PubMed] [Google Scholar]
- 37.Yamamoto Y, et al. Interaction between sulthiame and clobazam: sulthiame inhibits the metabolism of clobazam, possibly via an action on CYP2C19. Epilepsy Behav. 2014;34:124–6. doi: 10.1016/j.yebeh.2014.03.018. [DOI] [PubMed] [Google Scholar]
- 38.Pullar T, et al. The effect of cimetidine on the single dose pharmacokinetics of oral clobazam and N-desmethylclobazam. Br J Clin Pharmacol. 1987;23(3):317–21. doi: 10.1111/j.1365-2125.1987.tb03051.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bun H, et al. Effects of age and antiepileptic drugs on plasma levels and kinetics of clobazam and N-desmethylclobazam. Pharmacol Toxicol. 1990;67(2):136–40. doi: 10.1111/j.1600-0773.1990.tb00799.x. [DOI] [PubMed] [Google Scholar]
- 40.Monjanel-Mouterde S, et al. Pharmacokinetics of a single oral dose of clobazam in patients with liver disease. Pharmacol Toxicol. 1994;74(6):345–50. doi: 10.1111/j.1600-0773.1994.tb01371.x. [DOI] [PubMed] [Google Scholar]
- 41.Tolbert D, et al. An integrative population pharmacokinetics approach to the characterization of the effect of hepatic impairment on clobazam pharmacokinetics. J Clin Pharmacol. 2016;56(2):213–22. doi: 10.1002/jcph.586. [DOI] [PubMed] [Google Scholar]
- 42.Connolly CN, et al. Assembly and cell surface expression of heteromeric and homomeric gamma-aminobutyric acid type A receptors. J Biol Chem. 1996;271(1):89–96. doi: 10.1074/jbc.271.1.89. [DOI] [PubMed] [Google Scholar]
- 43.Wafford KA, et al. Functional comparison of the role of gamma subunits in recombinant human gamma-aminobutyric acidA/benzodiazepine receptors. Mol Pharmacol. 1993;44(2):437–42. [PubMed] [Google Scholar]
- 44.Olsen RW, et al. Effects of drugs on gamma-aminobutyric acid receptors, uptake, release and synthesis in vitro. Brain Res. 1978;139(2):277–94. doi: 10.1016/0006-8993(78)90929-0. [DOI] [PubMed] [Google Scholar]
- 45.Sigel E. Mapping of the benzodiazepine recognition site on GABA(A) receptors. Curr Top Med Chem. 2002;2(8):833–9. doi: 10.2174/1568026023393444. [DOI] [PubMed] [Google Scholar]
- 46.Khom S, et al. Pharmacological properties of GABAA receptors containing gamma1 subunits. Mol Pharmacol. 2006;69(2):640–9. doi: 10.1124/mol.105.017236. [DOI] [PubMed] [Google Scholar]
- 47.Nikas P, et al. Study of the Interaction of 1,4- and 1,5-Benzodiazepines with GABAA Receptors of Rat Cerebellum Granule Cells in Culture. J Mol Neurosci. 2015;56(4):768–72. doi: 10.1007/s12031-015-0495-8. [DOI] [PubMed] [Google Scholar]
- 48.Ogris W, et al. Affinity of various benzodiazepine site ligands in mice with a point mutation in the GABA(A) receptor gamma2 subunit. Biochem Pharmacol. 2004;68(8):1621–9. doi: 10.1016/j.bcp.2004.07.020. [DOI] [PubMed] [Google Scholar]
- 49.Jensen HS, et al. Clobazam and its active metabolite N-desmethylclobazam display significantly greater affinities for alpha(2)- versus alpha(1)-GABA(A)-receptor complexes. PLoS One. 2014;9(2):e88456. doi: 10.1371/journal.pone.0088456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ralvenius WT, et al. The clobazam metabolite N-desmethyl clobazam is an alpha2 preferring benzodiazepine with an improved therapeutic window for antihyperalgesia. Neuropharmacology. 2016;109:366–75. doi: 10.1016/j.neuropharm.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hammer H, et al. Functional characterization of the 1,5-benzodiazepine clobazam and its major active metabolite N-desmethylclobazam at human GABA(A) receptors expressed in Xenopus laevis oocytes. PLoS One. 2015;10(3):e0120239. doi: 10.1371/journal.pone.0120239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gielen MC, Lumb MJ, Smart TG. Benzodiazepines modulate GABAA receptors by regulating the preactivation step after GABA binding. J Neurosci. 2012;32(17):5707–15. doi: 10.1523/JNEUROSCI.5663-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gatta E, et al. New 1,5-benzodiazepine compounds: activity at native GABA(A) receptors. Neuroscience. 2010;166(3):917–23. doi: 10.1016/j.neuroscience.2010.01.013. [DOI] [PubMed] [Google Scholar]
- 54.Fisher JL. Interactions between modulators of the GABA(A) receptor: Stiripentol and benzodiazepines. Eur J Pharmacol. 2011;654(2):160–5. doi: 10.1016/j.ejphar.2010.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Doi T, et al. Molecular regulation of glutamate and GABA transporter proteins by clobazam during epileptogenesis in Fe(+++)-induced epileptic rats. Brain Res Mol Brain Res. 2005;142(2):91–6. doi: 10.1016/j.molbrainres.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 56.Koeppen D. Review of clinical studies on clobazam. Br J Clin Pharmacol. 1979;7(Suppl 1):139s–150s. doi: 10.1111/j.1365-2125.1979.tb04684.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Katsura M, et al. Up-regulation of L-type high voltage-gated calcium channel subunits by sustained exposure to 1,4- and 1,5-benzodiazepines in cerebrocortical neurons. J Neurochem. 2007;103(6):2518–28. doi: 10.1111/j.1471-4159.2007.04984.x. [DOI] [PubMed] [Google Scholar]
- 58.Zhou Y, Ingelman-Sundberg M, Lauschke VM. Worldwide Distribution of Cytochrome P450 Alleles: A Meta-analysis of Population-scale Sequencing Projects. Clin Pharmacol Ther. 2017 doi: 10.1002/cpt.690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hashi S, et al. Effect of CYP2C19 polymorphisms on the clinical outcome of low-dose clobazam therapy in Japanese patients with epilepsy. Eur J Clin Pharmacol. 2015;71(1):51–8. doi: 10.1007/s00228-014-1773-z. [DOI] [PubMed] [Google Scholar]
- 60.Caudle KE, et al. Standardizing terms for clinical pharmacogenetic test results: consensus terms from the Clinical Pharmacogenetics Implementation Consortium (CPIC) Genet Med. 2017;19(2):215–223. doi: 10.1038/gim.2016.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Saruwatari J, et al. Effects of CYP2C19 and P450 oxidoreductase polymorphisms on the population pharmacokinetics of clobazam and N-desmethylclobazam in japanese patients with epilepsy. Ther Drug Monit. 2014;36(3):302–9. doi: 10.1097/FTD.0000000000000015. [DOI] [PubMed] [Google Scholar]
- 62.Seo T, et al. Impact of CYP2C19 polymorphisms on the efficacy of clobazam therapy. Pharmacogenomics. 2008;9(5):527–37. doi: 10.2217/14622416.9.5.527. [DOI] [PubMed] [Google Scholar]
- 63.Kosaki K, et al. A major influence of CYP2C19 genotype on the steady-state concentration of N-desmethylclobazam. Brain Dev. 2004;26(8):530–4. doi: 10.1016/j.braindev.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 64.Contin M, et al. Evidence of polymorphic CYP2C19 involvement in the human metabolism of N-desmethylclobazam. Ther Drug Monit. 2002;24(6):737–41. doi: 10.1097/00007691-200212000-00009. [DOI] [PubMed] [Google Scholar]
- 65.Parmeggiani A, et al. Unusual side-effects due to clobazam: a case report with genetic study of CYP2C19. Brain Dev. 2004;26(1):63–6. doi: 10.1016/s0387-7604(03)00074-3. [DOI] [PubMed] [Google Scholar]
- 66.Aylett SE, Cross H, Berry D. Eye rolling as a manifestation of clobazam toxicity in a child with epilepsy. Dev Med Child Neurol. 2006;48(7):612–5. doi: 10.1017/S0012162206001289. [DOI] [PubMed] [Google Scholar]
- 67.Yamamoto Y, et al. Influence of CYP2C19 polymorphism and concomitant antiepileptic drugs on serum clobazam and N-desmethyl clobazam concentrations in patients with epilepsy. Ther Drug Monit. 2013;35(3):305–12. doi: 10.1097/FTD.0b013e318283b49a. [DOI] [PubMed] [Google Scholar]