

Abstract
Bispecific antibodies offer a clinically validated platform for drug discovery. In generating functionally active bispecific antibodies, it is necessary to identify a unique parental antibody pair to merge into a single molecule. However, technologies that allow high-throughput production of bispecific immunoglobulin Gs (BsIgGs) for screening purposes are limited. Here, we describe a novel bispecific antibody format termed tethered-variable CLBsIgG (tcBsIgG) that allows robust production of intact BsIgG in a single cell line, concurrently ensuring cognate light chain pairing and preserving key antibody structural and functional properties. This technology is broadly applicable in the generation of BsIgG from a variety of antibody isotypes, including human BsIgG1, BsIgG2 and BsIgG4. The practicality of the tcBsIgG platform is demonstrated by screening BsIgGs generated from FGF21-mimetic anti-Klotho-β agonistic antibodies in a combinatorial manner. This screen identified multiple biepitopic combinations with enhanced agonistic activity relative to the parental monoclonal antibodies, thereby demonstrating that biepitopic antibodies can acquire enhanced functionality compared to monospecific parental antibodies. By design, the tcBsIgG format is amenable to high-throughput production of large panels of bispecific antibodies and thus can facilitate the identification of rare BsIgG combinations to enable the discovery of molecules with improved biological function.
Keywords: biepitopic antibody, bispecific antibody, FGF21, FGFR1, high throughput screening, KLB, therapeutic mAb
Introduction
Bispecific antibodies, a single moiety derived from two monoclonal antibodies, each with a distinct binding specificity, offer a clinically validated platform for drug discovery (Garber, 2014; Spiess et al., 2015; Suryadevara et al., 2015). The simplest application of the bispecific antibody technology is the simultaneous blocking of two signaling pathways through the association of two ligand-blocking antibodies (e.g. anti-IL-12/IL-18, anti-VEGF/ANG2) (Kontermann, 2012). In addition, bispecific antibodies provide the opportunity to direct natural killer cells or cytotoxic T-cells to tumors by combining antibodies that bind to cell type specific surface antigens (e.g. anti-CD3/EpCAM, catumaxomab; anti-CD3/CD19, blinatumomab) (Frankel and Baeuerle, 2013; Grandjenette et al., 2015; Zhukovsky et al., 2016). Bispecific antibodies can also be used as cofactor mimetics to bring two proteins into a spatially appropriate position (e.g. anti-FIXa/FX, emicizumab) (Kitazawa et al., 2012) or as ligand mimetics to stimulate receptor signaling by inducing receptor clustering or conformational changes in the receptor complex (e.g. anti-FGFR1/Klotho-β (KLB)) (Kolumam et al., 2015). In addition to antibodies that target two different antigens, there is an increasing interest in antibodies that engage two different epitopes on the same target (biepitopic antibodies). Such biepitopic antibodies could provide higher affinity, superior receptor silencing, internalization and intracellular trafficking over traditional monoclonal antibodies, or serve as ligand mimetics (e.g. anti-EpoR/EpoR) (Zhang et al., 2012; Lewis et al., 2014; Fleetwood et al., 2016; Li et al., 2016). Bispecific antibodies can be classified into five distinct structural groups (Spiess et al., 2015). In particular, bispecific antibodies with immunoglobulin G like structure (BsIgG) represent a promising technology that combines the unique advantages of dual target engagement with the superb pharmacokinetic properties and immune-effector functions of IgG molecules.
In discovering bispecific or biepitopic antibodies, it is critical to identify the best parental antibody combination to merge into a single molecule. This is a fairly straight-forward process for bispecific antibodies that target two soluble ligands, such as cytokines and growth factors. In this case, antibodies towards the two targets can be developed independently and subsequently combined to generate the bispecific antibody as demonstrated by, e.g. anti-VEGF/ANG2 or anti-IL-12/IL-18 BsIgG (Wu et al., 2007; Schaefer et al., 2011). However, there are cases when the desired biological function of bispecific antibodies cannot be fully predicted by the activity of the individual Fabs. For example, the screening of ~40 000 bispecific combinations of antibodies towards FIXa and FX identified a unique BsIgG that acts as a FVIII–mimetic by facilitating FIXa-catalyzed FX activation (Kitazawa et al., 2012). In another example, a unique combination of non-agonistic Fabs towards two cell surface receptors, FGFR1 and KLB, was selected to identify a distinct BsIgG that acts as a FGF21 ligand–mimetic by selectively activating the FGFR1/KLB receptor complex (Kolumam et al., 2015). This anti-FGFR1/KLB BsIgG induced weight loss and other metabolic benefits in mice and cynomolgus monkeys, mimicking the activity of FGF21, the natural ligand for the FGFR1/KLB complex (Kolumam et al., 2015).
These studies elegantly demonstrate that BsIgG can acquire activities that cannot be predicted by the activity of each Fab, thus necessitating screening of BsIgG pairs to identify the best combination from a matrix of possible antibody pairs. Typically, a BsIgG is assembled from two different heavy (H1 + H2) and light (L1 + L2) chains. For the production of a BsIgG, efficient heterodimerization of the heavy chains can be achieved by amino acid substitutions in the antibody Fc region (e.g. knobs-into-holes or charge pairs) (Ridgway et al., 1996; Gunasekaran et al., 2010; Strop et al., 2012; Labrijn et al., 2013; Von Kreudenstein et al., 2013). An equally important aspect of the BsIgG production is the correct light and heavy chain pairing (L1:H1 and L2:H2). A commonly used strategy for this is the in vitro assembly of the BsIgG from two separately expressed component antibodies (Strop et al., 2012; Labrijn et al., 2013; Spiess et al., 2013). While in vitro assembly is an attractive and validated strategy for producing bispecific antibodies in large quantities (Williams et al., 2015), this process involves several steps that do not make this technology readily amenable to high-throughput production of small quantities of a larger panel of BsIgG. Alternatively, domain crossovers (CrossMAb) or engineered Fab domains have been used to produce BsIgG in a single cell line (Schaefer et al., 2011; Lewis et al., 2014; Liu et al., 2015; Mazor et al., 2015; Dillon et al., 2017). While the CrossMAb technology enabled the generation and screening of a small number of bispecific antibodies for superior HIV neutralization (Huang et al., 2016), the applicability of other single cell line technologies for screening has not yet been demonstrated so far. In addition, these single cell line technologies have only been described for human IgG1 antibodies, and their utility in generating BsIgG in other therapeutically relevant isotypes, namely IgG2 or IgG4, has not been demonstrated. Therefore, novel bispecific formats that allow high-throughput production of a matrix of bispecific antibodies for screening purposes is expected to facilitate BsIgG discovery.
Here, we describe a novel bispecific antibody format termed tethered-variable CLBsIgG (tcBsIgG) that allows the robust expression of intact BsIgGs in a single cell line, while ensuring cognate light chain pairing and preserving the key structural and functional properties of the parental antibodies. This technology can be extended to the production of human IgG2 and IgG4. Previously, a monospecific anti-KLB agonist antibody, called mimAb1, that specifically activates FGFR1/KLB complex and induces weight loss in obese cynomolgus monkeys was identified (Foltz et al., 2012). We have generated agonistic anti-KLB antibodies with similar properties with mimAb1 and tested the applicability of this new platform by screening a matrix of unique biepitopic combinations generated from five of such anti-KLB antibodies in a combinatorial fashion. This screen led to the identification of five antibody pairs that exhibit enhanced agonistic activity over their corresponding parental antibodies. Our work demonstrates that biepitopic anti-KLB antibodies can be utilized to improve agonistic activity of traditional monoclonal antibodies, a concept that may extend to identifying other agonistic antibodies.
Materials and Methods
Antibody constructs
The heavy and light chains of antibody genes were cloned by standard molecular biology techniques into separate pRK expression vectors, as described previously (Eaton et al., 1986; Polson et al., 2007). For the human IgG2 construct, the second hinge cysteine, C220 (EU numbering), was mutated to serine to enable efficient in vitro assembly. For BsIgG production, heterodimerization of the heavy chains was achieved by ‘knobs-into-holes’ mutations (knob: T366W; hole: T366S:L368A:Y407V) (Ridgway et al., 1996; Atwell et al., 1997). In vitro assembly of BsIgG from knob and hole half-antibodies was described previously (Shatz et al., 2013; Spiess et al., 2013). For expression of BsIgG in the tcIgG format, the antibody VL domain residues (1-R108) were genetically fused to the antibody heavy chain via a (G4S)4 linker. The CL domain (human kappa, T109-C214) was expressed from a separate plasmid. The Kabat and EU numbering schemes were used to designate residue position in the antibody variable and constant domains, respectively. For co-transfections, plasmids were transfected in the same mass ratio into mammalian cells. Antibodies were expressed in HEK293T, Expi293T™ or CHO cells at the 30 ml or 1 l scale (Wong et al., 2010; Bos et al., 2014) followed by MabSelectSure (GE Healthcare, USA) purification according to the manufacturer's protocol.
Characterization of purified antibodies by capillary electrophoresis
Samples were analyzed on a Caliper GX II microfluidic system (PerkinElmer Biotechnology, Waltham, MA, USA). All samples were prepared as described previously (Kim et al., 2016). The chip was prepared according to the manufacturer's instructions provided in the LabChip GXII User Guide.
Biacore kinetics analysis
For binding affinity determination of tcIgG and conventional IgG, surface plasmon resonance (SPR) measurements using a BIAcore T200 and ProteinA sensor chip were used and dilutions of monomeric human KLB as analyte were injected over the immobilized tcIgG or traditional IgG at 25°C to determine affinities. Association rates (kon) and dissociation rates (koff) were calculated using a simple one-to-one Langmuir binding model. The equilibrium dissociation constant (Kd) was calculated as the ratio koff/kon.
Development of anti-KLB agonistic antibodies
Wild-type BALB/c mice or Klb deficient mice in mixed BALB/c-C57BL/6 background (Kolumam et al., 2015) were immunized with HEK293 or 300.19 cells stably expressing human FGFR1 and human KLB protein or by hydrodynamic tail vein (HTV) injection of plasmid DNA encoding FGFR1 and KLB. FGFR1 used for immunization or elsewhere in this manuscripts are the c-isoform of FGFR1 that binds to KLB, rather than the b-isoform that does not bind to KLB (Kurosu et al., 2007). Cell immunizations were achieved by injecting stably transfected HEK293 or 300.19 cells expressing human FGFR1 and human KLB in PBS weekly via intraperitoneal (i.p.) injection for a total of 12 injections. For DNA immunizations, 50 μg each of either pRK or pCMV vectors expressing either KLB or FGFR1 separately, or with a pCMV.hKLB.IRES.hFGFR1 vector expressing KLB and FGFR1, with or without pORF-mFlt3 ligand and pORF-mGM-CSF (Genentech), diluted in lactated Ringer's solution via HTV injection at 1–4 week intervals for a total of 3–13 injections. Mice received a pre-fusion boost of either KLB and FGFR1 plasmid DNA via HTV or transfected cells along with 2 μg each of human and cynomolgus KLB proteins via i.p. injection. Splenocytes from these mice, all of whose sera demonstrated strong binding to HEK293 cells overexpressing KLB and/or KLB/FGFR1 complex by FACS, were fused with P3X63-Ag8U.1 mouse myeloma cells (American Type Culture Collection, Manassas, VA) via electrofusion (Cyto Pulse CEEF-50 apparatus; BTX Harvard Apparatus, USA). Fused cells were plated into semi-solid methylcellulose-based media (Clonacell-HY Medium D, Stemcell Technologies, Cambridge, MA) with an anti-mouse IgG Fc-FITC detection reagent according to the manufacturer's instructions. After 7–8 days, IgG positive colonies were picked and transferred to 96-well plates, and supernatants were screened for binding to HEK293 cells overexpressing KLB and/or KLB/FGFR1 complex by cell-based ELISA. Positive clones were confirmed for FACS binding and agonistic activity in the GAL-ELK1 luciferase assay using HEK293T cells expressing FGFR1 and/or KLB. The identified anti-KLB antibodies were kappa (Cκ) isotype.
Luciferase reporter assay
HEK293T cells were transiently transfected with expression vectors encoding Renilla luciferase (pRL-SV40, Promega), appropriate receptors (FGFR and/or KLB), a transcriptional activator (pFA2-Elk1, Stratagene), and a firefly luciferase reporter driven by GAL4 binding sites (pFR-luc, Stratagene), using FuGENE HD (Promega, Madison, WI). On the next day, the transfected cells were cultured for an additional 7.5–8 h in serum-free medium with FGF21 or IgG at various concentrations. The cellular luciferase activity was determined using the Dual-Glo Luciferase Assay System (Promega, USA) and EnVision Multilabel Reader (Perkin Elmer, USA). Firefly luciferase signal was normalized to Renilla luciferase signal to calculate relative luminescence units (RLU). All the experiments were run in triplicate. Data was analyzed using the GraphPad Prism software package.
Western blotting
Human primary subcutaneous pre-adipocytes were acquired from Lonza (Walkersville, MD). Cells were grown and differentiated according to the supplier's protocol. Briefly, cells were grown in preadipocyte basal medium-2 containing FBS, L-glutamine and GA-1000. Once confluent, cells were differentiated in growth media containing insulin, dexamethasone, indomethacin and 3-isobutyl-1-methylxanthine (IBMX). For ERK signaling analysis, cells were differentiated for 10 days, grown in serum-free medium for 3 h, and then further cultured for an additional hour with the indicated antibodies. Cell extracts were generated by lysing cells in buffer consisting of 150 mM NaCl, 20 mM Tris, pH 7.5, 1 mM EDTA, 1 mM EGTA and 1% Triton X-100, and containing protease and phosphatase inhibitor tablets (Roche, USA). Samples were used for western blot analysis by standard methods. Antibodies used for western blot analysis were from Cell Signaling Technology (Danvers, MA): anti-pERK1/2 (T202/204) (catalog #4370), anti-ERK1/2 (catalog #4695) and anti-HSP90 (catalog #4874).
Bio-layer interferometry epitope binning
Epitope binning experiments were performed in 8-channel or 16-channel mode using anti-mouse Fc or anti-human Fc capture biosensors on the Octet Red384 system (Pall Life Sciences, USA). The assay consisted of a seven-step binding cycle: (1) anti-mouse Fc biosensors were dipped into running buffer (1x kinetics buffer, ForteBio, USA; catalog # 18–5032) for 1 min to establish a baseline, (2) 20 µg/ml mouse IgG2a reference antibody was captured for 10 min, (3) another baseline was established for 1.5 min, (4) 100 nM of KLB was loaded for 10 min, (5) a third baseline was established for 1.5 min, (6) 5 µg/ml of the human IgG1 test antibodies were allowed to associate for 10 min and (7) dissociate for 10 min. Epitope binning with the anti-human Fc capture biosensors were done similarly. All antibodies were subjected to four redundant assays: as reference antibodies (step 2) and, in reciprocal assays, as test antibodies (step 5), both as human IgG1 and as mouse IgG2a.
Non-antibody proteins
Recombinant human KLB-ECD protein with a C-terminal tandem Flag-His6 tag was produced in stably transfected HEK293S cells and purified via affinity columns to monodispersity. Human FGF21 protein was acquired from R&D Systems (cat# 2539-FG-025/CF, Minneapolis, MN).
Results
VL-HC fusion and CL expression in trans solves cognate light chain pairing problem for BsIgG
A schematic diagram for the novel tethered-variable CL Bispecific IgG (tcBsIgG) format we designed is shown in Fig. 1A. In this novel platform, the heterodimerization of the heavy chains was accomplished by the previously described ‘knobs-into-holes’ mutations (Ridgway et al., 1996; Atwell et al., 1997). In order to achieve correct heavy-light chain pairing, we designed a VL-HC fusion (VLfH) using a short (Gly4Ser)4-linker to tether the C-terminal end of the antibody VL domain to the N-terminus of the heavy chain, similar to the design of an antibody single chain variable fragment (scFv) (Fig. 1A, left). We decided to only tether the VL domain instead of the entire light chain as described previously (Wranik et al., 2012; Schanzer et al., 2014) to minimize potential light chain mispairing due to the inherent spatial flexibility of a long linker sequence. We initially tested the feasibility of this new format with a monospecific antibody lacking the ‘knobs-into-holes’ mutations. The expression of VLfH alone without the CL in HEK293 cells resulted in little to no VLfH expression (‘V’ lanes, Fig. 1B, left panel), while co-expression of an isolated CL (kappa isotype) from a separate plasmid allowed productive secretion of the VLfH hybrid protein (‘T’ lanes, Fig. 1B, left panel). Since this new IgG format is comprised from two tethered variable domains and an isolated CL, we named it tethered-variable CLIgG (tcIgG) and its bispecific counterpart tcBsIgG (Fig. 1A, right). We recovered tcIgG in amounts that were comparable to the intact, parental antibody (‘I’ lanes, Fig. 1B, left panel). The lack of expression in the absence of CL is somewhat expected as it has been proposed that the folding of the antibody CH1 domain requires pairing with the CL domain (Feige et al., 2009) and serves as final quality control step to ensure that only properly folded antibody is secreted (Feige and Buchner, 2014). Finally, to ensure that the intact tcIgG included the two covalently linked polypeptide chains, VLfH and CL, the antibodies were analyzed under reduced conditions (Fig. 1B, right panel). Two bands corresponding to the molecular weights of both chains were detected, confirming the incorporation of the CL into the tcIgG. A similar banding pattern was observed for the other isotypes (data not shown).
Fig. 1.

tcBsIgG format enables bispecific antibody generation in a single cell line. (A) Schematic representation of the tcBsIgG format. The antibody VL domain is tethered via a (G4S)4 linker to the antibody heavy chain for the VLfH format (left). The folding defect of CH1 is complemented by CL expression in trans from a separate plasmid (right). (B) Capillary electrophoresis under non-reducing (left panel) and reducing conditions (right panel) of expression of IgG1, IgG2, and IgG4 as well as the aglycosylated (N297G) version of IgG1 and IgG4 after protein A affinity column purification recovery. Expression as VLfH alone (lane V) without the CL in HEK293 cells results in only trace amount of or no VLfH expression at all; VLfH co-expressed with CL (lane T) results in tcIgG expression that is comparable to a standard IgG expression (lane I). (C) Capillary electrophoresis of protein A affinity purified tcIgG for conventional IgG1, IgG2, and IgG4 as well as the tcIgG1, tcIgG2, and tcIgG4 counterparts as knob half-antibody (lane K), hole half-antibody (lane H), and knob and hole co-expressed IgG in a single cell (lane C).
While the human IgG1 isotype is currently the predominant antibody class for clinical development (Brezski and Georgiou, 2016), other isotypes are also leveraged to modulate antibody effector function and antibody activity (Chan and Carter, 2010; White et al., 2015). Thus, we evaluated the translatability of the tcIgG technology to other therapeutically relevant human isotypes, IgG2 and IgG4. The requirement of the CL domain was consistent across expression of VLfH in IgG2 and IgG4 and the common aglycosylated version of the IgG1 and IgG4 with the N297G mutation behaved similarly (Fig. 1B). Overall productivity after CL co-expression was comparable to the respective wild-type IgG isotype control. By electrophoretic analysis, we demonstrated that the inter-chain disulfide bond between CL and CH1 was fully formed. Thus, for productive folding and secretion of an antibody, the antibody VL and CL domains do not need to be connected as a single polypeptide chain.
Next, we assessed if the tcIgG format is compatible with the ‘knobs-into-holes’ mutations for efficient production of a bispecific antibody. We compared the expression and assembly of knob and hole half-antibodies either by themselves or when co-expressed. As observed previously (Shatz et al., 2013), expression of just the knob or hole antibody by itself resulted predominantly in half-antibody and some covalent homodimer (‘K’ and ‘H’ lanes, Fig. 1C). The co-expression of knob and hole half-antibodies in the same cell resulted in efficient assembly of the ~150 KDa species in all three major human isotypes, IgG1, IgG2 and IgG4 (Fig. 1C). The observed expression and product quality of human tcBsIgG2 and tcBsIgG4 as both monospecific bivalent tcIgG as well as bispecific tcIgG were comparable to their respective IgG isotypes.
Isolation and characterization of complex dependent anti-KLB agonistic antibodies
To address whether the tcBsIgG format is amenable to high-throughput BsIgG screening, we decided to use anti-KLB agonistic antibodies as a test case. Previously, the generation of an agonistic anti-KLB antibody that mimics the activity of the anti-diabetic hormone FGF21 by activating the FGFR1/KLB receptor complex was reported (Foltz et al., 2012). Such antibodies may have a therapeutic utility since recombinant FGF21-analogs and bispecific anti-FGFR1/KLB agonistic antibodies have shown metabolic benefits in obese mice, non-human primates or even obese humans (Kharitonenkov et al., 2007; Coskun et al., 2008; Xu et al., 2009; Wu et al., 2011; Foltz et al., 2012; Gaich et al., 2013; Kolumam et al., 2015). To generate agonistic antibodies for the FGFR1/KLB complex, mice were immunized either by injecting cells expressing both KLB and FGFR1 or by expressing KLB and FGFR1 in the liver via HTV injection of plasmid DNA. We identified a total of 320 hybridoma clones that showed binding to a stable HEK293T cell line expressing KLB. We found only two anti-FGFR1 clones, probably due to the high sequence identity between mouse and human FGFR1. No clone that binds only to the complex was identified. All the hybridoma supernatants were subsequently screened by GAL-ELK1 luciferase assay in HEK293T cells expressing FGFR1 with or without co-expression of KLB. In this cell-based assay system, activation of FGFR signaling pathway leads to an ERK-dependent phosphorylation of the transcription factor ELK1 fused to the GAL4 DNA binding domain and the consequential induction of luciferase reporter gene under the control of GAL-response elements (Wu et al., 2011). HEK293 cells do not endogenously express KLB protein, therefore, KLB-dependent FGFR activation can be studied by comparing the response between cells expressing KLB and those without KLB expression. This assay identified 16 unique anti-KLB clones that induced luciferase activity in a KLB-dependent manner. The results of the luciferase assay with 5 representative hybridoma clones (12B8, 2C12, 23B3, 4H7 and 28B7) are shown in Fig. 2A.
Fig. 2.

Characterization of agonistic anti-KLB monoclonal antibodies. (A) Agonistic activity of five anti-KLB hybridoma antibodies in GAL-ELK1 luciferase assay with HEK293T cells expressing either FGFR1 and KLB (left) or FGFR1 alone (right). Luciferase signal was expressed as fold induction over an irrelevant IgG control (IgG). The data represents means ± SEM (N = 3). (B) Schematic representation of the three epitope bins identified by cross-competition. (C) Schematic representation of KLB protein structure is shown at the top. Each bar represents human KLB (black), rat KLB (white), or chimeric constructs as color-coded. At right, qualitative binding of anti-KLB antibodies based on FACS with HEK293T cells transiently expressing each construct is shown. Note that anti-KLB antibodies do not bind to rat KLB ECD, but introduction of the human sequence conferred specific binding. (D) Agonistic activity of recombinant anti-KLB antibodies in GAL-ELK1 luciferase assay with HEK293T cell expressing either FGFR1 (orange) or FGFR2 (black) and KLB. Luciferase activity was expressed as fold induction over mock treatment. The data represents means ± SEM (N = 3).
To characterize the epitope of these antibodies, epitope binning was conducted using bio- layer interferometry. As shown in Fig. 2B, these antibodies bound to three distinct epitopes: one for each for 12B8 and 28B7, and a common epitope for 2C12, 23B3 and 4H7. Furthermore, FACS binding with HEK293T cells expressing human/rat KLB chimeric proteins indicated that the antibodies in bins 1 and 2 (12B8, 2C12, 23B3 and 4H7) bound to different subdomains of the C- terminal portion of human KLB, whereas 28B7 bound to the N-terminal portion of human KLB protein (Fig. 2C). Subsequently, these five clones were produced as chimeric mouse/human IgG1 recombinant antibodies. Signaling activity of the recombinant antibodies was confirmed in a GAL-ELK1 luciferase assay in HEK293T cells expressing human KLB, together with either FGFR1, or its most closely related receptor, FGFR2. As shown in Fig. 2D, all five anti-KLB antibodies induced luciferase activity in a manner dependent on FGFR1, while the natural ligand FGF21 worked with FGFR1 or FGFR2 as expected (Kurosu et al., 2007). Thus, all five anti-KLB antibodies are selective agonists for the FGFR1/KLB complex. The reason why these anti-KLB antibodies without measurable affinity to FGFR1 act as FGFR1-selective agonists is not clear; however, the previously reported mimAb1 also exhibited such FGFR1 selectivity. Because all our anti-KLB antibodies as well as mimAb1 were all generated by immunizing with a FGFR1/KLB complex, it is possible that they interact directly with FGFR1 with an affinity that is too low to measure or binds a unique conformation of KLB that is present only when bound to FGFR1 but not to other FGFRs. Although the heightened selectivity of agonistic anti-KLB antibodies over the natural ligand FGF21 was an attractive feature, their activities in the GAL-ELK1 luciferase assay were not as robust as that of the natural ligand FGF21. This led us to the idea that the synergy of two agonistic antibodies, facilitated by a biepitopic antibody, may exhibit an enhanced functionality closer to that of FGF21.
Production of anti-KLB biepitopic antibodies in the tcBsIgG format
In order to produce biepitopic antibodies derived from the five anti-KLB monoclonal antibodies described above, we first cloned each antibody as murine/human chimera into the VLfH.knob and VLfH.hole vectors. This enabled us to produce 25 different antibodies in the tcBsIgG format (Table I, blue shaded fields), including 10 possible biepitopic combinations in both heavy chain orientations (i.e. knob/hole and hole/knob) in HEK293T cells. Having both of the knob–hole orientations for each antibody combination provided us with independent replicates. In addition, we generated parental monospecific antibodies to serve as a benchmark by tcIgG.knob and tcIgG.hole co-expression (Table I; grey shaded fields). We also included a well-expressing antibody AbA (Table I; green shaded fields) and a poorly expressing antibody AbB (Table I; orange shaded fields) as additional controls.
Table I.
Resulting tcBsIgG combinations from seven parental antibodies (blue: biepitopic tcBsIgG; green: tcBsIgG paired with well-expressing AbA; orange: tcBsIgG paired with poorly expressing AbB; grey: monoepitopic tcIgG) and tcIgGs as bivalent controls (unshaded). Numerical values represent the monomer content (%) for each tcBsIgG and tcIgG
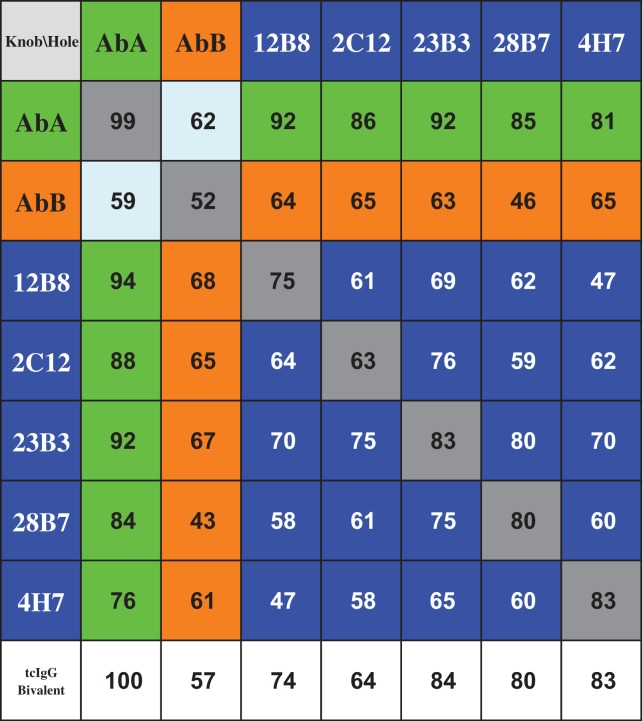 |
All 25 anti-KLB tcBsIgG antibodies were characterized by capillary electrophoresis and demonstrated comparable assembly efficiency. The predominant band for all antibodies was around ~150 kDa indicating correct domain pairing and disulfide bond formation (Fig. 3A). We noticed that the inefficient folding of one of the parental antibodies limits the overall expression of the resulting bispecific antibody. This can be explained as a consequence of the efficient heterodimerization by the knobs-into-holes mutations and the autonomous folding of the antibody Fab and Fc region. The misfolding of one of the Fab regions will impair the secretion of the entire BsIgG. For example, the pairing with the poorly expressed antibody AbB resulted in low productivity when expressed as bivalent as well as when co-expressed with any other antibody (Fig. 3A). At the same time, this ensured no or only minimal homodimer formation as verified by mass spectrometric analysis (data not shown).
Fig. 3.

Production of anti-KLB biepitopic antibodies in the tcBsIgG format. (A) Capillary electrophoresis of co-expression of the five anti-KLB antibodies and two control antibodies (AbA, highly expressing antibody; AbB, poorly expressing antibody) as tcBsIgGs. The indicated knob half-antibody was co-expressed in a single cell with the hole half-antibody (1: AbA.hole; 2: AbB.hole; 3: 12B8.hole; 4: 2C12.hole; 5: 23B3.hole; 6: 28B7.hole; 7: 4H7.hole) and purified by protein A chromatography prior to analysis. Co-expression with a poorly expressed AbB antibody leads to poor recovery, indicating efficient heterodimerization. (B) Analytical SEC of AbA tcBsIgG (green) and 23B3/28B7 tcBsIgG (blue) after protein A affinity chromatography.
To further characterize the tcBsIgGs, we analyzed them by analytical size-exclusion chromatography (SEC). After HTP expression and single column purification by protein A chromatography, the tcBsIgGs had monomer contents ranging from 43 to 100% which appeared to be related to the Fv properties of the parental IgGs (Table I) since the monomer contents were consistent across different antibodies for both tcBsIgG (Table I; grey shaded fields) and tcIgG (Table I; unshaded fields). When the well-expressing control antibody AbA was co-expressed as knob and hole tcBsIgG, we did not observe any high molecular weight (HMW) species (Fig. 3B). In contrast, the tcBsIgG of two relatively poorly expressing anti-KLB antibodies, 12B8 and 23B3, had much higher amounts of HMW species (Fig. 3B). Although HMW species could affect the activity of each antibody, we decided to proceed with characterization of the 25 antibodies in the luciferase reporter assay. An additional column step would reduce the throughput, so we decided to validate the activity with further purified materials at a later time.
In vitro screening of anti-KLB tcBsIgG biepitopic antibodies identifies five superior pairs that translate to linkerless BsIgG
Of the 10 possible biepitopic combinations, we identified four biepitopic antibodies with increased agonistic activity compared to the parental monoepitopic antibodies (Fig. 4A, four left panels). In addition, one combination (28B7/4H7) showed an equivalent activity to one of the parent antibody (4H7), but at a high concentration, showed a superior activity. As anticipated, the mirrored knob–hole and the hole–knob orientations exhibited an indistinguishable activity in each case. All these five combinations were the results of assembling two antibodies from different epitope bins (Fig. 2B). Not surprisingly, the combinations of two antibodies from the same epitope bin (e.g. 2C12/23B3, Fig. 4A, right panel) did not yield an improvement in agonistic activity compared to the parental monoclonal controls.
Fig. 4.

Identification of bispecific antibodies with higher agonistic activity compared to their parental monoclonal antibodies. (A) Screening of 25 anti-KLB tcBsIgG in the GAL-ELK1 luciferase assay with HEK293T cells expressing FGFR1 and KLB, carried out with protein A purified antibodies. The results of 5 combinations identified to have improved agonistic activity compared to the monospecific parental tcIgG are shown, with 28H7/4H7 having improved activity only at high concentrations. In addition, the results of the 2C12/23B3 combination are shown as a representative example for a pair in which the tcBsIgG did not display improved agonistic activity. (B and C) The five hits from (A) were further validated by tcBsIgG purified by protein A and size-exclusion chromatography to remove potential high molecular weight species (B), or in vitro assembled conventional BsIgG (C). (D) Comparison of the BsIgG with an equimolar mixture of the parental monovalent antibodies. Note that the 23B3/28B7 combination was the only combination that requires an obligate bispecific format. In (A–D), luciferase activity is expressed as relative luminescence units (RLU). The data represents means ± SEM (N = 3).
We subsequently produced the five biepitopic antibodies and the parental antibodies in the same tcBsIgG format, but removed the HMW species and other potential impurities after ProteinA recovery by size exclusion chromatography. These monodispersed tcBsIgG preparations confirmed the superior agonistic activity of the four antibodies compared to the monospecific parentals (Fig. 4B), confirming that an additional purification step was not necessary during the primary screening process in our situation. Subsequently, we produced biepitopic antibodies in the natural IgG1 architecture by conventional in vitro annealing of separately purified knob and hole half-antibodies. Like the tcBsIgG counterparts, the linkerless antibodies were more efficacious than their parental monoclonal antibodies (Fig. 4C). Thus, the tcBsIgG system predicts activity of biepitopic antibodies in a conventional BsIgG format. We also compared the activity of the biepitopic antibodies against an equimolar mixture of the two parental monospecific antibodies (Fig. 4D). While four of the antibody pairs performed either worse or as well as mixture and biepitopic format, one pair (23B3/28B7) clearly demonstrated improved activity in the biepitopic format, suggesting a potential utility of the tcBsIgG screening system in identifying specific bispecific pairs for a superior activity.
Lastly, to ensure the linker in the tcBsIgG format did not interfere with the binding affinity of the biepitopic tcIgG during the screen, binding kinetics of antibodies 4H7 and 28B7 with the highest relative agonistic activity was assessed by Biacore as both tcIgG1 as well as standard IgG1. No significant differences were observed in binding kinetics for the two formats, indicating that the tcIgG format preserves the binding kinetics of the parental antibody (Table II).
Table II.
Binding kinetics of anti-KLB antibodies 28B7 and 4H7 as IgG and tcIgG format. Values are mean ± range of two independent Biacore experiments
| Antibody clone and format | kon/105 (M−1 s−1) | koff/10−4(s−1) | Kd (nM) |
|---|---|---|---|
| 28B7 IgG | 1.04 ± 0.04 | 2.76 ± 0.08 | 2.7 ± 0.1 |
| 28B7 tcIgG | 1.17 ± 0.02 | 2.44 ± 0.09 | 2.1 ± 0.1 |
| 4H7 IgG | 5.7 ± 0.2 | 4.4 ± 0.1 | 0.78 ± 0.05 |
| 4H7 tcIgG | 3.32 ± 0.09 | 4.27 ± 0.07 | 1.29 ± 0.06 |
Impact of antibody isotype on antibody agonistic activity
It has been reported previously that the antibody isotype can modulate the agonistic or ligand mimetic activity of antibodies (Sampei et al., 2015; White et al., 2015). Consequently, we explored whether the antibody isotype can affect the activity of the 4H7/28B7 biepitopic antibody, which had the highest agonistic activity relative to other biepitopic pairs. We produced the BsIgG as human IgG1, IgG2 and IgG4 isotype and compared the activity of the different isotypes in our luciferase reporter assay (Fig. 5A). In this assay, the activity of the 4H7/28B7 biepitopic antibody as IgG1 isotype was approximately twice of that of IgG2 isotype, while IgG4 isotype had an intermediate level of activity. The EC50 of these three antibodies were similar, consistent with the idea that the antibody isotype does not affect binding affinity.
Fig. 5.

BsIgG 28B7/4H7 has agonistic properties as human IgG1, IgG2 and IgG4 isotype. (A) Agonistic activity of BsIgG 28B7/4H7 in human IgG1, IgG2 or IgG4 isotype backbones in GAL-ELK1 luciferase reporter assay. Luciferase activity was expressed as fold induction over an irrelevant IgG control. The data represents means ± SEM (N = 3). (B) Western blotting was conducted to test the activity of BsIgG 28B7/4H7 of human IgG1, IgG2 and IgG4 isotype on primary human adipocytes. Antibodies were used at the indicated concentrations (μg/ml) to assess a dose dependent phosphorylation of ERK (pERK). HSP90 serves as a loading control. (C) Agonistic activity of 28B7/4H7 BsIgG or an equimolar mixture of 28B7 and 4H7 over monovalent parental antibodies. Replicates of two independent experiments for each antibody are shown.
Next, we extended the evaluation to assessing ERK1/2 phosphorylation in human primary adipocytes. Human primary adipocytes express both KLB and FGFR1 endogenously and have been shown to increase phosphorylation of the signaling intermediate ERK1/2 in response to a treatment with recombinant FGF21 or a FGF21-mimetic antibody (Foltz et al., 2012; Kolumam et al., 2015). These primary cells are considered to represent a more physiologically relevant system compared to transfected HEK293 cells. For this assay, differentiated primary human adipocytes were treated with various monoclonal antibodies, and the levels of ERK1/2 phosphorylation was assessed by western blotting. The results of this assay using primary cells were consistent with the luciferase reporter assay; the 4H7/28B7 biepitopic antibody acts as an agonist as IgG1, IgG2 and IgG4 isotype (Fig. 5B and C). Furthermore, 4H7 and 28B7 antibodies exhibited superior activity when combined as an equimolar mixture or a single molecule biepitopic antibody as compared with the single parental molecule activity (Fig. 5C).
Discussion
It has been demonstrated previously that bispecific and biepitopic antibodies can serve as potent ligand mimetics. However, identifying these antibodies is often difficult, necessitating screening of up to thousands of combinations to identify a unique functional combination (Kitazawa et al., 2012; Zhang et al., 2012; Kolumam et al., 2015). The tcBsIgG format described here provides a novel system to simplify expression and production of BsIgG, enabling high-throughput screening of a large panel of BsIgG combinations. Using this strategy, we demonstrated that the activity of anti-KLB agonistic antibodies can be further enhanced by co-formulation or combination into a biepitopic antibody. We do not currently understand the mechanism by which anti-KLB antibodies activate the FGFR1/KLB complex or the mechanisms by which two anti-KLB antibodies exhibit an enhanced functionality as compared to a parental antibody. FGFR1 forms a ligand-independent homodimer via the transmembrane and kinase domains with or without KLB binding (Comps-Agrar et al., 2015) and is thought to be activated by a ligand-induced conformational change. Thus, the enhanced activity of biepitopic antibodies could be due to an induction of a more active FGFR1 dimer configuration, or perhaps by clustering of receptor complexes and an increased density of intracellular signaling molecules. In any case, the biepitopic combinations that exhibited enhanced activity could not be fully anticipated from the characterization of parental antibodies, demonstrating the need to experimentally identify the pairs and the utility of the tcBsIgG system. Our observation could be extended to other agonistic antibodies, for example to endow immunostimmulatory antibodies against OX40, CD40, CD27, GITR or DR5 with superagonistic properties (Mellman et al., 2011; Chen and Mellman, 2013; White et al., 2015).
The tcBsIgG format contains alterations in the antibody structure, in particular the addition of a linker and of a neoepitope at the CL N-terminus that may elicit an immune response in vivo. However, these issues are of no concern for in vitro screening activities. Once promising antibody pairs are identified, a limited number of BsIgG can be produced in a conventional BsIgG format that is more validated for downstream preclinical and clinical development. To this point, it is critical for any screening format that results obtained can be recapitulated in a conventional BsIgG format. The tcBsIgG format described here preserves the overall architecture of the antibody, and as we demonstrate, enables translation of results to a linkerless BsIgG that is more desirable for clinical applications. This is an advantage over other technologies such as fusions of scFv to a hetero-dimeric Fc (Moore et al., 2011) that may provide a similar throughput in producing bispecific molecules like our tcBsIgG format, but alter the geometry and distance between the target arms. In addition, another advantage of the tcBsIgG format over other BsIgG formats is that a reduced number of plasmids, i.e. only one plasmid for each half antibody, need to be cloned to produce a matrix of bispecific antibodies. Because we only produced tcBsIgG of the light chain kappa isotype, it remains untested if the design translates to tcBsIgG of lambda isotype. Beyond screening of antibodies in drug development, the tcBsIgG format has a potential to produce bispecific antibodies for diagnostic applications. The potential immunogenicity of the linker sequence or the neoepitope of the CL N-terminus for diagnostic antibodies is of little concern while the single cell line provides a cost-efficient way to produce the BsIgG.
Another major advantage of the tcIgG format is its applicability to IgG2 and IgG4 isotypes. It has been reported previously that human IgG2 antibodies against CD40 displayed superagonistic activity over human IgG1 and IgG4 isotypes (White et al., 2015) and the amplitude of Factor VIII ligand mimetic activity of a bispecific antibody towards Factors IXa/X was dependent on the antibody class (Sampei et al., 2015). Based on these observations, we compared the agonistic activity of our 4H7/28B7 lead biepitopic IgG as human IgG1, IgG2 and IgG4 isotype. While we observed significant differences between antibody isotypes, human IgG1 displayed the best agonistic activity for the 4H7/28B7 biepitopic IgG. This may be partially attributed to the fact that we initially screened the anti-KLB pairs as human IgG1. Potentially, a different pair and isotype may have been selected if the initial screen was done using BsIgG of all isotypes. The compatibility of the tcBsIgG format with other human isotypes would enable this screening. In addition, this system would allow detection of antibodies with different levels of engagement with Fc gamma receptors to form ternary complexes. While Fc engagement has no relevance to our anti-KLB model system, it might be important for tumor-targeting agonistic antibody discovery.
In conclusion, we believe that the tcBsIgG format provides an excellent strategy for BsIgG screening for broad applications. While in this study we apply our technology to only a limited number of bispecific antibodies, by design, the concept is readily scalable to produce thousands of bispecific antibody pairs that can be handled by any high-throughput antibody expression and purification platform. Further study is warranted to determine whether the tcBsIgG strategy could actually be utilized to discover therapeutic bispecific antibody with a rare activity.
Acknowledgments
We thank members of the Antibody Engineering, Protein Chemistry, and Biochemical and Cellular Pharmacology Departments, in particular Gabe Quinones, Jean-Michel Vernes and Avinash Gill, and the Research Materials group at Genentech for technical support.
Author Contributions
H.S.K., D.R.D., J.A.E., J.S., C.S. designed the experimental strategy, analyzed data, and wrote the article. A.Y., J.H. performed the hybridoma work. D.R.D. and J.S. conducted luciferase and phosphorylation assays. H.S.K. performed molecular biology and biochemistry experiments. F.F., I.K. and R.T. purified and characterized proteins. All authors read and approved the final article.
Funding
This work was funded by Genentech, Inc.
Conflict of interest
All authors are current or former employees of Genentech, Inc., a wholly owned subsidiary of F. Hoffmann-La Roche AG, and may hold stock and options.
References
- Atwell S., Ridgway J.B., Wells J.A. and Carter P. (1997) J. Mol. Biol., 270, 26–35. [DOI] [PubMed] [Google Scholar]
- Bos A.B., Duque J.N., Bhakta S., Farahi F., Chirdon L.A., Junutula J.R., Harms P.D. and Wong A.W. (2014) J. Biotechnol., 180, 10–16. [DOI] [PubMed] [Google Scholar]
- Brezski R.J. and Georgiou G. (2016) Curr. Opin. Immunol., 40, 62–69. [DOI] [PubMed] [Google Scholar]
- Chan A.C. and Carter P.J. (2010) Nat. Rev. Immunol., 10, 301–316. [DOI] [PubMed] [Google Scholar]
- Chen D.S. and Mellman I. (2013) Immunity, 39, 1–10. [DOI] [PubMed] [Google Scholar]
- Comps-Agrar L., Dunshee D.R., Eaton D.L. and Sonoda J. (2015) J. Biol. Chem., 290, 24166–24177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coskun T., Bina H.A., Schneider M.A., Dunbar J.D., Hu C.C., Chen Y., Moller D.E. and Kharitonenkov A. (2008) Endocrinology, 149, 6018–6027. [DOI] [PubMed] [Google Scholar]
- Dillon M., Yin Y., Zhou J. et al. (2017) MAbs, 9, 213–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton D.L., Wood W.I., Eaton D. et al. (1986) Biochemistry, 25, 8343–8347. [DOI] [PubMed] [Google Scholar]
- Feige M.J. and Buchner J. (2014) Biochim. Biophys. Acta, 1844, 2024–2031. [DOI] [PubMed] [Google Scholar]
- Feige M.J., Groscurth S., Marcinowski M., Shimizu Y., Kessler H., Hendershot L.M. and Buchner J. (2009) Mol. Cell., 34, 569–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleetwood F., Güler R., Gordon E., Ståhl S., Claesson-Welsh L. and Löfblom J. (2016) Cell. Mol. Life Sci, 73, 1671–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foltz I.N., Hu S., King C. et al. (2012) Sci. Transl. Med., 4, 162ra153–162ra153. [DOI] [PubMed] [Google Scholar]
- Frankel S.R. and Baeuerle P.A. (2013) Curr. Opin. Chem. Biol, 17, 385–392. [DOI] [PubMed] [Google Scholar]
- Gaich G., Chien J.Y., Fu H. et al. (2013) Cell. Metab., 18, 333–340. [DOI] [PubMed] [Google Scholar]
- Garber K. (2014) Nat. Rev. Drug. Discov., 13, 799–801. [DOI] [PubMed] [Google Scholar]
- Grandjenette C., Dicato M. and Diederich M. (2015) Curr. Pharm. Biotechnol, 16, 670–683. [DOI] [PubMed] [Google Scholar]
- Gunasekaran K., Pentony M., Shen M. et al. (2010) J. Biol. Chem., 285, 19637–19646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y., Yu J., Lanzi A. et al. (2016) Cell, 165, 1621–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharitonenkov A., Wroblewski V.J., Koester A., Chen Y.F., Clutinger C.K., Tigno X.T., Hansen B.C., Shanafelt A.B. and Etgen G.J. (2007) Endocrinology, 148, 774–781. [DOI] [PubMed] [Google Scholar]
- Kim H.S., Kim I., Zheng L., Vernes J.M., Meng Y.G. and Spiess C. (2016) MAbs, 8, 1536–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitazawa T., Igawa T., Sampei Z. et al. (2012) Nat. Med., 18, 1570–1574. [DOI] [PubMed] [Google Scholar]
- Kolumam G., Chen M.Z., Tong R. et al. (2015) EBioMedicine, 2, 730–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontermann R. (2012) MAbs, 4, 182–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Kreudenstein T.S., Escobar-Carbrera E., Lario P.I. et al. (2013) MAbs, 5, 646–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurosu H., Choi M., Ogawa Y. et al. (2007) J. Biol. Chem., 282, 26687–26695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrijn A.F., Meesters J.I., de Goeij B.E.C.G. et al. (2013) Proc. Natl. Acad. Sci. USA, 110, 5145–5150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis S.M., Wu X., Pustlinik A. et al. (2014) Nat. Biotechnol., 32, 191–198. [DOI] [PubMed] [Google Scholar]
- Li J.Y., Perry S.R., Muniz-Medina V. et al. (2016) Cancer Cell, 29, 117–129. [DOI] [PubMed] [Google Scholar]
- Liu Z., Leng E.C., Gunasekaran K. et al. (2015) J. Biol. Chem., 290, 7535–7562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazor Y., Oganesyan V., Yang C. et al. (2015) MAbs, 7, 377–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellman I., Coukos G. and Dranoff G. (2011) Nature, 480, 480–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore G.L., Bautista C., Pong E. et al. (2011) MAbs, 3, 546–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polson A.G., Yu S.-F., Elkins K. et al. (2007) Blood, 110, 616–623. [DOI] [PubMed] [Google Scholar]
- Ridgway J.B., Presta L.G. and Carter P. (1996) Protein Eng., 9, 617–621. [DOI] [PubMed] [Google Scholar]
- Sampei Z., Igawa T., Soeda T. et al. (2015) MAbs, 7, 120–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer W., Regula J.T., Bähner M. et al. (2011) Proc. Natl. Acad. Sci. USA, 108, 11187–11192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schanzer J.M., Wartha K., Croasdale R. et al. (2014) J. Biol. Chem., 289, 18693–18706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shatz W., Chung S., Li B., Marshall B., Tejada M., Phung W., Sandoval W., Kelley R.F. and Scheer J.M. (2013) MAbs, 5, 872–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiess C., Merchant M., Huang A. et al. (2013) Nat. Biotechnol., 31, 753–758. [DOI] [PubMed] [Google Scholar]
- Spiess C., Zhai Q. and Carter P.J. (2015) Mol. Immunol., 67, 95–106. [DOI] [PubMed] [Google Scholar]
- Strop P., Ho W.H., Boustany L.M. et al. (2012) J. Mol. Biol., 420, 204–219. [DOI] [PubMed] [Google Scholar]
- Suryadevara C.M., Gedeon P.C., Sanchez-Perez L., Verla T., Alvarez-Breckenridge C., Choi B.D., Fecci P.E. and Sampson J.H. (2015) Oncoimmunology, 4, e1008339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White A.L., Chan H.T.C., French R.R. et al. (2015) Cancer Cell, 27, 138–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams A.J., Giese G. and Persson J. (2015) Biotechnol. Prog., 31, 1315–1322. [DOI] [PubMed] [Google Scholar]
- Wong A.W., Baginski T.K. and Reilly D.E. (2010) Biotechnol. Bioeng., 106, 751–763. [DOI] [PubMed] [Google Scholar]
- Wranik B.J., Christensen E.L., Schaefer G., Jackman J.K., Vendel A.C. and Eaton D. (2012) J. Biol. Chem., 287, 43331–43339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu A.L., Kolumam G., Stawicki S. et al. (2011) Sci. Transl. Med., 3, 113ra126–113ra126. [DOI] [PubMed] [Google Scholar]
- Wu C., Ying H., Grinnell C. et al. (2007) Nat. Biotechnol., 25, 1290–1297. [DOI] [PubMed] [Google Scholar]
- Xu J., Lloyd D.J., Hale C. et al. (2009) Diabetes, 58, 250–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H., Wilson I.A. and Lerner R.A. (2012) Proc. Natl. Acad. Sci. USA, 109, 15728–15733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhukovsky E.A., Morse R.J. and Maus M.V. (2016) Curr. Opin. Immunol., 40, 24–35. [DOI] [PMC free article] [PubMed] [Google Scholar]